
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced reactivity of cationic Au(μ-H)2MCp2 complexes (M = Mo and W) enabled by bulky tris-biaryl phosphines†
Martina
Landrini
 ab,
Miquel
Navarro
bcd,
Jesús
Campos
*b and
Luca
Rocchigiani
*a
ab,
Miquel
Navarro
bcd,
Jesús
Campos
*b and
Luca
Rocchigiani
*a
aDepartment of Chemistry, Biology and Biotechnology, University of Perugia and CIRCC, Via Elce di Sotto 8, 06123, Perugia, Italy. E-mail: luca.rocchigiani@unipg.it
bInstituto de Investigaciones Quimicas (IIQ), Departamento de Quimica Inorganica and Centro de Innovacion en Quimica Avanzada (ORFEO-CINQA), CSIC-Universidad de Sevilla, Sevilla 41092, Spain. E-mail: jesus.campos@iiq.csic.es
cDepartamento de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Cantoblanco, Madrid 28049, Spain
dInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, Madrid 28049, Spain
First published on 5th December 2024
Abstract
[(L1)Au(μ-H)2MCp2][BF4] complexes (M = Mo and W) featuring cavity-shaped tris-2-(4,4′-di-tert-butylbi-phenylyl)phosphine (L1) have been isolated. The tungsten derivative showed a remarkably fast reactivity in photolytic hydride transfer to generate the mononuclear gold hydride (L1)AuH. Both bimetallic adducts trap Ag+ cations, forming unprecedented {![[upper bond 1 start]](https://www.rsc.org/images/entities/char_e010.gif) Au(μ-H)M(μ-H)Ag
Au(μ-H)M(μ-H)Ag![[upper bond 1 end]](https://www.rsc.org/images/entities/char_e011.gif) } trimetallic assemblies with destabilized Au–M interactions.
} trimetallic assemblies with destabilized Au–M interactions.
Heterobimetallic hydride complexes have been extensively investigated over the past few decades owing to their peculiar bonding features, as well as their applications in synthesis and catalysis.1 In the context of gold chemistry, bimetallic hydrides2 are interesting targets because they show increased thermal stability with respect to terminal mononuclear hydrides3 and can serve as platforms for broad structural investigations.4 More recently, these species have garnered renewed interest in connection with hydride transfer/insertion chemistry and different bimetallic combinations have been produced with the aim of unlocking novel reactivity.5
In this respect, we reported that Mo and W metallocene dihydrides are suitable donors for Au(I) and Au(III) cationic species, forming stable bridging hydride and bimetallic interactions that arise from the marked Lewis basicity of group VI metals in the +IV oxidation state.6 In the case of Au(I), heterobimetallic adducts of the type [LAu(μ-H)2MCp2][X] (M = Mo or W, L = N-heterocyclic carbene (NHC) or phosphine, and X = weakly coordinating anion) can be photolysed to produce mononuclear gold hydrides and cationic metallocenium species.7 The outcome of this reaction depends on multiple factors including ancillary ligands, group VI metals and incident wavelengths. Notably, quantitative hydride transfer is achieved exclusively when NHCs are used to bind the gold centre, as phosphine-based complexes tend to decompose rapidly.
This behaviour is not unexpected, given the low stability of (phosphine)Au(I) hydrides, which tend to decompose into Au(0) and homoleptic compounds of the type [AuL2]+.8 To address this limitation, we recently explored the encapsulating effect of the cavity-shaped tris-2-(4,4′-di-tert-butylbiphenylyl)phosphine ligand (L1, Fig. 1), which has already proven effective in stabilizing unusual Au(I) π–ethylene and π–acetylene adducts.9 Although the resulting Au(I) terminal hydride, (L1)AuH, still exhibited reduced thermal stability, it could be accessed.10
 | ||
| Fig. 1 Outline of this work. | ||
Capitalizing on these results, we now report that [(L1)Au]+ fragments form stable bimetallic adducts with Cp2MoH2 and Cp2WH2, despite their congested environment. Importantly, unlike other phosphine-based gold precursors, the resulting bimetallic adducts can be successfully used in assessing photolytic hydride transfer. Moreover, the presence of the dangling biphenyl rings has been exploited to introduce a third metal into the cavity, showcasing how the flexibility of this ligand impacts the reactivity of heterobimetallic bridging hydrides.
