
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum(II/IV) complexes with N-substituted carboxylate ethylenediamine/propylenediamine ligands: preparation, characterization and in vitro activity†
Stefan
Richter
a,
Peter
Lönnecke
 a,
Dijana
Bovan
b,
Nicoleta
Andrian
c,
Bianca
Stoean
c,
Maria
Lehene
c,
Radu
Silaghi-Dumitrescu
c,
Luiza
Gaina
c,
Sanja
Mijatović
b,
Danijela
Maksimović-Ivanić
b,
Goran N.
Kaluđerović
d and
Evamarie
Hey-Hawkins
*ce
a,
Dijana
Bovan
b,
Nicoleta
Andrian
c,
Bianca
Stoean
c,
Maria
Lehene
c,
Radu
Silaghi-Dumitrescu
c,
Luiza
Gaina
c,
Sanja
Mijatović
b,
Danijela
Maksimović-Ivanić
b,
Goran N.
Kaluđerović
d and
Evamarie
Hey-Hawkins
*ce
aInstitute of Inorganic Chemistry, Faculty of Chemistry and Mineralogy, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany
bDepartment of Immunology, Institute for Biological Research “Siniša Stanković” National Institute of Republic of Serbia, University of Belgrade, Bulevar despota Stefana 142, 11060 Belgrade, Serbia
cDepartment of Chemistry, Babeş-Bolyai University, Str. Arany Janos Nr. 11, RO-400028 Cluj-Napoca, Romania
dDepartment of Engineering and Natural Sciences, University of Applied Sciences Merseburg, Eberhard Leibnitz-Str. 2, 06217 Merseburg, Germany
eUniversität Leipzig, Faculty of Chemistry and Mineralogy, Institute of Bioanalytical Chemistry, BBZ, Deutscher Platz 5, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
First published on 26th December 2024
Abstract
The synthesis and characterization of novel platinum(II) and platinum(IV) complexes derived from unsymmetrical ethylene or propylenediamine derivatives are presented. IR spectroscopy and ESI mass spectrometry techniques were employed to characterize the complexes, revealing distinctive absorption bands and isotope patterns. Furthermore, the complexes were characterized by 1H and 13C NMR spectroscopy. Single-crystal X-ray structural analysis elucidated the coordination geometry and intermolecular interactions of complexes 3, 4 and 6. Cytotoxicity evaluation of the complexes on various cell lines highlighted complex 3 as the most active, realizing its tumoricidal activity through induction of apoptosis and increased total caspase activity in MCF-7 cells. Since its application is followed by cytoprotective autophagy, the effectiveness can be additionally empowered by concomitant inhibition of this process. Furthermore, the PtIV compound 3 induces oxidative stress in hemoglobin, and is reducible by glutathione, suggesting its potential as a carrier for the active PtII precursor 2a to cancer cells without increasing cytotoxicity. Cyclic voltammetry corroborates the ability of complex 3 to undergo reduction under physiological conditions.
Introduction
Platinum-based complexes, particularly cisplatin, have played a pivotal role in cancer treatment for nearly 60 years.1 Discovered by Barnett Rosenberg, the cytotoxic properties of cisplatin were a breakthrough in cancer treatment. Although initially synthesized in 1844, it only entered clinical use in the late 1970s. Cisplatin is widely employed against various tumors, achieving significant success, especially in testicular cancer, once considered fatal.2 However, its effectiveness is marred by severe side effects and the development of resistance by tumor cells.3,4 To address these challenges, research has focused on enhancing selectivity and developing new platinum-based compounds. Derivatives like carboplatin, oxaliplatin and platinum(IV) complex satraplatin (2nd–4th generation of platinum-based drugs)5 showcase advancements, aiming for improved efficacy and reduced side effects. Platinum(IV) complexes represent an innovative avenue in platinum-based chemotherapy.6 These complexes offer the potential for oral administration, addressing some limitations associated with intravenous delivery. Satraplatin, the first oral platinum-based drug, showed promising properties, demonstrating efficacy against cancer cells resistant to cisplatin, attributed to decreased platinum transport.7 Synergistic pairings involving satraplatin include erlotinib for ovarian cancer, bevacizumab, docetaxel, and prednisone for prostate cancer. Moreover, satraplatin exhibited synergy with gemcitabine in addressing a spectrum of cancers, including prostate cancer, pancreatic, hepatocellular carcinoma, gastric, bladder, and papillary renal carcinoma. Despite the success of platinum complexes, challenges such as resistance development and side effects persist. The goal in development of each platinum-based drug was to overcome limitations of cisplatin and refine platinum-based chemotherapy for better cancer treatment outcomes.Platinum complexes have been at the forefront of medicinal chemistry for decades, particularly in the field of cancer treatment.8 Among the various platinum complexes, those involving ethylenediamine (en) and propylenediamine (pn) ligands have demonstrated significant promise due to potential therapeutic applications. Platinum(II) complexes with en and pn ligands have been extensively studied. The groups of Nolan and Kaluđerović investigated platinum(II/IV) complexes with different ethylenediamine derivatives.9–14In vitro studies have demonstrated for instance that some of these complexes showed improved pharmacological profile under normoxic and hypoxic conditions in comparison to cisplatin.11 Also the selectivity toward tumor cells was enhanced, which is crucial for minimizing collateral damage to healthy tissues and reducing adverse effects during treatment. These findings were furthermore corroborated with in vivo experiments, showcasing the potential of platinum(IV) complexes as promising candidates for cancer therapy.13 Their ability to exhibit enhanced tumor-targeting properties while mitigating systemic toxicity underscores their potential as effective anticancer agents. On the other hand some transition metal ions (NiII, CrIII, CoIII) bearing 2-(2-aminoethylamino)acetate as ligand are well studied.15–18 Furthermore crystal structures were determined for ReI, CuII, CrIII, and CoIII complexes.19–24 The group of Gielen investigated different platinum(II) complexes with 4-[(2-aminoethylamino)methyl]phenyl derivatives.24
In the present work we have explored the synthetic pathways, structural characteristics, and biological implications of platinum(II) and platinum(IV) complexes incorporating ethylenediamine and propylenediamine ligands (Scheme 1).
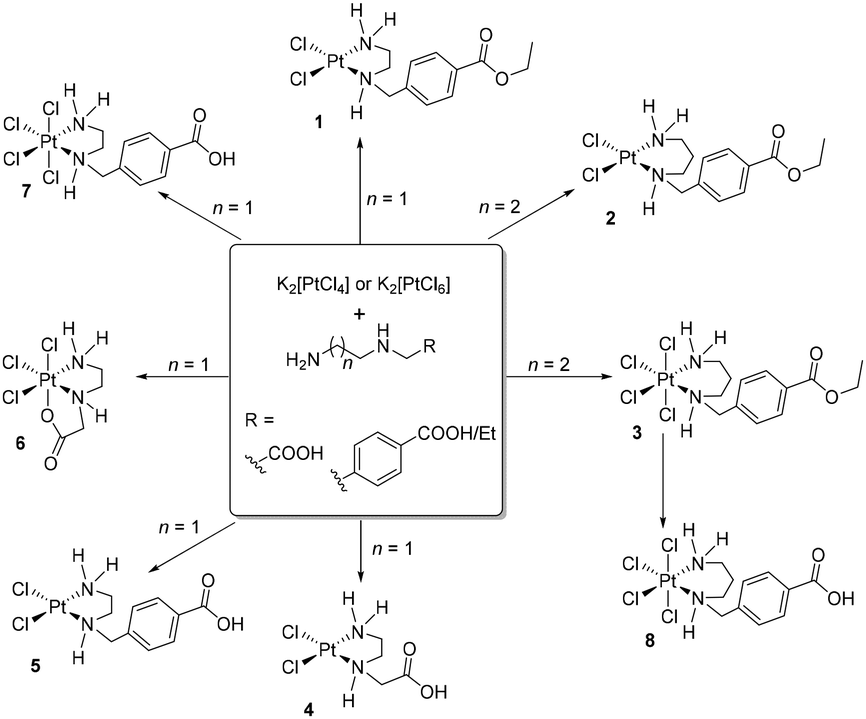 | ||
| Scheme 1 Synthesis of the PtII and PtIV complexes. | ||
Results and discussion
Synthetic procedures
As starting compounds for the synthesis of novel platinum(II) and platinum(IV) complexes, K2[PtCl4] or K2[PtCl6] and unsymmetrical ethylene- or propylenediamine derivatives were used (Scheme 1). The ester-protected ligands employed can be directly added as basic diamines or hydrochloride salts to an aqueous solution of the appropriate platinum complex. However, it is crucial to precisely control the pH milieu in this synthesis, as exceeding a pH value of 7 due to a high concentration of unreacted diamines leads to decomposition of the starting materials, platinum(II) or platinum(IV) complex, to metallic platinum. Complex 8 was obtained in good yield by reacting complex 3 with an aqueous HCl solution (0.1 M) for several hours. The isolated platinum(II) and platinum(IV) complexes were characterized by 1H, 13C NMR spectroscopy, IR spectroscopy, and mass spectrometry. The purity of the compounds was confirmed by elemental analysis. All complexes are obtained as racemates (chiral secondary nitrogen atom).Characterization
IR spectra of complexes 1–8 exhibit a prominently evident absorption band centered around 1700 cm−1, assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (Table 1), associated with either carboxylic acids or their ethyl esters. The symmetric COOH/COOEt stretching vibrations appear at ca. 1370 cm−1. For the platinum(II) and platinum(IV) complexes bearing N,N′-disubstituted ethylene- and propylenediamine ligands appropriate absorptions are found at similar wavelengths.25–27 The platinum(II) complex 4 was already reported by Nolan et al. and showed good agreement (e.g. ν(COOH) was observed at 1719 cm−1 (literature: 1720 cm−1)).9 Furthermore, O–H vibrational bands were detected at around 3440 cm−1, N–H vibrational band at approximately 3200 cm−1.
