
Ring-opening polymerization of ε-caprolactone with a macrocyclic tetracarbene indium complex†
Henry R.
Brothers
a,
Raju
Chambenahalli
 b,
Gary S.
Nichol
b,
Jennifer A.
Garden
*b and
David M.
Jenkins
*a
b,
Gary S.
Nichol
b,
Jennifer A.
Garden
*b and
David M.
Jenkins
*a
aDepartment of Chemistry, The University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: jenkins@ion.chem.utk.edu
bSchool of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3FJ, UK. E-mail: J.Garden@ed.ac.uk
First published on 17th December 2024
Abstract
The first chiral tetracarbene indium(III) complexes have been synthesized by employing a rigid dianionic macrocyclic tetra-NHC ligand. The macrocyclic indium tetra-NHC bromide and ethoxide complexes are structurally similar to analagous salen complexes. The indium ethoxide complex effectively promotes living ring-opening polymerization of ε-caprolactone at room temperature.
Neutral auxiliary ligands that are ubiquitous for transition-metal catalysts, such as N-heterocyclic carbenes (NHCs),1,2 have found limited application in p-block metal catalysis.3,4 Main group metals’ preference for ligands with anionic character has limited NHCs to a single donor ligand (as a Lewis base) in most cases,5–7 which has curtailed utilization of polydentate NHCs as supporting ligands.8 Indeed, to circumvent this mismatch, two primary strategies have been adopted to include an anionic component to the NHCs. Preparation of NHC ligand precursors which contain hydroxyl or amino functionality gives rise to main group metal–NHC complexes with anionic linkages to the NHC motif via alkoxy or amido tethers, respectively.9,10 Alternatively, inclusion of borate groups within NHC ligand precursors is an effective strategy towards main group metal–NHC complexes which are stabilized by the anionic contributions from distal borate moieties instead of anionic tethers.11,12 While these two strategies have been effectively employed for metals such as germanium and tin, there are very few reports of tethered or borate-containing NHCs on group 13 metals such as gallium,10,12 indium,11 or thallium.
This lack of effective NHC ligands for indium is disappointing since indium catalysis has been demonstrated to be effective and selective for organic transformations such as directed carbonyl–ene reactions,13 catalytic alcohol dehydration for SN1 reactions,14 and catalytic alkyne hydrofunctionalization reactions.15 Beyond these molecular processes, indium catalysis has particularly shined in the development of ring opening polymerization processes, especially for the polymerization of cyclic esters.16,17 Recently, the Williams,6,18 Rieger,19 Carpentier,20,21 and Mehrkhodavandi22–24 research groups have all independently developed effective indium catalysts for synthesis of polylactic acid (PLA) or polycaprolactone (PCL). Notably, the auxiliary ligands for these systems are often Schiff bases that are based on a tetradentate salen-type motif.
Our group has developed extensive chemistry for anionic tetradentate NHC ligands which are structurally analogous to salen-type ligands.25,26 These ligands are highly effective at supporting iron catalysts for catalytic C2 + N1 aziridination reactions.27 Recently, we reported an approach to synthesize a D2-symmetric variant of this ligand with enhanced structural rigidity.28 However, to our knowledge no anionic borate-based NHCs have been employed for catalysis with p-block metals.
In this manuscript, we showcase the syntheses of stable indium complexes supported by a chiral anionic tetracarbene macrocyclic ligand. The absolute stereochemistries of the complexes were confirmed by single crystal X-ray diffraction. An indium complex with ethoxide as an axial ligand efficiently promotes living polymerization of ε-caprolactone at room temperature.