Monitoring the reaction between [(L1)Au(NCMe)][BF4] and Cp2MH2 (M = W and Mo) by in situ NMR (CD2Cl2, 298 K) revealed that novel heterobimetallic complexes form immediately upon mixing.7 The presence of a single hydride signal in the 1H NMR spectrum at δH = −9.35 for Mo and δH = −11.08 ppm for W, coupled with 31P (2JHP = 29.6 and 37.8 Hz for W and Mo, respectively), was particularly indicative of the formation of [(L1)Au(μ-H)2MCp2][BF4] adducts 1 (M = W) and 2 (M = Mo; Scheme 1). Consistently, 1H NOESY NMR showed the presence of dipolar coupling between the metallocene dihydride signals and the aromatic protons of the biaryl units (Fig. S6, ESI†).
 | ||
| Scheme 1 Synthesis of bimetallic complexes 1 (Au–W) and 2 (Au–Mo). | ||
The bimetallic complexes are stable at RT and were isolated in good yields (74% and 88% for 1 and 2, respectively) by reaction scale-up and purification by recrystallisation. The spectroscopic fingerprints of the metallocene in 1, including the 1H–183W scalar coupling (1JWH = 74.0 Hz), appear very similar to those previously reported for analogous [(R3P)Au(μ-H)2MCp2]+ cations,7 as does the 31P NMR shift (δP = 46.0 ppm). The same NMR trends are observed for complex 2. Similarly, the spectroscopic properties of the phosphine ligand are unaffected by the coordination of the metallocene in both 1 and 2.
Slow diffusion of pentane into dichloromethane solutions of complexes 1 and 2 resulted in single crystals suitable for X-ray diffraction analysis. The solid-state molecular structures (Fig. 2 for 1 and Fig. S46† for 2) confirm that the metallocene donors perfectly fit into the [(L1)Au]+ cavity, establishing a quasi-linear P–Au–M arrangement (a P1–Au1–W1 angle of 176.76(4)° and a P1–Au1–Mo1 angle of 176.67(3)°). The two bimetallic adducts appear isostructural, with a metal–metal distance that is only marginally longer in 2 (Au1–W1 = 2.7523(5) Å and Au1–Mo1 = 2.7779(5) Å). Consistent with the NMR results in solution, there are no notable differences between the structural parameters of 1 and 2 and those previously reported for [(R3P)Au(μ-H)2MCp2]+.7 While this may appear surprising at first, the space filling representation of the structure of 1 (Fig. 2) clearly shows that the high flexibility of the biaryl arms of the phosphine encapsulates the metallocene fragment with an optimal geometry, fitting the cyclopentadienyl rings into the cone described by the tert-butyl phenyl groups and leaving enough space for the bridging hydride interactions.