O stretching vibration (Table 1), associated with either carboxylic acids or their ethyl esters. The symmetric COOH/COOEt stretching vibrations appear at ca. 1370 cm−1. For the platinum(II) and platinum(IV) complexes bearing N,N′-disubstituted ethylene- and propylenediamine ligands appropriate absorptions are found at similar wavelengths.25–27 The platinum(II) complex 4 was already reported by Nolan et al. and showed good agreement (e.g. ν(COOH) was observed at 1719 cm−1 (literature: 1720 cm−1)).9 Furthermore, O–H vibrational bands were detected at around 3440 cm−1, N–H vibrational band at approximately 3200 cm−1.
| Complex | IR data | NMR data | ||||
|---|---|---|---|---|---|---|
| ν(N–H) | ν(O–H) |
ν(CO) |
NHCH2C | NHCH2C | CO | |
| δ H | δ C | δ C | ||||
| 1 | 2962 | — | 1717 | 4.01 | 54.6 | 165.6 |
| 2 | 3264 | — | 1708 | 4.31 (br) | 55.8 | 165.9 |
| 3 | 3200 | — | 1716 | 4.31 (br) | 50.2 | 165.8 |
| 4 | 3141 | 3446 | 1719 | 3.45–4.09 | 56.9 | 170.1 |
| 5 | 3214 | 3433 | 1690 | 4.00–4.32 | 55.3 | 168.5 |
| 6 | 3183 | — | 1679 | 3.46–4.06 | 55.4 | 179.7 |
| 7 | 3181 | 3440 | 1686 | 4.32 (br) | 56.1 | 167.5 |
| 8 | 3240 | 3456 | 1701 | 4.19 | 49.7 | 166.9 |
The ESI mass spectra of complexes 5 and 7, representing platinum(II) and platinum(IV) complexes with two and four chlorido ligands, are discussed here as examples. Due to the natural abundance of various platinum (194Pt 32.90%, 195Pt 33.80%, 196Pt 25.3%, 198Pt 7.2%) and chlorine isotopes (35Cl 75.77%, 37Cl 24.23%), the isotope patterns of the presented platinum(II) complexes are comparatively rich in lines and informative regarding the number of chlorido ligands. From this, the elemental composition can be inferred by calculating the relative intensities of the isotope peaks. Fig. 1 shows excerpts of the ESI mass spectra in the range of the molecular ion peak of 5, namely [5 + Na]+, along with the calculated isotope pattern. Compared to the platinum(II) complexes, the depicted platinum(IV) complexes contain two additional chlorido ligands, and therefore the obtained mass spectra are considerably richer in lines. The mass spectrum of complex 7 shown in Fig. 1, measured in negative ion mode, closely matches the calculated isotope pattern in appearance and shape, particularly in the region of the molecular ion peak. In this case, the complex is negatively charged due to the release of a proton [7–H+]−.
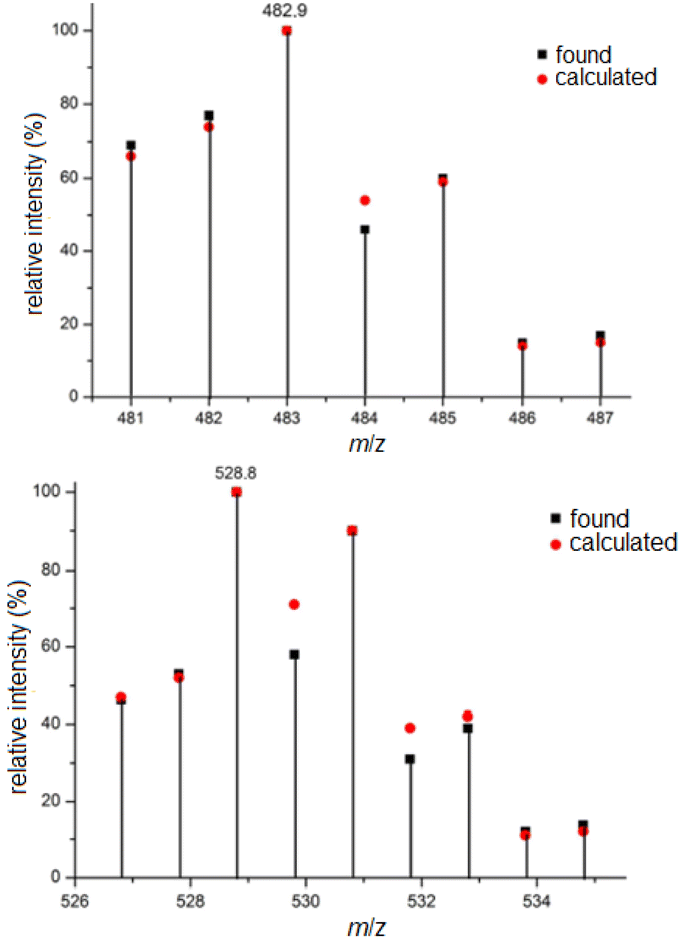 | ||
| Fig. 1 Molecular ion peak in the ESI mass spectrum of 5 (upper: [5 + Na]+) and 7 (lower: [7–H+]−). | ||
The ethylenediamine- or propylenediamine-containing ligands, bind in a κ2-N,N′ mode to the platinum(II) or platinum(IV) ions, forming five- or six-membered rings. A comparison of the chemical shifts in the 1H NMR spectra of the ligands and the appropriate platinum(II) and platinum(IV) complexes showed a coordination-induced shift of the proton resonances in the ligand backbone as well as multiplets, as the protons exhibit diastereotopic behavior upon coordination. This is particularly evident in the resonance pattern of the methylene bridge between the amine-containing chelate systems and the aromatic ring in the 1H NMR spectrum, as observed and shown exemplarily for complex 5 (Fig. 2). Upon coordination of 4-[(2-aminoethylamino)methyl]benzoic acid, the chemically identical hydrogen atoms H3 and H3′ become diastereotopic. Due to the resulting 2JH,H coupling, the chemical shift, originally observed as a singlet in the free ligand, changes to a complex, unsymmetrical multiplet, observed in the range of 4.00–4.32 ppm for complex 5.
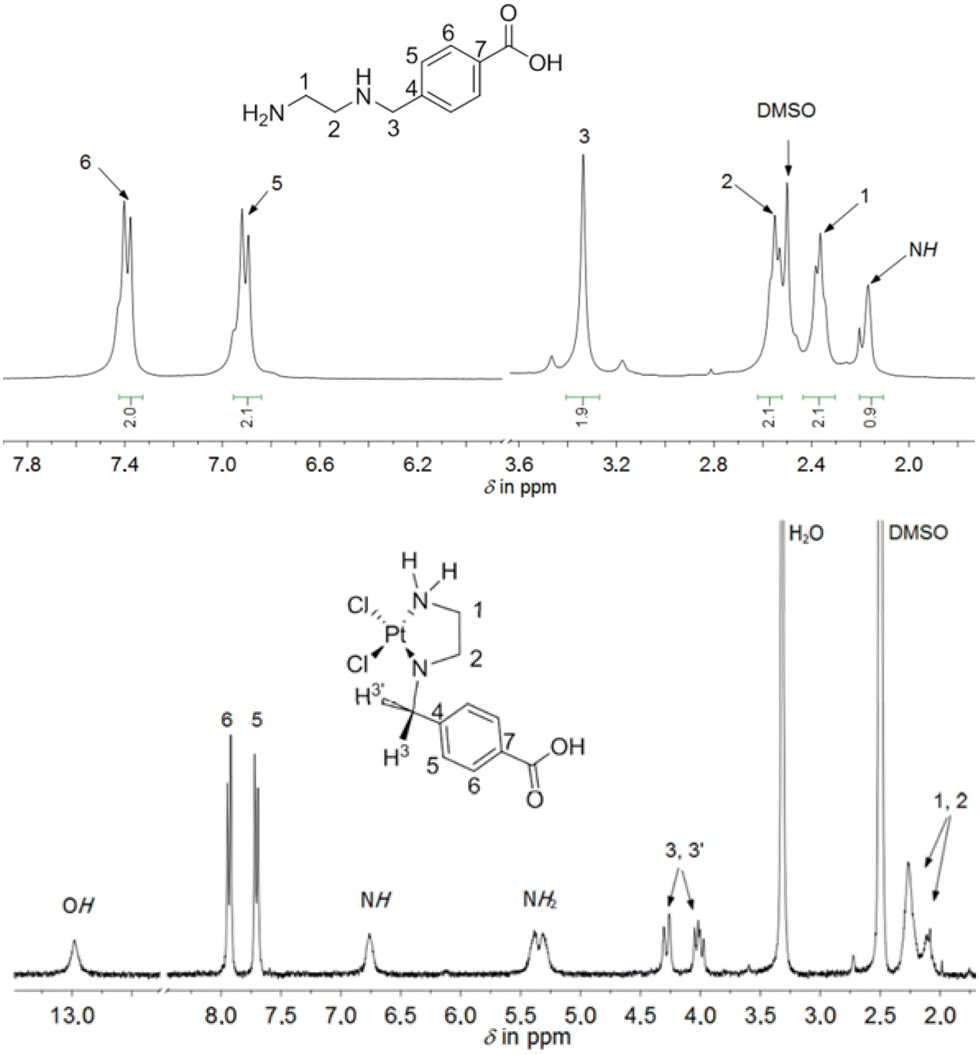 | ||
| Fig. 2 1H NMR spectrum of 4-[(2-aminoethylamino)methyl]benzoic acid and 5 (300 MHz, DMSO-d6). | ||
Moreover, a coordination-induced shift was observed in the ethylenediamine/propylenediamine chelating moiety. In the 1H NMR spectrum, exemplarily shown for complex 5, the four hydrogen atoms of the CH2 groups in the NHCH2CH2NH2 moiety (in the range of 2.10–2.30 ppm) were downfield shifted in comparison to the free ligand (Fig. 2). One reason might be the possible δ- and λ-conformation of the five-membered ring in complex 5. Yoshikawa et al. reported the 1H NMR spectra of platinum(II) complexes with coordinated N-substituted ethylenediamines.28 In that study, a spectrum analysis was conducted after ligand coordination at the platinum atom. Similar to the present work, upon coordination of the analogous chelating ligand N-benzylethylenediamine, the resonances of the methylene hydrogen atoms are observed as a multiplet, a similar pattern to what was found in our study. Similarly to complex 5, other platinum(II) and platinum(IV) complexes (1–4 and 6–8) showed multiplets or at least a broad resonance in the range of 3.43–4.32 ppm for the methylene hydrogen atoms. The chemical shifts of the respective CH2 groups in the appropriate complexes can be found in the 13C{1H} NMR spectra between 50–60 ppm. The hydrogen and carbon atoms within the Et moieties (from the ester function) of complexes 1–3 displayed resonances resembling those seen in the NMR spectra of their respective free ligands.
