
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Non-aggregated ruthenium naphthalocyanine enabling homogeneous carbene insertion into N–H bonds at low catalyst loading†
Andrey P. Kroitor ab,
Alexander G. Martynov*b,
Yulia G. Gorbunova*bc,
Aslan Yu. Tsivadzebc and
Alexander B. Sorokin*a
ab,
Alexander G. Martynov*b,
Yulia G. Gorbunova*bc,
Aslan Yu. Tsivadzebc and
Alexander B. Sorokin*a
aInstitut de Recherches sur la Catalyse et l'Environnement de Lyon IRCELYON, UMR 5256, CNRS - Université Lyon 1, 2 av. A. Einstein, 69626 Villeurbanne, France. E-mail: alexander.sorokin@ircelyon.univ-lyon1.fr
bFrumkin Institute of Physical Chemistry and Electrochemistry Russian Academy of Sciences, 31-4 Leninsky prospect, 119071, Moscow, Russia. E-mail: martynov@phyche.ac.ru
cKurnakov Institute of General and Inorganic Chemistry Russian Academy of Sciences, 31 Lenisky prospect, 119991 Moscow, Russia. E-mail: yulia@igic.ras.ru
First published on 6th February 2025
Abstract
The novel ruthenium octa-n-butoxy-naphthalocyanine complex was shown to retain an essentially monomeric state in dilute solutions. It was successfully applied as a homogeneous catalyst for carbene insertion into N–H bonds of amines with various substitution patterns, providing high yields of glycine derivatives.
A wide variety of complexes based on porphyrin,1–3 phthalocyanine,4 corrole,5 corrolazine6 and porphyrazine7,8 macrocyclic ligands are widely applied as catalysts for many reactions. In particular, these tetrapyrrolic complexes are robust and versatile catalysts for various carbene transfer reactions.9,10 For instance, carbene insertion into N–H bonds, considered as a challenging transformation owing to the high coordinating properties of amines leading to catalyst poisoning, can provide a wide range of bioactive compounds and pharmaceuticals. Compared to traditional Rh-based catalysts, which are usually required in amounts of 1 to 10 mol%,11 tetrapyrrolic complexes efficiently catalyze carbene transfer reactions at a rather low loading. While ruthenium12–16 and iron15,17–19 porphyrins mediated N–H carbene insertion when using 1 mol% catalyst loading, iridium porphyrin afforded high product yields in the presence of only 0.06 mol%.20 Furthermore, iron corroles were shown to be particularly efficient at 0.1 mol% loading providing a fast carbene insertion into N–H bonds of primary and secondary amines at high substrate concentration.21,22 In turn, iron and ruthenium phthalocyanines exhibited promising activity in cyclopropanation and carbene insertion into N–H bonds.23–29 Our groups have shown that ruthenium octa-n-butoxyphthalocyanines in amounts as low as 0.05–0.2 mol% catalyze the insertion of carbene into N–H bonds of aromatic and aliphatic amines under mild conditions.27,28 The high catalytic efficiency of different tetrapyrrolic complexes suggests that other overlooked porphyrin-like complexes deserve more careful evaluation in carbene transfer reactions. Indeed, modification of the tetrapyrrolic structure strongly influences their spectroelectrochemical properties30–32 and hence should affect their catalytic properties.
Naphthalocyanine complexes can be regarded as the closest phthalocyanine analogues derived from the annulation of benzene moieties onto the isoindoline fragments of a phthalocyanine core. To date, their application in catalysis has been very rare and limited to heterogeneous processes. For example, unsubstituted cobalt naphthalocyaninate CoNc deposited on a rotating disc electrode electrocatalytically reduced oxygen and oxidized hydrazine.33 Cathode-supported CoNc was superior to carbon black in the oxygen reduction reaction (ORR).34 Lithium naphthalocyaninate Li2Nc deposited on graphite catalyzed the electroreduction of CO2 in KHCO3 aqueous solution.35 However, to the best of our knowledge, naphthalocyanine complexes have never been used as catalysts in carbene transfer and other homogeneous reactions. In fact, they tend to strongly aggregate in solution and their aggregation properties are much more pronounced compared to their phthalocyanine counterparts which can hinder access to the metal site and therefore prevent the catalytic reaction. Although several strategies can be employed to suppress naphthalocyanine aggregation—such as dilution,36 heating,37,38 supramolecular interactions,38,39 attachment of appropriate groups to the macrocyclic core39 or addition of axial bulky ligands40—their homogeneous catalytic applications have yet to be demonstrated.
Herein, we report the first catalytic application of a naphthalocyanine complex in a carbene transfer reaction. The ruthenium complex [(γ-BuO)8NcRu](CO), bearing eight n-butoxy groups in the peripheral positions, was synthesized and characterized by UV-Vis, HR ESI MS and 1H NMR techniques. Unexpectedly, [(γ-BuO)8NcRu](CO) is non-aggregated at 2–20 μM concentrations, which allows its application in homogeneous catalysis exemplified by the reaction of amines with ethyl diazoacetate (EDA).
