
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of heptacoordinated molybdenum(II) complexes supported with 2,6-bis(pyrazol-3-yl)pyridine (bpp) ligands†
Arno
Estival
a,
Luis E.
Blancarte
 b,
Loïc
Pinto
a,
Romane
Pointis
a,
Nathan
Galas
a,
Alix
Sournia-Saquet
a,
Laure
Vendier
a,
Rosa
Santillan
c,
Norberto
Farfán
b,
Jean-Baptiste
Sortais
a,
Mary
Grellier
a and
Antoine
Simonneau
*a
b,
Loïc
Pinto
a,
Romane
Pointis
a,
Nathan
Galas
a,
Alix
Sournia-Saquet
a,
Laure
Vendier
a,
Rosa
Santillan
c,
Norberto
Farfán
b,
Jean-Baptiste
Sortais
a,
Mary
Grellier
a and
Antoine
Simonneau
*a
aLCC-CNRS, Université de Toulouse, CNRS, UPS, 205 route de Narbonne, BP44099 F-31077 Toulouse cedex 4, France. E-mail: antoine.simonneau@lcc-toulouse.fr
bFacultad de Química, Departamento de Química Orgánica, Universidad Nacional Autónoma de México, 04510 CDMX, Mexico
cDepartamento de Química, Centro de Investigación y de Estudios Avanzados del IPN, México D.F. Apdo. Postal 14–740, 07000, Mexico
First published on 6th January 2025
Abstract
Functional pincer ligands that engage in metal–ligand cooperativity and/or are capable of redox non-innocence have found a great deal of success in catalysis. These two properties may be found in metal complexes of the 2,6-bis(pyrazol-3-yl)pyridine (bpp) ligands. With this goal in mind, we have attempted the coordination of 2,6-bis(5-trifluoromethylpyrazol-3-yl)pyridine (LCF3) and its tBu analogue 2,6-bis(5-tert-butylpyrazol-3-yl)pyridine (LtBu) to Mo(0) by reactions with mixed phosphine/carbonyl complexes [Mo(CO)2(MeCN)n−1(PMe3−nPhn)5−n] 1–3 (1 ≤ n ≤ 3). These afforded mixtures of several Mo compounds among which low yields of heptacoordinated Mo(II) complexes [Mo(CO)2(bpp)(PMe3−nPhn)2] 4a–c (LCF3-supported) and 5a–c (LtBu-supported) bearing a doubly deprotonated bpp ligand were systematically produced. More selective syntheses of 4a–c and 5a–c were achieved by repeating these experiments in the presence of an oxidant (AgOAc or Ag2O), with moderate to good yields. 4a–c and 5a–c were characterized by means of NMR, IR and UV-Vis spectroscopies, sc-XRD and cyclic and square-wave voltammetries for 4a, 4b and 5b. The deprotonated LtBu ligand in 5a–c is re-protonated with 2 equiv. of HOTf to afford the dicationic [Mo(CO)2(LtBu)(PMe3−nPhn)2][OTf]2 complexes 6a–c. Acidic treatment of 4a–c led to the decomposition of the complexes.
Introduction
Tridentate ligands that enforce meridional coordination fall under the generic denomination of “pincer” ligands. By providing access to well-defined and easily tunable coordination spheres, pincer ligands have found widespread use in various fields of catalysis and inorganic and organometallic chemistry.1–6 By introduction of functions on the pincer ligand that may participate in metal–ligand cooperativity and/or offer the possibility to store electrons to grant it (photo)redox non-innocence, chemists are able to design pincer-supported metal complexes with specifically engineered reactivity.7–13 The introduction of nitrogenous groups or heterocycles remains a prominent strategy to provide either property, sometimes both. As an example, the bis(pyrazol-3-yl)pyridine (bpp) ligand family14 allows, thanks to its pyrazolyl arms, embedding of proton-responsive sites, while the conjugation of the three N-heterocycles provides a low-energy LUMO suited for electron storage or charge transfer. Substituents may easily be introduced on N1 or C5 of the pyrazoles to modulate the properties of the ligand. Historically, bpp ligands were first employed for the design of Fe-based spin-crossover (SCO) complexes and materials, and Fe(bpp) compounds continue to actively nurture this field of research.15–20 Nowadays, the bpp platform has been associated with many elements of the p, d and f blocks to provide complexes with a vast range of applications: luminescence,21–27 imaging,28 liquid–liquid extraction,29,30 medicinal chemistry,31–33 small molecule activation34–42 and catalysis.43–48 In the past few years, our group has been interested in the use of phosphine-supported molybdenum and tungsten complexes for small molecule activation.49–57 While phosphines remain generally spectator ligands, proton-responsiveness and redox non-innocence are two properties that are desirable for this field of research. Because the bpp platform potentially offers both, we have attempted its pairing with molybdenum, a metal curiously absent from the catalogue of the already-reported, bpp-supported complexes,58 fueled by the recent successes of Mo-pincer complexes in catalysis.59–69 In this article, we show that our initial goal of coordinating different bpp ligands to Mo(0) ended in failure, presumably because the bpp platform is not suited for this oxidation state when other electron-donating ligands are present, promoting uncontrolled redox chemistry that leads to non-selective reaction outcomes. However, variable amounts of bpp/phosphine-supported, hepta-coordinated Mo(II) complexes in which the bpp ligand is deprotonated could be collected. By adjusting the reaction conditions, we were able to isolate and characterize in good to high yields a series of these compounds, the first Mo complexes incorporating the bpp platform (Fig. 1). All were characterized by multi-nuclei NMR spectroscopy, IR and UV/Vis spectrophotometry, single crystal X-ray diffraction (sc-XRD) and cyclic voltammetry (CV). The proton-responsiveness of the deprotonated bpp ligand in these edifices is demonstrated.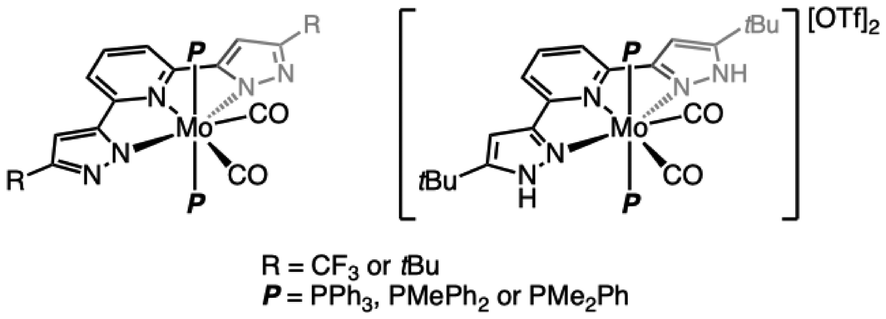 | ||
| Fig. 1 Overview of the new heptacoordinated Mo(II) complexes described in this article. | ||
Results and discussion
