
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Borane-tethered heteroscorpionate zinc catalysts for the hydroboration of carbon dioxide, isocyanates, esters and nitriles†
Tiago F. C. Cruz * and
Luís F. Veiros
* and
Luís F. Veiros
Centro de Química Estrutural, Institute of Molecular Sciences, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049 001 Lisboa, Portugal. E-mail: carpinteirocruz@tecnico.ulisboa.pt
First published on 22nd January 2025
Abstract
This work describes the synthesis, characterization and catalytic hydroboration activity of a family of zinc bis(κ2-borohydride) complexes containing borane-functionalized heteroscorpionate ligands. The new zinc bis(κ2-borohydride) complexes with R-substituted bis(3,5-dimethylpyrazolyl)methane ligands with or without borane functionalities [(L1–4)Zn(BH4)2] 1–4 (1, R = H; 2, R = CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2; 3, R = CH2CH2CH2(9-borabicyclo[3.3.1]nonane) or CH2CH2CH2(9-BBN); 4, R = CH2CH2CH2BCy2) were synthesized by reacting the corresponding dichloride complexes [(L1–4)ZnCl2] with two equivalents of NaBH4 in THF. The new complexes were characterized by NMR spectroscopy, FTIR spectroscopy and elemental analysis, while complex 3 was characterized by single-crystal X-ray diffraction. The borane-functionalized complexes 3 and 4 catalyzed the hydroboration of CO2 at 1 bar pressure, an array of isocyanates and esters and the dihydroboration of a set of nitriles with pinacolborane (HBPin) at catalyst loadings of 1 mol% and temperature of 60 °C in yields of 32–99%. In the cases of CO2 and isocyanates, the methylated products (along with PinBOBPin) were preferred after 16 h. The catalytic reactions utilizing 1 mol% of the unfunctionalized complexes 1 and 2 achieved yields in the range of 7–24%, an average 4-fold decrease in catalytic activities, confirming that the catalytic intermediates benefit from the intra- or intermolecular stabilization of incorporated boranes.
CH2; 3, R = CH2CH2CH2(9-borabicyclo[3.3.1]nonane) or CH2CH2CH2(9-BBN); 4, R = CH2CH2CH2BCy2) were synthesized by reacting the corresponding dichloride complexes [(L1–4)ZnCl2] with two equivalents of NaBH4 in THF. The new complexes were characterized by NMR spectroscopy, FTIR spectroscopy and elemental analysis, while complex 3 was characterized by single-crystal X-ray diffraction. The borane-functionalized complexes 3 and 4 catalyzed the hydroboration of CO2 at 1 bar pressure, an array of isocyanates and esters and the dihydroboration of a set of nitriles with pinacolborane (HBPin) at catalyst loadings of 1 mol% and temperature of 60 °C in yields of 32–99%. In the cases of CO2 and isocyanates, the methylated products (along with PinBOBPin) were preferred after 16 h. The catalytic reactions utilizing 1 mol% of the unfunctionalized complexes 1 and 2 achieved yields in the range of 7–24%, an average 4-fold decrease in catalytic activities, confirming that the catalytic intermediates benefit from the intra- or intermolecular stabilization of incorporated boranes.