Results and discussion
We have previously described the synthesis of the chiral macrocyclic ligand ((S,S)-1,2-Cy,BMe2TCH), 1, and its iron and palladium complexes.28 To prepare the indium complex of 1, we employed a similar ligation strategy. Compound 1 was deprotonated with nBuLi at −35 °C in THF before addition of indium(III) bromide at room temperature (Scheme 1). Repeated extraction and filtration of the crude product with benzene efficiently removes residual lithium bromide salts. ((S,S)-1,2-Cy,BMe2TCH)InBr, 2, was crystallized by diluting a concentrated benzene solution with n-pentane, affording colorless needles of 2 in 43% yield. Unlike our previously reported tetracarbene complex of indium, 2 is stable in air free environments, which is likely due to the improved steric protection of the chiral pocket.11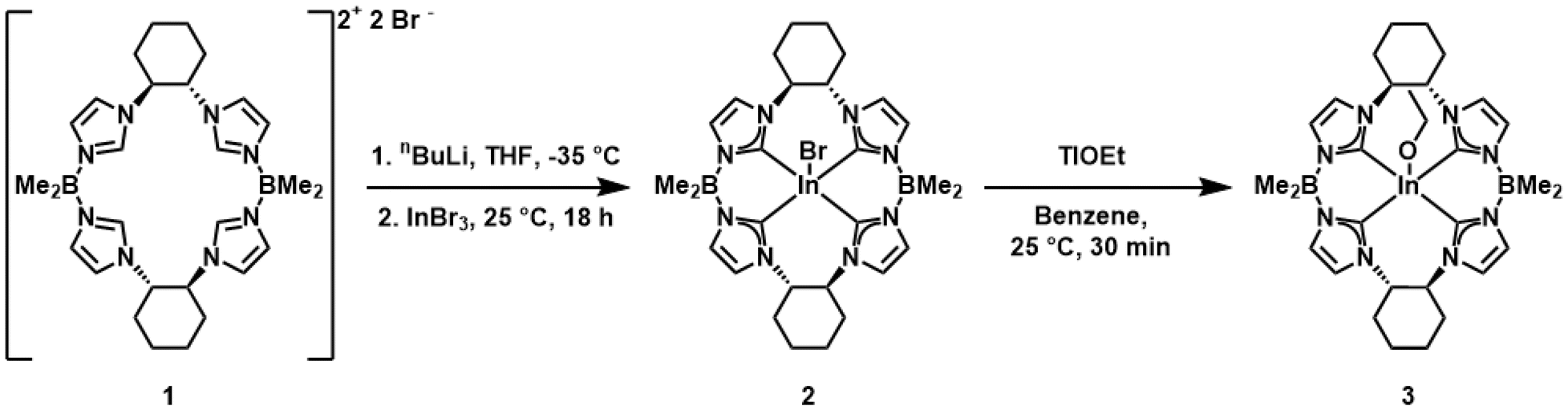 | ||
| Scheme 1 Synthesis of indium complexes 2 and 3. | ||
The 1H NMR spectrum for 2 shows a C2-symmetric complex with resonances broadly similar to square planar ((S,S)-1,2-Cy,BMe2TCH)Pd (Fig. S1†).28 Two separate 1H NMR signals are observed for protons bound to the chiral carbons and there are four vicinal doublets that correspond to the imidazolylidene protons. Like similar diamagnetic five coordinate tetracarbene complexes that we have reported previously, the protons from the methyl groups attached to the borate moieties are diastereotopic and locked into a single conformation.26,28,29 Finally, the 13C NMR spectrum for 2 gives two carbene resonances at 179.36 ppm and 178.15 ppm (Fig. S2†).28
Single crystal X-ray diffraction for ((S,S)-1,2-Cy,BMe2TCH)InBr shows that the complex adopts a slightly distorted square pyramidal geometry (Fig. 1A). The In–C bond distances are inequivalent with In–C1 (2.20(2) Å) and In–C3 (2.20(2) Å) being shorter than In–C2 (2.29(2) Å) and In–C4 (2.28(2) Å). Likewise, the trans angles across the macrocycle are also not equivalent, where C1–In–C3 is 153.3(7)° while C2–In–C4 is 140.0(8)°. These tight angles move the indium atom out of the macrocycle's plane much more than the achiral variant, (Et,BMe2TCH)InBr, which we previously reported.11
 | ||
| Fig. 1 X-Ray crystal structures of 2 (A) and 3 (B). Purple, olive, blue, orange, red, grey, and white ellipsoids (50% probability) represent indium, boron, nitrogen, bromine, oxygen, carbon, and hydrogen atoms, respectively. Solvent molecules and H-atoms on non-stereogenic carbons are omitted for clarity. One conformer from the asymmetric unit cell of both 2 and 3 are shown. Selected bond distances (Å) and angles (°) are as follows: for 2, In1–C1, 2.20(2); In1–C2, 2.29(2); In1–C3, 2.20(2); In1–C4, 2.28(2); In1–Br1, 2.583(3); C1–In1–C3, 153.3(7); C2–In1–C4, 140.0(8). For 3, In1–C1, 2.326(2); In1–C2, 2.221(2); In1–C3, 2.343(2); In1–C4, 2.235(2); In1–O1, 2.07(1); C1–In1–C3, 145.73(6); C2–In1–C4, 149.16(6). | ||
We were interested in synthesizing an indium alkoxide complex since alkoxide ligands are effective initiators for ring-opening polymerization of cyclic esters.6,18 Since 2 bears a single axial bromide ligand, we thought that ligand metathesis could allow for preparation of a macrocyclic tetra-NHC indium(III) alkoxide complex. Indeed, treating a benzene solution of 2 with thallium ethoxide resulted in immediate precipitation of pale yellow TlBr salt and afforded ((S,S)-1,2-Cy,BMe2TCH)In(OEt), 3, in 67% yield after crystallization (Scheme 1).