The syntheses of all ruthenium naphthalocyanines described in the literature were performed by the template condensation of the corresponding phthalonitrile in the presence of a ruthenium precursor.41–44 However, the template condensation of 6,7-dibutoxynaphthalene-2,3-dicarbonitrile with Ru3(CO)12 in refluxing o-dichlorobenzene (o-DCB) did not yield even trace amounts of the target ruthenium naphthalocyanine (Scheme 1). Next, we attempted the direct insertion of the ruthenium cation into the preformed macrocycle following a method previously used for the synthesis of ruthenium phthalocyanines (Scheme 1).25,29,45,46 The peripherally substituted octa-n-butoxy-naphthalocyanine H2[(γ-BuO)8Nc] was prepared from the corresponding phthalonitrile by Li-template condensation with subsequent acid treatment of the lithium complex.47 The reaction of H2[(γ-BuO)8Nc] with Ru3(CO)12 proceeded smoothly in refluxing o-DCB with the progressive disappearance of the broad Q-bands of H2[(γ-BuO)8Nc] at 713 nm and 783 nm and the appearance of the narrow Q band of the metal complex at 733 nm (Fig. S1†). The unreacted naphthalocyanine was separated by column chromatography on silica. Further purification by size-exclusion chromatography allowed for the isolation of the target ruthenium naphthalocyanine [(γ-BuO)8NcRu](CO) in a 25% yield. It should be noted that in a similar reaction of peripherally substituted phthalocyanines with Ru3(CO)12 in o-DCB, in addition to the expected mononuclear complexes [PcRu](CO), μ-carbido binuclear complexes [PcRu]2(μ-C) were also formed.25,45 In contrast, we did not observe any other naphthalocyanine complexes in the MALDI TOF spectra of all isolated fractions, indicating more selective ruthenium insertion in the naphthalocyanine case.
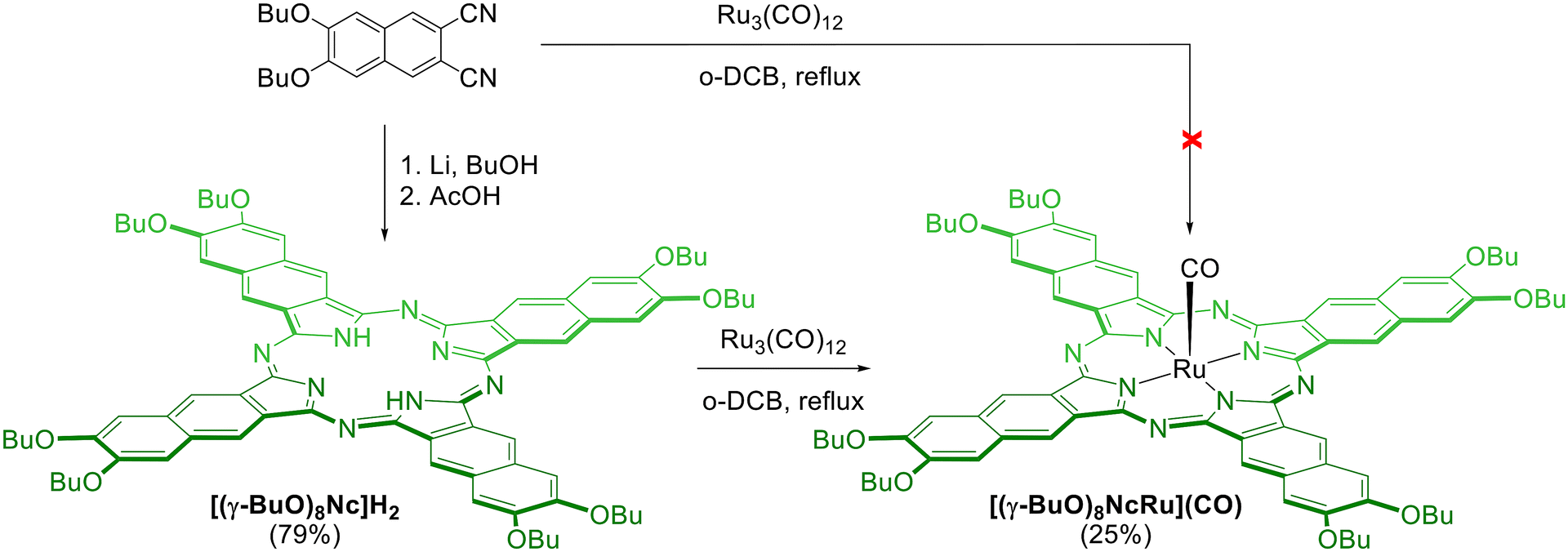 | ||
| Scheme 1 Synthesis of the ruthenium naphthalocyanine [(γ-BuO)8NcRu](CO). | ||
The obtained ruthenium complex was characterized by UV-Vis, 1H NMR, MALDI-TOF MS and HR ESI-MS techniques. The UV-Vis spectrum of naphthalocyanine [(γ-BuO)8NcRu](CO) in CH2Cl2 exhibits a narrow Q-band at 726 nm (Fig. S2†). The UV-vis spectra remained unchanged in CH2Cl2 and in C2H4Cl2 in the concentration range of 2–20 μM (Fig. 1 and Fig. S7†), showing a linear dependence of absorbance intensity on concentration at five wavelengths, which suggests the absence of aggregation. A comparison of the normalized UV-vis spectra of 2 μm and 20 μM solutions indicates very small differences in the Soret region as well as in the shoulders at 697 nm and around 800 nm (Fig. S8†), which might be compatible with a minor aggregation phenomenon. Thus, [(γ-BuO)8NcRu](CO) maintains essentially a monomeric state in dilute solutions, although the presence of trace amounts of aggregated species cannot be excluded. Previous studies showed that the addition of eight butoxy substituents at either peripheral or non-peripheral positions of phthalocyanine complexes prevents their aggregation.25,28 Consequently, the introduction of eight butoxy groups into the naphthalocyanine platform, which is more prone to aggregation due to its extended aromatic plane, should reduce the aggregation. The presence of the CO axial ligand also hinders the aggregation. Thus, the metal-free naphthalocyanine H2[(γ-BuO)8Nc] is strongly aggregated at 10−4–10−5 M concentrations, whereas [(γ-BuO)8NcRu](CO) is in the monomeric form (Fig. S1†).