Molecular structures
Three complexes 3, 4 and 6 have been characterized by single-crystal X-ray structural analysis. Clear yellow crystals of complex 3 suitable for single-crystal X-ray structural analysis were obtained from a saturated DMSO solution. The complex crystallizes in the monoclinic space group P21/n with four molecules of platinum(IV) complex and DMSO each, as well as two molecules of water in the asymmetric unit. With the exception of the COOEt substituent and solvent molecules, these complex molecules are symmetry related (pseudo symmetry; Fig. S1 (ESI)†). Platinum atoms are octahedrally coordinated (Fig. 3 and S2 (ESI)†). The six-membered ring formed by the coordination of nitrogen atoms adopts the typical chair conformation, with N–Pt–N bond angles ranging from 96.5(2)° to 97.2(2)° (Table 2). DMSO and water molecules, co-crystallized in “solvent channels” parallel to (100), interact via intermolecular hydrogen donor acceptor bonds with adjacent molecules and are arranged in a completely ordered manner (Fig. 3, bottom; Table S2, ESI†). The most important intermolecular hydrogen donor⋯acceptor distances are N1(H)⋯O9 (2.788(5) Å), N3(H)⋯O12 (2.754(5) Å), N5(H)⋯O11 (2.753(5) Å), N7(H)⋯O10 (2.796(5) Å), O13(H)⋯O11 (2.863(7) Å), O13(H)⋯Cl4 (3.445(6) Å), O14(H)⋯O12 (2.862(7) Å) and O14(H)⋯Cl16 (3.493(6) Å).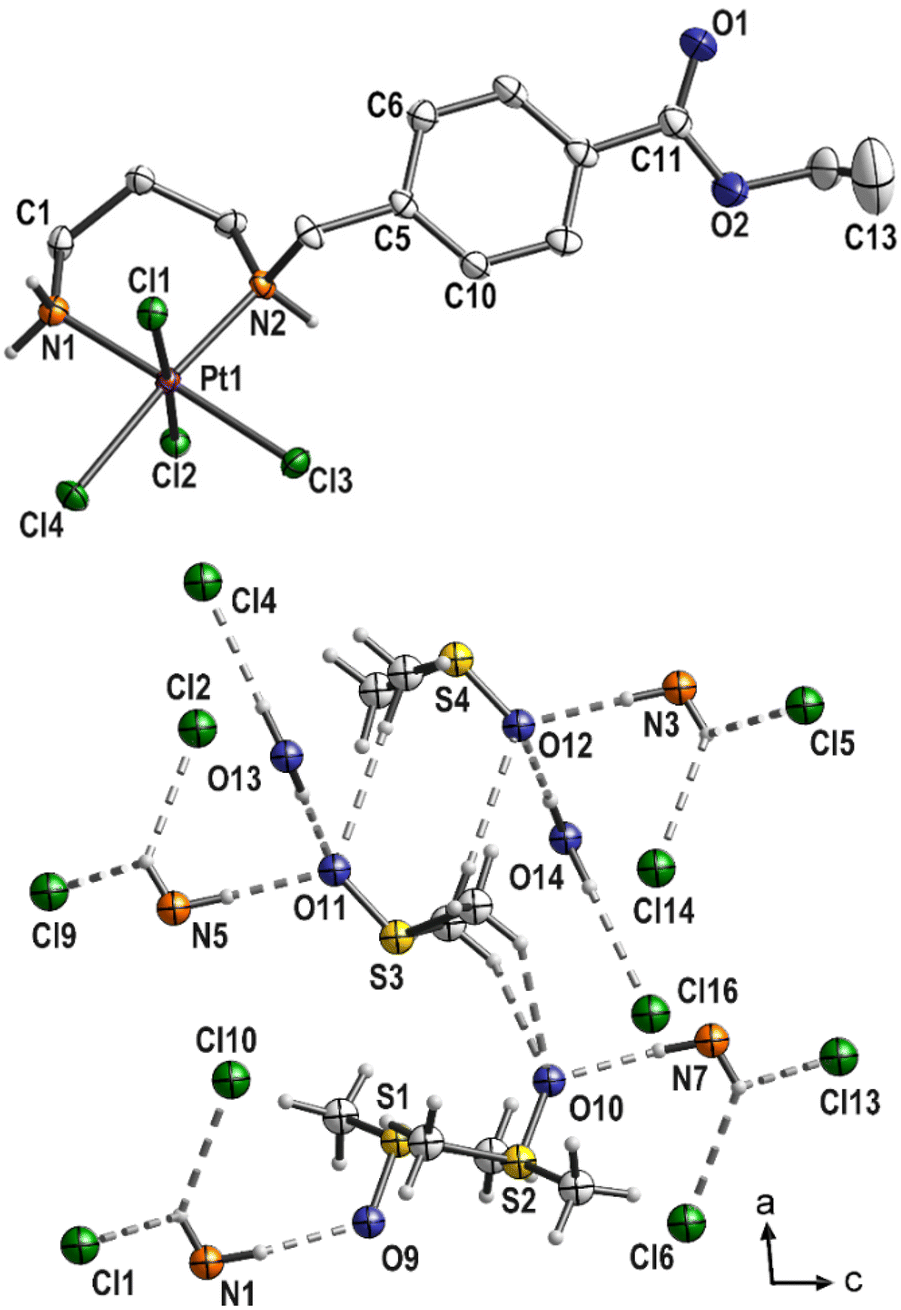 | ||
| Fig. 3 Molecular structure of 3 (top; displacement ellipsoids 50% probability) with co-crystallized solvent molecules (bottom; ball and stick). | ||
| a For the four independent molecules of 3 a range is given. b Disordered Cl3 overlapped with Br3F. | |||||
|---|---|---|---|---|---|
| 3 | 4 | 6 | |||
| Pt–N1 | 2.050–2.056(4) | Pt–N1 | 2.032(4) | Pt–N1 | 2.037(3) |
| Pt–N2 | 2.095–2.109(4) | Pt–N2 | 2.058(4) | Pt–N2 | 2.048(3) |
| Pt–Cl1 | 2.321–2.326(1) | Pt–Cl1 | 2.301(1) | Pt–Cl1 | 2.293(1) |
| Pt–Cl2 | 2.299–2.304(1) | Pt–Cl2 | 2.320(1) | Pt–Cl2 | 2.298(1) |
| Pt–Cl3 | 2.334–2.339(1) | N1⋯O3 | 2.871(6) | Pt–Cl3 | 2.295(8)b |
| Pt–Cl4 | 2.306–2.308(1) | O1⋯O2′ | 2.639(5) | Pt–O1 | 2.017(3) |
| NH–O(DMSO) | 2.753–2.796(5) | O3⋯O3′ | 2.941(7) | N1⋯O2′ | 2.843(5) |
| OH–O(DMSO) | 2.862–2.863(7) | N1–Pt–N2 | 84.0(2) | N1⋯O2′′ | 2.839(5) |
| CH–O | 3.211–3.580(7) | N1–Pt–Cl1 | 91.6(1) | N1–Pt–N2 | 84.6(1) |
| OH–Cl | 3.445–3.493(6) | N1–Pt–Cl2 | 177.9(1) | N1–Pt–Cl1 | 91.4(1) |
| N1–Pt–N2 | 96.5–97.2(2) | N2–Pt–Cl2 | 94.4(1) | N1–Pt–Cl2 | 89.8(1) |
| N1–Pt–Cl1 | 86.7–87.6(1) | Cl1–Pt–Cl2 | 90.07(4) | N2–Pt–Cl3 | 94.1(3)b |
| N1–Pt–Cl2 | 89.2–89.7(1) | N1–Pt–Cl2 | 177.9(1) | Cl1–Pt–Cl2 | 92.65(4) |
| N1–Pt–Cl4 | 86.5–87.2(1) | ||||
Complex 4 has already been described and characterized by Nolan et al. in 2001.9 In the present study, complex 4 was additionally characterized by single crystal X-ray diffraction analysis. Complex 4 crystallizes from a boiling aqueous solution by slow cooling to 4 °C as colorless needles in the monoclinic space group P21/c. The corresponding platinum(IV) complex 6 crystallizes also from a boiling aqueous solution by slow cooling to 4 °C as yellow needles in the monoclinic space group C2/c. The molecular structures of both complexes are depicted in Fig. 4, a selection of bond lengths and angles of the two complexes is compiled in Table 2 and Table S2, ESI†.
 | ||
| Fig. 4 Molecular structures of platinum(II) complex 4·H2O (upper) and platinum(IV) complex 6 (bottom). The depicted ellipsoids correspond to a probability of 50%. For the sake of clarity, disordered and hydrogen atoms, except NH and OH, are omitted. | ||
The platinum(II) complex 4 crystallizes with one equivalent of water. The platinum atom in the complex exhibits a typical square-planar coordination environment, with a total angle sum of 360.07°. In the solid state hydrogen donor⋯acceptor bonds are detectable (Fig. 5, top; Table S2, ESI†). Thus, the complex interacts with the water molecule O3 (N1(H)⋯O3 = 2.871(6) Å and O3′(H)⋯Cl1 = 3.282(5) Å), via N2 with Cl1′ (N2(H)⋯Cl1′ = 3.280(4) Å) and the COOH group of an adjacent molecule (O1(H)⋯O2′ = 2.639(5) Å), which is typical for carboxylic acids. In addition, the water molecules interact with each other as well (O3(H)⋯O3′ = 2.941(7) Å). As O3 and O3′ are related by symmetry (center of inversion), the hydrogen atoms H2O3 and H2OF are disordered by symmetry (Fig. 5, top). Overall, all these intermolecular interactions form a 3D polymer network. The five-membered Pt–N1–C1–C2–N2 ring adopts the typical δ and λ conformations, with the carbon atoms C1 and C2 protruding from the square-planar coordination sphere of the platinum atom (Fig. 4).