 | ||
| Fig. 2 Left: molecular structure of 1. Hydrogen atoms, solvent molecules and anions are omitted for clarity. Thermal ellipsoids are set at 50% probability. Selected bond lengths [Å] and angles [°]: Au1–W1: 2.7523(5), Au1–P1: 2.278(1), P1–Au1–W1: 176.76(4), and Cpcentroid–W1–Cpcentroid: 141.68. Right: CPK representation of the solid-state structure of [1]+. | ||
Photolysis experiments were performed by exposing diluted THF-d8 solutions of 1 and 2 to broadband UV light (280–385 nm, Xe lamp). These irradiation conditions were found to be the most effective in achieving a faster and more selective generation of terminal (NHC)AuH species.7 Unlike other phosphine-based [(R3P)Au(μ-H)2WCp2]+ species, the irradiation of a 0.6 mM solution of 1 does not lead immediately to reductive decomposition. Instead, monitoring the reaction by 1H NMR revealed that a clean photolysis of the bimetallic hydride occurs, leading to the generation of terminal (L1)AuH and [Cp2W(H)THF][BF4] (Scheme 2). The formation of the terminal gold hydride was corroborated by the presence of the characteristic doublet at δH = 3.27 ppm in the 1H NMR spectrum, with a 2JHP of 221.2 Hz (Fig. 3, right), and at δp = 34.0 ppm in the 31P NMR spectrum, which resolves into singlets upon selective decoupling from 31P and 1H, respectively.10
 | ||
| Scheme 2 Outcome of the photolysis of 1 (280–385 nm, THF-d8, and 298 K). | ||
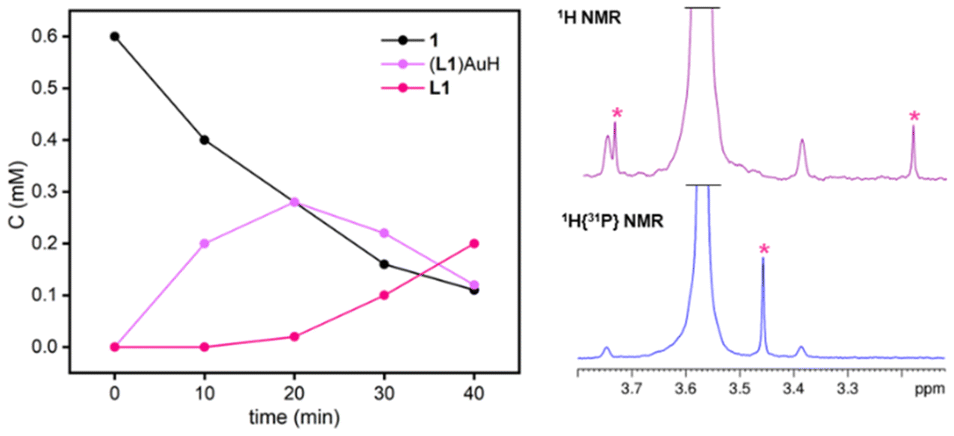 | ||
| Fig. 3 Concentration versus time profile for irradiation with UV light in THF-d8 of 1 (left); comparison between the 1H NMR and 1H{31P} NMR spectra of (L1)AuH, highlighting the hydride shift (right). | ||
Strikingly, the photolytic hydride transfer is over ten times faster than that reported for [(IPrMe)Au(μ-H)2WCp2][BF4] (IPrMe = C{(NiPrCMe)2}).7 After 20 min, 50% of 1 is converted into the terminal hydride (L1)AuH (Fig. 3, left), while (IPrMe)AuH formed in 45% yield only after 200 min under the same conditions. This is most likely ascribed to the steric congestion around the bimetallic core, which not only facilitates splitting, but also may disfavour any reversible association between metal fragments formed upon photolysis. (L1)AuH exhibits limited stability and therefore it does not accumulate over 50%. Its degradation leads to free phosphine (δP = −29.0 ppm), probably due to a reductive elimination pattern supported by the presence of a small amount of H2 (δH = 4.54 ppm). This is somewhat related to the decomposition of gold methyl complexes via ethane release and likely involves multimetallic intermediates.11 Kinetically, the mixture follows a typical consecutive reaction pathway, where 1 decreases and the free ligand increases over time, while (L1)AuH accumulates for the first 20 min and is completely consumed in less than 1 hour (Fig. 3, left).
Surprisingly, complex 2 proves to be photostable and no hydride transfer occurs upon irradiating a 0.6 mM THF-d8 solution over 30 min. After 2 hours of irradiation, the 1H NMR spectrum shows only 15% degradation, with no trace of gold-hydride formation. Instead, the generation of (L1)AuCl was observed, where the chloride atom probably derives from traces of dichloromethane (Fig. S35, ESI†).