Inspired by a recent body of work on pincer-supported molybdenum catalysts from the Beller group,63–65,67,70 we attempted the coordination of the bpp platform to the family of heteroleptic Mo(0) complexes [Mo(CO)2(MeCN)n−1(PMe3−nPhn)5−n] 1–3 (1 ≤ n ≤ 3).71–74 We selected two different bpp ligands bearing either CF3 (LCF3) or tBu (LtBu) groups at their C5 positions and exerting opposite electronic effects on the platform. Attempts at coordinating either LtBu or LCF3 to 1–3 under various conditions (refluxing toluene, 3–24 h) led to surprisingly unselective reaction mixtures, particularly messy in the LtBu complexation experiments. From the reaction of 2 with LCF3, we could isolate the major diamagnetic compound 4b from the reaction mixture (refluxing toluene, 3 h) bearing two PMePh2 ligands (31P{1H} NMR δ = 22.6 ppm, 19F NMR δ = −61.5 ppm) and retaining two carbonyl ligands (νsym = 1880 cm−1 and νasym = 1948 cm−1) in low yield (35%). Slow diffusion of hexane into a concentrated toluene solution allowed us to grow crystals suitable for single-crystal X-ray diffraction (sc-XRD) analysis. The molecular structure of 4b is shown in Fig. 2, showing a C2-symmetric, heptacoordinated molybdenum complex in which LCF3 is fully complexed to the Mo center. The C2 axis spreads along the Mo–Npy bond (py = pyridine), and in the equatorial plane are found the phosphines, in trans relationship to each other, and the two Npz atoms (pz = pyrazole). The carbonyl ligands complete the 7-coordination environment in an overall capped trigonal prismatic (CTP) geometry, occupying the edges as predicted for π-accepting ligands by molecular orbital analysis.75 Attempts to localize protons on the unbound nitrogen atoms of the pyrazole arms in the Fourier density map were unsuccessful, while their positioning generated negative electron density in their environment. This coupled with the fact that a 7-coordinated Mo(0) compound would have 20 valence electrons as well as the absence of detectable Npz–H proton resonances in the 1H NMR spectrum convinced us we isolated a Mo(II) compound supported by a deprotonated LCF3 ligand. Diamagnetism dismisses the non-innocent character of the ligand in this complex. We were unable to identify the other product of these reactions, but the isolation of 4b made us postulate that uncontrolled electron transfers may take place and that the redox active bpp platform76,77 was perhaps not suited to stabilize molybdenum in oxidation state 0. We therefore wondered whether it would be possible to obtain 4b selectively from 2 by adding two equivalents of a one electron oxidant. Indeed, in THF and in the presence of AgOAc, 4b was isolated as a red crystalline solid from the reaction mixture in a much higher yield (69%, see Scheme 1). Repeating the experiment with precursors 1 or 3 allowed us to prepare compounds 4a and 4c, respectively, isolated as red powders (Scheme 1). Unfortunately, these conditions were not optimal when attempting complexation of LtBu, as comparatively lower yields of the 5a–c congeners were recorded due to the formation of unidentified side-products. Reasoning that the acetate anion was probably assisting deprotonation of the LCF3 ligand in the reactions affording 4a–c, we evaluated silver(I) oxide (Ag2O) as oxidant having a more basic anion we thought would be more suited to the expectedly higher pKa of the LtBu ligand. This approach led to a substantially cleaner outcome after a comparatively longer reaction time for 5a and facilitated its isolation (51% yield), but no difference was observed for the synthesis of 5b and 5c, even when excess Ag2O was engaged (44 and 51% yield, respectively). Crystals generating sc-XRD data of sufficient quality could be grown for compounds 4a–c and 5b from concentrated THF solutions layered with pentane. In the case of 5a, and despite several crystallization attempts, only microcrystalline materials were obtained. For 5c, tiny, non-monocrystalline platelets were systematically obtained, some of them diffracting in the XRD apparatus. This brought confirmation of the atomic connectivity but the data were not of sufficient quality to comment on the metrics (Fig. 2). Pertinent structural parameters for 4a–c and 5b are gathered in Table 1. Akin to 4a, 4b and 4c and 5b and 5c all feature a capped trigonal prismatic geometry, with a C2 axis along the Mo–Npy bond and bisecting the OC–Mo–CO angle. Distortions from ideality occurring in the lattice are flagrant in the case of 4c and 5c for which the respective LCF3 and LtBu ligands appear convex and the P–Mo–P array deviates significantly from linearity. In 5b, the pyrazole arms are oppositely tilted away from the pyridine plane. All compounds of the LCF3 series crystallized in the monoclinic system, while 5b and 5c were found in the orthorhombic and triclinic ones, respectively. For the four complexes, the Mo atom is found at 2.16–2.18 Å from the nitrogen atoms of the pyrazol arms and 2.19–2.21 Å from the pyridine's N atom. No significant change is noticed when comparing the LCF3- and LtBu-supported complexes. For the LCF3 series, the shortest Mo–P bonds are found in 4b (PMePh2 ligand), a counter-intuitive observation since we would have expected the length of the Mo–P bonds would follow the trend of the phosphine's respective cone angles.78 However, the Mo–CO bond lengths correlate rather well with the Tolman electronic parameter78 (TEP) thereof, since a progressive shortening is observed as the TEP decreases (PPh3 > PMePh2 > PMe2Ph), but the same is not true when considering the C–O bond lengths. Again, PMePh2-supported 4b stands out with the longest recorded C–O bonds. | ||
| Fig. 2 Molecular structures of compounds 4a–c and 5b and 5c in the solid state. Thermal ellipsoids at the 25% probability level. For clarity, hydrogen atoms are omitted and phosphorus substituents appear as wireframe. Two molecules are present in the asymmetric unit of the crystal of 5c (Z′ = 2), and only one is represented. One of the CF3 groups of 4a presents a disorder. | ||
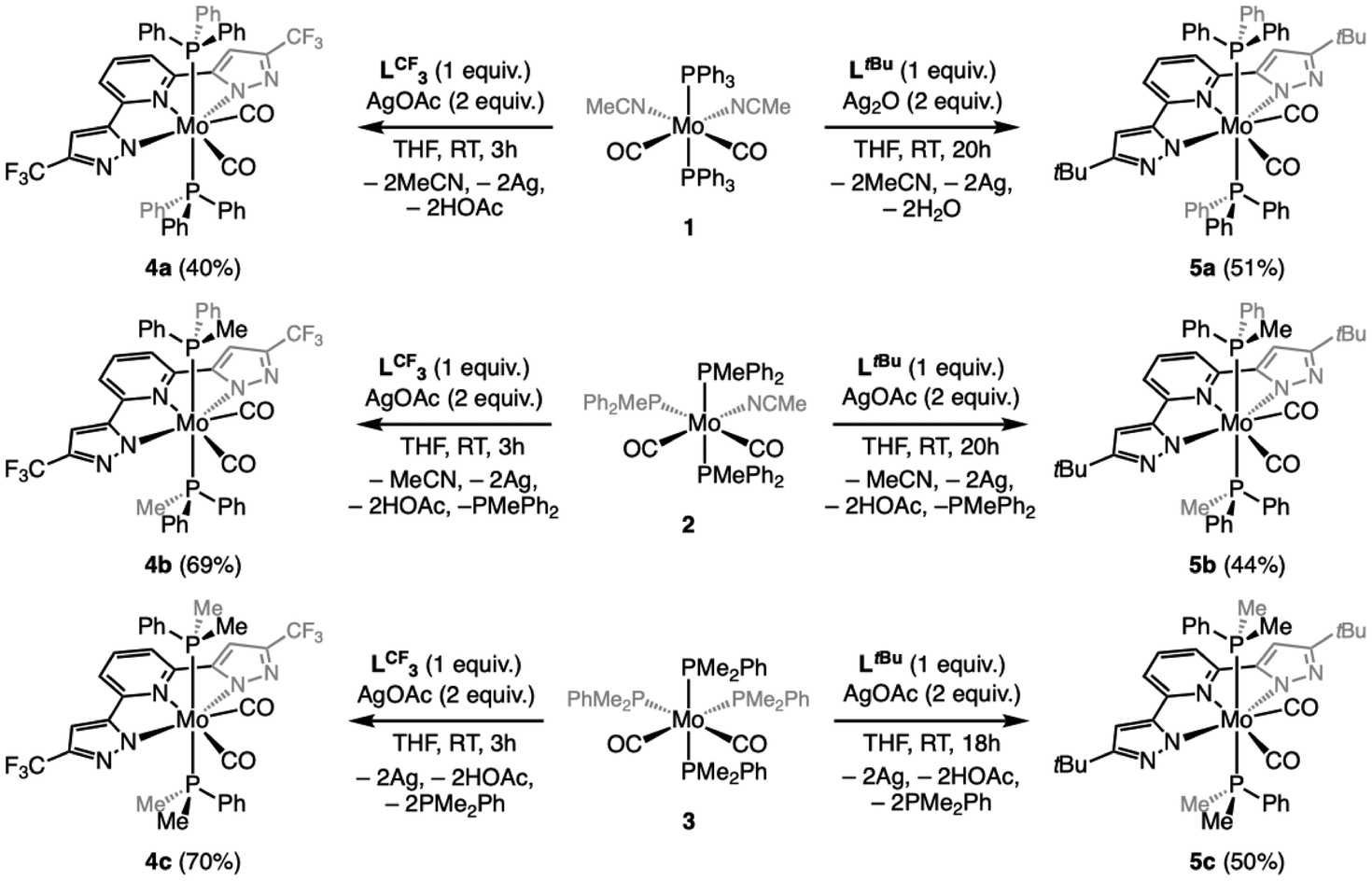 | ||
| Scheme 1 Synthesis of the LCF3-supported complexes 4a–c and LtBu-supported 5a–c. | ||
| 4a | 4b | 4c | 5b | |
|---|---|---|---|---|
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21/n | P21/n | P21/c | P b c a |
| Mo–N2 (Å) | 2.177(2) | 2.171(2) | 2.178(2) | 2.173(1) |
| Mo–N3 (Å) | 2.206(2) | 2.200(2) | 2.192(2) | 2.190(1) |
| Mo–N4 (Å) | 2.182(2) | 2.162(2) | 2.180(2) | 2.168(1) |
| Mo–P1 (Å) | 2.5711(6) | 2.5131(8) | 2.5540(7) | 2.5218(7) |
| Mo–P2 (Å) | 2.5442(6) | 2.5226(8) | 2.5190(7) | 2.5372(7) |
| Mo–C1 (Å) | 1.991(2) | 1.974(3) | 1.970(3) | 1.963(2) |
| Mo–C2 (Å) | 1.986(2) | 1.979(3) | 1.971(3) | 1.980(2) |
| C1–O1 (Å) | 1.137(3) | 1.159(3) | 1.144(4) | 1.152(2) |
| C2–O2 (Å) | 1.149(3) | 1.168(4) | 1.151(4) | 1.147(2) |
Comparison of NMR spectroscopy data does not reveal any singular feature. The aromatic signals of the ligand in 1H NMR are expectedly downfield shifted in the case of the LCF3 series. Of note, the bpp ligand exerts a limited influence on the chemical shift of the phosphorus resonances: 37.8 (4a) vs. 37.1 ppm (5a), 22.6 ppm for both 4b and 5b, and 9.6 (4c) vs. 11.1 ppm (5c).