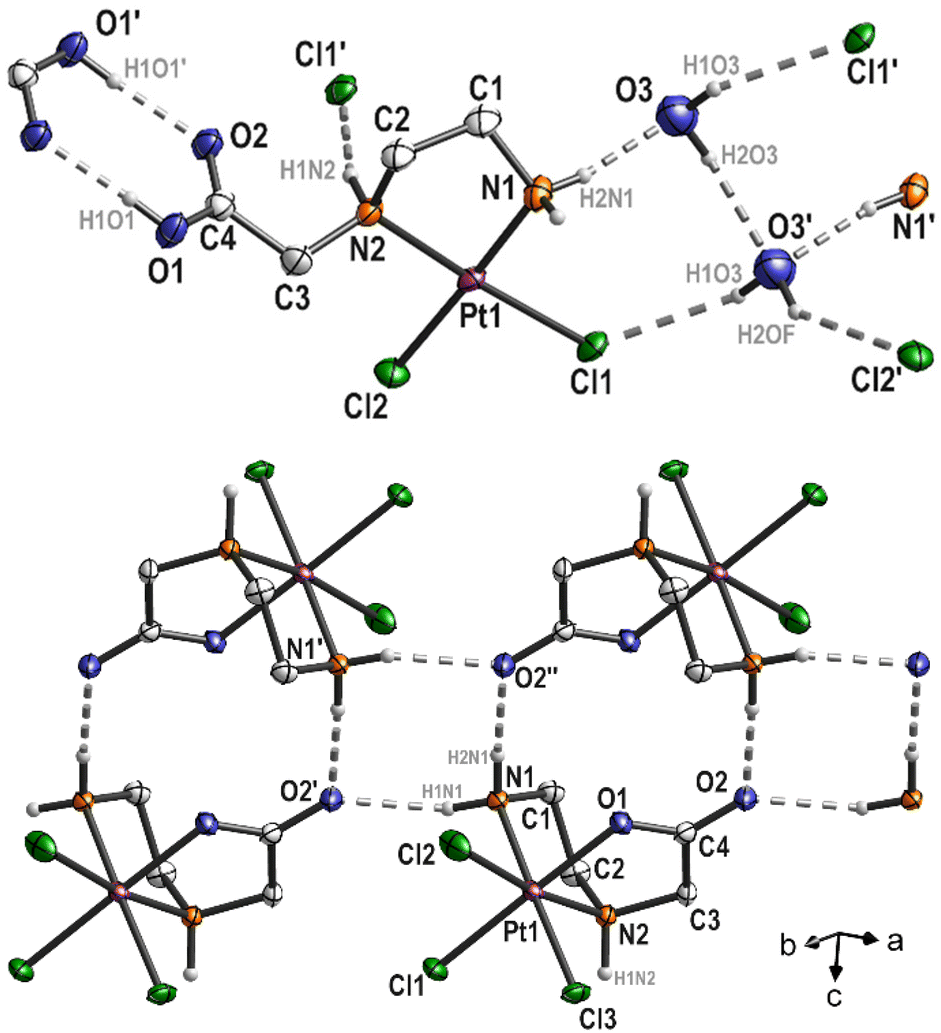 | ||
| Fig. 5 Intermolecular interactions in complexes 4 (top) and 6 (bottom). | ||
In the molecular structure of complex 6, bromine impurities are observed (Cl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Br3F disorder = 0.776(3):0.224(3)). It can be assumed that bromide was present as an impurity in the protonated ligand, as bromoacetic acid was used as a starting material for the synthesis. However, this impurity exhibits an interesting property as only the Cl3 substituent is affected. Through chelating coordination of the ethylenediamine ligand, a five-membered ring (Pt–N1–C1–C2–N2) is formed, which, similar to platinum(II) complex 4, adopts δ and λ conformations. Another feature of complex 6 is the coordination of the carboxylate group at the platinum atom, resulting in the formation of a second five-membered ring (Pt–N2–C3–C4–O1) in an envelope conformation (Fig. 4). Platinum(IV) complexes described in the literature with a comparable ethylenediamine derivative (ethylenediamine-N,N'-diacetic acid, H2edda) as a ligand also show coordination of the carboxylato groups at the central atom. In the platinum(IV) complex [PtCl2(edda)] described by Nolan et al.,9 water molecules form hydrogen bonds to the oxygen atom of one of the coordinated carboxylato groups in the solid state. However, the platinum(IV) complex 6 discussed in this study crystallizes without solvent, yet intermolecular hydrogen bonding occurs between N1 and O2 (N1(H)⋯O2′ = 2.843(5) and N1(H)⋯O2′′ = 2.839(5); Fig. 5 bottom) forming chains along (1
:Br3F disorder = 0.776(3):0.224(3)). It can be assumed that bromide was present as an impurity in the protonated ligand, as bromoacetic acid was used as a starting material for the synthesis. However, this impurity exhibits an interesting property as only the Cl3 substituent is affected. Through chelating coordination of the ethylenediamine ligand, a five-membered ring (Pt–N1–C1–C2–N2) is formed, which, similar to platinum(II) complex 4, adopts δ and λ conformations. Another feature of complex 6 is the coordination of the carboxylate group at the platinum atom, resulting in the formation of a second five-membered ring (Pt–N2–C3–C4–O1) in an envelope conformation (Fig. 4). Platinum(IV) complexes described in the literature with a comparable ethylenediamine derivative (ethylenediamine-N,N'-diacetic acid, H2edda) as a ligand also show coordination of the carboxylato groups at the central atom. In the platinum(IV) complex [PtCl2(edda)] described by Nolan et al.,9 water molecules form hydrogen bonds to the oxygen atom of one of the coordinated carboxylato groups in the solid state. However, the platinum(IV) complex 6 discussed in this study crystallizes without solvent, yet intermolecular hydrogen bonding occurs between N1 and O2 (N1(H)⋯O2′ = 2.843(5) and N1(H)⋯O2′′ = 2.839(5); Fig. 5 bottom) forming chains along (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0).
0).
Stability study
Despite the widespread use of DMSO as a solvent for the anticancer drug cisplatin in pharmacobiochemical experiments, until Sadler's investigations there were insufficient data on potential solvolysis products, primarily due to the limited methods available to detect and structurally characterize such products.29 Using 195Pt NMR spectroscopy, the Sadler group was first to identify up to six different solvolysis products of cisplatin in a DMSO solution, with cis-[PtCl(NH3)2(DMSO)]+ being the main product.As an example of the solvolysis susceptibility of the platinum(II) complexes synthesized in this study, we will use complex 5. Complex 5 was dissolved in DMSO-d6, and immediately afterward, a 1H NMR spectrum was recorded (Fig. 6), which is considered representative of the intact complex.
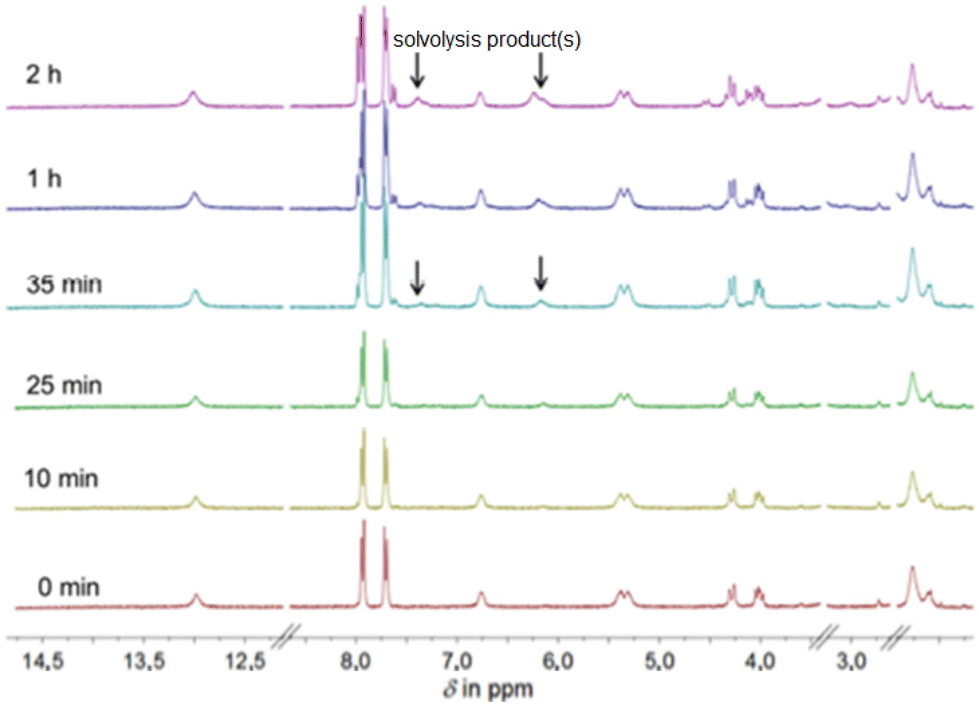 | ||
| Fig. 6 Time-resolved 1H NMR spectra of 5 (300 MHz, DMSO-d6). | ||
At 2.20 ppm, the proton signals of the ethylenediamine backbone are observed as a multiplet. The two diastereotopic protons of the methylene group NHCH2C split into a multiplet in the range of 4.15 ppm due to a geminal coupling. The proton resonances of the coordinated amine groups are observed as doublet and singlet at 5.30 ppm and 6.75 ppm, respectively. The center of gravity of the AB spin system of the four protons of the aromatic ring is found around 7.82 ppm. Likewise, the proton of the free carboxylic acid group appears as a broad singlet around 13.0 ppm. After immediately recording the proton spectrum, additional spectra of the same sample were recorded 10, 25, 35, 60, and 120 min after complex 5 was dissolved. Already 35 min after dissolution, in addition to the initial chemical shifts, further resonances in the range of the amine protons can be found (Fig. 6, 35 min). These are shifted to lower field compared to the amine proton resonances observed earlier. After 2 h, a series of new resonances appeared in the 1H NMR spectrum, making the overall spectrum difficult to interpret (Fig. 6, 2 h). The expected solvolysis product is shown in Scheme 2. However, the complexes do not undergo solvolysis in DMF for the time period relevant for in vitro tests (96 h).
