
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Push–pull effect – how to effectively control photoinduced intramolecular charge transfer processes in rhenium(I) chromophores with ligands of D–A or D–π–A structure†
Katarzyna
Choroba
 *,
Joanna
Palion-Gazda
,
Anna
Kryczka
,
Ewa
Malicka
and
Barbara
Machura
*
*,
Joanna
Palion-Gazda
,
Anna
Kryczka
,
Ewa
Malicka
and
Barbara
Machura
*
Institute of Chemistry, Faculty of Science and Technology, University of Silesia, Szkolna 9, 40-006 Katowice, Poland. E-mail: barbara.machura@us.edu.pl; katarzyna.choroba@us.edu.pl
First published on 7th January 2025
Abstract
Over the last five decades, diimine rhenium(I) tricarbonyl complexes have been extensively investigated due to their remarkable and widely tuned photophysical properties. These systems are regarded as attractive targets for design functional luminescent materials and performing fundamental studies of photoinduced processes in transition metal complexes. This review summarizes the latest developments concerning Re(I) tricarbonyl complexes bearing donor–acceptor (D–A) and donor–π–acceptor (D–π–A) ligands. Such compounds can be treated as bichromophoric systems with two close-lying excited states, metal-to-ligand charge transfer (MLCT) and intraligand-charge-transfer (ILCT). A role of ILCT transitions in controlling photobehaviour was discussed for Re(I) tricarbonyls with six different diimine cores decorated by various electron-rich amine, sulphur-based and π-conjugated aryl groups. It was evidenced that this approach is an effective tool for enhancement of the visible absorptivity, bathochromic emission shift and significant prolongation of the excited-state, opening up new possibilities in the development of more efficient materials and expand the range of their applications.
1. Background and introduction
Since the pioneering work of Wrighton and Morse in the middle of the 1970s,1 rhenium(I) tricarbonyl complexes with chelating α-diimine (N∩N) ligands have occupied a prominent position among photoluminescent molecular materials. The convenient and easy synthesis, thermal and photochemical stability, and outstanding photophysical behaviour make complexes [ReL(CO)3(N∩N)]n (L = ancillary ligand, n = 0 or 1+) appealing for employment in optoelectronics,2–8 photocatalysis9–18 and life science.19–40 The photophysical parameters of these systems, including the visible absorptivity, emission energy, quantum yield and triplet excited state lifetime, are adjusting to requirements of their potential applications by proper structural modifications of α-diimine core and alteration of the ancillary ligand.Along with promising functional features, Re(I) tricarbonyl complexes are perfectly suited for fundamental studies of photoinduced processes whose precise control is a key to success of designing compounds with pre-defined photophysical parameters. In this regard, the great advantage of [ReL(CO)3(N∩N)]n systems is the existence in the coordination sphere of the single diimine ligand, allowing to avoid the problem of excited-electron localization, and presence of carbonyl ligands, acting as efficient IR markers of redistribution of electron density upon photoexcitation. Excited-state dynamics of [ReL(CO)3(N∩N)]n have been widely investigated using ultrafast experimental methods like time-resolved infrared spectroscopy, fluorescence up-conversion methods, time-resolved emission spectroscopy and transient absorption.41–52
As demonstrated in ref. 53–60, the diverse photophysical behaviour of [ReL(CO)3(N∩N)]n is attributed to different triplet exited state nature metal-to-ligand charge transfer (MLCT), ligand-to-ligand charge transfer (LLCT), ligand-centered (IL), intraligand-charge-transfer (ILCT) nature or their superposition, controlled by the structural and electronic features of α-diimine and ancillary ligands. The molecules in different excited states brings different emission characteristics.
In this article, we summarized the latest developments concerning Re(I) tricabonyl complexes bearing donor–acceptor (D–A) and donor–π–acceptor (D–π–A) ligands. The attachment of the organic push–pull chromophore to the {Re(CO)3}+ core can results in new photophysical properties owing to the dominant role of ILCT transitions, with the charge flow from the electron-donating organic group (D) to the accepting diimine core (A). The efficiency of intramolecular charge transfer in the coordinated D–A and D–π–A ligands may be widely modified by the donor and acceptor strengths of electron-accepting and electron-donating components in the molecular diimine skeleton, their electronic coupling, as well as the environment – solvent polarity, hydrogen bonding with polar solvents.61–63
Regarding functional photophysical parameters of resulting complexes [ReL(CO)3(N∩N)]n, the incorporation of D–A and D–π–A into the coordination sphere of Re(I) tricarbonyl complexes may be an effective tool for enhancement of the visible absorptivity, bathochromic emission shift and significant prolongation of the excited-state. Photoactive red or NIR light emitting transition metal complexes with extended excited state lifetimes are strongly desirable for the photodynamic therapy (PDT),24,25,33,64,65 time-resolved bioimaging,66 and triplet–triplet annihilation up-conversion (TTA UC) techniques.67–69 Understanding and precise controlling of long-lived charge-separated excited states in these systems still remains a major challenge and is crucial for designing improved functional materials.
To better illustrate the impact of the incorporated donor group on the photobehavior Re(I) carbonyl complexes, excited-state dynamics and photophysics of the most representative prototypical chromophores [ReCl(CO)3(N∩N)] with 2,2′-bipyridine (bipy), 1,10-phenanthroline (phen), dipyrido[3,2-a:2′,3′-c]phenazine (dppz), imidazo[4,5-f][1,10]phenanthroline (imphen), 2-(2-pirydyl)benzimidazole (pybimd) and 2-(1-benzyl-1H-1,2,3-trizol-4-yl)pyridine (pytri-Bn) are firstly discussed. In the next sections, we concentrate on the optical properties and photoinduced processes of [ReL(CO)3(N∩N)]n bearing these diimine π-acceptor ligands functionalized with electron-rich amine, sulphur-based and π-conjugated aryl groups. The final part presents the most appealing applications of Re(I) complexes with D–A or D–π–A diimine ligands.
2. Prototypical Re(I) chromophores
Over five decades of intensive investigations, 2,2′-bipyridine (bipy), 1,10-phenanthroline (phen), dipyrido[3,2-a:2′,3′-c]phenazine (dppz), imidazo[4,5-f][1,10]phenanthroline (imphen), 2-(2-pirydyl)benzimidazole (pybimd) and 2-(1-benzyl-1H-1,2,3-trizol-4-yl)pyridine (pytri-Bn) and their derivatives have become the most recognizable and most extensively studied diimine ligands in the Re(I) coordination chemistry. As 2,2′:6′,2′′-terpyridine (terpy) may coordinate to the metal center of the {Re(CO)3}+ core in a bidentate mode and form [ReL(CO)3(terpy-κ2N)]n systems analogues to that with bipy, it has also been taken into considerations in this section (Scheme 1).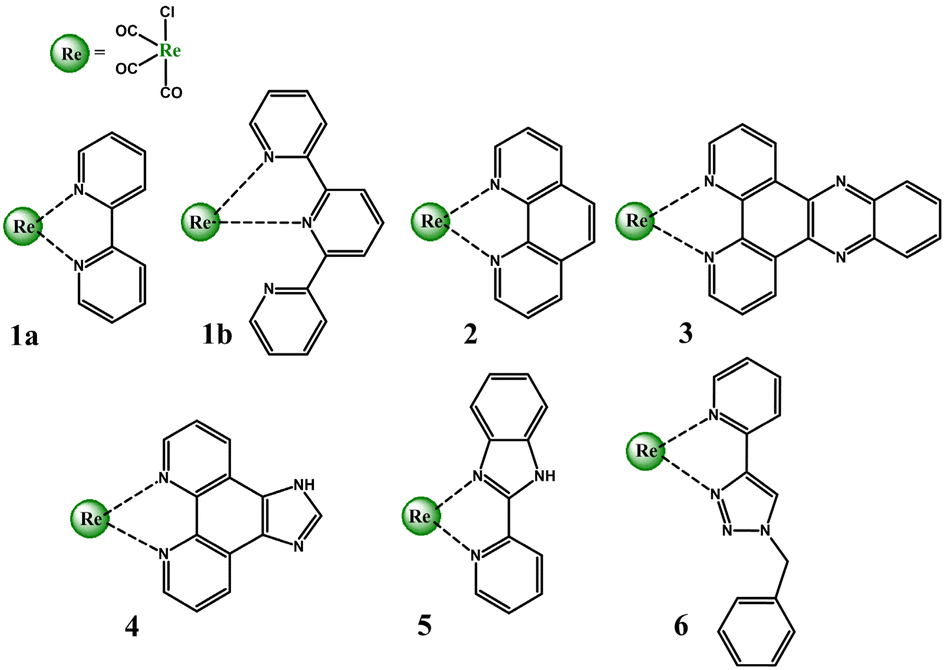 | ||
| Scheme 1 Re(I) tricabonyl complexes discussed in this section. | ||
Possessing low lying π* orbitals, bipy, terpy, phen, dppz, imphen, pybimd and pytri-Bn act as π-acceptor ligands upon attachment to the electron-rich {Re(CO)3}+ core. For all complexes, [ReCl(CO)3(bipy)] (1a), [ReCl(CO)3(terpy-κ2N)] (1b) [ReCl(CO)3(phen)] (2), [ReCl(CO)3(dppz)] (3), [ReCl(CO)3(imphen)] (4), [ReCl(CO)3(pybimd)] (5) and [ReCl(CO)3(pytri-Bn)] (6), the LUMO resides on the diimine core, while the HOMO, H-1 and H-2 are contributed by dπ rhenium orbitals in a bonding relation to the carbonyl π* orbitals and an antibonding arrangement to the chlorine occupied p orbitals (Fig. 1). Regarding the LUMO distribution, a more detailed comment is required for Re(I) complexes 1b, 3, 4 and 6. The LUMO of 1b is delocalized over the two coordinated rings – the bipy-like moiety, as in 1a. In the case of 3 bearing dppz, which can be considered as a fusion of phenanthroline (phen) and phenazine (phz), the DFT calculations indicate a predominant contribution of the phz component in the lowest unoccupied molecular orbital. The phenanthroline π-antibonding orbitals participate in the higher lying LUMO+1. The LUMO of 4 is solely contributed by the phen core, while the higher in energy LUMO+1 is delocalized over the whole imphen ligand. As the triazole ring acts as an electronic insulator,70 the benzyl (Bn) molecular orbitals show no contribution in LUMO of 6, signalling that substituents introduced in this part of the pytri ligand have negligible impact on optical properties (Fig. S1 and S2†). As demonstrated in Fig. 1, the HOMO–LUMO gap of 1a, 1b, 2, 4 and 5 varies in a small range 3.88–3.91 eV. The introduction of dppz into the Re(I) coordination sphere results in a noticeable decrease in the HOMO–LUMO gap (3.51 eV), contrary to pytri-Bn which induces its increase (4.32 eV).