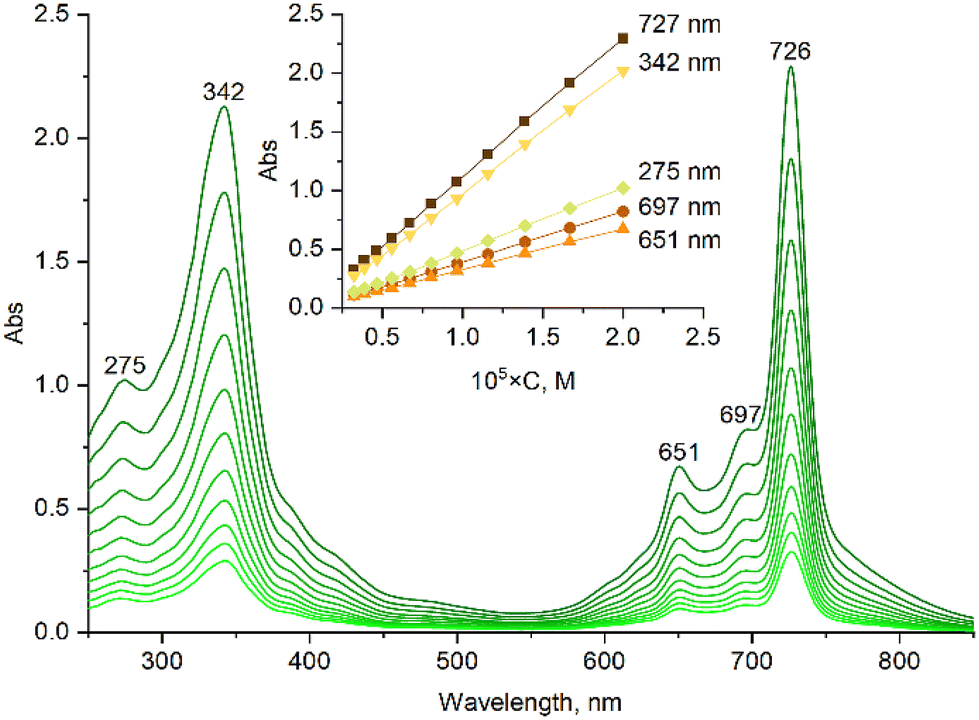 | ||
| Fig. 1 The concentration dependence of the UV-Vis spectra of the ruthenium naphthalocyanine [(γ-BuO)8NcRu](CO) in CH2Cl2. | ||
This is in sharp contrast to the metal-free H2[(β-BuO)8Pc] and its ruthenium complex [(β-BuO)8PcRu](CO) found in the monomeric state in this concentration range in nonpolar solvents such as CH2Cl2 and CHCl3.25
Compared to the peripherally substituted octa-n-butoxy phthalocyanine [(β-BuO)8PcRu](CO), the maximum of the Q band in the UV-vis spectrum of the naphthalocyanine complex bathochromically shifted to 71 nm (Fig. S2†). Since this absorption originates from the HOMO–LUMO transition, this shift can be explained by the destabilization of the HOMO upon annulation with the benzene rings.30
The 1H NMR spectrum of [(γ-BuO)8NcRu](CO) recorded in CDCl3 in the presence of 5 μL pyridine-d5 exhibits two singlet signals at 7.70 and 9.51 ppm attributed to the aromatic protons of the naphthalocyanine macrocycle and four signals at 4.35, 2.03, 1.67 and 1.11 ppm corresponding to the aliphatic chains of the n-butoxy groups (Fig. S3†). Upfield shifted shoulder signals of low intensity near the signals of HAr2 (7.70 ppm), α-CH2 (4.35 ppm), γ-CH2 (1.67 ppm) and CH3 (1.11 ppm) were also observed (Fig. S3†). Given the much higher [(γ-BuO)8NcRu](CO) concentration (∼10−3 M) used in 1H NMR measurements compared to UV-vis measurements and catalytic experiments (2–20 μM), this observation can be explained by minor aggregation occurring even in the presence of 5 μL of pyridine-d5 which was added to prevent the aggregation due to the axial coordination of pyridine. We have recorded the 1H NMR spectrum of [(γ-BuO)8NcRu](CO) in pure pyridine-d5 and observed no additional minor signals, thus confirming the purity of [(γ-BuO)8NcRu](CO) and its low tendency for aggregation.