The NMR spectra for 3 are similar to 2, but feature additional resonances related to the ethoxide ligand, with diagnostic 1H NMR resonances at 4.01 and 1.23 ppm for the ethoxide ligand (Fig. S6†). Furthermore, the triplet-of-doublet resonances related to diastereotopic protons on the stereogenic cyclohexane carbons are shifted upfield to 6.00 and 4.21 ppm relative to those in the 1H NMR spectrum for 2 (6.39 and 4.31 ppm). The 13C NMR spectrum for 3 gives two carbene carbon resonances at 181.77 ppm and 180.39 ppm in addition to two new resonances at 63.14 and 23.65 ppm which correspond to ethoxide carbons (Fig. S7†).
Crystals of suitable quality for SCXRD were obtained via vapor diffusion of n-pentane into a benzene solution of 3. Like 2, a distorted square pyramidal geometry is observed in the crystal structure for 3 with C1–In–C3 as 145.73(6)° while C2–In–C4 is 149.16(6)° (Fig. 1B). The In–CNHC bond lengths for 3 between 2.221(2) Å and 2.343(2) Å are longer compared to those for 2. The In–O1 bond distance (2.07(1) Å) is comparable to the few other examples of monomeric indium alkoxides.6,30
Ring opening polymerization (ROP) of ε-caprolactone (CL) promoted by 2 and 3 was examined to determine the effectiveness of 1 as an auxiliary ligand versus similarly structured tetradentate ligands.18,19,24 As such, the reactivities of complexes 2 and 3 with CL were tested under conditions similar to previous reported methods: 1 mmol CL, 0.01 mmol complex (7.5 mM, benzene), at 25 °C. The PCL products were analyzed using size exclusion chromatography (SEC). Reaction kinetics were monitored by quenching aliquots at specified time intervals before 1H NMR analysis.
Synthesis of PCL was attempted first with 2 as initiator. These reactions quickly produced extremely viscous mixtures that gave inconsistent kinetic data and afforded PCL products with unexpectedly high molecular weights and high dispersity values. These observations suggest poor initiation efficiency from the In–Br unit, which affects poor molecular weight control over PCL products.31 These results prompted the synthesis of 3 so that an efficient and unambiguous initiator for CL ROP could be prepared.
When complex 3 was used as the initiator for CL ROP, kinetic experiments showed rapid consumption of CL consistent with a first-order reaction rate (Fig. 2A), albeit with a slight induction period. The linear correlation between the polymer number-averaged molecular weight (Mn) and monomer conversion suggests that 3 promotes living CL ROP (Fig. 2B and Table S1†). Strict molecular weight control was maintained over the PCL products that were made using 3; dispersity values were narrowed from 1.24 to 1.08 as monomer conversion increased, which indicates single site activation (Fig. 2B). Efficient, living CL ROP by 3 is further evidenced by the monomodal molecular weight distributions observed throughout the reaction (Fig. 2C).