Infrared spectroscopy analyses were performed on solid samples by the ATR method (Fig. S41–49†). Two symmetric and asymmetric C–O stretching bands are observed between 1950 and 1870 cm−1 (Table 2). Lower stretching frequencies were expectedly recorded for the LtBu series, with differences of less than 10 wavenumbers from one series to the other. Only the symmetric C–O stretches follow the electronic parameter trend of the phosphine ligand. Electronic spectroscopy (Table 2, Fig. 3 and S50–S56†) did not allow detection of transitions in the visible region, even at high concentrations (10–3 M−1). All complexes strongly absorb below 275 nm, which is not surprising given the conjugated aromatic nature of the bpp ligand and the presence of aromatic substituents on the phosphines, both permitting symmetry-allowed intraligand (IL) π–π* transition.47 Shoulders are visible for 4a–c and 5a at 252, 247, 239 and 254 nm, respectively. The energetic cost of these transitions increases with the donating nature of the phosphine. In the LCF3 series, all complexes feature a less intense absorption at 291 nm, clearly visible for 4b and 4c but rather broad in the case of 4a. Two other bands can be found in the recorded electronic spectra when moving towards near UV, difficult to discern in the case of PPh3-supported 4a and 5a, but clearly visible for 4b and 4c and 5b and 5c. These two transitions are unchanging within a bpp series. The most blue-shifted ones have a higher energy cost in the case of the LtBu series. Although trends can be extracted from the electronic spectra, the particular CTP geometry combined with the complexity of the ligand sphere makes it difficult for any tentative assignment of these transitions through a qualitative molecular orbital (MO) analysis, but few comments can be made. Bai et al. have shown in a computational study on the spectroscopic properties of homoleptic bis(2-pyridylpyrazolate)-supported square planar platinum(II) complexes79 that the LUMO of the complexes is always a π* MO mainly localized on the pyridyl moiety, whatever electron-donating (tBu) or -withdrawing (CF3) group is carried by the pyrazolate arm. The absorptions with the largest molar absorptivity coefficient are mostly due to IL π → π* excitation, d → π* metal–ligand charge transfer (MLCT) or a mix thereof, in line with what is frequently observed for other pyridyl-azolate complexes,80 while ligand field (LF) transitions occur in the far-UV. Given the similarities in the UV-Vis spectra within a bpp series (flagrant when comparing 4b and 4c or 5b and 5c, Fig. 3) it is probable that the observed bands are the results of bpp-centered π → π* IL transitions, or d → π*bpp MLCT ones, in line with the recorded molar absorption coefficients (>1000 L mol−1 cm−1, Table 2). The d orbitals in a model C2v CTP ML7 complex (where L is σ-donating only) are all non-degenerate, with a set of two stabilized orbitals (dx2−y2 and dyz, both filled with an electron pair in the case of d4 Mo(II)) and three destabilized ones (dxz, dz2, and dxy) (Fig. 4).75 Both filled dx2−y2 and dyz orbitals overlap with π* (CO, bpp) or σ* (phosphines) orbitals located on the ligands, making various MLCT transitions possible. The dyz orbital is oriented for overlap with the pyridyl component of a π*bpp orbital while being unperturbed by the phosphines. If IL and dyz → π* transitions govern the near-UV part of the electronic spectrum of 4b and 4c and 5b and 5c, the phosphines should not exert a significant influence. Interestingly, the study by Bai et al. shows that the HOMO–LUMO gap grows larger when hydrogen is substituted by electron-withdrawing groups (–CF3 or –C3F7) at the 3-position of the pyrazole ring, resulting in an overall blue-shift of the absorption. The trend is reversed when a tBu group is present. In our case, considering the lowest energy absorption, 5b and 5c (LtBu-supported) exhibit transitions at 375 and 373 nm, respectively, red-shifted compared to the one of 4b and 4c (LCF3-supported, both 359 nm). We cannot exclude that other MLCT transitions such as d → π*CO81 or d → σ*P82–85 participate also in the overall spectrum.
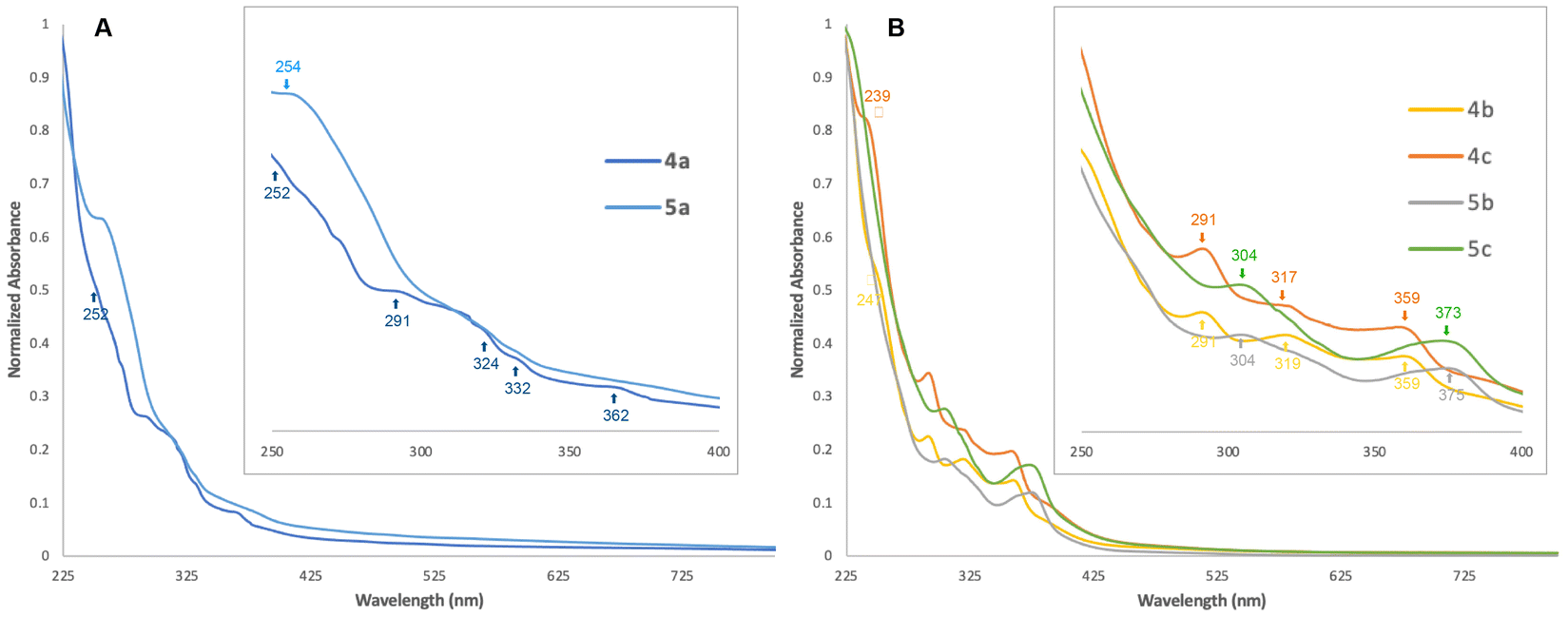 | ||
| Fig. 3 Normalized UV-Vis spectra recorded at ca. 10−5 M−1 with magnification of the 250–400 nm region of (A) 4a and 5a and (B) 4b and 4c and 5b and 5c. | ||
 | ||
| Fig. 4 d orbital splitting for a model C2v ML7 complex (left) and qualitative prediction of the frontier orbitals for the series of complexes 4a–c and 5a–c with depiction of possible overlap of filled d orbitals with ligands’ π-shaped MOs. | ||
| 4a | 4b | 4c | 5a | 5b | 5c | |
|---|---|---|---|---|---|---|
| ν(CO)as (cm−1) | 1944 | 1948 | 1934 | 1938 | 1940 | 1946 |
| ν(CO)sym (cm−1) | 1888 | 1880 | 1873 | 1886 | 1879 | 1877 |
| λ abs (nm) (ε [L mol−1 cm−1]) | 252 (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 417) 417) |
247 (9969) | 239 (12007) |
254 (19584) |
304 (3078) | 304 (4606) |
| 291 (7019) | 291 (4090) | 291 (5010) | 375 (2019) | 373 (2844) | ||
| 324 (4492) | 319 (3308) | 317 (3470) | ||||
| 332 (3623) | 359 (2588) | 359 (2859) | ||||
| 362 (2221) |
The electrochemical behavior of complexes 4a, 4b and 5b was examined in THF by cyclic and square wave voltammetries (Table 3, Fig. 5 and S63–84†), with tetra(n-butyl)ammonium hexafluorophosphate (TBA[PF6]) as the supporting electrolyte. Towards cathodic potentials, voltammograms (Fig. 5, bottom left and S73 and S77†) reveal an irreversible reduction wave for 4b and 5b occurring at Ep = −1.63 V and −2.00 V vs. SCE, respectively, and two very close reductions at Ep = −1.67 and −1.83 V for 4a. The free, protonated ligands LCF3 and LtBu both exhibit an irreversible reduction wave under the same conditions at slightly lower potentials (Ep = −1.93 and −2.21 V vs. SCE, respectively, see Fig. S63–68†). The more cathodically shifted potential in the case of the tBu-substituted compound (5b) is consistent with the inductive donor effects of the tBu groups resisting reduction. Irreversibility could thus stem from a ligand-centered reduction that compromises the stability of the complexes, which is further corroborated by equal first reduction peak potentials for 4a and 4b, both supported by LCF3. While a ligand-centered reduction indicates its possible non-innocent character, its irreversible nature suggests that practical application may be complicated. For both 4a and 4b, this event is followed by a second irreversible wave at Ep = −1.83 and −2.36 V, respectively. It is possible that the less σ-donating characteristic of PPh3 grants access to an irreversible Mo2+ → Mo1+ reduction event in 4a that is shifted cathodically for 4b but, within the swept voltage range, out of reach for 5b that features a more electron-rich ligand sphere. Upon return to anodic potentials, several oxidation waves between −1.5 and 0 V as well as an increase of capacitive current are noticed, suggesting the decomposition of the reduced complexes into several, unidentified species that may also adsorb on the working electrode. Beyond 0 V, CV curves of 4a, 4b, and 5b show a quasi-reversible wave at E1/2 = 0.97, 0.92, and 0.56 V vs. SCE, respectively, also observed when first scanning towards anodic potentials. This can reasonably be assigned to Mo2+ → Mo3+ oxidation (Fig. 5, bottom). Loss of reversibility is observed upon return of voltage sweeping from high potentials (Fig. 5, bottom right, 4b). This is followed by a second, smaller peak at Ep = 1.27, 1.19 and 0.94 V vs. SCE, respectively, more or less reversible. Depending on the product, other irreversible oxidation waves may be observed. The various mechanisms have not been identified, but we can consider the following hypotheses which are plausible among others: CO or phosphine loss as their ligand properties become a poor match for high oxidation state Mo, ligand degradation, or fluoride abstraction from the supporting electrolyte by a now-electrophilic metal center.