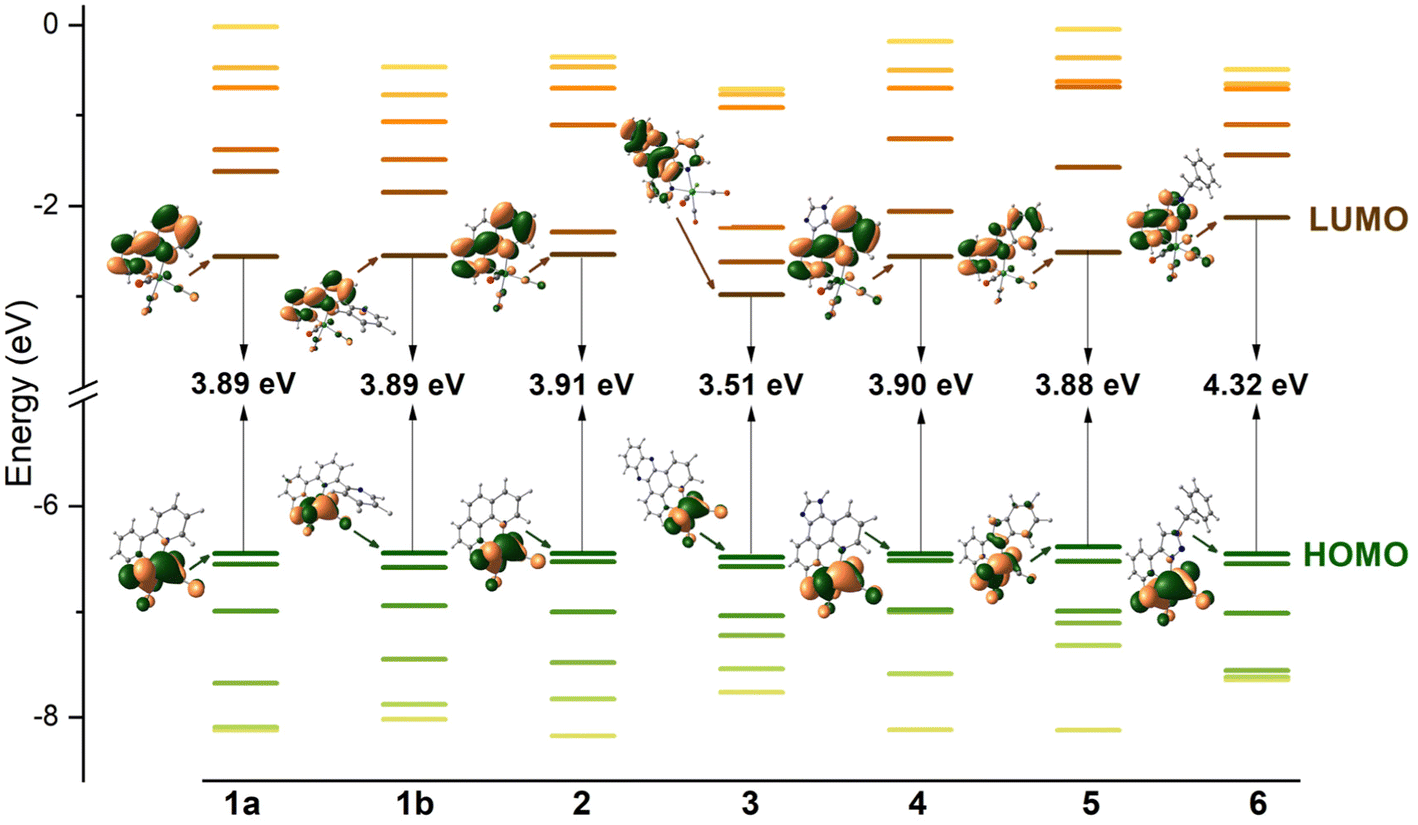 | ||
| Fig. 1 Frontier molecular orbitals energy diagram for complexes 1–6 along with electron density plots of the highest occupied and lowest unoccupied MOs. | ||
Theoretically assigned character of the frontier molecular orbitals of 1–6 was fully supported by electrochemical studies. For all complexes 1–6, the first oxidation wave is associated with the oxidation of metal centre Re(I/II), while the reduction occurs in the diimine core (Table S1†). For complex 3, the reduction potential is much closer to that of phz than that of bipy or phen, supporting the involvement of phz MO orbitals in the reduction process.71
Based on the frontier molecular energy diagram, an electron can be promoted from the dπ(Re) into π* diamine orbitals, giving rise to MLCT transitions, which determine the optical properties of these systems. For complex 3, the lowest singlet excited state is of MLCTphz nature.44,72,73 Due to participation of the halogen orbitals in the highest occupied molecular orbitals, the first excited state of 1–6 may also be classified as MLLCT (metal–ligand-to-ligand charge transfer). However, regarding that MLCT predominates over XLCT in chloride diamine Re(I) tricarbonyls,41,46,74 the simplified notation is used in this article.
All complexes 1–6 are characterized by the weak absorptivity in the region 320–400 nm covering 1MLCT spin-allowed transitions (Table S2†). The reason of their weak absorptivity is the poor spatial overlap of HOMOs contributed by dπ metal orbitals and LUMO localized on π* diimine molecular orbitals. In most cases, the MLCT absorption lies at low energy edge of the intense band attributed to π–π* intraligand (IL) transitions.
As demonstrated by fluorescence up-conversion (FlUC), UV-Vis transient absorption (TA), or time-resolved infrared (TRIR) spectroscopies,43,45,47,50–52,75,76 the optically populated 1MLCT state of 1a, 1b, 2 and 4 undergoes the efficient intersystem crossing (ISC) in tens of femtoseconds, populating an interligand-localized excited state (3IL) and vibrationally hot 3MLCT excited states. On a picosecond timescale, depending on the solvent and ancillary ligand, 3IL is converted into the 3MLCT excited state. The vibrationally relaxed 3MLCT state decays then to the ground state. TA spectra of these systems are characterized by intense band in the near UV region due to π–π* transitions localized at the formally reduced diimine ligand and the broad excited state absorption (ESA) across the entire visible region (400–650 nm) originating from Cl/diamine˙− → Re (ligand-to-metal charge-transfer, LMCT) triplet–triplet transitions. Consistent with the removal of electron density from the π* antibonding orbital of the carbonyl groups after the MLCT photoexcitation, the TRIR spectra of 1a and 2 show a shift of νCO to higher energies, with the magnitude of the positive νCO band shifts of 40–60 cm−1.41,57,77 For complexes 5 and 6, the formation of 3MLCT excited state was postulated on the basis of steady-state and time-resolved spectroscopic data accompanied with theoretical calculations, as well as more advanced techniques for related systems.70,78–83 Using the TRIR technique, 3MLCT character was evidenced for the bromide analogue of the complex 6.70
The introduction of the dppz ligand into the Re(I) coordination sphere resulted in a greater triplet excited state diversity, as a result of possessing of close-lying MLCT and IL excited states.44,50 The excited state dynamics of 3 in solvents of different polarity was explored by TRIR spectroscopy. The photophysics of this complex in polar MeCN and C3H7CN was found to be govern by 3MLCTphz, 3MLCTphen and 3ILπ–π* excited states, while the formation of only 3MLCTphen and 3ILπ–π* was revealed in non-polar CH2Cl2. The increased solvent polarity was evidenced to lead to a higher population of 3MLCTphz excited state. Contrary to 3MLCTphen, 3MLCTphz excited state is dark. In general, two sets of transient bands are observed in TRIR spectra of 3, that are shifted to higher and lower energy relative to the parent νCO absorptions, respectively. The former ones are characteristic of the 3MLCT excited state, while those with negative shifts represents 3ILπ–π*. The difference between 3MLCTphz, 3MLCTphen concerns the magnitude of the positive shift. The phz-based MLCT excited state are represented by νCO transient absorptions shifted to higher wavenumber in comparison to those of 3MLCTphen.44,50
Typically of 3MLCT emitters, the emission spectra of 1a, 1b, 2, 4 exhibit broad and unstructured band in both solution and rigid-glass matrix (77 K). Consistent with the rigidochromic effect, the frozen-state emission bands of these systems are noticeably blue-shifted and show extended lifetime relative to those at room temperature.52,84,85 The emission properties of 5 and 6 were largely studied only in solution at RT, and broad structureless 3MLCT emission band was evidenced for both of them.80–82,86–88 The complex 3 was found to possess more complicated emission features due to an interplay between two, close in energy, 3MLCT and 3IL excited states. At temperature >145 K, the complex 3 behaves as 3MLCT emitter, showing unstructured emission band. With decreasing temperature, it becomes a π–π* emitter, with long lifetime and structured profile of the emission band.72,89 The wavelength emission maxima of 1–6 in solutions at RT generally cover the yellow-orange spectral region. The triplet state lifetimes of these systems, albeit noticeably longer than fluorescence lifetimes of organic chromophores, are usually shorter than 500 ns (Table S2†), which is rationalized by the fact that the metal parentage in a triplet excited state increases the radiative decay rate constant. The excited-state lifetimes of 1–6 are usually insufficiently long enough for efficient intermolecular photoinduced energy- or electron-transfer processes, required for such applications as photodynamic therapy, time-resolved bioimaging, or triplet–triplet annihilation up-conversion. Regarding above-mentioned applications, also the weak visible absorptivity of 1–6 is a significant disadvantage.
3. Rhenium(I) chromophores with D–A or D–π–A diimine ligands
Functionalization of diimines with electron-rich donors leads to the increase in the level of complexity in the photophysical behavior of complexes [ReL(CO)3(N∩N)]n. In such systems, two distinct photoinduced charge flow processes are possible, that are from the peripheral donor substituents to the diimine core, and from metal center to the π-acceptor diimine framework, as illustrated in Scheme 2. Such systems can be regarded as bichromophoric systems with two close in energy MLCT and ILCT states.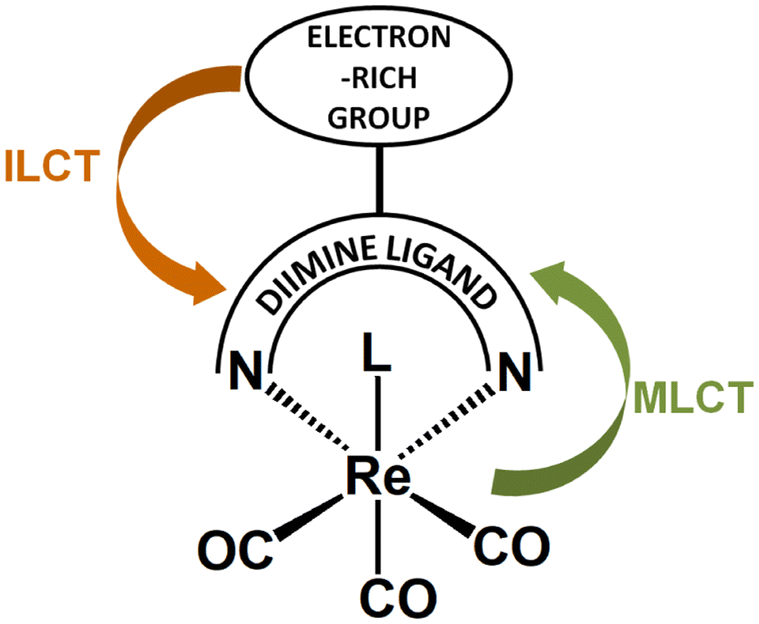 | ||
| Scheme 2 Schematic structure of the Re(I) carbonyl chromophore with D–A diimine ligand, along with two possible photoinduced charge flow processes. | ||
The interplay between MLCT and ILCT excited states, as well as perturbations in photophysics of [ReL(CO)3(N∩N)]n due to the ILCT involvement are discussed below for Re(I) bearing the diimine π-acceptor framework functionalized with the amine, sulphur-based and π-conjugated aryl groups, regarded as most recognizable and most extensively studied donors.