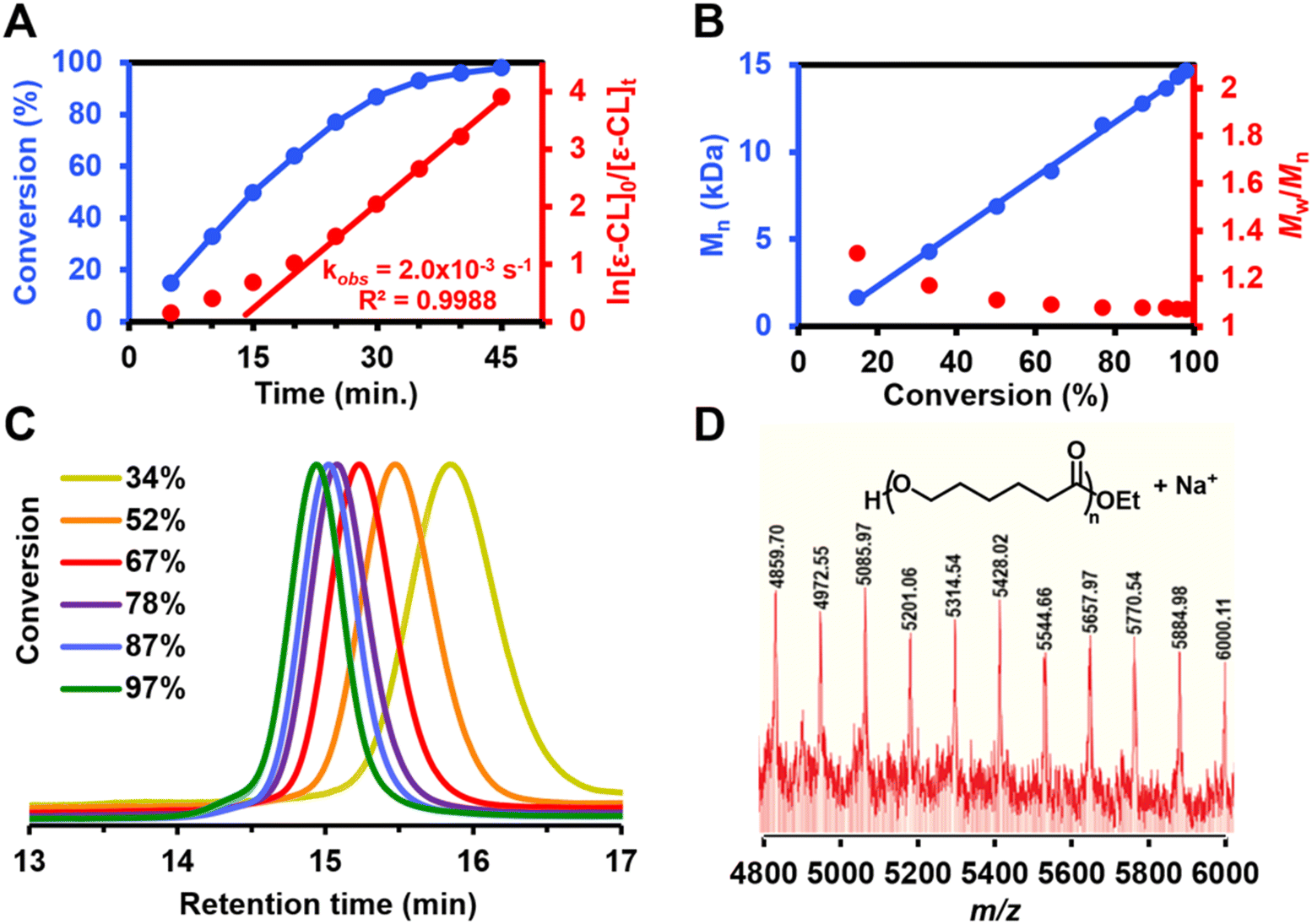 | ||
Fig. 2 (A) Kinetic plots for polymerization of ε-caprolactone (CL) using 3 as initiator ([initiator]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[CL] = 1:100, [M] = 0.075 M in benzene). (B) Molecular weight (Mn,SEC) and dispersity correlation with CL conversion for polycaprolactone (PCL) synthesized using 3. (C) SEC traces for PCL synthesized using 3 at different CL conversions. (D) MALDI-TOF mass spectrum of PCL synthesized using 3. :[CL] = 1:100, [M] = 0.075 M in benzene). (B) Molecular weight (Mn,SEC) and dispersity correlation with CL conversion for polycaprolactone (PCL) synthesized using 3. (C) SEC traces for PCL synthesized using 3 at different CL conversions. (D) MALDI-TOF mass spectrum of PCL synthesized using 3. | ||
Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF-MS) analysis of PCL samples prepared using 3 showed only one series, which was identified as PCL with ethoxy end groups (Fig. 2D). The identity of the end groups is further evidenced in the 1H NMR spectrum for purified PCL samples prepared using 3 (Fig. S12†), which gives signals of requisite integration as a quartet (4.20 ppm) and a triplet (1.32 ppm). These signals are consistent with reported spectra for PCL produced with metal ethoxide initiators.32,33 Considering the first-order reaction kinetics, narrow molecular weight distribution, and observed end group fidelity, we propose an ethoxy-initiated coordination–insertion mechanism for CL ROP with 3 as initiator (Fig. 3).32,33
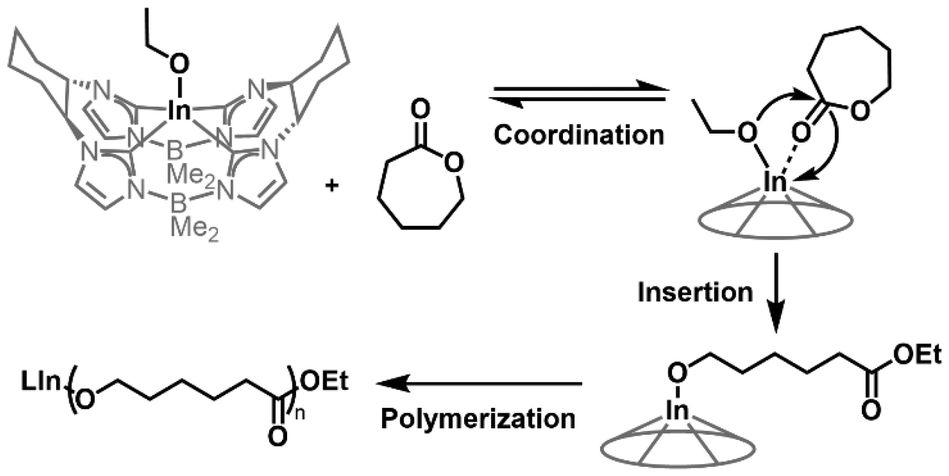 | ||
| Fig. 3 Proposed coordination–insertion mechanism for CL ROP promoted by 3. | ||
Conclusions
We have synthesized two C2-symmetric indium complexes that are supported by chiral macrocyclic tetracarbenes. These complexes were characterized by multinuclear NMR, HRMS, and SCXRD. Switching from a bromide to an ethoxide axial ligand enabled living polymerization of ε-caprolactone at room temperature with preserved ethoxy end groups. These results highlight a new platform for indium catalysis that