Introduction
The selective reduction of inexpensive unsaturated molecules containing heteroatoms such as oxygen or nitrogen is of great importance in organic chemistry, as it paves the way to the synthesis of alcohols or amines, respectively, being often of added value to the fine chemistry sector. Substrates such as carbon dioxide (CO2), isocyanates, esters and nitriles, which are also readily available, fall into such a category.Among the ways to achieve chemical reduction, hydroboration is one of the most sought after, because not only does it avoid the harsh reaction conditions of hydrogenation and hydrosilylation processes, but also formally incorporates a borane moiety on the respective unsaturation products, thus creating molecules containing new heteroatom–boron bonds. Such synthons are very important classes of reagents because they pave the way to carbon–heteroatom bond formation via stereospecific tandem cross-coupling or oxidation processes (Chart 1A).1 CO2 hydroboration using pinacolborane (HBPin) as the borane source has been catalyzed by several systems based on transition metals such as Mn,2 Fe,3 Co,4 Ni,5 Cu,6 Mo,7 Ru,8 and Pd9 or main group elements.10 Among these, only particular cases have selectively produced the methyl boronate ester product H3COBPin11 resulting from the triple reduction of CO2. The hydroboration of isocyanates has been catalyzed by transition metals such as Co,12 Cu,13 and Ag,14 main group elements15 and rare earth16 elements, in which, depending on the catalytic conditions used, formamide or methylamide levels of chemical reduction can be achieved. Regarding the hydroboration of esters with HBPin, relevant works have been very few, with most catalysts reported being those based on molecular compounds of the main group elements, such as aluminum and lithium.17 Lastly, the dihydroboration of nitriles with HBPin has been widely covered in the literature, being catalyzed by complexes containing transition metals such as Mn,18 Fe,19 Co,20 Ni,21 Ru,22 and Ag23 or main group24 and rare-earth elements.25 Among all the systems discussed above, only selected complexes have served as multitasking catalysts, in which multiple families of substrates were readily converted: KHBEt3 has been known to hydroborate isocyanates as well as esters,26 while some other systems catalyzed the hydroboration of both nitriles and esters (Chart 1B).27
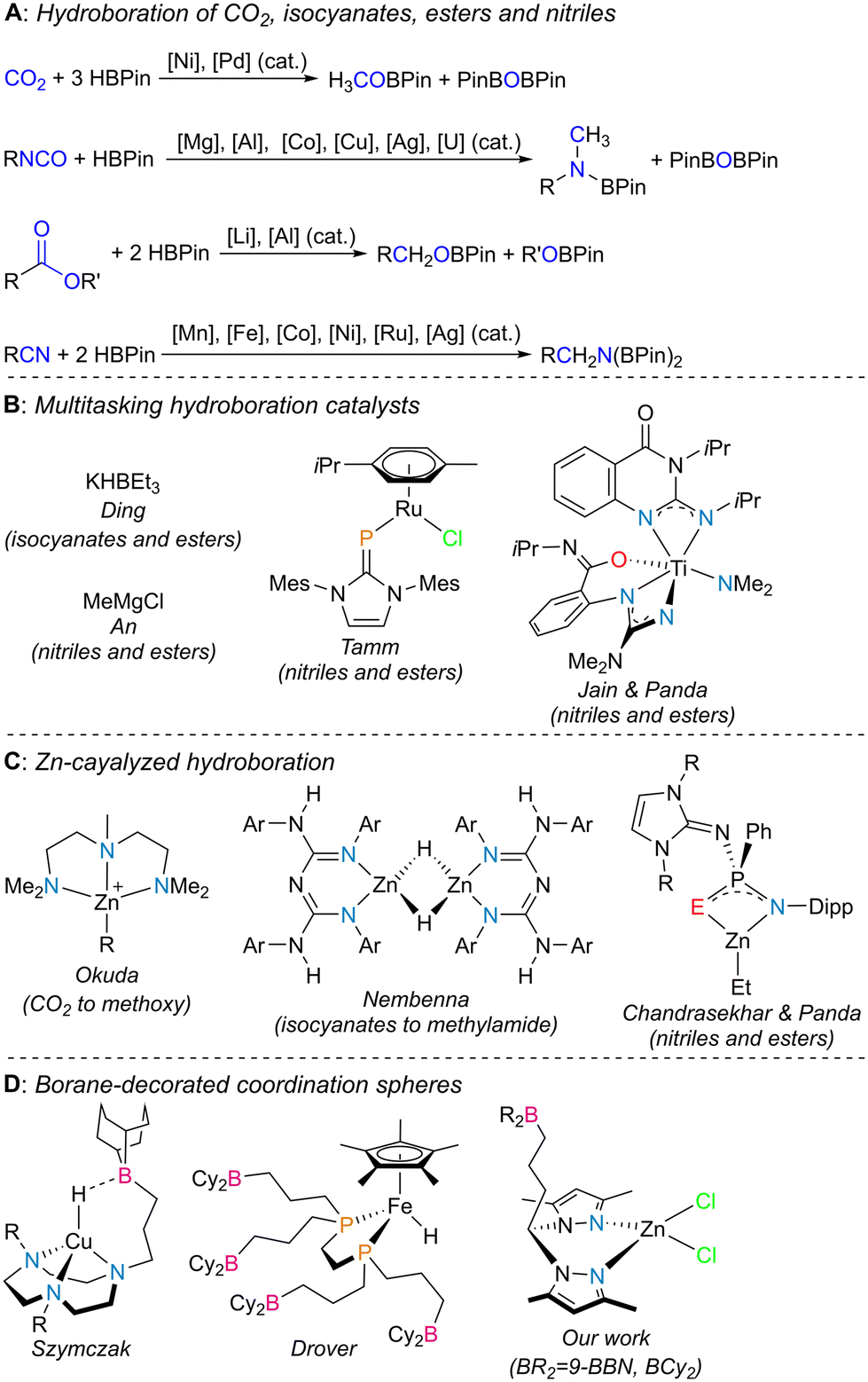 | ||
| Chart 1 Catalyst systems reported for the hydroboration of CO2 (methoxy level of reduction), isocyanates (methylamide level of reduction), esters and nitriles with HBPin (A), catalyst systems used in the reduction of multiple families of substrates with HBPin (B), selected cases of zinc-catalyzed hydroboration with HBPin (C), and examples of borane-functionalized coordination spheres (D). Abbreviations Mes, Dipp and 9-BBN denote mesityl (2,4,6-trimethylphenyl), 2,6-diispropylphenyl and 9-borabicyclo[3.3.1]nonane, respectively. | ||