These results highlight that the hydride transfer reaction is affected by the nature of group VI metals, consistent with our previous findings.7 In particular, the efficiency of hydrogen transfer is related to the nature of the HOMO–LUMO transition and, specifically, to the presence of partial σ*(M–H) character in the excited state. It is reasonable that in the molybdocene complex, such antibonding character is somehow depressed and the complex does not undergo photolysis. These observations showcase the peculiar role played by the bulky phosphine, which allows us to observe both complete and arrested hydride transfer in this series of compounds.
Encouraged by these results, we also investigated whether this ligand would stabilise multimetallic interactions with a third metal. To this end, Ag+ was chosen as an electrophilic cation prone to forming metallophilic interactions with both Au and W. Besides, it has potential for triggering electron transfer reactions due to its high reduction potential and to enable cooperative effects within the context of gold catalysis.12
The addition of solid AgBF4 to a CD2Cl2 solution of 1 or 2 leads to the formation of new trimetallic bridging hydride adducts (3 and 4, Fig. 4, left) over 10 minutes at RT, as suggested by in situ NMR spectroscopy. However, these species rapidly decompose, resulting in a black precipitate along with the known trimetallic [(L1)Au(μ3-Cl)Ag2]2+ species (Fig. S25, ESI†).9 The reaction was replicated by adding 0.5 equiv. of AgBF4 at a time, revealing that 2 equiv. of silver are required to achieve the quantitative formation of the trimetallic compounds (Fig. S17, ESI†), possibly owing to the presence of an equilibrium reaction.
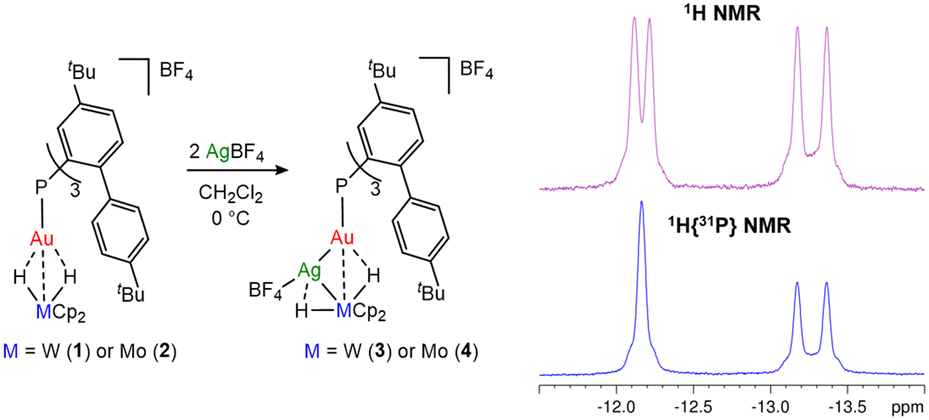 | ||
| Fig. 4 Synthesis of trimetallic Au–Ag–M bridging hydrides 3 (M = W) and 4 (M = Mo) (left); 1H and 1H{31P} NMR spectra in CD2Cl2 of 3 at −80 °C zoomed in on the hydride shift (right). | ||
To limit decomposition effects, the reaction was performed at 273 K. The spectroscopic data show that the hydride signals of the trimetallic adducts are low-frequency shifted with respect to those of 1 and 2 (δH = −12.51 ppm and Δδ = −1.43 for 3 and δH = −11.79 ppm and Δδ = −2.44 for 4). JHP values are also lower (19.3 and 27.9 Hz for 3 and 4, respectively), while the 31P NMR chemical shift is high-frequency shifted with respect to those of the precursors (δP = 53.8 ppm and Δδ = 7.8 for 3 and δP = 41.0 ppm and Δδ = 4.6 for 4). Upon lowering the temperature to 193 K, the hydride resonance splits into two distinct doublet signals (δH = −12.17 and −13.27 ppm for 3 (Fig. S11, ESI†) and δH = −11.98 and −12.32 ppm for 4 (Fig. S20, ESI†)) that show two significantly different coupling constant values. Only the high-frequency shifted signal resolved into a singlet upon performing a 1H{31P} NMR experiment (JPH = 40.3 and 53.4 Hz for 3 (Fig. 4, right) and 4, respectively), while the second resonance retained its multiplicity. This observation is attributed to scalar coupling between the hydride and 107Ag/109Ag nuclei (JAgH = 75.6 for 3 and 78.9 Hz for 4) and indicates that the two hydride moieties have an asymmetric configuration, where one is interacting with 31P via the Au centre and the other is bound to Ag only. Most likely, this configuration is highly fluxional, leading to averaging in the 1H NMR spectrum at higher temperatures. Consistently, also the Cp and the ortho-H of the 4-tert-butyl-aryl ring split into two different resonances at 193 K (see the ESI†), confirming that the reaction with Ag+ removes the symmetry of the bridging hydrides.