3.1. Amine-substituted ligands
Extensive research has been done to study ground and excited states of Re(I) complexes with diimine ligands functionalized with strong electron-donating triphenylamine (TPA) group, demonstrated in Scheme 3 and Table S3.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5,52,70,90–105 Investigations in this field were largely performed by Gordon's research group with the use of a wide spectrum of spectroscopic techniques, including transient absorption and emission, time-resolved IR and Raman spectroscopies.70,93,95–100,102–105
5,52,70,90–105 Investigations in this field were largely performed by Gordon's research group with the use of a wide spectrum of spectroscopic techniques, including transient absorption and emission, time-resolved IR and Raman spectroscopies.70,93,95–100,102–105
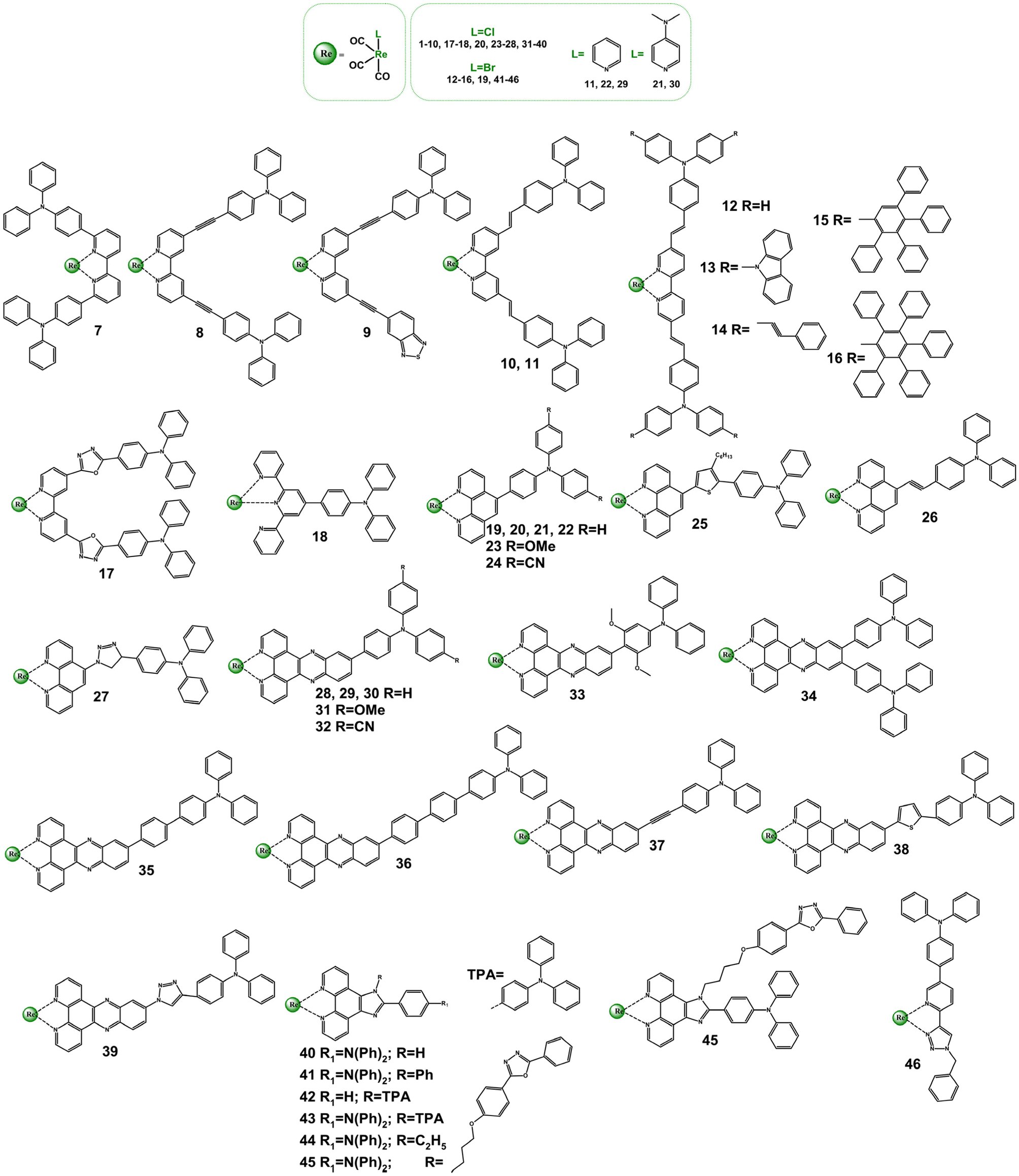 | ||
| Scheme 3 Re(I) carbonyl complexes bearing diimine ligands functionalized with the triphenylamine (TPA) motive. | ||
The structural modification of bipy with TPA motive leads to noticeably perturbations in the photophysics of all complexes 7–16 relative to the parent chromophore 1. However, only for systems 12–16, ILCT excited states become dominated in controlling their photobehaviour. Consistent with the contribution of 1ILCT transitions, the lowest absorption band of these systems shows a batochromic shift and experiences a large increase in the molar absorptivity compared to the reference chromophore 1. The decreased contribution of 1ILCT transitions in the lowest energy absorption was indicated for 7 and 9, which was rationalized by the large TPA–bipy dihedral angle for 7 and replacement of one of TPA groups by electron-withdrawing benzothiadiazole for 9.103,104
The complexes 7–9 show dual fluorescence–phosphorescence emission, relatively rare observed for diamine Re(I) carbonyl complexes.103,104 The lowest triplet emitting state of 7, represented by the low energy component in the emission spectrum, was confirmed to possess 3MLCT nature. The energy and lifetime of the phosphorescence band of 7 is only slightly lower than those for the parent chromophore 1.103 The relative intensities of the fluorescence–phosphorescence components of 8 and 9 were found to be strongly dependent on the polarity environment, showing a significant drop in triplet emission intensities in more polar solvents. Gordon's research group evidenced that triplet excited states of these systems vary in nature, depending on the solvent polarity. In less polar CH2Cl2, the emissive state is of 3MLCT nature. In polar MeCN, the lowest triplet state is of 3ILCT character. It is a dark state, populated via internal conversion from 3MLCT104 (Fig. 2).
 | ||
| Fig. 2 Relative energies of excited states and transitions for 8 in CH2Cl2 and MeCN. Reprinted with permission from ref. 104 Copyright © 2021, American Chemical Society. | ||
The variable contribution of 3ILCT in the lowest triplet state depending on the solvent polarity was also postulated for 18.101 The complexes 10 and 11 show the single emission band, with maximum at ∼520 nm and lifetime shorter than 10 ns, assigned to prompt florescence from 1π → π* state. In similarity to 8 and 9 in polar environment, the lowest triplet state of 10 and 11 is dark, but its character was assigned as a mixed 3ILCT and 3IL. The 3IL contribution was rationalized by the large overlapping of the donor and acceptor orbitals. Compared to 8 in MeCN, the complex 10 shows a noticeably elongated lifetime, 140 ns for 8 and 600 ns for 10.93 Outstanding emissive properties were revealed for 12–16. All these systems exhibit broad unstructured emission band in a deep red to near-infrared range (680–708 nm), red-shifted with the increasing π-conjugation of the TPA unit, and originated from 3ILCT excited state. Importantly, the luminescence quantum yields of the systems with bulky amine groups (15 and 16) were found to be comparable to that of the non-substituted 12, despite their red shift in emission energies.94 The introduction of the oxadiazole linkage between bipy and TPA units resulted in the formation of Re(I) carbonyl complex (17) with the emitting state of 3MLCT nature, which was clearly evidenced by transient DC photoconductivity technique.90
In a series of Re(I) complexes bearing 1,10-phenanthroline with appended TPA donor group at the 5 position, the relative energies and ordering of MLCT and ILCT charge transfer excited states were tuned by (i) changing the electron withdrawing ability of the ancillary ligand (19–22), (ii) modulating the donating ability of the TPA group through the introduction of additional methoxy and cyano groups (23 and 24), (iii) using various bridging groups between the TPA and diimine units (25–27).99,102,105
Independent on the electron withdrawing ability of the ancillary ligand, for all four complexes 19–22, the lowest-energy absorption region is dominated by 1ILCT transitions, and the emissive excited state remains 3ILCT. All these systems are characterized by strong visible absorptivity, red shifted with increasing the electron withdrawing ability of the ancillary ligand. Outstanding emissive photophysical properties was revealed for the halide species 19 and 20. Their excited-state lifetimes (6 μs for 19 and 3.9 μs for 20) were found to be longer by 2 orders of magnitude in relation to the non-halide systems (44 ns for 21 and 27 ns 22) and parent chromophore 2 (Table S2†). To explore different behaviour of the halide complexes (19 and 20) to non-halide ones (21 and 22), transient absorption and emission, transient resonance Raman, and time-resolved infra-red spectroscopy in combination with TD-DFT calculations were utilized. The complexes 19 and 20 were found to possess spectral signatures from both 3MLCT and 3ILCT states, contrary to the non-halide systems with contribution of only 3ILCT. The impact of the ancillary ligand on the excited state lifetimes of 19–22 was explained in terms of energy-gap law.99 The introduction of additional electron-donating methoxy groups into TPA moiety leads to the stabilization of the 3ILCT state in 23 compared to 20, but complex 23 becomes non-emissive in solution at RT, in contrast to 20. Typically of Re(I) systems with diimine ligands functionalized with the TPA group, TA spectra of 23 show an excited-state absorptions consistent with TPA˙+ (750–800 nm) and phen˙− (330–390 nm). The excited-state lifetime of 3ILCT, obtained from transient absorption measurements of 23 (28 ns), was found to be shorter by 2 orders relative to that of 20 (3.9 μs). For complex 24 with additional electron-withdrawing cyano groups in TPA motive, the 3ILCT excited states is noticeably destabilized in comparison to that of 20, becoming near-isoenergetic with 3MLCT excited state. Competitive excited-state relaxation pathways with contribution of 3ILCT and 3MLCT was postulated for 24. The complex emits at ∼600 nm at RT, and shows a relatively long excited-state lifetime (290 ns).102 The incorporation of an electronically conducting linker (thiophene-based in 25 and ethynyl unit in 26) was found to facilitate the TPA–phen communication, leading to the formation of 3ILCT/3IL dark excited states with long biexponential lifetimes, up to 45 μs. Such long-lived excited states were firstly confirmed for transition metal complexes with phen-TPA derivatives. Contrastingly, the 1,2,3-triazole bridge, a well-known electronic insulator, disrupts the TPA–phen communication in 27. Unlike 25 and 26, the complex 27 exhibits weak emission at ∼600 nm, with excited-state lifetime of 60 ns. The lowest triplet state of 27 was postulated to be 3MLCT in nature.105
The impact of the ancillary ligand, additional groups in the TPA motive and D–A linkers on the interplay between MLCT and ILCT was also widely explored for Re(I) carbonyl complexes with TPA-appended dppz ligands (28–39). As reported in a previous section, the photophysics of Re(I) systems with dppz-based ligand may be more complex compared to complexes with other diimine ligands, owing to the presence of close-lying MLCTphen, MLCTphz and IL excited states.
The Frank-Condon photophysics of all systems 28–39 was found to be predominately contributed by 1ILCT transitions. The introduction of additional electron-donating groups into TPA motive and replacement of the halide ancillary ligand by pyridine or dimethylaminopyridine result in a bathochromic shift of the broad intense ILCT band relative to 28. Contrastingly, the extended donor–acceptor distance induces a blue-shift of the ILCT absorption, while the increased D–A torsion angle, leading to the poorer overlap of the donor and acceptor molecular orbitals, is manifested by a noticeably drop in molar absorptivity of the ILCT band. Excited-state photophyscics of all complexes 28–39 is also controlled by ILCT states.95–98,100 The compounds 28–30 were evidenced to exhibit a weak emission, originating from the triplet state of ILCT character and showing a linear correlation between the Stokes shift and solvent polarity. All these systems are characterised by the increase in the emission intensity with increasing temperature, indicated the thermal equilibrium with a dark triplet excited state of lower energy. By TRIR spectroscopy, this dark state was assigned as phz-based 3ILCT, with lifetimes from 1.4 to 10.1 μs.95 The incorporation of additional groups into the TPA motive results in formation of complexes, which show fluorescence occurring from 1ILCT excited state, strongly dependent on the substituent and solvent polarity (31 and 32). Contrastingly, the lifetimes of the lowest triplet dark states 3ILCT of 31 and 32, confirmed through TA spectroscopy, are only slightly affected by the appended substituents: 3.8 μs for 28, 3.6 μs for 31 and 3.0 μs for 32.97 The excited-state photophysics of other Re(I) with dppz-based ligand (33–39) is also governed by 3ILCT state, populated from 1ILCT. However, variations in the donor–acceptor communication, achieved by the modulation of D–A torsion angle, D–A distance and linker nature, lead to changes of 3ILCT lifetimes, which range from 3.9 μs for 34 to 600 ns for 39.98,100 All complexes 33–39 show short-lived emission (<6 ns).