The MALDI TOF mass spectrum shows two molecular clusters corresponding to mononuclear {[(γ-BuO)8NcRu] + H}+ and binuclear {[(γ-BuO)8NcRu]2 + H}+ species without axial ligands (Fig. S6†).
However, the presence of the coordinated CO was evidenced by the HR ESI-MS technique, which showed the molecular cluster of [(γ-BuO)8NcRu](CO) (Fig. S9†). Previously, the CO axial ligand was also found in ruthenium phthalocyanine complexes.29,45,48–50 Although a range of ruthenium naphthalocyanines have been prepared,41–44,51–53 complexes containing a coordinated CO ligand have not yet been published.
The lack of [(γ-BuO)8NcRu](CO) aggregation at 2–20 μM concentrations prompted us to probe its catalytic activity in the benchmark carbene transfer reaction between ethyl diazoacetate (EDA) and aniline using low catalyst loadings (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalystb (mol%) | Reaction time, h | Aniline conversion % | Yield of double insertion product, % | Yield of single insertion product, % | TON |
| a Reaction conditions: aniline (0.5 mmol), EDA (1.05 mmol), catalyst, C2H4Cl2 (0.5 mL), 40 °C, and Ar. Yields and aniline conversions based on aniline were determined by the 1H NMR method.b A = [(γ-BuO)8NcRu](CO) and B = [(β-BuO)8PcRu](CO). | ||||||
| 1 | A (0.1) | 1 | 100 | 96 | 4 | 1960 |
| 2 | A (0.05) | 3 | 100 | 92 | 8 | 3840 |
| 3 | A (0.025) | 5 | 96 | 91 | 5 | 7480 |
| 4 | A (0.01) | 6 | 100 | 41 | 59 | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
| 5 | A (0.002) | 16 | 40 | 1 | 39 | 20050 |
| 6 | B (0.002) | 16 | 26 | <1 | 26 | 13250 |
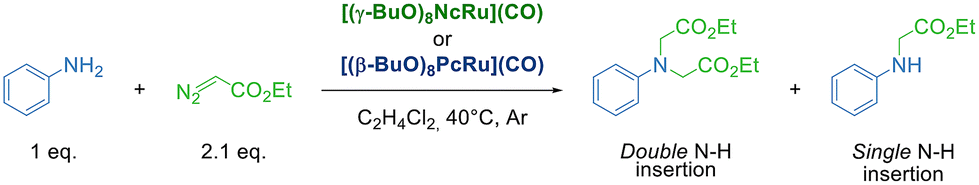
We have initially used 2 equiv. of EDA in order to determine whether [(γ-BuO)8NcRu](CO) is capable of performing single and double N–H insertions. The complete aniline conversion and a 96% yield of the tertiary derivative were obtained using 0.1 mol% [(γ-BuO)8NcRu](CO) (Table 1, entry 1). A decrease in the catalyst loading to 0.025 mol% slightly reduced the yield of the double N–H insertion product (Table 1, entry 3). Further reduction to 0.01 mol% resulted in comparable yields of mono- and disubstituted glycine derivatives (59% and 41% yields, respectively) (Table 1, entry 4). It is noteworthy that even with a 0.002 mol% catalyst loading, a 39% yield of the single N–H insertion product was obtained, although the reaction required 16 h (Table 1, entry 5). The very high turnover number (TON) of 20050 obtained under these conditions indicates the high efficiency of [(γ-BuO)8NcRu](CO) as a catalyst for carbene insertion reactions. Interestingly, when the previously reported phthalocyanine counterpart [(γ-BuO)8PcRu](CO)28 was used under the same reaction conditions, only a 26% yield of the single insertion product and less than 1% yield of the double insertion product were obtained (TON = 13250) (Table 1, entry 6). Previously, iron porphyrin and iron corrole complexes were reported as efficient catalysts for N–H insertion, affording a TON of up to 970 with 0.1 mol% catalyst loading.22 Iridium porphyrin catalyzed both single and double EDA insertion to aromatic and aliphatic amines with a total TON of up to 3180 in the presence of 0.06 mol% catalyst.20 In turn, engineered variants of myoglobin mediated asymmetric N–H carbene insertion of aromatic amines with 2-diazopropanoate to achieve a TON of up to 2470 with 0.02 mol%.54 Thus, the high efficiency of [(γ-BuO)8NcRu](CO) in carbene insertion into amines places this complex among the most efficient catalysts for this reaction.