Single crystals of 3 were obtained by slow diffusion of pentane into a concentrated dichloromethane solution. The X-ray diffraction analysis confirmed the assignment proposed by NMR and showed that an Ag+ cation binds both Au and W at the same time, with Ag⋯Au and Ag⋯W distances of 2.7150(9) and 3.090(1) Å, respectively. The original linearity of the P–Au–W arrangement is not perturbed, with a P1–Au1–W1 angle of 175.63(6)°. The silver cation also establishes a weak interaction with a [BF4]− anion (d(Ag2–F1) = 2.532(8) Å) and a phenyl ring of the ligand (d(Ag2–C11) = 2.65(1) Å and d(Ag2–C12) = 2.45(1) Å). Evidently, the presence of the dangling diphenyl ring acts as a stabilizing factor for Ag coordination, showcasing again the unique properties of the cavity-shaped ligand L1.13
The presence of the three metals in close proximity makes locating the hydride moieties by X-ray diffraction extremely difficult. For this reason, we performed a conformational analysis starting from the X-ray structures (see the ESI† for details). Taking into account the NMR fingerprints, we hypothesized two possible scenarios (Fig. 5): (i) an outside approach of the Ag atom to the bimetallic core, where the pre-existing Au(μ-H)W bridging hydride interactions are not broken or (ii) an inside approach where Ag inserts into one of the Au bridging hydride bonds. Our calculations suggest that the inside structure is more stable by over 5.6 kcal mol−1 (Fig. 5), and therefore we propose that the new trimetallic species 3 and 4 arise from the insertion of an Ag cation into one bridging hydride, in full agreement with the observed spectroscopic pattern.
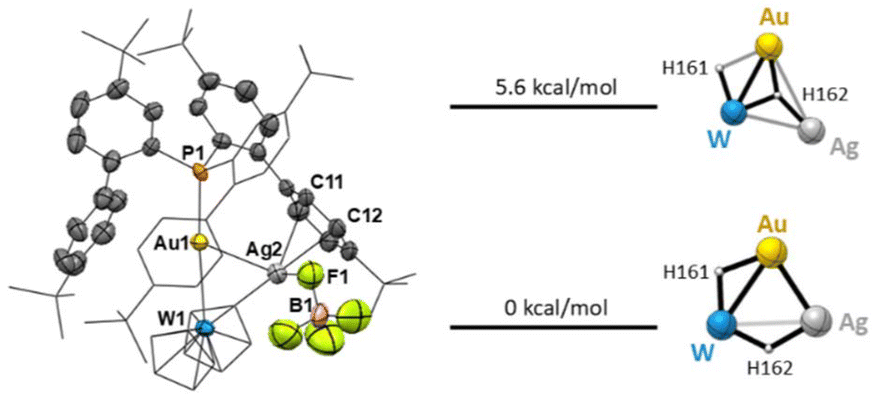 | ||
| Fig. 5 Left: molecular structure of 3. Hydrogen atoms, solvent molecules and anions are omitted for clarity. Thermal ellipsoids are set at 50% probability. Selected bond lengths [Å] and angles [°]: Au1–Ag2: 2.7150(9), W1–Ag2: 3.090(1), Au1–W1: 2.8541(5), Au1–P1: 2.311(2), Ag2–F1: 2.532(8), Ag2–C11: 2.65(1), Ag2–C12: 2.45(1), P1–Au1–W1: 175.63(6), and Cpcentr–W2–Cpcentr: 138.28. Right: calculated conformational energies for the “outside” silver insertion (+5.6 kcal mol−1) or insertion into a bridging hydride bond (0 kcal mol−1) structure. | ||
Next we performed preliminary studies to assess the implications of silver insertion on the reactivity of 3 and 4 compared to 1 and 2. In line with previous studies, 1 and 2 were unreactive towards H2 (or D2), MeCN, pyridine (py), C2H4, CO and CO2 under relatively mild conditions (25 °C; 1 equiv. or 1 bar). On the other hand, trimetallic species 3 and 4 are much more reactive (Fig. 6), even though no bimetallic cooperativity could be triggered. In particular, the addition of H2 (1 bar) to a CD2Cl2 solution of 3 or 4 leads to the regeneration of 1 or 2, along with traces of (L1)AuCl and other unidentified species. In the case of 1, the formation of [Cp2WH3][BF4] was observed, suggesting that a heterolytic H2 splitting pathway may be active (Fig. S36, ESI†). However, no gold hydride was intercepted. The addition of an excess of acetonitrile regenerates instead of [(L1)AuNCMe][BF4] (Fig. S39, ESI†) and other bimetallic Ag/M species that undergo degradation.