So far, little attention has been paid to Re(I) complexes with TPA-appended imphen ligands. The photophysics of 40–45 has been not explored with the use of transient absorption and emission, time-resolved IR and Raman spectroscopies. The attachment of TPA does not result in an efficient improvement of the visible-light absorptivity of 40–45. The relatively weak lowest energy absorption of 40–45, tailing up to 500 nm, was tentatively assigned to 1MLCT excited state. All these systems exhibit phosphorescence at ∼600 nm, tentatively attributed to 3MLCT admixed with 3IL/3ILCT. Regarding good photophysical functional parameters (quantum yields and excited-state lifetimes), excited-state dynamics in these systems deserves more scientific interest, especially that some of these compounds are efficient emitters in electrophosphorescent devices.5,52,91,92
As mentioned in the previous section, substituents appended to the triazole ring have a limited impact on photobeahaviour of Re(I) complexes bearing pytri-based ligands. On the contrary, the incorporation of TPA on the triazole ring of the pytri-Bn ligand has been proven to be a good way of controlling the ground and excited properties of this type of complexes. The TPA donor group efficiently improves the visible absorptivity of 46, consistent with an involvement of 1ILCT transitions in the lowest energy absorption. The complex 46 shows emission at 573 nm, red-shifted compared to the unsubstituted chromophore (549 nm) and most likely occurring from 1ILCT excited state (τ < 10 ns). The TA and TRIR results are consistent with the formation of the 3ILCT dark excited state, with a lifetime of 2.7 μs.70
There have been several other interesting reports on the impact of other cyclic and acylic amine substituents on the photophyscics of diimine Re(I) carbonyl complexes (Scheme 4 and Table S4†).
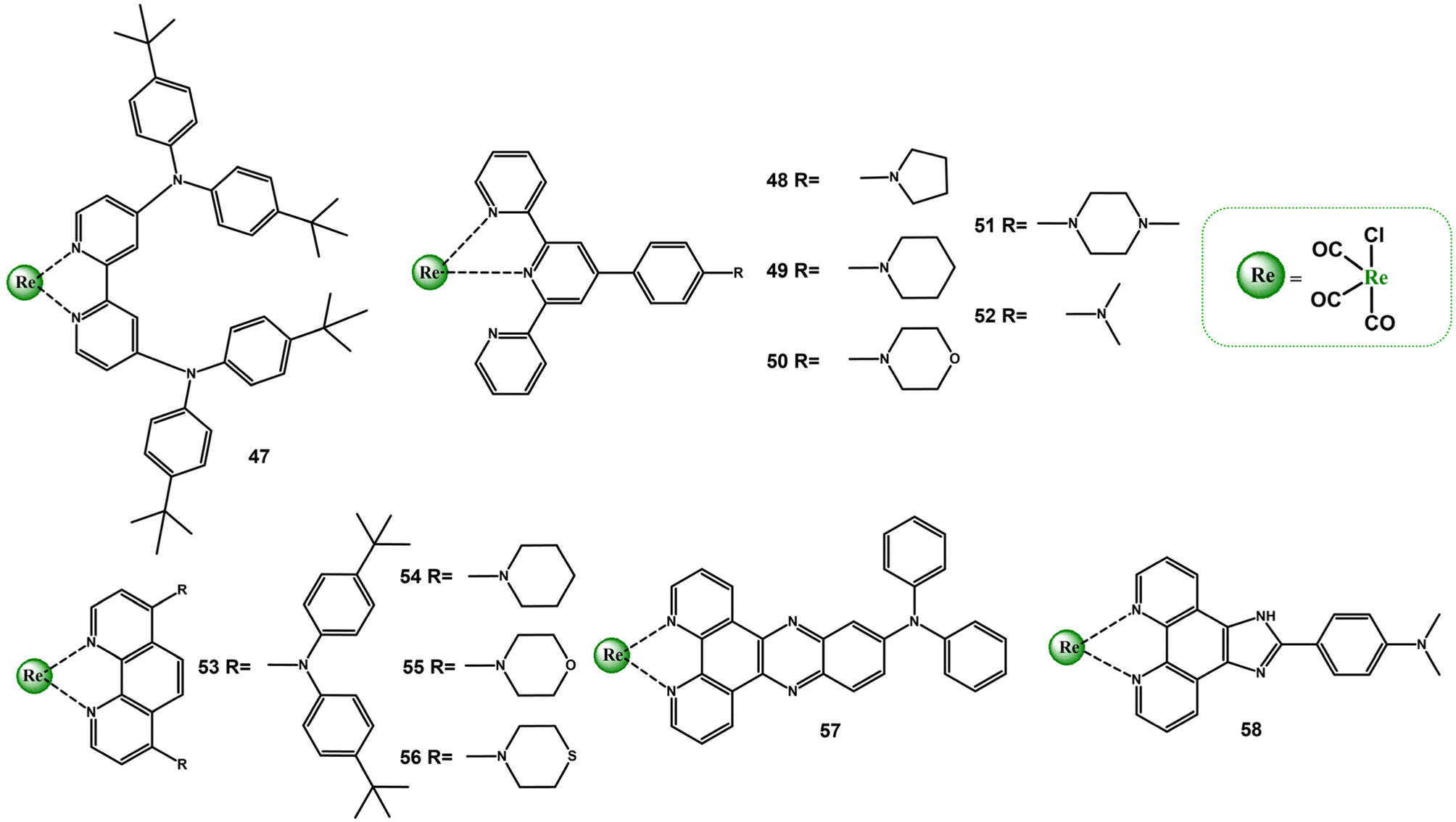 | ||
| Scheme 4 Re(I) carbonyl complexes bearing diimine ligands substituted with different types of amines. | ||
A comparative analysis of photophysical properties of complexes 47, 53 and 57 confirms a crucial role of the diimine core in controlling photobehaviour of Re(I) carbonyl complexes. Contrary to complexes 47 and 53, which possess absorption and emission spectral features from both MLCT and IL/ILCT states, affected additionally by the polarity environment, the singlet and triplet excited states of 57 are in predominately ILCT nature. Worthy of note, the intense visible absorption of 57 is bathochromically shifted compared to the complex with TPA motive (28), and its triplet excited-state lifetime is two times longer than that of 28.100,106 Outstanding absorption and emission properties were revealed for the complex 52. The attachment of –NMe2 resulted in an appearance of a very intense visible absorption band and dramatic elongation of the emissive triplet excited state lifetime. Compared to the reference chromophore [ReCl(CO)3(Ph-terpy-κ2N)], the lowest energy absorption band is red-shifted by ∼100 nm, and the triplet excited-sate lifetime is 260 times longer. These distinct absorption and emission properties of 52 were assigned to 1ILCT and 3ILCT excited states, supported by TA and TRIR spectroscopies in accompany with TD-DFT calculations.107–109 In turn, the introduction of –NMe2 group into the Ph-imphen does not result in the switch from 3MLCT to 3ILCT in 59. The lowest energy absorption state of 59 is predominantly MLCT in nature, and its emission in non-polar solvents occurs from the triplet excited state of the mixed IL/ILCT/MLCT nature. The 3IL/3ILCT character of the emitting triplet state is manifested by the red-shift of the emission band and increase in the lifetime relative to the reference chromophore [ReCl(CO)3(Ph-imphen)], as well as an appearance of the vibronic progression in frozen emission spectra profile of 59.52
The photoluminescence properties of Re(I) complexes with Ph-terpy decorated with cyclic amines (48–51) were found to be strongly dependent on the polarity environment. It was evidenced that polar solvents facilitate the formation of 3ILCT state, showing elongated lifetime relative to that of the model chromophore. The triplet excited state 3ILCT of 48 was found to be sufficiently long for singlet oxygen generation.101,109–111 The absorption and emission properties of Re(I) complexes with 1,10-phenantroline decorated with piperidine (54), morpholine (55) and thiomorpholine (56) were attributed to configurationally mixed MLCT/IL excited states. The inclusion of cyclic amine groups into the phen core was found to be effective at improving molar absorptivity and extending excited-state lifetimes. The most pronounced increase in the lifetime was confirmed for 56 in CHCl3 (6.67 μs).85
3.2. Diimines with sulphur-based donor groups
The interplay between MLCT and ILCT charge processes in Re(I) tricarbonyl complexes with diiimnes bearing sulphur-based donor groups has received relatively little scientific attention so far. More detailed spectroscopic investigations were performed for only a few systems (Scheme 5 and Table S5†), mainly tetrathiafulvalene-, thiophene/oligothiophene- and phenothiazine-substituted diimine Re(I) cabonyl complexes.71,85,112–118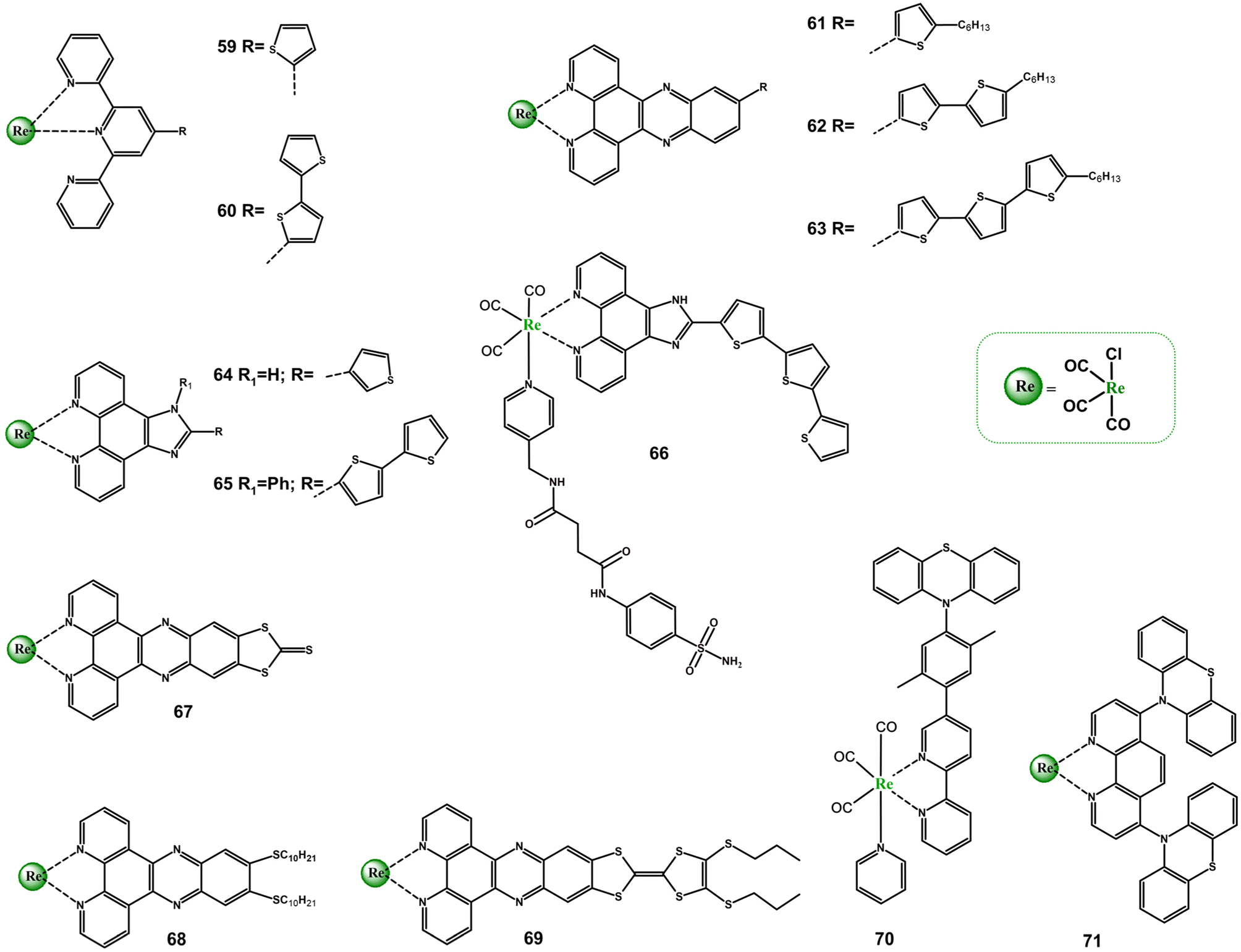 | ||
| Scheme 5 Re(I) carbonyl complexes bearing diimine ligands substituted with sulphur-based donor groups. | ||