Zinc-catalyzed CO2 hydroboration with HBPin has featured formate and methoxy selectivities.28 The hydroboration of isocyanates catalyzed by zinc complexes has been reported by Nembenna and coauthors with a conjugated bis(guanidinate) zinc hydride complex, in which the catalytic products resulting from mono, double and triple reduction of the isocyanates were selectively obtained by using 1, 2 or 3 equivalents of HBPin as the borane source, respectively.29 Zinc-catalyzed dihydroboration of nitriles has also been featured in the literature,30 while Panda and coworkers have reported tri-coordinated zinc alkyl complexes with N^E (E = S and Se) frameworks capable of catalyzing the dihydroboration of nitriles and the hydroboration of esters (Chart 1C).31
The manipulation of secondary coordination spheres in transition metal complexes has led to important influences on their catalytic ability and, therefore, is of paramount importance in the field of homogeneous catalysis.32 Manipulating secondary coordination spheres with boranes has the advantage of achieving push–pull reactivity,33 in which the borane acts as a Lewis acid and a ligand within the coordination sphere of a transition metal complex acts as a Lewis base, thus being further activated (Chart 1D). Bercaw or Klankermayer and respective coworkers used borane–phosphine ligands to activate hydride/carbonyl ligands in some transition metal complexes.34 Drover and coworkers extended this approach by reporting borane-functionalized 1,2-bis(phosphino)ethanes, which have been featured in the preparation of numerous transition metal complexes,35 one such system catalyzing the hydroboration of benzonitrile with HBPin.36 Szymczak and coworkers functionalized pyridinepyrazolyl,37 1,4,7-triazacyclononane38 or cyclic (alkyl)(amino)carbene39 derivatives with boranes and studied the chemistry of their respective transition metal coordination complexes, with applications in stoichiometric and catalytic small-molecule activation. Werlé and coworkers functionalized a phosphino triazine chelate with borane and its respective transition metal complexes were used as catalysts in the hydrogenation of nitroarenes or the synthesis of silyl enol ethers.40
In previous work, we reported new bis(3,5-dimethylpyrazolyl)methane ligands functionalized with pendant boranes and the chemistry of their respective coordination processes with zinc.41 In that work, we realized that the dichloride complexes [(L1–4)ZnCl2] (see Scheme 1), upon activation with 2 equivalents of KHBEt3, were active catalysts for the hydroboration of CO2 with HBPin, with complete selectivity to H3COBPin in a maximum yield of 86%. In that work, the complexes that exhibited the best results were those adorned with borane functionalities (that is, complexes [(L3,4)ZnCl2]). In those cases, the intramolecular stabilization of the hydride/formate catalytic intermediates by the pendant borane functionalities was behind their amplified catalytic activities.