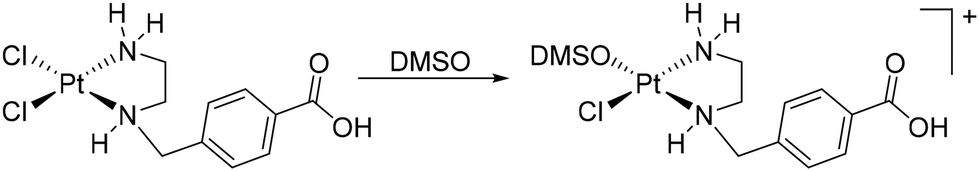 | ||
| Scheme 2 Solvolysis of 5 in DMSO. | ||
In vitro activity
Evaluation of the cytotoxic effects of five platinum(II) and platinum(IV) complexes on a panel of four distinct cell lines, namely 8505C, MCF-7, 518A2, and SW-480, was performed. The cells were treated for 96 h and the impact of the compounds on cell viability was determined via a sulforhodamine B (SRB) assay.30,31 The results obtained from these experiments, as well as literature values for cisplatin,32 are summarized in Table 3, while dose-dependent graphs can be found in Fig. S5 (ESI).†| Complex | 8505C | MCF-7 | 518A2 | SW-480 |
|---|---|---|---|---|
| 2 | 38 ± 2 | 17 ± 3 | 19 ± 4 | 8 ± 1 |
| 3 | 20 ± 1 | 13 ± 3 | 14 ± 4 | 13 ± 4 |
| 5 | >100 | >100 | >100 | 50 ± 8 |
| 7 | >100 | >100 | >100 | 84 ± 10 |
| 8 | 32 ± 5 | 29 ± 3 | 16 ± 3 | 10 ± 2 |
| Cisplatin32 | 5 ± 0 | 2 ± 0 | 2 ± 0 | 3 ± 0 |
Complexes 5 and 7 demonstrated lower cytotoxic activity compared to the other investigated platinum(II) and platinum(IV) complexes as well as cisplatin. Both 5 and 7 were inactive against the tested cell lines except SW-480 cells, suggesting a weaker cytotoxic potential and a moderate sensitivity of certain cell type. Of particular interest was the sensitivity of the SW-480 cell line to all platinum complexes. This underscores the importance of structural variations in platinum complexes and their impact on biological activity. Notably, while complex 3 exhibited significant antitumor activity across multiple cell lines, it did not surpass the efficacy of cisplatin. On the other hand, when the investigated complexes were initially dissolved in DMSO the viability experiments showed that solvolyzed products mostly lose initial in vitro activity showing clearly that solvolytic products exhibit mostly lower activity (Table S3, ESI†). Complex 3 emerged as the most active among all investigated complexes and was selected for further investigation. Primarily, the examination of the effect of 3 on human transformed MRC5 lung fibroblasts (Fig. S6, ESI†) revealed lower sensitivity comparing to MCF-7 cells, outlining a tendency to recognize tumor phenotypes over normal cells (the selectivity index (SI) is 4.6).
Further, an examination was conducted to determine whether 3 induces apoptosis in the MCF-7 cell line (Fig. 7). It was revealed that 3 causes an increase in the early and late apoptotic cell population. Upon identifying the cause of cell death in MCF-7 cells, further investigation was carried out to determine if complex 3 induces caspase activation. Despite the fact that the MCF-7 cell line is deficient in caspase 3, 3 was able to boost total caspase activation and consequently realization of the complete apoptotic program.33
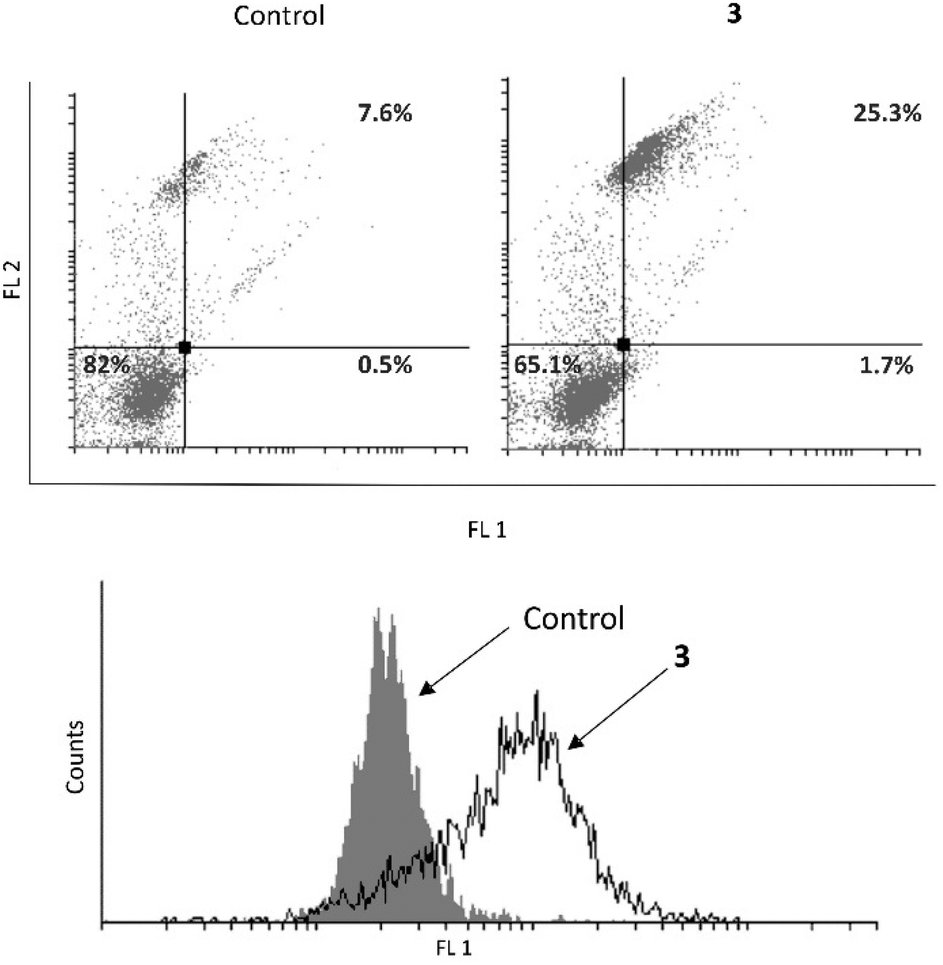 | ||
| Fig. 7 Apoptosis and caspase activation induced by complex 3. | ||
Subsequently, the impact of 3 on inducing autophagy in treated MCF-7 cells was examined (Fig. 8). It was detected that treatment with 3 led to extensive autophagy in MCF-7 cells. As autophagy initiation can serve as both, a cell destruction and a cytoprotective mechanism,34 the role of autophagy in MCF-7 cells was investigated in the presence of the autophagy inhibitor 3-methyladenine (3-MA) along with 3 (Fig. 8). It was demonstrated that in this case, autophagy induction influenced by 3 was exclusively of a cytoprotective nature.
 | ||
| Fig. 8 Autophagy and its cytoprotective role. | ||
Biologically-relevant reactivity
Since PtIV drugs or drug candidates are generally expected to be reduced to PtII by the thiol pools inside living cells, the reactivity of compound 3 towards a representative thiol, glutathione (GSH),35,36 was investigated. At concentrations ranging up to 1 mM and pH 7, which are biologically relevant,37 GSH caused a reduction in the UV absorbance characteristic of the compound (Fig. S7, ESI†). This reduction occurs at concentrations of 3 as low as 15 μM (and up to the highest tested concentration, 75 μM), and with thiol concentrations ranging from 1.5 μM to 1 mM. These concentrations span not only the values seen inside cells, but also those in blood. As such, one may expect that partial reduction of 3 would already occur in blood, before reaching the target tissues. Alternative reduction pathways for PtIV compounds in blood have also been evidenced, e.g. with ascorbate.38Serum albumin, which is a major reservoir of thiol groups in the blood and reaches concentrations of ca. 600 μM, was found not to affect the UV-vis spectrum of 3 upon mixing. Incubation of 3 at 37 °C in phosphate buffer for 4 h resulted in only a minor decrease in absorbance, similar to the one induced by GSH; serum albumin did not affect this phenomenon (Fig. S8, ESI†).
Another major component in blood, to which any therapeutic agent would be exposed, is hemoglobin (Hb). A range of compounds with therapeutic action or potential (e.g. anticancer, but also others), including PtII-based compounds, have been shown to affect the oxidative reactivity of Hb – either acting as antioxidants or as agents of oxidative stress that favor Hb autoxidation and ensuing pro-oxidant cascades.39,40 The latter situation may be of relevance in understanding the potential side effects or, in limiting cases, the modulation of the therapeutic effects of those agents. The sources of reactivity towards Hb may either be the redox-active groups in the therapeutic agents (e.g., aromatic rings, or metal centers) that interact with Hb directly or with Hb-derived oxidative stress agents, or simple allosteric interactions, or both. Compound 3 would in this context not be expected to be an efficient source of electrons – due to the oxidation state of the metal (PtIV) and the rather low redox reactivity of the pyridine ring in the ligand. Unlike some other anticancer drugs, compound 3 indeed showed no ability to reduce the high-valent ferryl form of Hb (Fig. S10, ESI†) known to be involved in physiologically relevant processes.40 Instead, 3 clearly induces autoxidation of oxy Hb even at low-micromolar concentrations (Fig. S9, ESI†) that are at the upper limits of physiologically attainable concentrations during intravenous administration of anticancer drugs.41
Binding of 3 to serum albumin as well as to Hb was confirmed by fluorescence measurements (Fig. S11, ESI†). The intrinsic fluorescence of these two proteins, due to the aromatic tryptophan and tyrosine residues, is quenched effectively by 3 at concentrations of 50 μM and higher (Fig. S11, ESI†), suggesting that the ability of 3 to bind to proteins is relatively limited. The latter would be an advantage for a therapeutically useful platinum(IV) complex whose goal is to deliver the Pt and its axial ligands into cells without remaining trapped on blood proteins.
Overall, in terms of biologically relevant reactivity, these data confirm that PtIV compounds of the class synthesized and described in the present study (1) show moderate affinities to proteins at physiologically relevant concentrations, (2) can act as inducers of oxidative stress towards a major metalloprotein in the blood, hemoglobin, and (3) can be efficiently reduced by a representative thiol, glutathione.
Electrochemistry
Because PtIV compounds function as prodrugs, the reduction potential represents a pivotal pharmacological parameter, enabling the assessment of drug activity toward tumoral cells. The correlation between the reduction potential of PtIV conjugates and the biostability of the compound in the bloodstream was mentioned in previous investigations;42 in case of a high reduction potential (tetraplatin Ered = −0.090 V, Ag/AgCl or −0.638 V FcH/FcH+), the reduction is too fast in the blood and the toxicity expressed by the compound is too high, while a low reduction potential (iproplatin Ered = −0.730 V, Ag/AgCl, or −1.278 V FcH/FcH+) diminishes the possibility of PtIV to be reduced to the biologically active PtII species in the tumor cells.43,44Due to the low solubility of PtIV derivative 3 in water, the redox potential was evaluated by cyclic voltammetry in DMF solution (Fig. S12, ESI†) and the potential was expressed against that of the ferrocene/ferrocenium (FcH/FcH+) redox couple. In the cyclic voltammogram 3 shows three oxido-reduction processes, where peak I is quasi reversible (ΔEp > 59 mV), while peak II and peak III are reversible (ΔEp < 59 mV).