A broad range of aromatic and aliphatic amines were then evaluated for N–H insertion with 1.1 equiv. of EDA with respect to the substrate using a catalyst loading of 0.0125 mol%. Aniline and its derivatives bearing electron-donating groups (p-Me and p-MeO) reacted smoothly, affording single N–H insertion products with 91–95% yields (Table 2, entries 1–3). Similarly, high product yields were obtained with halogen-substituted substrates (90, 98, 96 and 94% for p-F, p-Cl, o-F and o-Br derivatives) (Table 2, entries 4–7).
a
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Reaction time | Yield of single N–H insertion product, % | Yield of double N–H insertion product, % | TON |
| a Reaction conditions: amine (0.5 mmol), EDA (0.55 mmol), [(γ-BuO)8NcRu](CO) (0.0125 mol%), C2H4Cl2 (0.5 mL), Ar, and 40 °C. | |||||
| 1 |  |
1 h | 95 | 5 | 8400 |
| 2 |  |
1 h | 94 | 6 | 8480 |
| 3 |  |
2 h | 91 | 9 | 8720 |
| 4 |  |
1 h | 90 | 10 | 8800 |
| 5 |  |
1 h | 98 | 2 | 8160 |
| 6 |  |
4 h | 96 | 4 | 8320 |
| 7 | 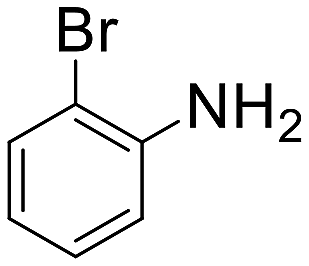 |
1 h 10 min | 94 | Traces | 7520 |
| 8 | 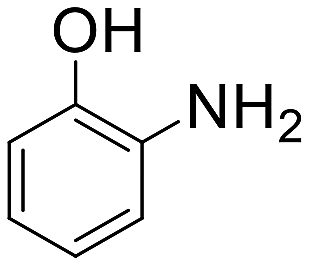 |
30 min | 88 | 2 | 7360 |
| 9 | 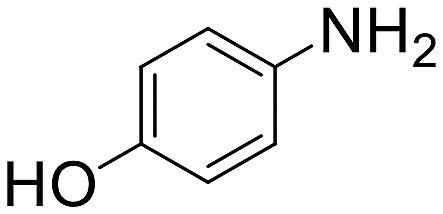 |
30 min | 78 | 20 | 9440 |
| 10 |  |
3 h 40 min | 84 | 2 | 7040 |
| 11 | 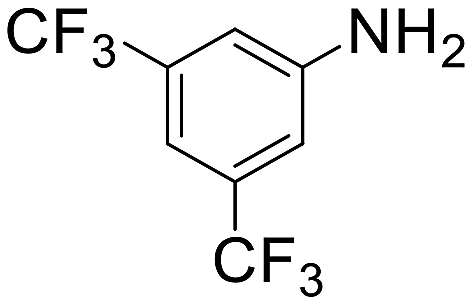 |
3 h 40 min | 93 | 3 | 7920 |
| 12 | 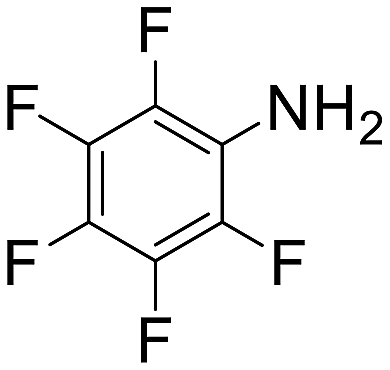 |
4 h 30 min | 73 | 0 | 5840 |
| 13 | 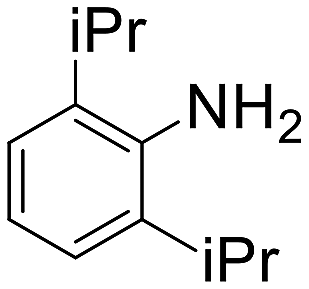 |
4 h | 52 | Traces | 4160 |
| 14 | 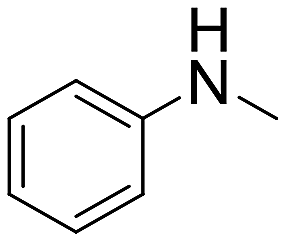 |
21 h | >99 | — | 7920 |
| 15 | 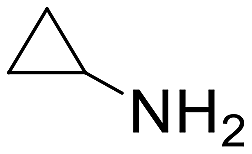 |
19 h | 16 | 0 | 1280 |
| 16 | 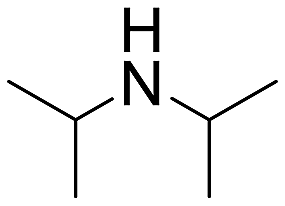 |
21 h | 69 | — | 5520 |
The introduction of strong electron-withdrawing groups, such as p-nitro and 3,5-bis-(trifluoromethyl), was tolerated, giving product yields of 84 and 93%, respectively, after longer reaction times (Table 2, entries 10 and 11). Even such an electron-deficient amine as pentafluoroaniline furnished the corresponding glycine derivative with a 73% yield (Table 2, entry 12). The presence of bulky substituents in the ortho-positions of 2,6-diisopropylamine did not prevent the reaction, though it led to a decrease in the product yield to 52% (Table 2, entry 13). Aliphatic amines were also suitable substrates for carbene insertion but required longer reaction times to achieve satisfactory yields. For instance, cyclopropylamine reacted slowly to furnish a 16% yield after 19 h (Table 2, entry 15). This decrease in the reactivity can be explained by the coordination of the amine to the ruthenium center, thus inhibiting the formation of active metal–carbene species. However, diisopropylamine underwent N–H insertion, giving a 69% product yield, most probably due to the bulky substituents hindering the coordination of this amine to the metal site (Table 2, entry 16). Finally, secondary amines can also be amenable to N–H carbene insertion. For example, N-methylaniline provided a quantitative yield of the target tertiary amine after 21 h (Table 2, entry 14).