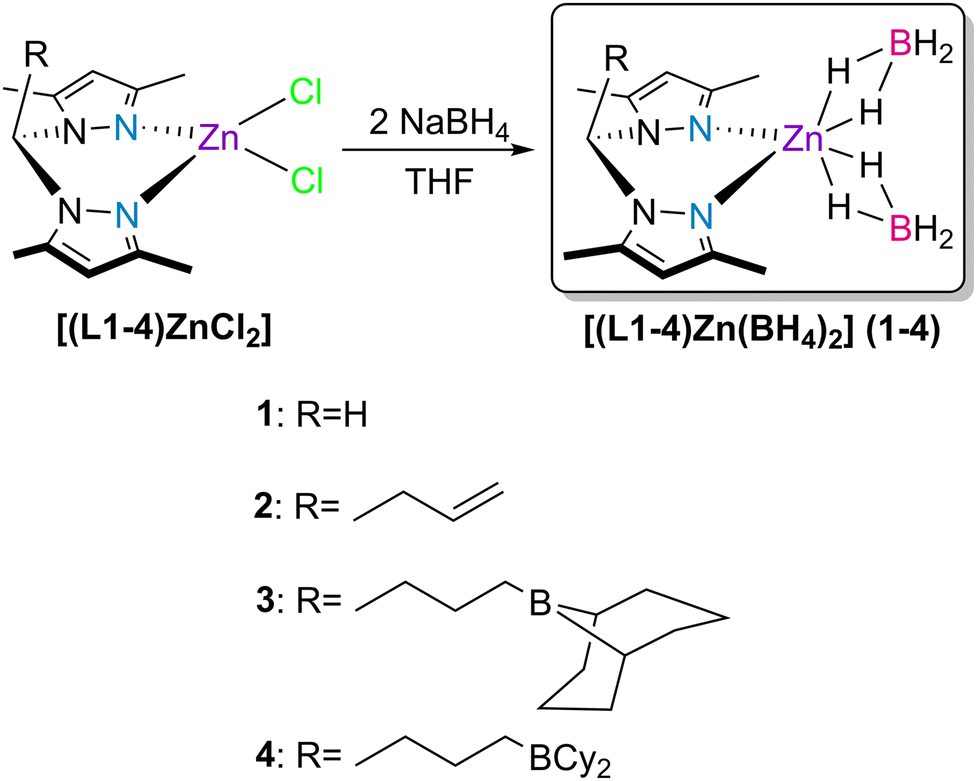 | ||
| Scheme 1 Synthesis of the bis(κ2-borohydride) zinc complexes 1–4. | ||
Picking up from our previous findings, and to avoid the pre-activation of the dichloride complexes with KHBEt3, we detail in the present report the synthesis and characterization of a new family of zinc bis(κ2-borohydride) complexes [(L1–4)Zn(BH4)2] 1–4 (see Scheme 1), and their activity as multitasking catalysts in the hydroboration of CO2, isocyanates, esters and nitriles.
Results and discussion
The bis(κ2-borohydride) zinc complexes [(L1–4)Zn(BH4)2] 1–4 bearing bis(3,5-dimethylpyrazolyl)methane (L1, R = H) or its allyl (L2, R = CH2CHCH2) or borane (L3, R = CH2CH2CH2(9-borabicyclo[3.3.1]nonane) or CH2CH2CH2(9-BBN) and L4, R = CH2CH2CH2BCy2) functionalized derivatives were prepared by the reaction of the previously isolated dichloride complexes [(L1–4)ZnCl2]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41,42 with two equivalents of NaBH4 in THF (Scheme 1).
41,42 with two equivalents of NaBH4 in THF (Scheme 1).
Complexes 3 and 4 were also isolated in good yields via post-anti-Markovnikov hydroboration of the allylated complex 2 with the 9-BBN dimer or dicyclohexylborane, respectively, in dichloromethane for 2 h and at 25 °C. The post-hydroboration reactions of 2 with diphenylborane, dimesitylborane or bis(pentafluorophenyl)borane (Piers’ borane) resulted in untraceable mixtures. Complexes 1–4 are insoluble in n-hexane and diethyl ether and soluble in toluene, THF and dichloromethane and were characterized by NMR and FTIR spectroscopies and elemental analysis. The spectra of all complexes are presented in Fig. S1–S16 in the ESI.† The NMR spectra of the complexes display the expected resonances for the moieties present in compounds L1–4. The CH resonance of the 3,5-dimethylpyrazolyl rings in the ligands are downfield-shifted upon coordination to zinc, going from 5.76–5.77 ppm to 6.08–6.15 ppm. Similarly, the methyl resonances in the 3,5-dimethylpyrazolyl moieties go from being seemingly overlapped at 2.19 ppm in ligands L1–4 to two distinct, upfield-shifted resonances at 2.41–2.52 ppm upon coordination to zinc. This latter fact is in line with the decrease in the symmetry of ligands L1–4 caused by the coordination strain of the respective bis(3,5-dimethylpyrazolyl)methyl moieties. The complexes exhibit respective proton borohydride resonances centered at 0.42–0.49 ppm as broad quartets, arising from coupling of the 1H and 11B nuclei (1JBH = 83–84 Hz). Additionally, the 11B resonances in these complexes are quintets at −42.9 to −45.7 ppm, with 1JBH = 83–85 Hz. These observations are indicative of the existence of borohydride moieties in the complexes.43 Complexes 3 and 4 also present 11B NMR resonances in the range of 82.9–87.4 ppm, characteristic of tricoordinate boranes simultaneously containing alkyl and 9-BBN/BCy2 fragments.44