is not based on salen/half-salen derivatives for CL ROP, but instead on NHCs as supporting ligands. This study increases the diversity of ligand structures that can be used to support In-catalysed CL polymerisation. Future research will focus on developing other cyclic ester polymerizations that take advantage of the chiral pocket on these main group complexes.Author contributions
H. R. B. synthesized the ligands and indium complexes and conducted polymerization reactions. H. R. B. conducted NMR studies on all molecules, including kinetic studies on polymers, and SCXRD analysis for 3. R. C. conducted polymer characterization including SEC measurements and MALDI-TOF MS. G. S. N. conducted SCXRD analysis for 2. J. A. G. and D. M. J. designed and supervised the project. The manuscript was written by H. R. B. and D. M. J. with additional editing provided by R. C. and J. A. G.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. R. B. and D. M. J. thank the National Science Foundation (NSF CHE-2154697) for support. Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. R. C. and J. A. G. gratefully acknowledge the EPSRC (EP/V049003/1 and EP/X028208/1), and J. A. G. also thanks the UKRI Future Leaders Fellowship (MR\T042710\1) and the Royal Society (Royal Society (RSG\R1\180101)).References
- G. G. Zámbó, J. F. Schlagintweit, R. M. Reich and F. E. Kühn, Catal. Sci. Technol., 2022, 12, 4940–4961 RSC.
- C. Romain, S. Bellemin-Laponnaz and S. Dagorne, Coord. Chem. Rev., 2020, 422, 213411 CrossRef.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef.
- S. Goswami, P. Mandal, S. Sarkar, M. Mukherjee, S. Pal, D. Mallick and D. Mukherjee, Dalton Trans., 2024, 53, 1346–1354 RSC.
- I. Jain and P. Malik, Eur. Polym. J., 2021, 150, 110412 CrossRef.
- N. Yuntawattana, T. M. McGuire, C. B. Durr, A. Buchard and C. K. Williams, Catal. Sci. Technol., 2020, 10, 7226–7239 RSC.
- C. Fliedel, G. Schnee, T. Avilés and S. Dagorne, Coord. Chem. Rev., 2014, 275, 63–86 CrossRef.
- A. Neshat, P. Mastrorilli and A. Mousavizadeh Mobarakeh, Molecules, 2022, 27, 95 CrossRef.