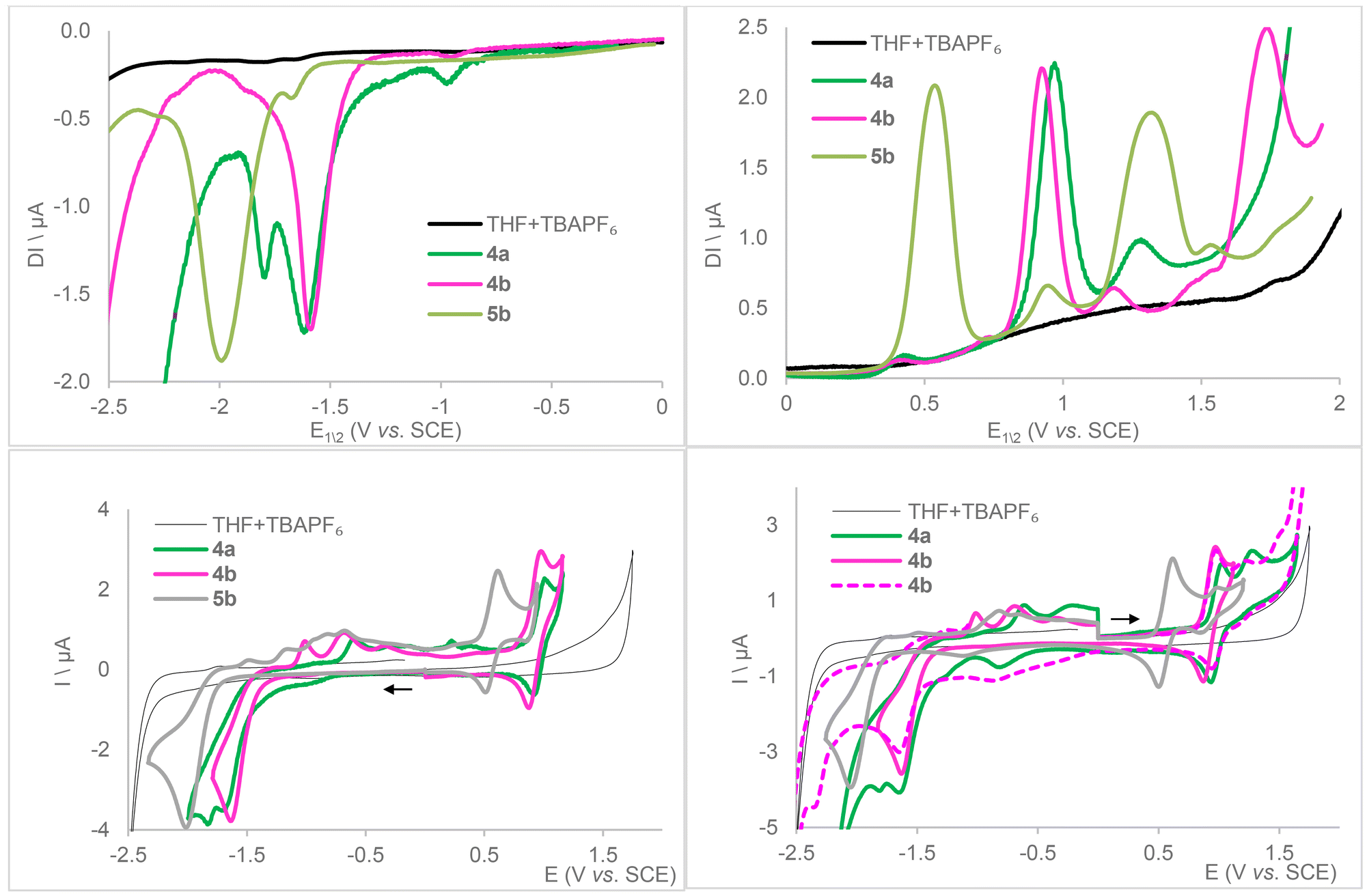 | ||
| Fig. 5 Square wave (top) and cyclic voltammograms on a glassy carbon electrode for compounds 4a, 4b and 5b ([C] = 1 mM) recorded under Ar in dry THF with 0.1 M TBA[PF6] supporting electrolyte (cyclic voltammetry: scan rate 0.2 V s−1; square wave voltammetry parameters: modulation amplitude 20 mV, frequency 20 Hz, and step potential 5 mV or 0.1 V s−1). | ||
Proton-responsiveness of the complexes was evaluated by treating solutions of 4a–c and 5a–c in THF with two equivalents of trific acid (HOTf) (Scheme 2).46 In the case of the LCF3 series, intractable mixtures containing paramagnetic compounds as well as the free ligand were obtained. It is likely that protonation of the electron-poor bpp ligand substantially labilizes it, compromising the stability of the thus-generated species. Conversely, the LtBu-supported complexes 5a–c were readily protonated to afford the dicationic products 6a–c precipitating out of the reaction mixture as yellow solids in moderate (5a, 5c) to good yields (5b). Product 6b could be crystallized from acetonitrile, affording a material suitable for an sc-XRD study. The solid-state structure of 6b is shown in Fig. 6. It crystallizes in the monoclinic crystal system and the C2/c space group. A two-fold rotation axis spreads again along the Mo–Npy bond. Despite several attempts, a high-quality structure could not be obtained either for 6b (Rint > 0.12) or for 6a and 6c. The presence of the protons on the pyrazole arms of 6b could be ascertained from the Fourier difference map.
 | ||
| Scheme 2 Reactions of the heptacoordinated Mo complexes with HOTf. | ||
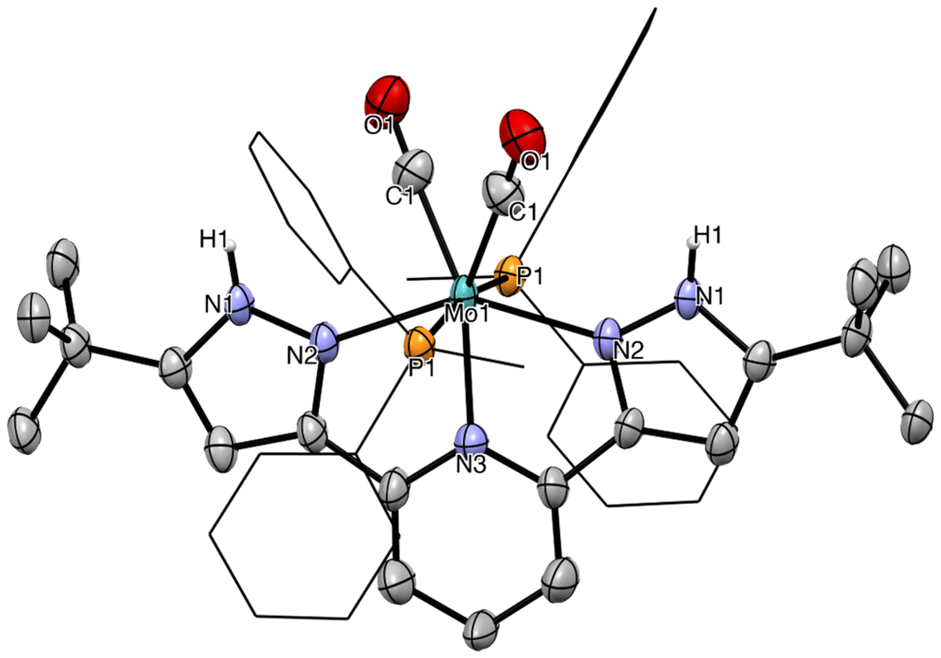 | ||
| Fig. 6 Molecular structure of compound 6b in the solid state. Thermal ellipsoids at the 25% probability level. For clarity, counterions and hydrogen atoms (except those bound to the Npz) are omitted, and phosphorus substituents appear as wireframe. | ||
Compared to 5b, 6b features elongated Mo–CO bonds (2.030(11) vs. 1.972(2) on average for 5b) and Mo–Npz bonds (2.192(6) vs. 2.171(1) on average for 5b), reflecting the overall diminished donating character of the pyrazole arms. This is counter-balanced by a shortened Mo–Npy bond (2.162(9) vs. 2.190(1) for 5b). Other metrics are roughly similar. IR data point to a diminished back donation to the CO ligands, in line with the protonated LtBu becoming less σ-donating and more π-accepting. Bands related to CO bond elongation are all hypsochromically shifted, more drastically for 6b by comparison with 6a and 6c (Δνas = 15, 22 and 7 cm−1 and Δνsym = 14, 29 and 10 cm−1 for 6a–c, respectively).