The FTIR spectra of complexes 1–4 revealed the presence of two strong broad bands at 2450–2200 cm−1 and 2200–2000 cm−1, respectively, attributed to terminal and bridging B–H stretching in the borohydride ligands. In addition, strong bands in the range of 1100–1110 cm−1 were also observed in all complexes, corresponding to BH2 deformation. These results are in line with the spectral observations made for complexes containing κ2-BH4 ligands.45 Furthermore, the presence of 9-BBN or BCy2 and propylenic moieties in complexes 3 and 4 was characterized by an increase of the intensity of C–H bond vibration modes in the 2750–3000 cm−1 range with respect to the unfunctionalized complexes 1 and 2.
Complex 3 was analyzed by single-crystal X-ray diffraction, having crystallized in the monoclinic system, in the P21 space group. Owing to the poor quality of the single crystal obtained for complex 3, its molecular structure is only presented as a proof of connectivity in Fig. 1 while its selected bond lengths and angles and crystallographic data are displayed in Tables S1 and S2 in the ESI.†
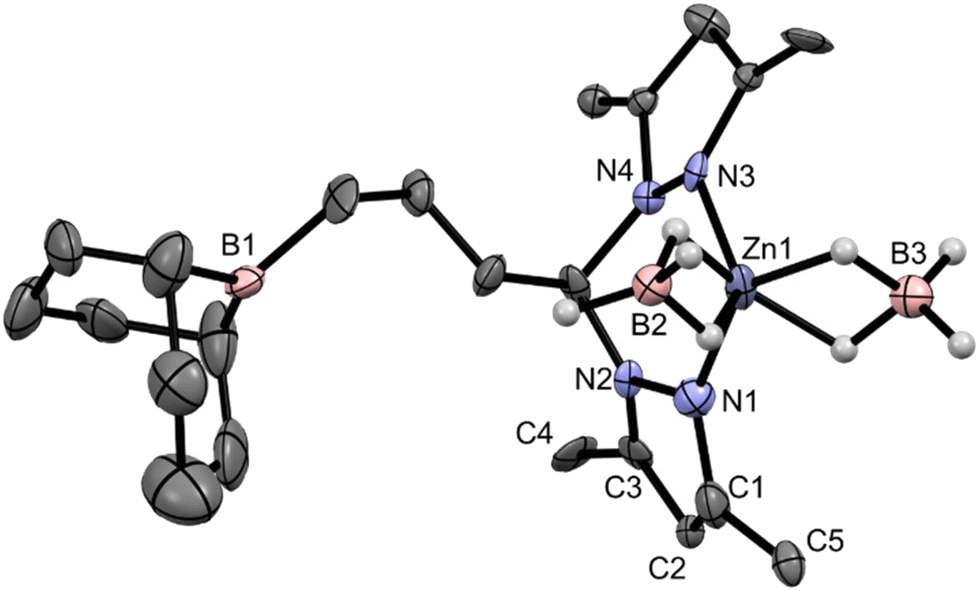 | ||
| Fig. 1 Proof of the molecular connectivity of complex 3 determined by single-crystal X-ray diffraction, showing 30% probability ellipsoids. All hydrogen atoms, except those of the borohydride ligands, have been omitted for clarity. | ||
Complex 3 demonstrates a κ2-N,N coordination mode of ligand L3, as well as two κ2-BH4 co-ligands. The borane arm is also evident in its molecular structure, where the three-carbon hydrocarbyl bridge between the bis(pyrazolyl)methane and the 9-BBN moieties adopt a staggered conformation. The two fused 6-membered rings of the 9-BBN moiety in complex 3 adopt a boat–boat configuration. The Zn1–N bond lengths are in the range of 2.017(18)–2.052(15) Å and the Zn1–B distances in complex 3 are in the range of 2.250(19)–2.260(20) Å. The coordination geometry of complex 3 is a distorted pseudo-tetrahedron; the τ4 parameter46 is equal to 0.92 when considering the N1, N3, B2 and B3 atoms as the edges of the tetrahedron centered on the Zn1 atom. The chelation ring in complex 3 adopts a boat configuration, where the α and β ring puckering parameters,47 defined as the angles between the {N1, N2, N3, N4} and {C11, N2, N4} and {N1, N2, N3, N4} and {Zn1, N1, N3} planes, are equal to 53.56° and 7.89°, respectively. Complex 3 is, to the best of our knowledge, the first crystallographically reported zinc bis(κ2-borohydride) complex with a bis(pyrazolyl) ligand motif. Three other zinc bis(κ2-borohydride) complexes have been reported in the literature, and their Zn–B distances are in the range of 2.252–2.355 Å,48 which are within those found for complex 3. The X-ray structure of complex 3 was accurately reproduced by DFT calculations, as highlighted by the respective structure superimpositions presented in Fig. S17 in the ESI.†