 | ||
| Fig. 6 Reactivity overview of trimetallic bridging hydrides (3 and 4). | ||
In the case of pyridine, 3 and 4 react with a stoichiometric amount of the base, affording [(L1)Au(py)][BF4] (5) and [pyH][BF4] (δH = 14.48 ppm), possibly arising from partial hydride oxidation (Fig. S40 and S41, ESI†).14 Finally, 3 and 4 also react with C2H4 and CO, affording the known gold(I) π-C2H4 or CO adducts (Fig. S43 and S45, ESI†). In the case of ethylene, small traces of an aqueous silver complex [(L1)Ag(H2O)]+ (6) were also found, probably due to the presence of adventitious water. Overall, these studies demonstrate the activating effect of the occluded silver atom within the bimetallic structures 1 and 2, offering an alternative approach for increasing the reactivity of otherwise inert bimetallic structures.
In summary, we have synthesized and characterized heterobimetallic bridging hydride compounds of the type [(L1)Au(μ-H)2MCp2][BF4] (M = W(1) and Mo(2)) by using the exceptionally bulky tris-2(4,4′-ditert-butylbiphenylyl)phosphine ligand. These complexes proved highly reactive under UV light irradiation, with hydride transfer reaction rates that are 10 times faster than previous NHC-based analogues. Besides, the addition of AgBF4 affords exotic trimetallic adducts upon silver insertion into one bridging hydride. These trimetallic species unlocked the reactivity towards small molecules of the otherwise inert bimetallic adducts, paving the way for the generation of functional multimetallic cooperative systems beyond two metals.
Data availability
All data are available within the manuscript and ESI.†Crystallographic data for 1, 2, 3, 5 and [Ag(py)2][SbF6] have been deposited at the CCDC under 2392134–2392138.†
All other datasets supporting this article have been uploaded as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. R. acknowledges the financial support from the University of Perugia (Fondo Ricerca di Base di Ateneo 2022), the Royal Society of Chemistry (R21-1292108967), and the European Union–NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041-VITALITY. J. C. acknowledges the European Research Council (ERC Starting Grant, CoopCat, Project 756575) and the Spanish Ministry of Science and Innovation (PID2022-139782NB-I00). M. N. acknowledges Junta de Andalucía for the post-doctoral program (DOC_00149).References
- A. Maity and T. S. Teets, Chem. Rev., 2016, 116, 8873–8911 CrossRef CAS PubMed.