To demonstrate the synthetic value of this protocol, we have carried out the cyclopropanation of p-chlorostyrene on a 10 mmol scale with 0.0125 mol% [(γ-BuO)8NcRu](CO) loading. After a 3 h reaction, 1.76 g (83% yield) of the pure target cyclopropanation product was isolated (Fig. S10†), thus confirming the scalability of this method.
The versatility of [(γ-BuO)8NcRu](CO) was further explored using different diazo compounds in the cyclopropanation of styrene (Table 3).
a
|
|||||
|---|---|---|---|---|---|
| Entry | Diazo compound | Reaction time, h | Yield of single N–H insertion product, % | Yield of double N–H insertion product, % | TON |
| a Reaction conditions: aniline (0.5 mmol), diazo compound (0.55 mmol), [(γ-BuO)8NcRu](CO), and C2H4Cl2 (0.5 mL).b 0.0125 mol% catalyst and 40 °C.c 0.02 mol% catalyst and 60 °C. | |||||
| 1b | 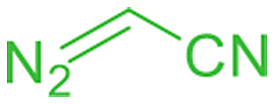 |
17 h | 86 | 13 | 8960 |
| 2b | 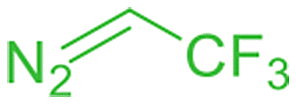 |
1 h | 29 | 0 | 2320 |
| 3c |  |
4 h | 88 | 7 | 4750 |
| 4c |  |
4 h | 84 | 7 | 4550 |
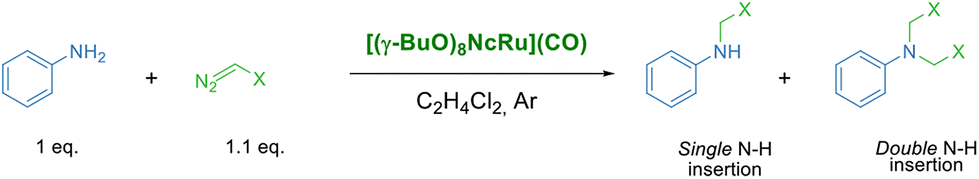
When we used t-butyl diazoacetate and benzyl diazoacetate as carbene precursors at 40 °C, we observed lower activity compared to EDA, most probably because of the bulkiness of t-butyl and benzyl groups. However, a smooth carbene N–H insertion occurred at 60 °C, affording single insertion products with 88 and 84% yields, respectively (Table 3, entries 3 and 4). Diazoacetonitrile was also effectively activated by [(γ-BuO)8NcRu](CO) providing a single insertion product containing the cyano group with an 86% yield (Table 3, entry 1). The introduction of a fluorinated fragment was achieved using 1,1,1-trifluorodiazoethane, albeit with an inferior product yield (Table 3, entry 2). After a 1 h reaction, the reaction mixture was bleached, indicating degradation of [(γ-BuO)8NcRu](CO), leading to a 29% yield of the secondary amine bearing a CF3 group.