Except for 61, the absorption and emission behaviour of thiophene-subsitiuted Re(I) species (59, 64) is dominated by MLCT/LLCT excited states.113,114 The lowest energy singlet–singlet electronic transition of 61 was found to be ILCT in nature.115 Independent on the diimine core, a degree of ILCT character in the excited states of Re(I) complexes increases when the number of appended thiophene units becomes greater. An extension of the donor π-conjugation, promoting the charge-separated excited states in 60, 62, 63, 65 and 66, results in their noticeably red-shift and enhanced light absorption in the visible light range compared to corresponding thiophene-subsitiuted or parent Re(I) systems.114,115,117,118 The emission of 61–63 was shown to occur from 1ILCT absorbing state. In agreement with TA studies and triplet state DFT calculations, the dark 3ILCT/3IL triplet excited state of 61–63 is the result of a 1n,π → 3π,π transition, and the increased number in thiophene units induces higher contribution of 3IL character in the excited state.115 The TA spectra of 66 were found to possess signatures of either 3MLCT and 3ILCT/3IL excited states, and the 3ILCT/3IL was postulated to participate in intermolecular charge transfer process between 66 and oxygen or biomolecules to generate reactive oxygen species (ROS).117
For complexes 67 and 68, the absorption properties were analysed using TD-DFT calculations and resonance Raman spectroscopy, which confirm the occurrence of both MLCT and ILCT transitions.71 A spectacular photobehaviour was revealed for the compound 69 possessing the lowest energy 1ILCT absorption (λmax = 580 nm) well-separated from the higher lying 1MLCT band (λmax = 375 nm). The direction of photo-induced charge-transfer processes in 69 can be controlled by the excitation wavelength. The excitation of 69 at 560 nm results in formation of the charge-separated state ILCT, representing by ESA of radicals TTF˙+ and dppz˙−, decaying in the tens of ps regime, typically of TTF-based D–A systems. In turn, fs-TA spectra recorded upon 400 nm excitation exhibit features of both ILCT and MLCT transitions, with the predominant contribution of the latter ones. After 80 ps, only 3MLCT is observed. The long-lived 3MLCT was found to form via intersystem crossing 1MLCT → 3MLCT.116
Regarding the phenothiazine-substituted systems (70 and 71), the formation of the charge separated excited state was directly evidenced only for 71. The excited-state absorptions corresponding to the phenanthroline radical anion phen˙− (340 nm) and phenothiazine radical cation ptz˙+ (530) were observed in TA spectra of 71 immediately after its laser excitation. On contrary, efforts to detect ptz˙+ in 70 were failed. It was postulated that the photoinduced charge separation process is significantly slower than the thermal charge recombination in this system. Importantly, the complex 71 was found to be a rare example of NIR light-emitting system (785 nm in CHCl3 with τ = 150 ns).85,112
3.3. ILCT contribution in Re(I) carbonyl complexes with diimines bearing π-conjugated aryl groups
The incorporation of π-conjugated aryl groups into the diimine cores facilitates a population of the triplet ligand-centered excited state. When 3MLCT and 3IL states sharing similar energy, an excited state equilibrium is established, meaning that the organic chromophore repopulates the luminescent 3MLCT excited state and may play a role of the energy “reservoir” or excited-state storage element. Such compounds are generally emissive and show significantly extended excited state lifetimes in solution at RT. The prolonged lifetimes are also expected when the triplet excited state localized on the organic chromophore lies significantly lower in energy than 3MLCT, but these bichromophore systems are usually non-emissive in solution at RT.119–124 The room-temperature phosphorescence attributable to the triplet excited state of π-conjugated aryl chromophore is extremely rarely observed.125–128 The chromophoric units (organic and inorganic) maintains independent when they are perpendicularly arranged and the mixing of orbitals of both chromophores is minimized. Reducing the dihedral angle between the substituent and core of the organic ligand results in partial “mixing” of orbitals of both chromophores, and may lead to a noticeable contribution of ILCT excited states in the photobahaviour of resulting chromophore systems as π-conjugated aryl groups have potential to act as electron-donating substituents due to the high π-electron density.Among Re(I) complexes with diimine ligands substituted with electron rich π-conjugated aryl groups 72–86, demonstrated in Scheme 6, a noticeable role of ILCT transitions in controlling photophysical properties was evidenced for 76, 77, 79, 80, and 82.37,129–139
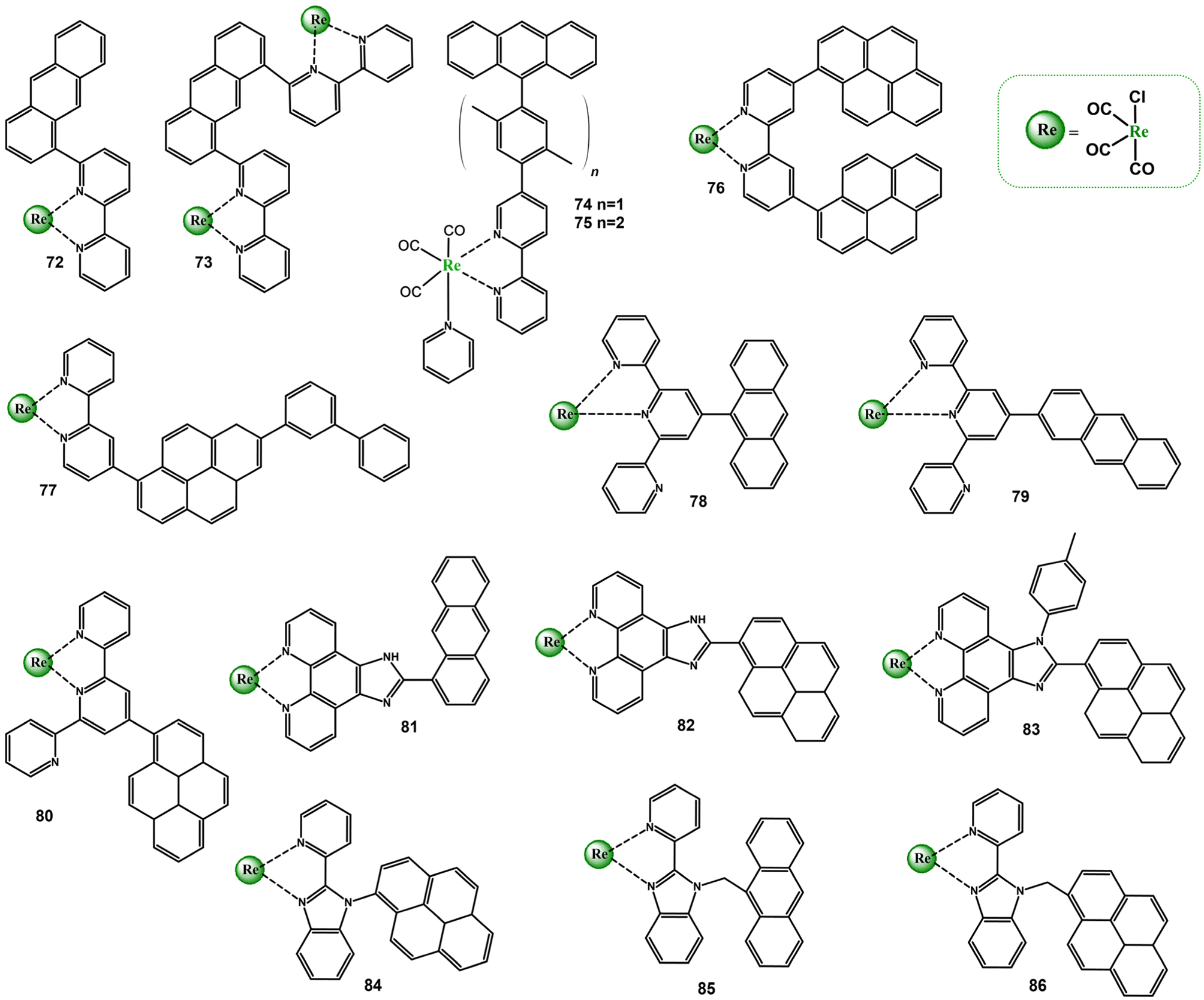 | ||
| Scheme 6 Re(I) carbonyl complexes bearing diimine ligands substituted with electron-rich pyrenyl π-conjugated aryl groups. | ||
Comprehensive investigations of the mutual chromophore orientation on photophyscical properties were performed for the pairs 78–79 and 82–83, in which the electronic coupling between the organic and metal-based chromophores were controlled by the anthryl linkage (78 and 79) and steric hindrance of the 4-(methyl)phenyl substituent introduced into the imidazole ring at 1H-position (82 and 83).
For complexes 78 and 83, with the high dihedral angle between the aryl substituent and diimine planes, πarene–πarene* absorptions with characteristic vibronic progression are well resolved from the weak 1MLCT band occurring in the lower energy region. On contrary, compounds 79 and 82 with a more coplanar ligand geometry display the unstructured, noticeably red-shifted and more intense lowest energy absorption in comparison to 78 and 83 (Fig. 3). Such findings, that are red-shift and significant absorptivity enhancement, along with the lack of characteristic anthracene/pyrene vibronic progression, are strongly supportive of the contribution of intra-ligand charge transfer transitions from the electron-rich aryl group to the π-accepting diamine core.