Cyclic voltammetry run on a solution of 6b in THF with TBA[PF6] as the electrolyte expectedly contrasted with that of 5b, with the presence of acidic protons complicating the electrochemistry (Fig. 7 and S81–S84† for square wave voltammetry). Four irreversible reduction waves are observed at Ep = −0.63, −1.16, −1.59 and −1.96 V and an irreversible oxidation one at 1.45 V vs. SCE. The latter occurs at a higher potential than for neutral 5b (reversible wave at 0.94 V) but matches its second oxidation wave. The fourth reduction wave occurs at a very close potential to the reduction wave recorded for 5b (−2.00 V), so we assume this could be the result of regenerated 5b within the electrochemical cell. A two-electron reduction would be sufficient to get 5b from 6b by evolution of H2. However, three redox events are recorded before reaching the most cathodic one, two of them could reasonably be the result of sequential reductions of the acidic protons carried by the pyrazoles. Further investigations regarding the behavior of compounds 6a–c are necessary to understand the fate thereof under reducing conditions.
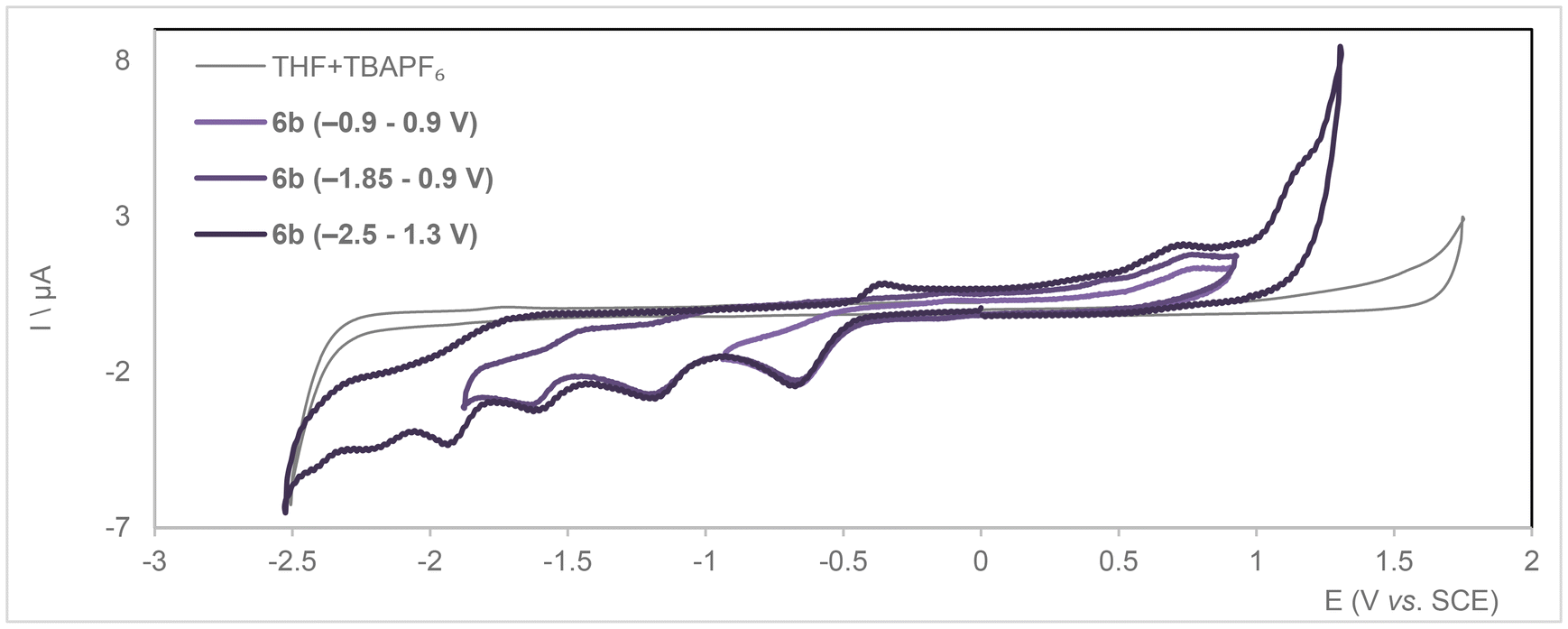 | ||
| Fig. 7 Cyclic voltammograms for compound 6b (bottom) ([C] = 1 mM) recorded over different voltage ranges under Ar in dry THF with 0.1 M TBA[PF6] supporting electrolyte (ν = 0.2 V s−1). | ||
Conclusions
In this article, we have attempted the coordination of tridentate, pincer-type 2,6-bis(pyrazol-3-yl)pyridine (bpp) ligands LCF3 (bearing a CF3 group at the C5 position of the pyrazole arm) and LtBu (bearing instead a tBu group) to molybdenum(0) precursors. With the chosen precursors [Mo(CO)2(MeCN)n−1(PMe3−nPhn)5−n] 1–3 (1 ≤ n ≤ 3), unclean reactions produced among other unidentified products heptacoordinated diamagnetic Mo(II) compounds [Mo(CO)2(bpp)(PMe3−nPhn)2] 4a–c (LCF3-supported) and 5a–c (LtBu-supported) in which the bpp ligand is deprotonated. We managed to obtain these new complexes selectively in moderate to good isolated yields by adding 2 equiv. of an oxidant (Ag+) to the reaction mixture. These were characterized by means of NMR, IR and UV-Vis spectroscopies, sc-XRD as well as circular and square-wave voltammetries for 4a, 4b and 5b. UV-Vis spectra of the series, especially those of 4b and 4c and 5b and 5c that show well-defined absorptions in the near-UV that are not influenced by the nature of the phosphines, point to IL and MLCT being presumably the least energetic transitions. In CV, the complexes investigated have in common the irreversibility of their first reduction wave (occurring below −1.5 V vs. SCE), being probably ligand-centered, and the reversibility of their first oxidation wave (occurring above 0.5 V) that we assigned to the Mo3+/Mo2+ couple. The deprotonated LtBu ligand in 5a–c can be readily re-protonated with 2 equiv. of HOTf to afford the dicationic [Mo(CO)2(LtBu)(PMe3−nPhn)2][OTf]2 complexes, but the same reaction in the case of the LCF3 series resulted in decomposition of intractable mixtures of compounds. This study provides further insights into the interaction of low-oxidation molybdenum with ligands that can potentially develop a non-innocent character. Our attempts with Mo(0) strongly suggest the non-innocence of the bpp platform, and the electrochemistry of Mo(II) complexes hints that an electron can be stored on the ligand, but at the cost of stability. Further engineering of low-valent Mo complexes is therefore needed to control and exploit the non-innocence of the surrounding bpp platform.Experimental methods
General considerations
All reactions were performed in flame- or oven-dried glassware with rigorous exclusion of air and moisture, using Schlenk line techniques or Jacomex (argon and dinitrogen) glove boxes (O2 < 1.0 ppm, H2O < 1 ppm). All of the experiments were carried out under an argon atmosphere. Solvents used were pre-dried by passing through a Puresolv MD 7 solvent purification machine, degassed by freeze–pump–thaw cycles (except for acetonitrile degassed by bubbling argon inside the solvent), dried over molecular sieves and stored in an argon glove box. The deuterated solvent (purchased from Eurisotop) was degassed by freeze–pump–thaw cycles, dried over molecular sieves and stored in an argon glove box. 1H, 13C, 19F and 31P NMR spectra were recorded using NMR tubes equipped with J. Young valves on a Bruker Avance III 400 spectrometer. Chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to the most upfield residual solvent resonance as the internal standard in 1H NMR (THF-d8: δ reported = 1.72 ppm; CD3CN: δ reported = 1.94 ppm). 19F and 31P NMR spectra were calibrated according to the IUPAC recommendation using a unified chemical shift scale based on the proton resonance of tetramethylsilane as the primary reference.86 Data are reported as follows: chemical shift, multiplicity (b = broad, s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet), coupling constant (Hz), and integration. Infrared (IR) spectra were recorded in a dinitrogen glove box (O2 < 1.0 ppm, H2O < 1 ppm) on an Agilent Cary 630 FT-IR spectrophotometer equipped with ATR or transmission modules and are reported in wavenumbers (cm−1) with (s), (m), and (w) indicating strong, medium, and weak absorption respectively. UV-Vis spectra were recorded on a PerkinElmer LAMBDA 950 UV/vis spectrophotometer equipped with a dual-beam setup. The UV cells (quartz cells equipped with airtight Teflon caps, 10 mm light path) were prepared under air with the solvent dried and degassed (as explained previously). Elemental analyses were performed on samples sealed in tin capsules under Ar or N2 by the Analytical Service of the Laboratoire de Chimie de Coordination; the results are the average of two independent measurements.Syntheses
The syntheses of complexes 1–3 have been done as reported in the literature from unpurified, commercially available chemicals and stored in a glove box.71–74 Also, the syntheses of both ligands LCF3 or LtBu have been done as reported in the literature.42,87 For all the heptacoordinated complexes (4a–c and 5b–c) except for 5a, a common procedure was used for the syntheses.4a–c: In a 25 mL vial, a solution containing LCF3 (1.0 mmol, 1.0 equiv.) in 5 mL of THF is added under stirring to a solution of the metallic precursor (1/2/3, 1.0 mmol, 1.0 equiv.) solubilized in 5 mL of THF. Then, a solution of AgOAc (2.1 mmol, 2.1 equiv.) in 10 mL of THF is added dropwise over the course of 5 minutes. The solution is left under stirring at room temperature for 3 h, during which dark red/orange coloration appears. The crude product is filtered to separate metallic silver that precipitated, and the volatiles are removed under vacuum. The orange/brown oil recovered is washed two times with pentane (2 × 5 mL) and two times with diethyl ether (2 × 5 mL). The precipitate is separated from the solution by filtration with a filtrating cannula, to obtain 4a–c as red solids in 40%, 69% and 70% yields, respectively.