Conclusions
Ruthenium naphthalocyanine [(γ-BuO)8NcRu](CO) containing peripheral butoxy substituents was prepared by the direct reaction of the metal-free ligand with Ru3(CO)12 for the first time. The strong tendency for aggregation in solution is a significant drawback of naphthalocyanine complexes, preventing their catalytic applications. In contrast to published naphthalocyanine complexes, [(γ-BuO)8NcRu](CO) shows no appreciable aggregation at 2–20 μM concentrations. Taking advantage of this property, we have demonstrated the first catalytic application of the naphthalocyanine complex exemplified by the carbene transfer reaction. The complex [(γ-BuO)8NcRu](CO) was shown to be a very efficient catalyst for the insertion of ethyl diazoacetate into N–H bonds of sixteen aromatic and aliphatic amines at low catalyst loading (0.0125 mol%) and a quasi-equimolar EDA/substrate ratio. A wide range of monosubstituted glycine derivatives was obtained with up to quantitative yields and up to 9440 turnover numbers. Importantly, a catalyst loading as low as 0.002 mol% can be successfully used in the reaction between aniline and EDA to afford the single N–H insertion product with a TON of 20050. The generality of this methodology was further demonstrated using several diazo compounds as carbene precursors. The promising results obtained in this work on the carbene insertion into N–H bonds suggest that the yet overlooked naphthalocyanine complexes might be efficient catalysts for other types of reactions.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CNRS and RFBR through the International Emerging Action 2021 and the research project 21-53-15004 and by the Agence Nationale de Recherche (ANR), project ANR-23-CE07-0039.References
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- M. Costas, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS.
- H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899–1909 RSC.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- A. Mahammed and Z. Gross, J. Am. Chem. Soc., 2023, 145, 12429–12445 CrossRef CAS PubMed.
- D. P. Goldberg, Acc. Chem. Res., 2007, 40, 626–634 CrossRef CAS PubMed.
- L. P. Cailler, M. Clémancey, J. Barilone, P. Maldivi, J. M. Latour and A. B. Sorokin, Inorg. Chem., 2020, 59, 1104–1116 Search PubMed.
- M. S. Yusubov, C. Celik, M. R. Geraskina, A. Yoshimura, V. V. Zhdankin and V. N. Nemykin, Tetrahedron Lett., 2014, 55, 5687–5690 CrossRef CAS.
- C. Damiano, P. Sonzini and E. Gallo, Chem. Soc. Rev., 2020, 49, 4867–4905 RSC.
- D. Intrieri, D. M. Carminati and E. Gallo, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2016, vol. 38, pp. 1–99 Search PubMed.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef CAS PubMed.
- E. Galardon, P. Le Maux and G. Simonneaux, J. Chem. Soc., Perkin Trans. 1, 1997, 1, 2455–2456 RSC.
- P. Le Maux, I. Nicolas, S. Chevance and G. Simonneaux, Tetrahedron, 2010, 66, 4462–4468 CrossRef CAS.
- C. M. Ho, J. L. Zhang, C. Y. Zhou, O. Y. Chan, J. J. Yan, F. Y. Zhang, J. S. Huang and C. M. Che, J. Am. Chem. Soc., 2010, 132, 1886–1894 CrossRef CAS PubMed.
- H. F. Srour, P. Le, S. Chevance, D. Carrié, N. Le and G. Simonneaux, J. Mol. Catal. A: Chem., 2015, 407, 194–203 Search PubMed.
- L. Chen, H. Cui, Y. Wang, X. Liang, L. Zhang and C. Su, Dalton Trans., 2018, 47, 3940–3946 Search PubMed.
- I. Aviv and Z. Gross, Chem. Commun., 2006, 4477 RSC.
- L. K. Baumann, H. M. Mbuvi, G. Du and L. K. Woo, Organometallics, 2007, 26, 3995–4002 Search PubMed.
- J. Li, D. Zhang, J. Chen, C. Ma and W. Hu, ACS Catal., 2020, 10, 4559–4565 Search PubMed.
- B. J. Anding and L. K. Woo, Organometallics, 2013, 32, 2599–2607 CrossRef CAS.
- I. Aviv and Z. Gross, Synlett, 2006, 951–953 Search PubMed.
- I. Aviv and Z. Gross, Chem. – Eur. J., 2008, 14, 3995–4005 CrossRef CAS PubMed.
- H.-H. Liu, Y. Wang, Y.-J. Shu, X.-G. Zhou, J. Wu and S.-Y. Yan, J. Mol. Catal. A: Chem., 2006, 246, 49–52 CrossRef CAS.
- V. B. Sharma, S. L. Jain and B. Sain, Catal. Commun., 2006, 7, 454–456 Search PubMed.
- A. P. Kroitor, L. P. Cailler, A. G. Martynov, Y. G. Gorbunova, A. Y. Tsivadze and A. B. Sorokin, Dalton Trans., 2017, 46, 15651–15655 RSC.
- L. P. Cailler, A. G. Martynov, Y. G. Gorbunova, A. Y. Tsivadze and A. B. Sorokin, J. Porphyrins Phthalocyanines, 2019, 23, 497–506 Search PubMed.
- L. P. Cailler, A. P. Kroitor, A. G. Martynov, Y. G. Gorbunova and A. B. Sorokin, Dalton Trans., 2021, 50, 2023–2031 RSC.
- A. P. Kroitor, A. A. Dmitrienko, A. G. Martynov, Y. G. Gorbunova and A. B. Sorokin, Org. Biomol. Chem., 2023, 21, 69–74 Search PubMed.