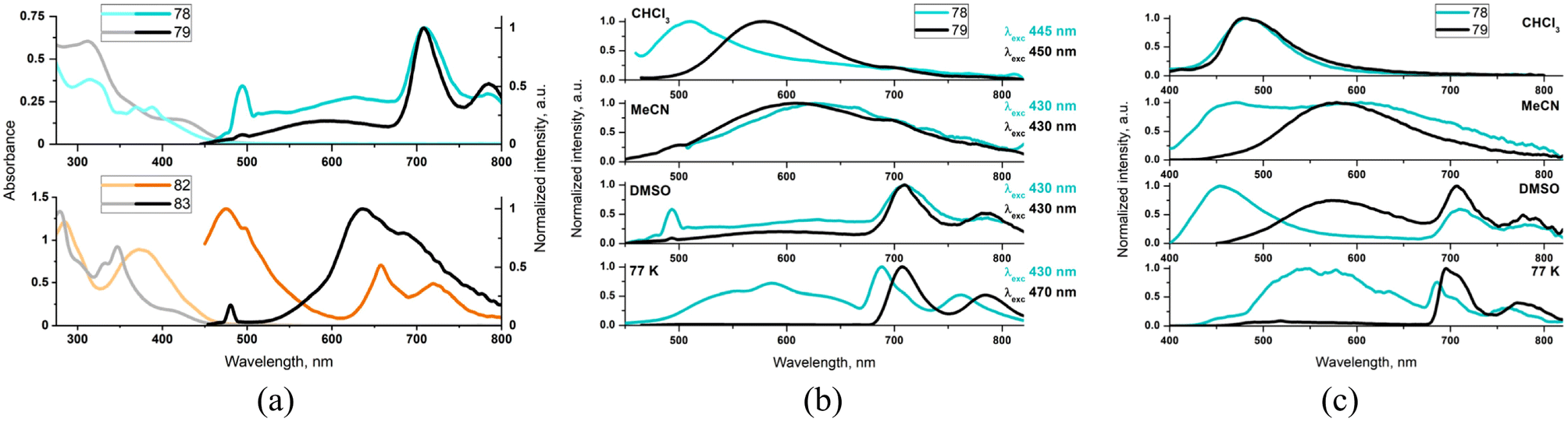 | ||
| Fig. 3 Photophysical data of 78–79 and 82–83: absorbance (lighter lines) and emission (darker lines) in DMSO (a); emission spectra of 78 and 79 in different environments upon excitation at the red side of the lowest energy absorption (b) and 355 nm (c). Adapted from ref. 136 and 137 Copyright © 2022, 2023, American Chemical Society. | ||
The TA studies 78–79 and 82–83 revealed that a more planar ligand geometry in 78 and 82 facilitates the population of the long-lived triplet ligand-centered excited state. In general, it is populated via the triplet–triplet energy transfer from 3MLCT excited state. For the complex 82, the contribution of the another 1ILpyrene/1ILCT → 3ILpyrene/3ILCT pathway was also evidenced. Compared to 79 and 83, T1 → Tn absorptions of 78 and 82 are noticeable red-shifted and broadening, in agreement with the contribution of the photo-induced charge transfer processes occurring from the electron-rich aryl group to the π-accepting diamine core. The lowest triplet excited state was found to be of 3ILarene/3ILCT for 79 and 82 and 3ILarene for 78 and 83. The complexes 78, 79 and 83 were found to be rare examples of coordination compounds that show aryl-based phosphorescence in solution at RT. As demonstrated by steady-state emission studies, the population of 3MLCT and 3ILarene of 78 and 79 can be controlled by the solvent polarity and excitation wavelengths (Fig. 3). In DMSO, compounds 78 and 79 upon excitations λ > 410 nm show a dual 3MLCT and 3An emission, extremely rarely observed.
Having long excited-state lifetimes (Table S6†), the complexes 78–79 and 82–83 were found to be suitable for transferring the excited triplet-state energy to molecular oxygen and generating singlet oxygen. The pyrenyl-subsitiuted complexes, with record-high triplet excited-state lifetimes at RT (∼1800 μs for 82 and ∼1500 μs for 83) act also as photosensitizers in triplet–triplet annihilation upconversion.136,137 Using TA spectroscopy, the complex 80 was demonstrated to exhibit ‘ping-pong’ energy transfer 1ILCT → 1MLCT → 3MLCT → 3IL/3ILCT. In agreement with the triplet-state equilibrium between3MLCT → 3IL/3ILCT, the complex 80 shows an extended triplet excited-state lifetime at RT (4.4 μs).135 The combined spectroscopic data of 77 were found to be consistent with lowest triplet excited state being either of 3IL or 3ILCT character.129
Importantly, the incorporation of π-conjugated aryl groups into the diimine cores of Re(I) carbonyl complexes, which facilitates a population of the triplet ligand-centered excited state and enhances photoinduced energy transfer processes, were found to be a powerful strategy to design more efficient catalysts for the conversion of CO2 into CO using solar energy (see next section).
4. Appealing applications of Re(I) complexes with D–A or D–π–A diimine ligands
The rhenium(I) tricarbonyl complexes bearing π-extended pyrene and anthracene chromophores attached to the diimne ligand 73, 76, and 84 were garnered substantial attention for the light-driven CO2-to-CO reduction.132–134 The appended π-extended aryl groups were demonstrated to promote the Re(I) catalyst's ability, which was rationalized by their increased visible light-harvesting abilities and long-lived excited states corresponding to the ligand-localized triplet state compared to the appropriate reference Re(I) complexes (see section 3.3). Most importantly, the complex 76 was the first example of a single-molecule catalyst investigated directly under the sun. Relative to the model chromophore [ReCl(CO)3(bipy)] (TONCO = 22 ± 2), the turnover number TONCO of the complex 76 was 40-fold higher (350 ± 36) under these conditions.133The terpyridine-based complex [ReCl(CO)3(Me2N-C6H4-terpy-κ2N)] (52) was the first Re(I) carbonyl system, which was tested as photosensitizer in the photocatalytic hydrogen production. Its 3ILCT excited-state reactivity was analysed relative to [ReCl(CO)3(C6H5-terpy-κ2N)], possessing a much shorter-lived 3MLCT excited state. For [ReCl(CO)3(Me2N-C6H4-terpy-κ2N)], the hydrogen evolution was found to occur dramatically faster and with a noticeably higher final turnover number. Also, 10-fold-smaller concentration of [ReCl(CO)3(Me2N-C6H4-terpy-κ2N)] was needed compared to the model chromophore. A striking difference between [ReCl(CO)3(Me2N-C6H4-terpy-κ2N)] and [ReCl(CO)3(C6H5-terpy-κ2N)] was evidenced using TRIR spectroscopy. While [ReCl(CO)3(Me2N-C6H4-terpy-κ2N)] was found to transfer the electron directly to the cobalt catalyst, the model chromophore underwent the reductive quenching in a bimolecular fashion by triethanolamine, used as the sacrificial donor.108
The terpyridine Re(I) complexes [ReCl(CO)3(R-terpy-κ2N)] with appended π-extended aryl and electron-donating amine groups (52, 78, 80) were also emerged as a novel category of anticancer agents. Possessing abilities for both 1O2 generation and releasing CO under ultrasound irradiation, they could be regarded as combined agents for photodynamic therapy and photo-activated chemotherapy.37,140
Last but not least, the Re(I) compounds bearing diimine ligand of D–A or D–π–A type deserved an attention in view of their potential applications as phosphorescent emitting materials in OLED technology. Excellent device performances were achieved for OLEDs with Re(1) complexes 41–43 as dopant emitters. The last of them (43) showed the performance with a maximum current efficiency of 36.5 cd A−1, maximum power efficiency of 31.7 lm W−1 and maximum external quantum efficiency of 12.0%, which were among the best data for the Re(I)-based OLEDs reported so far.3,5
5. Summary and future prospects
The growth of many technologies, including photocatalysis, photodynamic therapy and triplet–triplet annihilation for energy up-conversion, requires efficient photosensitizers with strong absorption in the visible range, good solubility and sufficiently long excited state lifetimes. Within this review, we summarized the latest developments concerning the bichromophoric effect in diimine Re(I) tricarbonyls functionalized with electron-rich donors. It was demonstrated that the formation of intraligand charge transfer (ILCT) excited states, due to incorporation of the organic push–pull chromophore to the {Re(CO)3}+ core, may lead to enhancement of visible absorptivity, bathochromic shift of the emission and significant prolongation of the excited-state lifetimes of these systems. The role of ILCT transitions in controlling of excited state properties was discussed for Re(I) tricarbonyls bearing six different diimine cores (bipy, terpy, phen, dppz, imphen, pybimd and pytri), decorated by various electron-rich amine, sulphur-based and π-conjugated aryl groups.Although a huge progress has been made in understanding photoinduced processes occurring in bichromophoric Re(I) complexes bearing donor–acceptor ligands, a precise control of long-lived charge-separated excited states in these systems still remains a major challenge, and more advanced studies in this field are strongly desired to establish structure–property relationships and make further progress in the development of more efficient materials. The current review clearly highlights that Re(I) complexes with imphen-, pybimd- and pytri-based ligands of the donor–acceptor structure is noticeably underdeveloped compared to those bearing derivatives of bipy, terpy, phen and dppz. In particular, the research with the use of highly advanced and time-resolved spectroscopic measurement techniques are most required to provide a more complete picture of excited state dynamics and rate of formation of triplet excited states in these systems. Regarding a type of a donor part, Re(I) tricarbonyl complexes with diiimnes bearing sulphur-based electron-rich groups deserves more scientific attention, especially that the Ru(II) complex with terthiophene-subsitiuted imidazo[4,5-f][1,10]phenanthroline (TLD1433)64 is the first organometallic photosensitizers introduced into a human clinical trial, and Re(I) tricarbonyls bearing imphen-based ligands have recently emerged as promising anticancer agents.117,141 An important advantage of imidazo[4,5-f][1,10]phenanthrolines is their convenient synthesis along with the possibility of introduction all types of into the imidazole ring at 1H- and C2-positions, directly (via the covalent bond) or through an aryl/heterocycle bridge, giving the possibility to establish reliable structure–property relationships. Furthermore, as demonstrated in section 3.3, the mutual chromophore arrangement in [ReL(CO)3(R1,R2-imphen)]n with π-conjugated aryl groups, controlled by steric hindrance of the substituent introduced into the imidazole ring at 1H-position, impacts the efficiency of intermolecular photoinduced energy triplet state transfers. Explorations of spatial effects in these systems seem to be interesting future research direction. Regarding Re(I) tricarbonyl complexes with bipy- and phen-based ligands of the donor–acceptor structure, a research gap concerns investigations of the effect of different ligand substitution patterns. The introduction of donor groups into different positions of 2,2′-bipyridine and 1,10-phenanthroline skeleton is expected to provide different electron density delocalization, which may be essential in controlling photoinduced charge-transfer processes. Finally, to facilitate population of ILCT excited states, a scientific attention should be directed towards Re(I) complexes with well-separated 1ILCT and 1MLCT absorption bands. Photo-induced charge-transfer processes in such systems could be precisely controlled by the excitation wavelength.
Author contributions
The manuscript was written through the contributions of all authors. KC: formal analysis, visualization, writing – original draft. JPG: data curation, writing – review & editing. AK: formal analysis, visualization. EM: project administration, supervision, validation. BM: conceptualization, funding acquisition, project administration, supervision, writing – original draft, writing – review & editing. All authors have given approval to the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financed by the Research Excellence Initiative of the University of Silesia in Katowice.References
- M. Wrighton and D. L. Morse, J. Am. Chem. Soc., 1974, 96, 998–1003 CrossRef.
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef.
- G.-W. Zhao, J.-H. Zhao, Y.-X. Hu, D.-Y. Zhang and X. Li, Synth. Met., 2016, 212, 131–141 CrossRef.
- Y.-X. Hu, G.-W. Zhao, Y. Dong, Y.-L. Lü, X. Li and D.-Y. Zhang, Dyes Pigm., 2017, 137, 569–575 CrossRef.
- X. Li, G.-W. Zhao, Y.-X. Hu, J.-H. Zhao, Y. Dong, L. Zhou, Y.-L. Lv, H.-J. Chi and Z. Su, J. Mater. Chem. C, 2017, 5, 7629–7636 RSC.
- M. R. Gonçalves, A. R. V. Benvenho and K. P. M. Frin, Opt. Mater., 2019, 94, 206–212 CrossRef.
- C. Bizzarri, F. Hundemer, J. Busch and S. Bräse, Polyhedron, 2018, 140, 51–66 CrossRef.
- S. Gazzari, P. Dreyse, D. Cortés-Arriagada and I. A. González, Mater. Sci. Semicond. Process., 2022, 147, 106733 CrossRef.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef.
- J. Agarwal, E. Fujita, H. F. I. Schaefer and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef PubMed.
- A. Zarkadoulas, E. Koutsouri, C. Kefalidi and C. A. Mitsopoulou, Coord. Chem. Rev., 2015, 304–305, 55–72 CrossRef.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef PubMed.
- L.-Q. Qiu, Z.-W. Yang, X. Yao, X.-Y. Li and L.-N. He, ChemSusChem, 2022, 15, e202200337 CrossRef PubMed.