4a: 1H NMR (400 MHz, THF-d8) δ 7.70 (t, J = 7.9 Hz, 1H), 7.28 (t, J = 7.5 Hz, 6H), 7.09 (t, J = 7.6 Hz, 12H), 6.99–6.92 (m, 13H overlapping), 6.36 (s, 2H); 31P{1H} NMR (162 MHz, THF-d8) δ 37.75 (s, 2P); 19F NMR (377 MHz, THF-d8) δ −61.61; 13C{1H} NMR (101 MHz, THF-d8) δ 267.12 (t, J = 18.2 Hz), 134.65 (d, J = 6.0 Hz), 130.98, 129.04 (t, J = 4.6 Hz), 117.00, 104.29; IR (ATR, solid): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) 1944 cm−1 (s, C
1944 cm−1 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O as); 1888 cm−1 (s, CO sym); anal. calcd for C51H35F6MoN5O2P2; C 59.95, H 3.45, N 6.85; found C 59.63, H 3.34, N 6.94.
O as); 1888 cm−1 (s, CO sym); anal. calcd for C51H35F6MoN5O2P2; C 59.95, H 3.45, N 6.85; found C 59.63, H 3.34, N 6.94.
4b: 1H NMR (400 MHz, THF-d8) δ 7.45 (t, 1H), 7.21 (t, J = 7.4 Hz, 4H), 7.00 (t, 8H), 6.87 (d, J = 7.9 Hz, 2H), 6.74 (s, 2H), 6.68 (q, 8H), 1.63 (t, J = 3.5 Hz, 2H); 31P{1H} NMR (162 MHz, THF-d8) δ 22.60 (s, 2P); 19F NMR (377 MHz, THF-d8) δ −61.55 (s, 6F); 13C{1H} NMR (101 MHz, THF-d8) δ 267.48, 152.50, 150.80, 139.95, 132.70 (t, J = 5.5 Hz), 130.62, 129.08 (t, J = 4.6 Hz), 116.24, 104.31, 11.70; IR (ATR, solid): 1948 cm−1 (s, CO as); 1880 cm−1 (s, CO sym); anal. calcd for C41H31F6MoN5O2P2: C 54.86, H 3.48, N 7.80 found C 53.93, H 3.53, N 7.50.
4c: 1H NMR (400 MHz, THF-d8) δ 7.53 (t, J = 7.8 Hz, 1H), 7.12 (t, J = 7.4 Hz, 2H), 7.04 (d, J = 7.8 Hz, 2H), 6.92 (t, J = 7.6 Hz, 4H), 6.87 (s, 2H), 6.52–6.42 (m, 4H), 1.23 (t, 12H); 31P{H} NMR (162 MHz, THF-d8) δ 9.64 (s, 2P); 19F NMR (377 MHz, THF-d8) δ −61.50 (s, 6F); 13C{1H} NMR (101 MHz, THF-d8) δ 264.01, 151.02, 149.78, 138.34, 128.65 (t, J = 4.9 Hz), 128.40, 127.41 (t, J = 4.4 Hz), 114.46, 102.28, 10.90 (t, J = 13.1 Hz); IR (ATR, solid): 1934 cm−1 (s, CO as); 1873 (s, CO sym); anal. calcd for C41H31F6MoN5O2P2: C 48.14, H 3.52, N 9.05 found C 48.79, H 3.77, N 8.53.
5a: In a 25 mL vial, a solution containing LtBu (1.0 mmol, 1.0 equiv.) in 5 mL of THF is added under stirring to a solution of the metallic precursor (1, 1.0 mmol, 1.0 equiv.) solubilized in 5 mL of THF. Then, a solution of Ag2O (2.1 mmol, 2.1 equiv.) in 10 mL of THF is added dropwise over the course of 5 minutes. The solution is left under stirring at room temperature for 20 h, during which dark red/orange coloration appears. The crude product is filtered to separate metallic silver that precipitated, and the volatiles are removed under vacuum. The orange/brown oil recovered is washed three times with cold (−40 °C) pentane (3 × 5 mL) and three times with cold (−40 °C) diethyl ether (3 × 5 mL). The precipitate is separated from the solution by filtration with a filtrating cannula, to obtain a red/brown solid in 51% yield. However, the calculated yield is not accurate because this compound could not be isolated in pure form. 1H NMR (400 MHz, THF-d8) δ 7.22 (t, J = 7.3 Hz, 1H), 7.05 (t overlapping, J = 7.5 Hz), 6.63 (d, J = 7.8 Hz, 2H), 5.91 (s, J = 0.8 Hz, 2H), 1.40 (s, 18H); 31P{1H} NMR (162 MHz, THF-d8) δ 37.14 (s, 2P) free PPh3 at δ −4.86 (s, 1P); 13C{1H} NMR (101 MHz, THF-d8) δ 134.83, 134.63, 134.06 (t, J = 6.2 Hz), 130.27, 129.69 (d, J = 3.8 Hz), 129.44 (dd, J = 7.2, 2.7 Hz), 129.08 (q, J = 4.1 Hz), 114.52 (d, J = 4.3 Hz), 101.01, 31.93; IR (ATR, solid): 1938 cm−1 (s, CO as); 1886 (s, CO sym).
5b and 5c: In a 25 mL vial, a solution containing LtBu (1.0 mmol, 1.0 equiv.) in 5 mL of THF is added under stirring to a solution of the metallic precursor (2 or 3, 1.0 mmol, 1.0 equiv.) solubilized in 5 mL of THF. Then, a solution of AgOAc (2.1 mmol, 2.1 equiv.) in 10 mL of THF is added dropwise over the course of 5 minutes. The solution is left under stirring at room temperature for 20 h, during which a dark red/orange coloration appears. The crude product is filtered to separate metallic silver that precipitated, and the solution is dried under vacuum. The orange/brown oil recovered is washed twice with pentane (2 × 5 mL) and twice with diethyl ether (2 × 5 mL). The precipitate is separated from the solution by filtration with a filtrating cannula, to obtain 5b and 5c as red solids in 44% and 50% yields, respectively.
5b: 1H NMR (40 MHz, THF-d8) δ 7.45 (t, 1H), 7.21 (t, J = 7.4 Hz, 4H), 7.00 (t, 8H), 6.87 (d, J = 7.9 Hz, 2H), 6.74 (s, 2H), 6.68 (q, 8H), 1.63 (t, J = 3.5 Hz, 2H); 31P{1H} NMR (162 MHz, THF-d8) δ 22.60 (s, 2P); 19F NMR (377 MHz, THF-d8) δ −61.55 (s, 6F); 13C{1H} NMR (400 MHz, THF-d8) δ 267.48, 152.50, 150.80, 139.95, 132.70 (t, J = 5.5 Hz), 130.62, 129.08 (t, J = 4.6 Hz), 116.24, 104.31, 11.70; IR (ATR, solid): 1940 cm−1 (s, CO as); 1879 (s, CO sym); anal. calcd for C47H49MoN5O2P2; C 64.60, H 5.65, N 5.65, found C 63.73, H 5.76, N 6.62.
5c: 1H NMR (400 MHz, THF-d8) δ 7.34 (t, 1H), 7.10 (t, J = 7.5, 5.9, 1.8, 0.9 Hz, 2H), 6.94 (t, J = 8.7, 7.6, 1.2 Hz, 4H), 6.79 (d, J = 7.8 Hz, 2H), 6.63 (dtt, J = 8.4, 5.0, 1.5 Hz, 4H), 6.43 (s, 2H), 1.36 (s, 19H), 1.12 (t, 12H); 31P{1H} NMR (162 MHz, THF-d8) δ 11.11 (s, 2P); 13C{1H} NMR (101 MHz, THF) δ 266.93 (t, J = 17.2 Hz), 167.52, 130.91, 129.60, 129.16 (d, J = 5.3 Hz), 128.60 (t, J = 4.3 Hz), 113.38, 100.22, 31.77, 12.57 (t, J = 12.2 Hz); IR (ATR, solid): 1946 cm−1 (s, CO as); 1877 (s, CO sym).
6a–c: A common synthetic protocol was used for these complexes as described below.