Analyzing the values of the reduction potentials of compound 3 (Table 4) and comparing them with previously reported data, it can be said that the values obtained are much lower than those reported for iproplatin, whose reduction in the body to PtII is too slow, and higher than those for tetraplatin, whose reduction in the body is too rapid. This gives the PtIV derivative 3 the potential to act as a carrier of the active cytotoxic PtII precursor 2 to cancer cells when reduced by the cellular environment. Cyclic voltammetry alone is insufficient to guarantee the behavior of the platinum(IV) complex in the bloodstream and intracellular environment. However, when combined with biological investigations, it was observed that compound 3 can be reduced inside cells and in blood.
| Peak position | Potential/V (vs. Ag/AgCl) | Potential/V (vs. FcH/FcH+) | ΔEp/V | ||
|---|---|---|---|---|---|
| Oxidation (anodic) | Reduction (cathodic) | Oxidation (anodic) | Reduction (cathodic) | ||
| ΔEp = Epeak anodic − Epeak cathodic | |||||
| I | −0.064 | −0.305 | −0.612 | −0.853 | 0.241 |
| II | −0.755 | −0.800 | −1.303 | −1.348 | 0.045 |
| III | −1.340 | −1.390 | −1.888 | −1.938 | 0.050 |
Experimental
Materials and methods
K2[PtCl4], K2[PtCl6], ethanol, diethyl ether, CH3Cl, CH2Cl2, DMF, and DMSO are commercially available. Bovine hemoglobin (oxy Hb) was obtained as previously described and converted to the met form using potassium ferricyanide; the excess ferricyanide was removed through the desalting column.45 Proteins were manipulated in 137 mM NaCl, 2.7 mM KCl, and 12 mM NaH2PO4, pH 7.4 (phosphate buffered saline, PBS). Bovine serum albumin (BSA fraction V, Sigma-Aldrich, Germany), hydrogen peroxide 30% (ChemPUR), L-ascorbic acid (99%, A.C.S. reagent, Sigma, Germany), L-glutathione reduced (Sigma-Aldrich, Germany), dimethyl sulfoxide (DMSO, VWR United States) were used as provided without further purification. 1H and 13C NMR spectra (at 300 MHz and 75 MHz, as well as at 400 MHz and 100 MHz, respectively) were recorded on a Bruker Fourier-300 and Avance DRX-400 spectrometer, using tetramethylsilane (TMS) or the deuterated solvent as the internal reference (see Fig. S13–S19 (ESI)†). Melting points were determined with a Gallenkamp capillary apparatus. Elemental analysis was conducted using a Thermo Scientific Flash Smart CHNS analyzer (Italy). IR spectra were obtained with a PerkinElmer Spectrum 2000 FT-IR spectrometer, covering the range from 4000 to 400 cm−1, using KBr pellets. ESI mass spectra, in both positive and negative modes, were recorded on a Bruker Daltonics FT-ICR mass spectrometer, with isotope distribution calculations performed using Molecular Weight Calculator 6.45.46 Ligand precursors 2-(2-aminoethylamino)acetic acid hydrochloride (L3·HCl)26,47 and 4-[(2-aminoethylamino)methyl]benzoic acid hydrochloride (L4·HCl)48 were prepared according to modified published protocols (see ESI†). For other ligand precursors (4-[(2-aminoethylamino)methyl]benzoic acid ethyl ester (L1·HCl), 4-[(3-aminopropylamino)methyl]benzoic acid ethyl ester (L2·HCl), 4-[(3-aminopropylamino)methyl]benzoic acid hydrochloride (L5·HCl)), details are given in ESI.† UV-vis spectra were performed on a Lambda 25 spectrophotometer (PerkinElmer, Singapore), in the range of 200–1000 nm, in 1 mL quartz cuvettes under conditions indicated in the legends of the figures. Fluorescence emission spectra were obtained with a PerkinElmer LS55 spectrophotometer, in the range of 200–900 nm, 280 nm excitation wavelength, in 0.4 mL quartz cuvettes.Synthetic procedures
Yield: 165 mg (94%). Properties: light yellow solid; soluble in DMSO, DMF; insoluble in water, diethyl ether. Decomposition temperature: 275 °C (from light yellow to black). Elemental analysis for C12H18Cl2N2O2Pt (488.3), found (calc.): C 30.01 (29.52), H 3.81 (3.72), N 5.98 (5.74). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 1.31 (s, br, 3H, CH3a), 2.10–2.26 (m, 3,6H, CH2bCH2c), 3.85–4.02 (m, 2H, CH2d), 4.30 (s, 2H, CH2e), 5.34 (m, 2H, NH2), 6.77 (s, 1H, NH), 7.73–7.94 (m, 4H, CHf, CHh). 13C{1H} NMR (100 MHz, DMSO-d6, in ppm): δ 14.2 (s, CH3a), 46.5 (s, CH2b), 54.6 (s, CH2c), 54.9 (s, CH2d), 60.9 (s, CH2e), 129.2 (s, CHf), 129.6 (s, Cg), 130.5 (s, CHh), 140.6 (s, Cj), 165.6 (s, CO). ESI-MS (neg. mode): m/z: 523.1 ([PtCl3L1]−). IR: ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1): 3447 (s, br), 2962 (w), 2358 (w), 1868 (w), 1717 (s), 1647 (s), 1636 (s), 1616 (s), 1507 (m), 1473 (m), 1457 (m), 1418 (m), 1280 (s), 1181 (w), 1105 (s), 1021 (s).
(cm−1): 3447 (s, br), 2962 (w), 2358 (w), 1868 (w), 1717 (s), 1647 (s), 1636 (s), 1616 (s), 1507 (m), 1473 (m), 1457 (m), 1418 (m), 1280 (s), 1181 (w), 1105 (s), 1021 (s).
Yield: 120 mg (66%). Properties: beige solid; soluble in DMSO, DMF; poorly soluble in chloroform, dichloromethane; insoluble in water, diethyl ether. Decomposition temperature: 200 °C (from ochre to dark brown). Elemental analysis for C13H20Cl2N2O2Pt (501.1), found (calc.): C 31.42 (31.13), H 3.97 (4.02), N 4.96 (5.58). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 1.31–1.40 (m, br, 5.4H, CH3a, CHb), 1.59–2.05 (m, 2H, CH2c), 2.56–2.97 (m, 2.4H, CH2d), 4.31 (s, br, 2H, CH2e), 4.80–5.40 (m, 1.5H, NH2), 5.90–6.30 (s, br, 0.8H, NH), 7.40–7.93 (m, 4H, CHg, CHj). 13C{1H} NMR (100 MHz, DMSO-d6, in ppm): δ 14.4 (s, CH3a), 23.5 (s, CH2b), 42.8 (s, CHc), 48.3 (s, CH2d), 55.8 (s, CH2e), 61.1 (s, CH2f), 129.4 (s, CHg), 129.7 (s, Ch), 130.7 (s, CHj), 140.5 (s, Ck), 165.9 (CO). 195Pt NMR (85 MHz, DMSO-d6, in ppm): δ −2243 (s). ESI-MS (neg. mode): m/z: 537.1 ([PtCl3L2]−). IR: (cm−1): 3434 (s), 3264 (m), 2936 (w), 1708 (s), 1614 (w), 1454 (w), 1367 (w), 1283 (s), 1178 (w), 1108 (m), 1021 (w).
Yield: 80 mg (68%). Properties: light yellow powder; soluble in DMSO, DMF; insoluble in water, diethyl ether. Decomposition temperature: 189 °C (from light yellow to dark brown). Elemental analysis for C13H20Cl4N2O2Pt·H2O (591.2), found (calc.): C 26.48 (26.40), H 3.85 (3.70), N 4.69 (4.74). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 1.31 (s, br, 3H, CH3a), 1.91–2.11 (m, br, 2H, CH2b), 2.75–2.95 (m, br, 4H, CH2c, CH2d), 4.31 (m, br, 4H, CH2e, CH2f), 6.15 (s, br, 1H, NH), 7.63 (s, br, 2H, CHg), 8.00 (s, br, 2H, CHh), 8.94 (s, br, 2H, NH2). 13C{1H} NMR (100 MHz, DMSO-d6, in ppm): δ 14.6 (s, CH3a), 25.2 (s, CH2b), 26.3 (s, solvolysis product), 43.7 (s, CH2c), 45.2 (s, CH2d), 50.2 (s, CH2e), 61.4 (s, CH2f), 127.9 (s, solvolysis product), 129.8 (s, CHg), 130.4 (s, solvolysis product t), 130.7 (s, CHh), 131.9 (s, Cj), 137.4 (s, Ck), 165.8 (s, CO). ESI-MS (neg. mode): m/z: 609.0 ([PtCl5L2]−). IR: (cm−1): 3447 (s, br), 3200 (m), 2964 (m), 2363 (w), 1868 (w), 1844 (w), 1716 (s), 1653 (s), 1647 (s), 1616 (s), 1558 (m), 1541 (s), 1507 (s), 1457 (m), 1367 (w), 1281 (s, br), 1183 (w), 1106 (s), 1020 (s).
Yield: 45 mg (24%). Properties: colorless needles; soluble in DMSO; moderately soluble in water; insoluble in diethyl ether. Decomposition temperature: 230 °C (from light yellow to dark brown). Elemental analysis for C4H12Cl2N2O2Pt (384.1), found (calc.): C 12.87 (12.51), H 2.44 (2.62), N 7.18 (7.29). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 2.36 (s, br, 3H, CH2CH2), 2.75 (s, br, 1H, CH2CH2), 3.45–4.09 (m, 2.5H, NHCH2C), 5.36 (s, br, 2H, NH2), 6.40 (s, 1H, NH), 13.1 (s, br, 1H, OH). 13C{1H} NMR (100 MHz, DMSO-d6, in ppm): δ 46.5 (s, NH2CH2CH2), 53.1 (s, CH2CH2NH), 56.9 (s, NHCH2C), 170.1 (s, CO). IR: (cm−1): 3446 (s, br), 3301 (w), 3250 (w), 3141 (s), 2963 (w), 2934 (w), 1719 (s), 1568 (m), 1456 (w), 1432 (w), 1405 (w), 1355 (w), 1283 (w), 1261 (m).