- A. P. Kroitor, A. A. Sinelshchikova, M. S. Grigoriev, G. A. Kirakosyan, A. G. Martynov, Y. G. Gorbunova and A. B. Sorokin, Dyes Pigm., 2024, 222, 111830 CrossRef CAS.
- Y. Rio, M. S. Rodríguez-Morgade and T. Torres, Org. Biomol. Chem., 2008, 6, 1877–1894 Search PubMed.
- V. Novakova, P. Reimerova, J. Svec, D. Suchan, M. Miletin, H. M. Rhoda, V. N. Nemykin and P. Zimcik, Dalton Trans., 2015, 44, 13220–13233 RSC.
- R. V. Belosludov, D. Nevonen, H. M. Rhoda, J. R. Sabin and V. N. Nemykin, J. Phys. Chem. A, 2019, 123, 132–152 CrossRef CAS PubMed.
- M. Isaacs, M. J. Aguirre, A. Toro-Labbé, J. Costamagna, M. Páez and J. H. Zagal, Electrochim. Acta, 1998, 43, 1821–1827 CrossRef CAS.
- J. R. Kim, J. Y. Kim, S. B. Han, K. W. Park, G. D. Saratale and S. E. Oh, Bioresour. Technol., 2011, 102, 342–347 Search PubMed.
- T. V. Magdesieva, K. P. Butin, T. Yamamoto, D. A. Tryk and A. Fujishima, J. Electrochem. Soc., 2003, 150, E608 CrossRef CAS.
- A. V. Yagodin, A. G. Martynov, Y. G. Gorbunova and A. Y. Tsivadze, Macroheterocycles, 2021, 14, 130–134 CrossRef CAS.
- M. T. M. Choi, P. P. S. Li and D. K. P. Ng, Tetrahedron, 2000, 56, 3881–3887 Search PubMed.
- E. A. Safonova, M. A. Polovkova, A. G. Martynov, Y. G. Gorbunova and A. Y. Tsivadze, Dalton Trans., 2018, 47, 15226–15231 RSC.
- E. A. Safonova, A. G. Martynov, M. A. Polovkova, E. A. Ugolkova, V. V. Minin, Y. G. Gorbunova and A. Y. Tsivadze, Dyes Pigm., 2020, 180, 108484 CrossRef CAS.
- T. Keleş, B. Barut, A. Özel and Z. Biyiklioglu, Bioorg. Chem., 2021, 107, 104637 Search PubMed.
- M. Hanack, S. Knecht and R. Polley, Chem. Ber., 1995, 128, 929–933 CrossRef CAS.
- T. Rawling, A. M. McDonagh and S. B. Colbran, Inorg. Chim. Acta, 2008, 361, 49–55 Search PubMed.
- M. Hanack and R. Polley, Inorg. Chem., 1994, 33, 3201–3204 Search PubMed.
- J. F. Vollano, G. E. Bossard, S. A. Martellucci, M. C. Darkes, M. J. Abrams and R. C. Brooks, J. Photochem. Photobiol., B, 1997, 37, 230–235 Search PubMed.
- A. P. Kroitor, A. G. Martynov, Y. G. Gorbunova, A. Y. Tsivadze and A. B. Sorokin, Eur. J. Inorg. Chem., 2019, 1923–1931 Search PubMed.
- M. V. Cruz, M. Cyr, H. Boughanmi, J. García-Calvo, J. Brusso, T. Torres and B. H. Lessard, Polym. Chem., 2024, 15, 3475–3479 Search PubMed.
- A. G. Martynov, G. S. Berezhnoy, E. A. Safonova, M. A. Polovkova, Y. G. Gorbunova and A. Y. Tsivadze, Macroheterocycles, 2019, 12, 75–81 CrossRef CAS.
- L.-C. Song, F.-X. Luo, B.-B. Liu, Z.-C. Gu and H. Tan, Organometallics, 2016, 35, 1399–1408 CrossRef CAS.
- J. Fernández-Ariza, R. M. Krick Calderón, M. S. Rodríguez-Morgade, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2016, 138, 12963–12974 CrossRef PubMed.
- C. A. Enow, C. Marais and B. C. B. Bezuidenhoudt, J. Porphyrins Phthalocyanines, 2012, 16, 403–412 CrossRef CAS.
- M. Hanack and Y.-G. Kang, Chem. Ber., 1991, 124, 1607–1612 CrossRef CAS.
- S. Knecht, R. Polley and M. Hanack, Appl. Organomet. Chem., 1996, 10, 649–660 CrossRef CAS.
- R. R. Dasari, M. M. Sartin, M. Cozzuol, S. Barlow, J. W. Perry and S. R. Marder, Chem. Commun., 2011, 47, 4547–4549 RSC.
- V. Steck, D. M. Carminati, N. R. Johnson and R. Fasan, ACS Catal., 2020, 10, 10967–10977 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data of compounds. See DOI: https://doi.org/10.1039/d4dt03263b |
| This journal is © The Royal Society of Chemistry 2025 |