- P. L. Arnold and A. C. Scarisbrick, Organometallics, 2004, 23, 2519–2521 Search PubMed.
- P. Horeglad, O. Ablialimov, G. Szczepaniak, A. M. Dabrowska, M. Dranka and J. Zachara, Organometallics, 2014, 33, 100–111 CrossRef.
- S. A. Cramer, F. L. Sturgill, P. P. Chandrachud and D. M. Jenkins, Dalton Trans., 2014, 43, 7687–7690 RSC.
- L. P. Ho, L. Anders and M. Tamm, Chem.– Asian J., 2020, 15, 845–851 CrossRef PubMed.
- J.-F. Zhao, H.-Y. Tsui, P.-J. Wu, J. Lu and T.-P. Loh, J. Am. Chem. Soc., 2008, 130, 16492–16493 Search PubMed.
- M. Yoshimatsu, H. Goto, R. Saito, K. Iguchi, M. Kikuchi, H. Wasada and Y. Sawada, Commun. Chem., 2023, 6, 279 CrossRef PubMed.
- A. Da Lama, J. Pérez Sestelo, L. A. Sarandeses and M. M. Martínez, J. Org. Chem., 2024, 89, 16015–16021 CrossRef PubMed.
- E. Fazekas, P. A. Lowy, M. Abdul Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC.
- D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 CrossRef PubMed.
- J. Bruckmoser, D. Henschel, S. Vagin and B. Rieger, Catal. Sci. Technol., 2022, 12, 3295–3302 RSC.
- M. Normand, V. Dorcet, E. Kirillov and J.-F. Carpentier, Organometallics, 2013, 32, 1694–1709 CrossRef.
- S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 CrossRef.
- C. Goonesinghe, H.-J. Jung, H. Roshandel, C. Diaz, H. A. Baalbaki, K. Nyamayaro, M. Ezhova, K. Hosseini and P. Mehrkhodavandi, ACS Catal., 2022, 12, 7677–7686 CrossRef.
- H.-J. Jung, K. Nyamayaro, H. A. Baalbaki, C. Goonesinghe and P. Mehrkhodavandi, Inorg. Chem., 2023, 62, 1968–1977 CrossRef PubMed.
- I. Yu, A. Acosta-Ramírez and P. Mehrkhodavandi, J. Am. Chem. Soc., 2012, 134, 12758–12773 CrossRef CAS PubMed.
- J. F. DeJesus, R. W. Kerr, D. A. Penchoff, X. B. Carroll, C. C. Peterson, P. L. Arnold and D. M. Jenkins, Chem. Sci., 2021, 12, 7882–7887 RSC.
- M. R. Anneser, G. R. Elpitiya, J. Townsend, E. J. Johnson, X. B. Powers, J. F. DeJesus, K. D. Vogiatzis and D. M. Jenkins, Angew. Chem., 2019, 131, 8199–8202 CrossRef.
- P. P. Chandrachud, H. M. Bass and D. M. Jenkins, Organometallics, 2016, 35, 1652–1657 CrossRef.
- J. F. DeJesus and D. M. Jenkins, Chem. – Eur. J., 2020, 26, 1429–1435 CrossRef PubMed.
- M. R. Anneser, G. R. Elpitiya, X. B. Powers and D. M. Jenkins, Organometallics, 2019, 38, 981–987 CrossRef.
- D. C. Aluthge, J. M. Ahn and P. Mehrkhodavandi, Chem. Sci., 2015, 6, 5284–5292 RSC.
- J. W. J. Hughes, D. J. Babula, F. Stowers-Veitch, K. Yuan, M. Uzelac, G. S. Nichol, M. J. Ingleson and J. A. Garden, Dalton Trans., 2023, 52, 17767–17775 RSC.
- F. Peprah, G. E. Tarantola, A. S. Plaman, E. L. Vu, A. B. Huynh and C. B. Durr, Dalton Trans., 2024, 53, 7073–7080 RSC.
- A. Buchard, M. G. Davidson, G. Gobius du Sart, M. D. Jones, G. Kociok-Köhn, S. N. McCormick and P. McKeown, Inorg. Chem., 2023, 62, 15688–15699 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and characterization for all compounds is provided, including NMR spectra, IR spectra, UV-Vis spectra, and X-ray diffraction data. CCDC 2401889 and 2401890. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03198a |
| This journal is © The Royal Society of Chemistry 2025 |