In a 50 mL Schlenk tube connected to a Schlenk line, HOTf (0.4 mmol, 2.0 equiv.) is added under stirring to a cold (273 K) solution of 1/2/3 (0.2 mmol, 1.0 equiv.) in 20 mL of THF. Then, the solution is left under stirring at room temperature for 4 h. The pale-yellow precipitate formed is filtered and dried under vacuum to afford complexes 6a–c in 30%, 77% and 33% yields, respectively.
6a: 1H NMR (400 MHz, CD3CN) δ 11.69 (s, 2H), 7.94 (t, 0H), 7.49 (t, 6H), 7.45–7.36 (m, 2H), 7.29 (t, J = 7.8 Hz, 2H), 6.75 (q, J = 6.7, 6.3 Hz, 12H), 1.26 (s, 18H); 31P{1H} NMR (162 MHz, CD3CN) δ 39.60; 19F NMR (377 MHz, CD3CN) δ −79.20; IR (ATR, solid): 1953 cm−1 (s, CO as); 1900 (s, CO sym).
6b: 1H NMR (400 MHz, CD3CN) δ 12.09 (s, 2H), 7.94 (t, J = 7.9, 1.2 Hz, 1H), 7.50–7.38 (m, 6H), 7.20 (dt, J = 23.4, 7.6 Hz, 9H), 6.84–6.74 (m, 6H), 6.62–6.52 (m, 4H), 1.60 (t, J = 3.7 Hz, 6H), 1.42 (s, 18H); 31P{1H} NMR (162 MHz, CD3CN) δ 23.16 (s, 2P); 19F NMR (377 MHz, CD3CN) δ −79.22 (s, 6F); 13C{1H} NMR (101 MHz, CD3CN) δ 264.30 (t, J = 20.9 Hz), 165.26, 153.03, 148.17, 143.33, 132.77–132.13 (m), 130.26 (q, J = 5.4 Hz), 122.99, 118.32, 105.74, 32.98, 29.94, 11.81 (t, J = 14.8 Hz); IR (ATR, solid): 1962 cm−1 (s, CO as); 1908 (s, CO sym); anal. calcd for C49H51F6MoN5O8P2S2; C 50.13, H 4.38, N 5.97; found C 49.85, 4.39, H 5.98.
6c: 1H NMR (400 MHz, CD3CN) δ 12.02 (s, 2H), 7.97 (t, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.26 (t, 2H), 6.99 (t, J = 8.7, 7.5, 1.3 Hz, 4H), 6.78 (d, J = 2.0 Hz, 2H), 6.40–6.30 (m, 4H), 1.41 (s, 18H), 1.34 (t, J = 4.4 Hz, 12H); 31P{1H} NMR (162 MHz, CD3CN) δ 8.31 (s, 2P); 19F NMR (377 MHz, CD3CN) δ −79.18 (s, 6F); 13C{1H} NMR (101 MHz, CD3CN) δ 263.94 (t, J = 20.1 Hz), 164.30, 152.84, 148.38, 142.89, 131.27, 129.93 (t, J = 4.6 Hz), 129.56 (t, J = 4.6 Hz), 122.42, 118.32, 105.04, 29.91; IR (ATR, solid): 1953 cm−1 (s, CO as); 1887 (s, CO sym).
Author contributions
A. E., L. E. B., L. P., R. P. and N. G. ran the experiments and analysed the data. A. S.-S. performed the electrochemical measurements. L. V. performed the X-ray diffraction studies and solved the structures. A. S. and M. G. conceived the project and managed it. A. S., M. G., R. S. and N. F. acquired funding. A. S., M. G., R. S., N. F. and J.-B. S. supervised the work. A. S. wrote the manuscript. A. E., L. E. B. and A. S. compiled the ESI.† All co-authors were involved in manuscript and ESI† proof-reading.Data availability
The datasets are available on request from the authors. Results of data treatment (NMR, infrared and UV-Vis spectra, crystallographic details, cyclic and square wave voltammograms) have been compiled in a .pdf file freely available as part of the ESI.† X-ray diffraction data have been deposited on the Cambridge Crystallographic Data Centre (CCDC) under the deposition numbers 2403147–2403152.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
A. E. and A. S. acknowledge the Région Occitanie and Pôle RHyO for funding (grant CataLOHC). L. E. B., R. S., N. F. and A. S. have received funding from a CNRS “International Research Project”. All authors thank their respective institutions for providing access to facilities.References
- Pincer Compounds, ed. D. Morales-Morales, Elsevier, 2018 Search PubMed.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS.
- H. Valdés, M. A. García-Eleno, D. Canseco-Gonzalez and D. Morales-Morales, ChemCatChem, 2018, 10, 3136–3172 CrossRef.
- Organometallic Pincer Chemistry, ed. G. van Koten and D. Milstein, Springer, 2012 Search PubMed.
- G. van Koten, Pure Appl. Chem., 1989, 61, 1681–1694 CrossRef CAS.
- A. Kasera, J. P. Biswas, A. Ali Alshehri, S. Ahmed Al-Thabaiti, M. Mokhtar and D. Maiti, Coord. Chem. Rev., 2023, 475, 214915 CrossRef CAS.
- K. K. Manar, J. Cheng, Y. Yang, X. Yang and P. Ren, ChemCatChem, 2023, 15, e202300004 CrossRef CAS.
- Metal-Ligand Co-operativity: Catalysis and the Pincer-Metal Platform, ed. G. van Koten, K. Kirchner and M.-E. Moret, Springer International Publishing, 2021 Search PubMed.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 CrossRef CAS PubMed.
- M. A. Halcrow, Coord. Chem. Rev., 2005, 249, 2880–2908 CrossRef CAS.
- T. Buchen, P. Guetlich and H. A. Goodwin, Inorg. Chem., 1994, 33, 4573–4576 CrossRef CAS.
- L. A. Barrios, C. Bartual-Murgui, E. Peyrecave-Lleixà, B. Le Guennic, S. J. Teat, O. Roubeau and G. Aromí, Inorg. Chem., 2016, 55, 4110–4116 CrossRef CAS.
- V. Jornet-Mollá, Y. Duan, C. Giménez-Saiz, Y.-Y. Tang, P.-F. Li, F. M. Romero and R.-G. Xiong, Angew. Chem., Int. Ed., 2017, 56, 14052–14056 CrossRef PubMed.
- A. Djemel, O. Stefanczyk, M. Marchivie, E. Trzop, E. Collet, C. Desplanches, R. Delimi and G. Chastanet, Chem. – Eur. J., 2018, 24, 14760–14767 CrossRef CAS PubMed.
- S. P. Vallone, A. N. Tantillo, A. M. dos Santos, J. J. Molaison, R. Kulmaczewski, A. Chapoy, P. Ahmadi, M. A. Halcrow and K. G. Sandeman, Adv. Mater., 2019, 31, 1807334 CrossRef.
- M. A. Halcrow, I. Capel Berdiell, C. M. Pask and R. Kulmaczewski, Inorg. Chem., 2019, 58, 9811–9821 CrossRef CAS PubMed.
- C.-Y. Kuei, S.-H. Liu, P.-T. Chou, G.-H. Lee and Y. Chi, Dalton Trans., 2016, 45, 15364–15373 RSC.
- J. Yan, M. Song, D.-Y. Zhou, S.-M. Yiu, L.-S. Liao, Y. Chi and M. Xie, Organometallics, 2023, 42, 2070–2078 CrossRef CAS.
- D. A. Bardwell, J. C. Jeffery, P. L. Jones, J. A. McCleverty, E. Psillakis, Z. Reeves and M. D. Ward, J. Chem. Soc., Dalton Trans., 1997, 2079–2086 RSC.
- Y.-W. Yip, H. Wen, W.-T. Wong, P. A. Tanner and K.-L. Wong, Inorg. Chem., 2012, 51, 7013–7015 CrossRef CAS PubMed.
- A. Galstyan, A. R. Naziruddin, C. Cebrián, A. Iordache, C. G. Daniliuc, L. De Cola and C. A. Strassert, Eur. J. Inorg. Chem., 2015, 2015, 5822–5831 CrossRef CAS.
- C.-C. Chou, K.-L. Wu, Y. Chi, W.-P. Hu, S. J. Yu, G.-H. Lee, C.-L. Lin and P.-T. Chou, Angew. Chem., Int. Ed., 2011, 50, 2054–2058 CrossRef CAS PubMed.
- T. Kono, N. Masaki, M. Nishikawa, R. Tamura, H. Matsuzaki, M. Kimura and S. Mori, ACS Appl. Mater. Interfaces, 2016, 8, 16677–16683 CrossRef CAS PubMed.
- P. Delcanale, A. Galstyan, C. G. Daniliuc, H. E. Grecco, S. Abbruzzetti, A. Faust, C. Viappiani and C. A. Strassert, ACS Appl. Mater. Interfaces, 2018, 10, 24361–24369 CrossRef CAS.
- C. Adam, B. B. Beele, A. Geist, U. Müllich, P. Kaden and P. J. Panak, Chem. Sci., 2015, 6, 1548–1561 RSC.
- J. Stracke, P. Weßling, T. Sittel, C. Adam, F. Rominger, A. Geist and P. J. Panak, Inorg. Chem., 2024, 63, 13214–13222 CrossRef CAS PubMed.