- L. Wang, Catalysts, 2022, 12, 919 CrossRef.
- N. Nandal and S. L. Jain, Coord. Chem. Rev., 2022, 451, 214271 CrossRef.
- A. V. Müller, L. A. Faustino, K. T. de Oliveira, A. O. T. Patrocinio and A. S. Polo, ACS Catal., 2023, 13, 633–646 CrossRef.
- S. Hostachy, C. Policar and N. Delsuc, Coord. Chem. Rev., 2017, 351, 172–188 CrossRef.
- L. C.-C. Lee, K.-K. Leung and K. K.-W. Lo, Dalton Trans., 2017, 46, 16357–16380 RSC.
- C. C. Konkankit, S. C. Marker, K. M. Knopf and J. J. Wilson, Dalton Trans., 2018, 47, 9934–9974 RSC.
- E. B. Bauer, A. A. Haase, R. M. Reich, D. C. Crans and F. E. Kühn, Coord. Chem. Rev., 2019, 393, 79–117 CrossRef.
- P. Collery, D. Desmaele and V. Vijaykumar, Curr. Pharm. Des., 2019, 25, 3306–3322 CrossRef.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CrossRef.
- H. S. Liew, C.-W. Mai, M. Zulkefeli, T. Madheswaran, L. V. Kiew, N. Delsuc and M. L. Low, Molecules, 2020, 25, 4176 CrossRef PubMed.
- K. Schindler and F. Zobi, Chimia, 2021, 75, 837–837 CrossRef PubMed.
- C.-P. Tan, Y.-M. Zhong, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2021, 12, 2357–2367 RSC.
- J. Gong and X. Zhang, Coord. Chem. Rev., 2022, 453, 214329 CrossRef.
- S. Pete, N. Roy, B. Kar and P. Paira, Coord. Chem. Rev., 2022, 460, 214462 CrossRef.
- K. Schindler and F. Zobi, Molecules, 2022, 27, 539 CrossRef PubMed.
- A. Sharma S., N. Vaibhavi, B. Kar, U. Das and P. Paira, RSC Adv., 2022, 12, 20264–20295 RSC.
- L. E. Enslin, K. Purkait, M. D. Pozza, B. Saubamea, P. Mesdom, H. G. Visser, G. Gasser and M. Schutte-Smith, Inorg. Chem., 2023, 62, 12237–12251 CrossRef CAS.
- B. Kar, U. Das, N. Roy and P. Paira, Coord. Chem. Rev., 2023, 474, 214860 CrossRef CAS.
- C. Olelewe and S. G. Awuah, Curr. Opin. Chem. Biol., 2023, 72, 102235 CrossRef CAS PubMed.
- Q. Qi, Q. Wang, Y. Li, D. Z. Silva, M. E. L. Ruiz, R. Ouyang, B. Liu and Y. Miao, Molecules, 2023, 28, 2733 CrossRef CAS.
- K. Schindler, J. Horner, G. Demirci, Y. Cortat, A. Crochet, O. Mamula Steiner and F. Zobi, Inorganics, 2023, 11, 139 CrossRef.
- R. Kushwaha, A. Upadhyay, S. Saha, A. K. Yadav, A. Bera, A. Dutta and S. Banerjee, Dalton Trans., 2024, 53, 13591–13601 RSC.
- R. Kushwaha, V. Singh, S. Peters, A. K. Yadav, T. Sadhukhan, B. Koch and S. Banerjee, J. Med. Chem., 2024, 67, 6537–6548 CrossRef PubMed.
- A. Marco, P. Ashoo, S. Hernández-García, P. Martínez-Rodríguez, N. Cutillas, A. Vollrath, D. Jordan, C. Janiak, F. Gandía-Herrero and J. Ruiz, J. Med. Chem., 2024, 67, 7891–7910 CrossRef PubMed.
- T. Neumann, V. Ramu, J. Bertin, M. He, C. Vervisch, M. P. Coogan and H. C. Bertrand, Inorg. Chem., 2024, 63, 1197–1213 CrossRef PubMed.
- M. W. George and J. J. Turner, Coord. Chem. Rev., 1998, 177, 201–217 CrossRef.
- A. Vlček and M. Busby, Coord. Chem. Rev., 2006, 250, 1755–1762 CrossRef.
- A. Cannizzo, A. M. Blanco-Rodríguez, A. El Nahhas, J. Šebera, S. Záliš, A. Vlček Jr. and M. Chergui, J. Am. Chem. Soc., 2008, 130, 8967–8974 CrossRef PubMed.
- M. K. Kuimova, W. Z. Alsindi, A. J. Blake, E. S. Davies, D. J. Lampus, P. Matousek, J. McMaster, A. W. Parker, M. Towrie, X.-Z. Sun, C. Wilson and M. W. George, Inorg. Chem., 2008, 47, 9857–9869 CrossRef PubMed.
- A. El Nahhas, A. Cannizzo, F. van Mourik, A. M. Blanco-Rodríguez, S. Záliš, A. Vlček Jr. and M. Chergui, J. Phys. Chem. A, 2010, 114, 6361–6369 CrossRef PubMed.
- A. Vlček, in Photophysics of Organometallics, ed. A. J. Lees, Springer, Berlin, Heidelberg, 2010, pp. 115–158 Search PubMed.
- A. El Nahhas, C. Consani, A. M. Blanco-Rodríguez, K. M. Lancaster, O. Braem, A. Cannizzo, M. Towrie, I. P. Clark, S. Záliš, M. Chergui and A. Vlček, Inorg. Chem., 2011, 50, 2932–2943 CrossRef PubMed.
- Y. Yue, T. Grusenmeyer, Z. Ma, P. Zhang, T. T. Pham, J. T. Mague, J. P. Donahue, R. H. Schmehl, D. N. Beratan and I. V. Rubtsov, J. Phys. Chem. B, 2013, 117, 15903–15916 CrossRef CAS PubMed.
- L. M. Kiefer, J. T. King and K. J. Kubarych, Acc. Chem. Res., 2015, 48, 1123–1130 CrossRef CAS PubMed.
- R. Horvath, G. S. Huff, K. C. Gordon and M. W. George, Coord. Chem. Rev., 2016, 325, 41–58 CrossRef CAS.
- K. Choroba, S. Kotowicz, A. Maroń, A. Świtlicka, A. Szłapa-Kula, M. Siwy, J. Grzelak, K. Sulowska, S. Maćkowski, E. Schab-Balcerzak and B. Machura, Dyes Pigm., 2021, 192, 109472 CrossRef CAS.
- A. Szłapa-Kula, J. Palion-Gazda, P. Ledwon, K. Erfurt and B. Machura, Dalton Trans., 2022, 51, 14466–14481 RSC.
- D. R. Striplin and G. A. Crosby, Coord. Chem. Rev., 2001, 211, 163–175 CrossRef CAS.
- A. M. Blanco Rodríguez, A. Gabrielsson, M. Motevalli, P. Matousek, M. Towrie, J. Šebera, S. Záliš and A. Vlček, J. Phys. Chem. A, 2005, 109, 5016–5025 CrossRef.
- A. Vlček and S. Záliš, Coord. Chem. Rev., 2007, 251, 258–287 CrossRef.
- A. Coleman, C. Brennan, J. G. Vos and M. T. Pryce, Coord. Chem. Rev., 2008, 252, 2585–2595 CrossRef.
- A. Kumar, S.-S. Sun and A. J. Lees, in Photophysics of Organometallics, ed. A. J. Lees, Springer, Berlin, Heidelberg, 2010, pp. 37–71 Search PubMed.
- R. Baková, M. Chergui, C. Daniel, A. Vlček and S. Záliš, Coord. Chem. Rev., 2011, 255, 975–989 CrossRef.
- C. Daniel, Coord. Chem. Rev., 2015, 282–283, 19–32 CrossRef.
- J. R. Dilworth, Coord. Chem. Rev., 2021, 436, 213822 CrossRef.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899–4032 CrossRef.
- R. Misra and S. P. Bhattacharyya, in Intramolecular Charge Transfer, ed. R. Misra and S. P. Bhattacharyya, 2018, pp. 115–148 Search PubMed.
- A. Ito, M. Iwamura and E. Sakuda, Coord. Chem. Rev., 2022, 467, 214610 CrossRef.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef PubMed.
- T. C. Pham, V.-N. Nguyen, Y. Choi, S. Lee and J. Yoon, Chem. Rev., 2021, 121, 13454–13619 CrossRef PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef PubMed.
- W. Ahmad, J. Wang, H. Li, Q. Ouyang, W. Wu and Q. Chen, Coord. Chem. Rev., 2021, 439, 213944 CrossRef.
- R. Pérez-Ruiz, Top. Curr. Chem., 2022, 380, 23 CrossRef.
- G. S. Huff, W. K. C. Lo, R. Horvath, J. O. Turner, X.-Z. Sun, G. R. Weal, H. J. Davidson, A. D. W. Kennedy, C. J. McAdam, J. D. Crowley, M. W. George and K. C. Gordon, Inorg. Chem., 2016, 55, 12238–12253 CrossRef.
- M. G. Fraser, A. G. Blackman, G. I. S. Irwin, C. P. Easton and K. C. Gordon, Inorg. Chem., 2010, 49, 5180–5189 CrossRef PubMed.
- M. R. Waterland, K. C. Gordon, J. J. McGarvey and P. M. Jayaweera, J. Chem. Soc., Dalton Trans., 1998, 609–616 RSC.
- M. I. J. Polson, S. L. Howell, A. H. Flood, A. K. Burrell, A. G. Blackman and K. C. Gordon, Polyhedron, 2004, 23, 1427–1439 CrossRef.
- B. D. Rossenaar, D. J. Stufkens and A. Vlček, Inorg. Chem., 1996, 35, 2902–2909 CrossRef.
- D. J. Liard, M. Busby, P. Matousek, M. Towrie and A. Vlček, J. Phys. Chem. A, 2004, 108, 2363–2369 CrossRef.
- M. Chergui, Acc. Chem. Res., 2015, 48, 801–808 CrossRef.
- M. K. Kuimova, D. C. Grills, P. Matousek, A. W. Parker, X.-Z. Sun, M. Towrie and M. W. George, Vib. Spectrosc., 2004, 35, 219–223 CrossRef.
- K. Wang, L. Huang, L. Gao, L. Jin and C. Huang, Inorg. Chem., 2002, 41, 3353–3358 CrossRef PubMed.
- R. Czerwieniec, A. Kapturkiewicz, J. Lipkowski and J. Nowacki, Inorg. Chim. Acta, 2005, 358, 2701–2710 CrossRef.
- T. Y. Kim, A. B. S. Elliott, K. J. Shaffer, C. John McAdam, K. C. Gordon and J. D. Crowley, Polyhedron, 2013, 52, 1391–1398 CrossRef.
- W. K. C. Lo, G. S. Huff, J. R. Cubanski, A. D. W. Kennedy, C. J. McAdam, D. A. McMorran, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2015, 54, 1572–1587 CrossRef PubMed.
- S. Sinha, E. K. Berdichevsky and J. J. Warren, Inorg. Chim. Acta, 2017, 460, 63–68 CrossRef.
- A. M. Mansour, RSC Adv., 2019, 9, 15108–15114 RSC.
- J. Palion-Gazda, K. Choroba, A. M. Maroń, E. Malicka and B. Machura, Molecules, 2024, 29, 1631 CrossRef PubMed.
- J. Palion-Gazda, K. Choroba, M. Penkala, P. Rawicka and B. Machura, Inorg. Chem., 2024, 63, 1356–1366 CrossRef PubMed.
- O. D. C. C. De Azevedo, P. I. P. Elliott, C. D. Gabbutt, B. M. Heron, D. Jacquemin, C. R. Rice and P. A. Scattergood, Dalton Trans., 2021, 50, 830–834 RSC.