Yield: 100 mg (45%). Properties: beige powder; soluble in DMSO, DMF; moderately soluble in water; insoluble in diethyl ether. Decomposition temperature: 290 °C (from beige to brown). Elemental analysis for C10H14Cl2N2O2Pt (460.2), found (calc.): C 26.21 (26.01), H 3.16 (3.07), N 5.74 (6.09). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 2.10–2.30 (m, 4H, CH2aCHb), 4.00–4.32 (m, 2H, CH2c), 5.33 (s, 1H, NH2), 5.40 (s, 1H, NH2), 6.78 (s, 1H, NH), 7.72 (d, 3JH,H = 8.1 Hz, 2H, CHd), 7.95 (d, 3JH,H = 8.1 Hz, 2H, CHe). 13C{1H} NMR (100 MHz, DMSO-d6): δ 46.9 (s, CH2a), 55.0 (s, CH2b), 55.3 (s, CH2c), 129.7 (s, CHd), 130.7 (s, CHe), 130.9 (s, Cf), 140.2 (s, Cg), 168.5 (s, CO). 1H NMR (300 MHz, DMF-d7, in ppm): δ 2.36–2.71 (m, 4H, CH2aCH2b), 4.22–4.56 (m, 2H, CH2c), 5.47 (s, 2H, NH2), 6.68 (s, 1H, NH), 7.84 (d, 3JH,H = 7.8 Hz, 2H, CHd), 8.04 (d, 3JH,H = 7.6 Hz, CHe). 13C{1H} NMR (75 MHz, DMF-d7, in ppm): δ 47.8 (s, CH2a), 55.6 (s, CH b), 56.0 (s, CH2c), 130.2 (s, CHd), 131.0 (s, CHe), 131.5 (s, Cf), 141.2 (s, Cg), 167.9 (s, CO). ESI-MS (pos. mode): m/z: 482.9 ([PtCl2L4+Na]+). IR: (cm−1): 3433 (s), 3214 (s), 1690 (s), 1614 (m), 1420 (w), 1280 (m), 1180 (w), 1108 (w), 1059 (w), 1017 (w).
Yield: 80 mg (44%). Properties: light yellow powder; soluble in DMSO; poorly soluble in water; insoluble in diethyl ether. Decomposition temperature: 210 °C (from light yellow to brown). Elemental analysis for C4H9Cl3N2O2Pt (418.6), found (calc.): C 11.55 (11.48), H 2.21 (2.17), N 6.55 (6.69). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 2.73–3.25 (m, 3H, CH2CH2), 3.46–4.06 (m, 3H, CH2CH2, NHCH2CO), 7.80 (s, br, 3H, NH2, NH). 13C{1H} NMR (100 MHz, DMSO-d6, in ppm): δ 35.4 (s, NH2CH2), 46.2 (s, solvolysis product), 50.4 (s, NHCH2CH2), 55.4 (s, NHCH2C), 55.7 (s, solvolysis product), 59.3 (s, solvolysis product), 179.7 (s, CO), 181.3 (s, solvolysis product). ESI-MS (neg. mode): m/z: 452.9 ([PtCl4L3]−). IR: (cm−1): 3427 (s, br), 3183 (s, br), 2974 (w), 2928 (w), 1679 (s), 1666 (s), 1580 (m), 1497 (w), 1450 (m), 1411 (m), 1357 (m), 1295 (m).
Yield: 150 mg (69%). Properties: light yellow solid; very soluble in DMSO, DMF; moderately soluble in water; insoluble in diethyl ether. Decomposition temperature: 285 °C (from light yellow to brown). Elemental analysis for C10H14Cl4N2O2Pt (531.1), found (calc.): C 22.45 (22.60), H 2.81 (2.66), N 5.19 (5.27). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 3.20 (m, 4H, CH2aCHb), 4.32 (s, 2H, CH2c), 7.66 (d, 3JH,H = 7.8 Hz, 2H, CHd), 7.94 (s, 2H, NH2), 8.01 (d, 3JH,H = 7.7 Hz, 2H, CHe), 9.22 (s, 1H, NH), 13.14 (s, br, 0,5H, OH). 13C{1H} NMR (75 MHz, DMSO-d6, in ppm): δ 47.4 (s, CHa), 56.1 (s, br, CHb, CHc), 129.8 (s, CHd), 131.4 (s, Ce), 131.8 (s, CHf), 138.1 (s, Cg), 167.5 (s, CO). ESI-MS (neg. mode): m/z: 528.8 ([PtCl4L4-H]−). IR: (cm−1): 3440 (s), 3181 (s), 3131 (s), 2553 (w), 1686 (s), 1616 (m), 1562 (m), 1427 (m), 1294 (s), 1185 (w).
Yield: 45 mg (23%). Properties: light yellow powder; soluble in DMSO; poorly soluble in water; insoluble in diethyl ether. Decomposition temperature: 230 °C (from light yellow to dark brown). 1H NMR (400 MHz, DMSO-d6, in ppm): δ 2.13 (m, 2H, CH2a), 2.77 (m, 2H, CHb), 3.00 (m, 2H, CH2c), 4.19 (m, 2H, CH2d), 6.45 (t, 1JN,H = 52 Hz, 2H, NH2), 7.62 (d, 3JH,H = 8.0 Hz, 2H, CHe), 7.99 (d, 3JH,H = 8.3 Hz, 2H, CHf), 9.02 (s, 1H, NH). 13C{1H} NMR (100 MHz, DMSO-d6, in ppm): δ 23.7 (s, CH2a), 36.2 (s, CH2b), 43.9 (s, CH2c), 49.7 (s, CH2d), 129.5 (s, CHe), 130.3 (s, CHf), 131.2 (s, Cg), 136.6 (s, Ch), 166.9 (s, CO). ESI-MS (neg. mode): m/z: 580.8 ([PtCl5(L2-Et+H)]−). IR: (cm−1): 3456 (s), 3240 (s), 3197 (s), 1701 (s), 1617 (s), 1563 (m), 1423 (w), 1262 (w), 1114 (w).
Crystallography
The single crystal X-ray structure analyses of compounds 3, 4 and 6 were conducted using a Gemini diffractometer (Rigaku Oxford Diffraction) with Mo-Kα radiation and ω-scan rotation. Data reduction was performed using CrysAlis Pro,49 which included the SCALE3 ABSPACK program for empirical absorption correction and an analytical numeric absorption correction based on a multifaceted crystal model as described by Clark and Reid.50 The structures were determined by “direct methods” with SHELXS-2013 (3, 4) or “dual-space methods” with SHELXT-2014 (6) and refined with SHELXL-2018.51 Crystallographic parameters are summarized in Table S1 (ESI).† Structural images were generated and processed using the Diamond program.52 Supplementary crystallographic data can be accessed at https://www.ccdc.cam.ac.uk/structures/ with the following CCDC: 2390314 (3), 2390315 (4) and 2390316 (6).†Biological studies
Human colorectal (SW-480), breast (MCF-7) and thyroid (8505C) cancer cell lines, melanoma (518A2) and non-malignant fibroblast cell line (MRC5) were purchased from the American Type Culture Collection (ATCC, Manassas, Virginia, USA). Cells were grown in HEPES-buffered RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 0.01% sodium pyruvate and antibiotics (penicillin 100 units per mL and streptomycin 100 μg mL−1) at 37 °C in a humidified atmosphere with 5% CO2. For viability assays, cells were seeded in following densities per well: SW-480 and 8505C: 1.5 × 103; MCF-7: 2 × 103; 518A2: 1 × 103; and MRC5: 7 × 103. For flow cytometry, MCF-7 cells were seeded at 2 × 105 density per well in 6-well plates.
DMF stock solutions of complexes were prepared in 100 mM concentrations and kept at −20 °C. Desired final concentrations were diluted in culture medium.
Cyclic voltammetry
Cyclic voltammetry (CV) measurements were performed using an Autolab computer-controlled potentiostat (Metrohm, The Netherlands). For this purpose, a conventional three-electrode cell was employed, equipped with a glassy carbon electrode (GCE, 3 mm diameter) as the working electrode, an Ag/AgCl wire as the reference electrode, and a platinum plate as the auxiliary electrode. Before the voltammetric experiments, the working electrode was polished with 0.3 μm alumina on a clean polishing cloth (Buehler, USA), rinsed with water, sonicated in water thoroughly to remove alumina, rinsed successively with water and ethanol, and finally dried under argon gas. The electrochemical measurements were performed in a 1 mM solution of compound 3 dissolved in 0.1 M [n-Bu4N][BF4]/DMF, which was purged with argon before each measurement, in order to remove the oxygen. The same measurement was performed in 10−3 M ferrocene in the same 0.1 M [n-Bu4N][BF4]/DMF, and the potential of the studied compound was expressed against that of the ferrocene/ferrocenium(FcH/FcH+) redox couple (+0.75 V vs. SHE).Conclusions
The synthesis and characterization of novel platinum(II) and platinum(IV) complexes have provided valuable insights into their structural properties and cytotoxicity profiles. Complex 3, from all tested compounds herein, emerged as a standout candidate, demonstrating significant cytotoxic effect in MCF-7 cells. 3 demonstrated high antitumor potential, however lower than cisplatin, including induction of total caspase activity and apoptosis despite caspase 3 deficiency of MCF-7 cells. While 3 also triggered cytoprotective autophagy in MCF-7 cells, co-treatment with an autophagy inhibitor led to enhanced antitumor activity. These findings underscore the potential of complex 3 as a lead compound for further studies. Notably, PtIV compound 3 demonstrated limited protein binding, oxidative stress induction in hemoglobin, and reducibility by glutathione, indicating its potential as a non-cytotoxic carrier for the active cytotoxic PtII complex 2 to cancer cells. Cyclic voltammetry supported these findings by showing the reduction capability of the PtIV complexes under physiological conditions.Data availability
The data supporting this article have been included as part of the ESI.†Supplementary crystallographic data can be accessed at https://www.ccdc.cam.ac.uk/structures/ with the following CCDC: 2390314 (3), 2390315 (4) and 2390316 (6).†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the Graduate School BuildMoNa (S. R.) is gratefully acknowledged. The research was funded by the Ministry of Science, Technological Development and Innovation of the Republic of Serbia (No. 451-03-66/2024-03/20007). Support from the project “Targeted Tumor Therapy with multifunctional platinum(IV)-drug conjugates, T3-Pt”, PNRR-III-C9-2023-I8-CF76, contract no. 760240/28.12.2023 funded by the European Union – NextGenerationEU and the Romanian Government, under the National Recovery and Resilience Plan for Romania, through the Romanian Ministry of Research, Innovation and Digitalization, within Component 9, Investment I8 is gratefully acknowledged.References
- M. Duan, S. Leng and P. Mao, Pharmacol. Ther., 2024, 258, 108642 CrossRef CAS PubMed.