- S. Radisavljević, I. Bratsos, A. Scheurer, J. Korzekwa, R. Masnikosa, A. Tot, N. Gligorijević, S. Radulović and A. R. Simović, Dalton Trans., 2018, 47, 13696–13712 RSC.
- D. Ćoćić, S. Jovanović, S. Radisavljević, J. Korzekwa, A. Scheurer, R. Puchta, D. Baskić, D. Todorović, S. Popović, S. Matić and B. Petrović, J. Inorg. Biochem., 2018, 189, 91–102 CrossRef PubMed.
- M. M. Milutinović, J. V. Bogojeski, O. Klisurić, A. Scheurer, S. K. C. Elmroth and Ž. D. Bugarčić, Dalton Trans., 2016, 45, 15481–15491 RSC.
- N. S. Labrum, C.-H. Chen and K. G. Caulton, Chem. – Eur. J., 2019, 25, 7935–7940 CrossRef CAS PubMed.
- N. S. Labrum, A. C. Cabelof and K. G. Caulton, Chem. – Eur. J., 2020, 26, 13915–13926 CrossRef CAS PubMed.
- B. J. Cook, C.-H. Chen and K. G. Caulton, Chem. – Eur. J., 2018, 24, 5962–5966 CrossRef CAS PubMed.
- J. J. Kiernicki, M. Zeller and N. K. Szymczak, Inorg. Chem., 2020, 59, 9279–9286 CrossRef CAS PubMed.
- J. L. Kuo and K. I. Goldberg, J. Am. Chem. Soc., 2020, 142, 21439–21449 CrossRef CAS PubMed.
- K. Umehara, S. Kuwata and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 6754–6757 CrossRef CAS PubMed.
- J. J. Kiernicki, M. Zeller and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 18194–18197 CrossRef CAS PubMed.
- B. J. Cook, A. V. Polezhaev, C.-H. Chen, M. Pink and K. G. Caulton, Inorg. Chim. Acta, 2018, 483, 510–515 CrossRef CAS.
- A. Yoshinari, A. Tazawa, S. Kuwata and T. Ikariya, Chem. – Asian J., 2012, 7, 1417–1425 CrossRef CAS PubMed.
- T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2017, 56, 8651–8660 CrossRef CAS PubMed.
- Y. Nakahara, T. Toda and S. Kuwata, Polyhedron, 2018, 143, 105–110 CrossRef CAS.
- P. Weingart and W. R. Thiel, ChemCatChem, 2018, 10, 4844–4848 CrossRef.
- Y. Nakahara, T. Toda, A. Matsunami, Y. Kayaki and S. Kuwata, Chem. – Asian J., 2018, 13, 73–80 CrossRef CAS PubMed.
- C. Wu, B. Liu, X. Geng, Z. Zhang, S. Liu and Q. Hu, Polyhedron, 2019, 158, 334–341 CrossRef CAS.
- Y. Chen, T. Cui, H. Chen, X. li Zheng, H. Fu and R. Li, Dalton Trans., 2023, 52, 12368–12377 RSC.
- A. Simonneau, R. Turrel, L. Vendier and M. Etienne, Angew. Chem., Int. Ed., 2017, 56, 12268–12272 CrossRef CAS PubMed.
- A. Coffinet, D. Specklin, L. Vendier, M. Etienne and A. Simonneau, Chem. – Eur. J., 2019, 25, 14300–14303 CrossRef CAS PubMed.
- A. Bouammali, C. Bijani, L. Vendier, M. Etienne and A. Simonneau, Eur. J. Inorg. Chem., 2020, 2020, 1423–1427 CrossRef CAS.
- A. Coffinet, D. Zhang, L. Vendier, S. Bontemps and A. Simonneau, Dalton Trans., 2021, 50, 5582–5589 RSC.
- D. Specklin, A. Coffinet, L. Vendier, I. del Rosal, C. Dinoi and A. Simonneau, Inorg. Chem., 2021, 60, 5545–5562 CrossRef CAS PubMed.
- A. Bouammali, A. Coffinet, L. Vendier and A. Simonneau, Dalton Trans., 2022, 51, 10697–10701 RSC.
- D. Specklin, M.-C. Boegli, A. Coffinet, L. Escomel, L. Vendier, M. Grellier and A. Simonneau, Chem. Sci., 2023, 14, 14262–14270 RSC.
- N. Queyriaux, N. Durvin, D. Leon, M.-C. Boegli, L. Vendier and A. Simonneau, Eur. J. Inorg. Chem., 2023, 26, e202300426 CrossRef CAS.
- Q. Le Dé, F. Orbay, L. Vendier and A. Simonneau, J. Organomet. Chem., 2023, 986, 122604 CrossRef.
- W. R. Thiel and J. Eppinger, Chem. – Eur. J., 1997, 3, 696–705 CrossRef CAS.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- S. Kuriyama, K. Arashiba, K. Nakajima, H. Tanaka, N. Kamaru, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2014, 136, 9719–9731 CrossRef CAS PubMed.
- S. Chakraborty and H. Berke, ACS Catal., 2014, 4, 2191–2194 CrossRef CAS.
- Y. Zhang, P. G. Williard and W. H. Bernskoetter, Organometallics, 2016, 35, 860–865 CrossRef CAS.
- T. Leischner, A. Spannenberg, K. Junge and M. Beller, Organometallics, 2018, 37, 4402–4408 CrossRef CAS.
- T. Leischner, L. A. Suarez, A. Spannenberg, K. Junge, A. Nova and M. Beller, Chem. Sci., 2019, 10, 10566–10576 RSC.
- T. Leischner, A. Spannenberg, K. Junge and M. Beller, ChemCatChem, 2020, 12, 4543–4549 CrossRef CAS.
- T. Itabashi, K. Arashiba, A. Egi, H. Tanaka, K. Sugiyama, S. Suginome, S. Kuriyama, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2022, 13, 6161 CrossRef CAS PubMed.
- N. F. Both, A. Spannenberg, K. Junge and M. Beller, Organometallics, 2022, 41, 1797–1805 CrossRef CAS PubMed.
- P. Viereck, G. Hierlmeier, P. Tosatti, T. P. Pabst, K. Puentener and P. J. Chirik, J. Am. Chem. Soc., 2022, 144, 11203–11214 CrossRef CAS PubMed.
- A. F. Ibrahim, P. Garrido-Barros and J. C. Peters, ACS Catal., 2023, 13, 72–78 CrossRef CAS PubMed.
- N. F. Both, J. Thaens, A. Spannenberg, H. Jiao, K. Junge and M. Beller, ACS Catal., 2024, 14, 4082–4092 CrossRef CAS.
- F. Hohmann and H. T. Dieck, J. Organomet. Chem., 1975, 85, 47–71 CrossRef CAS.
- F. Hohmann, J. Organomet. Chem., 1977, 137, 315–328 CrossRef CAS.
- F. Hohmann, H. tom Dieck, C. Krüger and Y.-H. Tsay, J. Organomet. Chem., 1979, 171, 353–364 CrossRef CAS.
- B. J. Brisdon, D. A. Edwards and K. E. Paddick, Transition Met. Chem., 1981, 6, 83–86 CrossRef CAS.
- R. Hoffmann, B. F. Beier, E. L. Muetterties and A. R. Rossi, Inorg. Chem., 1977, 16, 511–522 CrossRef CAS.
- B. J. Cook, M. Pink, K. Pal and K. G. Caulton, Inorg. Chem., 2018, 57, 6176–6185 CrossRef CAS PubMed.
- N. S. Labrum, G. M. Curtin, E. Jakubikova and K. G. Caulton, Chem. – Eur. J., 2020, 26, 9547–9555 CrossRef CAS PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- F.-Q. Bai, X. Zhou, T. Liu, G.-J. Zhao, J.-P. Zhang and H.-X. Zhang, Int. J. Quantum Chem., 2009, 109, 308–319 CrossRef CAS.
- Y. Chi, B. Tong and P.-T. Chou, Coord. Chem. Rev., 2014, 281, 1–25 CrossRef CAS.
- M. Wrighton, Chem. Rev., 1974, 74, 401–430 CrossRef CAS.
- W. A. Fordyce and G. A. Crosby, Inorg. Chem., 1982, 21, 1455–1461 CrossRef CAS.
- P. D. Harvey, W. P. Schaefer and H. B. Gray, Inorg. Chem., 1988, 27, 1101–1104 CrossRef CAS.
- H. Tominaga, K. Sakai and T. Tsubomura, J. Chem. Soc., Chem. Commun., 1995, 2273–2274 RSC.
- H. Kunkely and A. Vogler, Inorg. Chem. Commun., 2000, 3, 143–144 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- E. F. Khmara, D. L. Chizhov, A. A. Sidorov, G. G. Aleksandrov, P. A. Slepukhin, M. A. Kiskin, K. L. Tokarev, V. I. Filyakova, G. L. Rusinov, I. V. Smolyaninov, A. S. Bogomyakov, D. V. Starichenko, Yu. N. Shvachko, A. V. Korolev, I. L. Eremenko and V. N. Charushin, Russ. Chem. Bull., 2012, 61, 313–325 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: A .pdf file compiling NMR, infrared and UV-Vis spectra, cyclic and square wave voltammograms, and crystallographic details. CCDC 2403147–2403152. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03264k |
| This journal is © The Royal Society of Chemistry 2025 |