- (a) H. Lehner, D. Matt, P. S. Pregosin, L. M. Venanzi and A. Albinati, J. Am. Chem. Soc., 1982, 104, 6825–6827 CrossRef CAS; (b) B. D. Alexander, B. J. Johnson, S. M. Johnson, P. D. Boyle, N. C. Kann, A. M. Mueting and L. H. Pignolet, Inorg. Chem., 1987, 26, 3506–3513 CrossRef CAS; (c) B. D. Alexander, M. P. Gomez-Sal, P. R. Gannon, C. A. Blaine, P. D. Boyle, A. M. Mueting and L. H. Pignolet, Inorg. Chem., 1988, 27, 3301–3308 CrossRef CAS.
- (a) A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS; (b) L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364–8451 CrossRef CAS PubMed.
- A. Albinati and L. M. Venanzi, Coord. Chem. Rev., 2000, 200–202, 687–715 CrossRef CAS.
- (a) M. J. Butler and M. R. Crimmin, Chem. Commun., 2016, 53, 1348–1365 RSC; (b) A. Hicken, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2017, 56, 15127–15130 CrossRef CAS.
- (a) L. Rocchigiani, W. T. Klooster, S. J. Coles, D. L. Hughes, P. Hrobárik and M. Bochmann, Chem. – Eur. J., 2020, 26, 8267–8280 CrossRef CAS; (b) M. Landrini, E. De Paolis, A. Macchioni, L. Tensi, P. Hrobárik and L. Rocchigiani, Eur. J. Inorg. Chem., 2022, e202200373 CrossRef CAS.
- M. Landrini, R. Patel, J. Tyrrell-Thrower, A. Macchioni, D. L. Hughes, L. Tensi, P. Hrobárik and L. Rocchigiani, Inorg. Chem., 2024, 63, 13525–13545 CrossRef CAS.
- (a) R. J. Harris and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2014, 53, 9369–9371 CrossRef CAS PubMed; (b) N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 5982–5993 CrossRef CAS PubMed.
- (a) M. Navarro, J. Miranda-Pizarro, J. J. Moreno, C. Navarro-Gilabert, I. Fernández and J. Campos, Chem. Commun., 2021, 57, 9280–9283 RSC; (b) C. L. Johnson, D. J. Storm, M. A. Sajjad, M. R. Gyton, S. B. Duckett, S. A. Macgregor, A. S. Weller, M. Navarro and J. Campos, Angew. Chem., 2024, 136, e202404264 CrossRef.
- M. Navarro, M. Holzapfel and J. Campos, Inorg. Chem., 2023, 62, 10582–10591 CrossRef CAS.
- J. Miranda-Pizarro, Z. Luo, J. J. Moreno, D. A. Dickie, J. Campos and T. B. Gunnoe, J. Am. Chem. Soc., 2021, 143, 2509–2522 CrossRef CAS PubMed.
- D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012–9019 CrossRef CAS PubMed.
- (a) P. Dotta, P. G. A. Kumar, P. S. Pregosin, A. Albinati and S. Rizzato, Organometallics, 2004, 23, 4247–4254 CrossRef CAS; (b) M. Yamashita, I. Takamiya, K. Jin and K. Nozaki, J. Organomet. Chem., 2006, 691, 3189–3195 CrossRef CAS.
- (a) Y. Zhang, T. J. Woods and T. B. Rauchfuss, J. Am. Chem. Soc., 2021, 143, 10065–10069 CrossRef CAS; (b) J. Y. Yang, R. M. Bullock, W. J. Shaw, B. Twamley, K. Fraze, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 5935–5945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and analytical data, and NMR, X-ray and computational data. CCDC 2392134–2392138. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03150d |
| This journal is © The Royal Society of Chemistry 2025 |