- K. M. Sadek, H. Y. AbdEllatief, S. F. E. Mahmoud, A. Alexiou, M. Papadakis, M. Al-Hajeili, H. M. Saad and G. E.-S. Batiha, Cancer Rep., 2024, 7, e2052 CrossRef CAS PubMed.
- C. A. Oliveira, É. A. B. Mercês, F. S. Portela, L. F. L. Malheiro, H. B. L. Silva, L. M. De Benedictis, J. M. De Benedictis, C. C. d’Á. E. Silva, A. C. L. Santos, D. P. Rosa, H. S. Velozo, T. de Jesus Soares and L. S. de Brito Amaral, Clin. Exp. Nephrol., 2024, 28, 711–727 CrossRef CAS PubMed.
- X. Chen, Y. Gan, N. P. B. Au and C. H. E. Ma, Front. Mol. Neurosci., 2024, 17, 1345811 CrossRef CAS.
- G. N. Kaluđerović and R. Paschke, Curr. Med. Chem., 2011, 18, 4738–4752 CrossRef PubMed.
- S. Gómez-Ruiz, D. Maksimović-Ivanić, S. Mijatović and G. N. Kaluđerović, Bioinorg. Chem. Appl., 2012, 2012, 140284 Search PubMed.
- D. Tsvetkova and S. Ivanova, Molecules, 2022, 27, 2466 CrossRef CAS.
- H. K. Sheikh, C. J. C. Ortiz, T. Arshad, J. M. Padrón and H. Khan, Eur. J. Med. Chem., 2024, 271, 116438 CrossRef CAS PubMed.
- J. N. Jolley, A. I. Yanovsky, L. R. Kelland and K. B. Nolan, J. Inorg. Biochem., 2001, 83, 91–100 CrossRef CAS PubMed.
- G. N. Kaluđerović, D. Miljkovic, M. Momcilovic, V. M. Djinovic, M. M. Stojkovic, T. J. Sabo and V. Trajkovic, Int. J. Cancer, 2005, 116, 479–486 CrossRef PubMed.
- M. Bulatović, M. R. Kaluđerović, M. Mojić, B. B. Zmejkovski, E. Hey-Hawkins, M. Vidaković, N. Grdović, G. N. Kaluđerović, S. Mijatović and D. Maksimović-Ivanić, Eur. J. Pharmacol., 2015, 760, 136–144 CrossRef.
- G. N. Kaluđerović, S. A. Mijatović, B. B. Zmejkovski, M. Z. Bulatović, S. Gómez-Ruiz, M. K. Mojić, D. Steinborn, D. M. Miljković, H. Schmidt, S. D. Stošić-Grujičić, T. J. Sabo and D. D. Maksimović-Ivanić, Metallomics, 2012, 4, 979–987 CrossRef PubMed.
- D. Maksimović-Ivanić, S. Mijatovic, I. Mirkov, S. Stosic-Grujicic, D. Miljković, T. J. Sabo, V. Trajković and G. N. Kaluđerović, Metallomics, 2012, 4, 1155–1159 CrossRef PubMed.
- B. B. Krajčinović, G. N. Kaluđerović, D. Steinborn, H. Schmidt, C. Wagner, Ž. Žižak, Z. D. Juranić, S. R. Trifunović and T. J. Sabo, J. Inorg. Biochem., 2008, 102, 892–900 CrossRef.
- J.-H. Choi, J. Appl. Spectrosc., 2015, 82, 360–366 CrossRef CAS.
- T. Yasui, T. Ama, H. Kawaguchi, K. Okamoto and J. Hidaka, Bull. Chem. Soc. Jpn., 1992, 65, 1920–1926 CrossRef CAS.
- Y. FuJii, E. Kyuno and R. Tsuchiya, Bull. Chem. Soc. Jpn., 1970, 43, 786–789 CrossRef CAS.
- T. Ama, H. Kawaguchi, T. Yasui, K. Matsumoto and S. Ooi, Bull. Chem. Soc. Jpn., 1985, 58, 2561–2568 CrossRef CAS.
- L. P. Battaglia, A. B. Corradi and M. E. V. Tani, Acta Cryst., 1974, C3, 331 Search PubMed.
- T. Ama, H. Kawaguchi, T. Yasui and K. Matsumoto, Bull. Chem. Soc. Jpn., 1985, 58, 2564 CrossRef.
- M. Lipowska, R. Cini, G. Tamasi, X. Xu and A. T. Taylor, Inorg. Chem., 2004, 43, 7774 CrossRef CAS PubMed.
- H. Masuda, A. Odani, T. Yamazaki, T. Yajima and O. Yamauchi, Inorg. Chem., 1993, 32, 1111–1118 CrossRef CAS.
- J.-H. Choi, T. Suzuki, Md. A. Subhan, S. Kaizaki and Y. C. Park, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m409–m412 CrossRef.
- M. V. de Almeida, J. D. S. Chaves, A. P. S. Fontes, E. T. César and M. Gielen, J. Braz. Chem. Soc., 2006, 17, 1266–1273 CrossRef CAS.
- G. N. Kaluđerović, V. M. Đinović, Z. D. Juranić, T. P. Stanojković and T. J. Sabo, J. Coord. Chem., 2006, 59, 815–819 CrossRef.
- G. N. Kaluđerović, H. Kommera, S. Schwieger, A. Paethanom, M. Kunze, H. Schmidt, R. Paschke and D. Steinborn, Dalton Trans., 2009, 10720–10726 RSC.
- B. B. Krajčinović, G. N. Kaluderović, D. Steinborn, H. Schmidt, C. Wagner, Z. Žižak, Z. D. Juranić, S. R. Trifunović and T. J. Sabo, J. Inorg. Biochem., 2008, 102, 892–900 CrossRef.
- M. Goto, M. Sumimoto, T. Matsumoto, M. Iwasaki, Y. Tanaka, H. Kurosaki, K. Yuto and Y. Yoshikawa, Bull. Chem. Soc. Jpn., 2000, 73, 1589–1598 CrossRef CAS.
- S. J. S. Kerrison and P. J. Sadler, Chem. Commun., 1977, 861–863 RSC.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
- G. Ludwig, I. Ranđelović, D. Dimić, T. Komazec, D. Maksimović-Ivanić, S. Mijatović, T. Rüffer and G. N. Kaluđerović, Biomolecules, 2024, 14, 420 CrossRef CAS PubMed.
- R. U. Jänicke, Breast Cancer Res. Treat., 2009, 117, 219–221 CrossRef PubMed.
- G. Mariño, M. Niso-Santano, E. H. Baehrecke and G. Kroemer, Nat. Rev. Mol. Cell Biol., 2014, 15, 81–94 CrossRef PubMed.
- Y. Shi, S.-A. Liu, D. J. Kerwood, J. Goodisman and J. C. Dabrowiak, J. Inorg. Biochem., 2012, 107, 6–14 CrossRef CAS PubMed.
- S. Chen, K.-Y. Ng, Q. Zhou, H. Yao, Z. Deng, M.-K. Tse and G. Zhu, Dalton Trans., 2022, 51, 885–897 RSC.
- S. Fenk, E. V. Melnikova, A. A. Anashkina, Y. M. Poluektov, P. I. Zaripov, V. A. Mitkevich, Y. V. Tkachev, L. Kaestner, G. Minetti, H. Mairbäurl, J. S. Goede, A. A. Makarov, I. Y. Petrushanko and A. Bogdanova, Redox Biol., 2022, 58, 102535 CrossRef CAS PubMed.
- C. K. J. Chen, P. Kappen and T. W. Hambley, Metallomics, 2019, 11, 686–695 CrossRef CAS PubMed.
- C. Zagrean-Tuza, I. Igescu, A. Lupan and R. Silaghi-Dumitrescu, Inorg. Chim. Acta, 2024, 567, 122053 CrossRef CAS.
- C. Puscas, L. Radu, F. Carrascoza, A. C. Mot, D. Amariei, O. Lungu, F. Scurtu, P. Podea, R. Septelean, A. Matei, M. Mic, A. A. Attia and R. Silaghi-Dumitrescu, Free Radicals Biol. Med., 2018, 124, 260–274 CrossRef CAS PubMed.
- C. Puscas, A. Mircea, M. Raiu, M. Mic, A. A. A. Attia and R. Silaghi-Dumitrescu, Chem. Res. Toxicol., 2019, 32, 1402–1411 Search PubMed.
- S. Choi, C. Filotto, M. Bisanzo, S. Delaney, D. Lagasee, J. L. Whitworth, A. Jusko, C. Li, N. A. Wood, J. Willingham, A. Schwenker and K. Spaulding, Inorg. Chem., 1998, 37, 2500–2504 CrossRef CAS.
- A. Escolà, M. Crespo, C. López, J. Quirante, A. Jayaraman, I. H. Polat, J. Badía, L. Baldomà and M. Cascante, Bioorg. Med. Chem., 2016, 24, 5804–5815 CrossRef PubMed.
- M. R. Reithofer, A. K. Bytzek, S. M. Valiahdi, C. R. Kowol, M. Groessl, C. G. Hartinger, M. A. Jakupec, M. S. Galanski and B. K. Keppler, J. Inorg. Biochem., 2011, 105, 46–51 CrossRef CAS.
- D. Hathazi, A. C. Mot, A. Vaida, F. Scurtu, I. Lupan, E. Fischer-Fodor, G. Damian, D. M. Kurtz Jr. and R. Silaghi-Dumitrescu, Biomacromolecules, 2014, 15, 1920–1927 CrossRef CAS.
- M. Monroe, Molecular Weight Calculator (version 6.41) https://alchemistmatt.com/mwtwin.html, 2011.
- E. p. Heimer, H. e. Gallo-Torres, A. m. Felix, M. Ahmad, T. j. LamBros, F. Scheidl and J. Meienhofer, Int. J. Pept. Protein Res., 1984, 23, 203–211 CrossRef CAS PubMed.
- L. I. Shevchenko, P. S. Pel'kis, M. O. Lozinskii and V. N. Kalinin, Ukr. Khim. Zh., 1984, 50, 301 CAS.
- Rigaku Oxford Diffraction, CrysAlisPro Software system, Rigaku Corporation, Wroclaw, Poland Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A:Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef; G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- K. Putz and K. Brandenburg, Diamond-4 (version 4.6.8)Crystal Impact GbR, Bonn Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2390314–2390316. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03041a |
| This journal is © The Royal Society of Chemistry 2025 |