- B.-C. Tzeng, B.-S. Chen, C.-K. Chen, Y.-P. Chang, W.-C. Tzeng, T.-Y. Lin, G.-H. Lee, P.-T. Chou, Y.-J. Fu and A. H.-H. Chang, Inorg. Chem., 2011, 50, 5379–5388 CrossRef.
- M. Obata, A. Kitamura, A. Mori, C. Kameyama, J. A. Czaplewska, R. Tanaka, I. Kinoshita, T. Kusumoto, H. Hashimoto, M. Harada, Y. Mikata, T. Funabiki and S. Yano, Dalton Trans., 2008, 3292–3300 RSC.
- J. R. Schoonover, W. D. Bates and T. J. Meyer, Inorg. Chem., 1995, 34, 6421–6422 CrossRef.
- Y. Kim, F. W. M. Vanhelmont, C. L. Stern and J. T. Hupp, Inorg. Chim. Acta, 2001, 318, 53–60 CrossRef.
- C. Liu, J. Li, B. Li, Z. Hong, F. Zhao, S. Liu and W. Li, Appl. Phys. Lett., 2006, 89, 243511 CrossRef.
- C. B. Liu, J. Li, B. Li, Z. R. Hong, F. F. Zhao, S. Y. Liu and W. L. Li, Chem. Phys. Lett., 2007, 435, 54–58 CrossRef.
- R. Horvath, M. G. Fraser, S. A. Cameron, A. G. Blackman, P. Wagner, D. L. Officer and K. C. Gordon, Inorg. Chem., 2013, 52, 1304–1317 CrossRef.
- T. Yu, D. P.-K. Tsang, V. K.-M. Au, W. H. Lam, M.-Y. Chan and V. W.-W. Yam, Chem. – Eur. J., 2013, 19, 13418–13427 CrossRef PubMed.
- C. B. Larsen, H. van der Salm, C. A. Clark, A. B. S. Elliott, M. G. Fraser, R. Horvath, N. T. Lucas, X.-Z. Sun, M. W. George and K. C. Gordon, Inorg. Chem., 2014, 53, 1339–1354 CrossRef PubMed.
- H. van der Salm, C. B. Larsen, J. R. W. McLay, G. S. Huff and K. C. Gordon, Inorg. Chim. Acta, 2015, 428, 1–7 CrossRef.
- C. B. Larsen, H. van der Salm, G. E. Shillito, N. T. Lucas and K. C. Gordon, Inorg. Chem., 2016, 55, 8446–8458 CrossRef PubMed.
- B. S. Adams, G. E. Shillito, H. van der Salm, R. Horvath, C. B. Larsen, X.-Z. Sun, N. T. Lucas, M. W. George and K. C. Gordon, Inorg. Chem., 2017, 56, 12967–12977 CrossRef PubMed.
- G. E. Shillito, T. B. J. Hall, D. Preston, P. Traber, L. Wu, K. E. A. Reynolds, R. Horvath, X. Z. Sun, N. T. Lucas, J. D. Crowley, M. W. George, S. Kupfer and K. C. Gordon, J. Am. Chem. Soc., 2018, 140, 4534–4542 CrossRef.
- J. E. Barnsley, G. E. Shillito, C. B. Larsen, H. van der Salm, R. Horvath, X. Z. Sun, X. Wu, M. W. George, N. T. Lucas and K. C. Gordon, Inorg. Chem., 2019, 58, 9785–9795 CrossRef.
- T. Klemens, A. Świtlicka, A. Szlapa-Kula, Ł. Łapok, M. Obłoza, M. Siwy, M. Szalkowski, S. Maćkowski, M. Libera, E. Schab-Balcerzak and B. Machura, Organometallics, 2019, 38, 4206–4223 CrossRef.
- G. E. Shillito, D. Preston, P. Traber, J. Steinmetzer, C. J. McAdam, J. D. Crowley, P. Wagner, S. Kupfer and K. C. Gordon, Inorg. Chem., 2020, 59, 6736–6746 CrossRef PubMed.
- D. A. W. Ross, J. I. Mapley, A. P. Cording, R. A. S. Vasdev, C. J. McAdam, K. C. Gordon and J. D. Crowley, Inorg. Chem., 2021, 60, 11852–11865 CrossRef.
- J. J. Sutton, D. Preston, P. Traber, J. Steinmetzer, X. Wu, S. Kayal, X.-Z. Sun, J. D. Crowley, M. W. George, S. Kupfer and K. C. Gordon, J. Am. Chem. Soc., 2021, 143, 9082–9093 CrossRef.
- G. E. Shillito, D. Preston, J. D. Crowley, P. Wagner, S. J. Harris, K. C. Gordon and S. Kupfer, Inorg. Chem., 2024, 63, 4947–4956 CrossRef PubMed.
- A. M. Maroń, A. Szlapa-Kula, M. Matussek, R. Kruszynski, M. Siwy, H. Janeczek, J. Grzelak, S. Maćkowski, E. Schab-Balcerzak and B. Machura, Dalton Trans., 2020, 49, 4441–4453 RSC.
- T. Klemens, A. Świtlicka-Olszewska, B. Machura, M. Grucela, H. Janeczek, E. Schab-Balcerzak, A. Szlapa, S. Kula, S. Krompiec, K. Smolarek, D. Kowalska, S. Mackowski, K. Erfurt and P. Lodowski, RSC Adv., 2016, 6, 56335–56352 RSC.
- R. Fernández-Terán and L. Sévery, Inorg. Chem., 2021, 60, 1334–1343 CrossRef.
- J. Palion-Gazda, A. Szłapa-Kula, M. Penkala, K. Erfurt and B. Machura, Molecules, 2022, 27, 7147 CrossRef CAS PubMed.
- T. Klemens, A. Świtlicka, A. Szlapa-Kula, S. Krompiec, P. Lodowski, A. Chrobok, M. Godlewska, S. Kotowicz, M. Siwy, K. Bednarczyk, M. Libera, S. Maćkowski, T. Pędziński, E. Schab–Balcerzak and B. Machura, Appl. Organomet. Chem., 2018, 32, e4611 CrossRef.
- K. Choroba, A. Maroń, A. Świtlicka, A. Szłapa-Kula, M. Siwy, J. Grzelak, S. Maćkowski, T. Pedzinski, E. Schab-Balcerzak and B. Machura, Dalton Trans., 2021, 50, 3943–3958 RSC.
- D. Hanss and O. S. Wenger, Inorg. Chim. Acta, 2009, 362, 3415–3420 CrossRef.
- J.-X. Wang, H.-Y. Xia, W.-Q. Liu, F. Zhao and Y. Wang, Inorg. Chim. Acta, 2013, 394, 92–97 CrossRef.
- T. Klemens, A. Świtlicka-Olszewska, B. Machura, M. Grucela, E. Schab-Balcerzak, K. Smolarek, S. Mackowski, A. Szlapa, S. Kula, S. Krompiec, P. Lodowski and A. Chrobok, Dalton Trans., 2016, 45, 1746–1762 RSC.
- J. R. W. McLay, J. J. Sutton, G. E. Shillito, C. B. Larsen, G. S. Huff, N. T. Lucas and K. C. Gordon, Inorg. Chem., 2021, 60, 130–139 CrossRef PubMed.
- E. J. Rohwer, Y. Geng, M. Akbarimoosavi, L. M. L. Daku, O. Aleveque, E. Levillain, J. Hauser, A. Cannizzo, R. Häner, S. Decurtins, R. J. Stanley, T. Feurer and S.-X. Liu, Chem. – Eur. J., 2021, 27, 5399–5403 CrossRef.
- X. Su, W.-J. Wang, Q. Cao, H. Zhang, B. Liu, Y. Ling, X. Zhou and Z.-W. Mao, Angew. Chem., Int. Ed., 2022, 61, e202115800 CrossRef PubMed.
- Y.-Q. Li and K.-Z. Wang, Molecules, 2023, 28, 3229 CrossRef PubMed.
- W. E. Ford and M. A. J. Rodgers, J. Phys. Chem., 1992, 96, 2917–2920 CrossRef.
- N. D. McClenaghan, Y. Leydet, B. Maubert, M. T. Indelli and S. Campagna, Coord. Chem. Rev., 2005, 249, 1336–1350 CrossRef.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- X. Cui, J. Zhao, Z. Mohmood and C. Zhang, Chem. Rec., 2016, 16, 173–188 CrossRef PubMed.
- X. Zhang, Y. Hou, X. Xiao, X. Chen, M. Hu, X. Geng, Z. Wang and J. Zhao, Coord. Chem. Rev., 2020, 417, 213371 CrossRef.
- X. Zhao, Y. Hou, L. Liu and J. Zhao, Energy Fuels, 2021, 35, 18942–18956 CrossRef.
- I. E. Pomestchenko, C. R. Luman, M. Hissler, R. Ziessel and F. N. Castellano, Inorg. Chem., 2003, 42, 1394–1396 CrossRef CAS.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953–1963 RSC.
- W. Wu, J. Sun, S. Ji, W. Wu, J. Zhao and H. Guo, Dalton Trans., 2011, 40, 11550–11561 RSC.
- Q. Li, H. Guo, L. Ma, W. Wu, Y. Liu and J. Zhao, J. Mater. Chem., 2012, 22, 5319–5329 RSC.
- A. Del Guerzo, S. Leroy, F. Fages and R. H. Schmehl, Inorg. Chem., 2002, 41, 359–366 CrossRef.
- N. M. Shavaleev, Z. R. Bell, T. L. Easun, R. Rutkaite, L. Swanson and M. D. Ward, Dalton Trans., 2004, 3678–3688 RSC.
- M. E. Walther and O. S. Wenger, Dalton Trans., 2008, 6311–6318 RSC.
- N. P. Liyanage, W. Yang, S. Guertin, S. S. Roy, C. A. Carpenter, R. E. Adams, R. H. Schmehl, J. H. Delcamp and J. W. Jurss, Chem. Commun., 2019, 55, 993–996 Search PubMed.
- L.-Q. Qiu, K.-H. Chen, Z.-W. Yang and L.-N. He, Green Chem., 2020, 22, 8614–8622 RSC.
- L.-Q. Qiu, K.-H. Chen, Z.-W. Yang, F.-Y. Ren and L.-N. He, Chem. – Eur. J., 2021, 27, 15536–15544 CrossRef PubMed.
- A. Szlapa-Kula, M. Małecka, A. M. Maroń, H. Janeczek, M. Siwy, E. Schab-Balcerzak, M. Szalkowski, S. Maćkowski, T. Pedzinski, K. Erfurt and B. Machura, Inorg. Chem., 2021, 60, 18726–18738 CrossRef PubMed.
- M. Małecka, A. Szlapa-Kula, A. M. Maroń, P. Ledwon, M. Siwy, E. Schab-Balcerzak, K. Sulowska, S. Maćkowski, K. Erfurt and B. Machura, Inorg. Chem., 2022, 61, 15070–15084 CrossRef.
- K. Choroba, M. Penkala, J. Palion-Gazda, E. Malicka and B. Machura, Inorg. Chem., 2023, 62, 19256–19269 CrossRef PubMed.
- R. Das and P. Paira, Dalton Trans., 2023, 52, 15365–15376 RSC.
- L. T. Babu, U. Das, R. Das, B. Kar and P. Paira, Dalton Trans., 2024, 53, 5993–6005 RSC.
- Y. Li, N. Lu, Q. Lin, H. Wang, Z. Liang, Y. Lu and P. Zhang, Chin. Chem. Lett., 2023, 34, 107653 CrossRef CAS.
- K. S. Kisel, J. R. Shakirova, V. V. Pavlovskiy, R. A. Evarestov, V. V. Gurzhiy and S. P. Tunik, Inorg. Chem., 2023, 62, 18625–18640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03237c |
| This journal is © The Royal Society of Chemistry 2025 |