With fully characterized complexes 1–4 in hand, they were tested in the hydroboration of different families of substrates. The first substrate to be tested in a hydroboration reaction with HBPin catalyzed by complexes 1–4 was CO2, given our previous results using complexes [(L1–4)ZnCl2] as precatalysts,41 the results of which are summarized in Table 1.
a
|
|||
|---|---|---|---|
| Entry | Cat. | Solvent | Conversionb (%) |
| a Conditions: 1 mol% cat.; 1 mmol of HBPin; 1 bar of CO2.b Conversions were determined by 1H NMR spectroscopy utilizing 1,3,5-trimethoxybenzene as an internal standard and based on HBPin.c 2 equivalents of KHBEt3 added.d Ref. 41. |
|||
| 1 | 1 | C6D6 | 18 |
| 2 | 2 | C6D6 | 24 |
| 3 | 3 | C6D6 | 34 |
| 4 | 4 | C6D6 | 32 |
| 5 | 3 | THF-d8 | 57 |
| 6 | [(L3)ZnCl2]c |
THF-d8 | 42d |
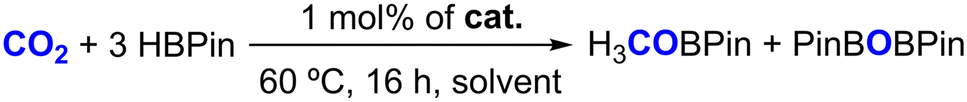
As indicated in Table 1, performing the hydroboration of CO2 in C6D6 at 1 bar and 60 °C catalyzed by 1 mol% of complexes 1–4 led to HBPin conversion rates of 18–34%, increasing in the following order: 1 < 2 < 3 ≈ 4 (Table 1, entries 1–4). The hydroboration of CO2 catalyzed by the currently reported complexes selectively gave rise to the formation of the methyl boronate ester H3COBPin, along with PinBOBPin, with their formation detected by 1H NMR spectroscopy, matching the data found in the literature.49 This result indicates that the borane functionalities present in complexes 3 and 4 are beneficial for their catalytic activity. Furthermore, when performing the hydroboration of CO2 in THF-d8 using complex 3 as a catalyst, the HBPin conversion increased to 57%, somewhat improving the previously obtained result using the catalyst system composed of [(L3)ZnCl2]/2KHBEt3 (Table 1, entries 5 and 6).
Given the success of the single-component system catalysts 1–4 in converting CO2 with HBPin, other isoelectronic heteroallenes were sought after, such as N,N′-dicyclohexylcarbodiimide and tert-butyl isocyanate. When using 1 mol% of complexes 1–4 at 60 °C, tert-butyl isocyanate was hydroborated with HBPin, as summarized in Table 2.
a
|
||||
|---|---|---|---|---|
| Entry | Cat. | Time (h) | Conversionb (%) | A:B:C ratiob |
| a Conditions: 1 mol% cat.; 0.5 mmol of HBPin; 0.5 mmol of tert-butyl isocyanate.b Conversions and product ratios were determined by 1H NMR spectroscopy by relative integration of the resonances of the HBPin and products and were based on HBPin. | ||||
| 1 | 1 | 1 | 9 | 1:0:0 |
| 2 | 2 | 1 | 10 | 1:0:0 |
| 3 | 3 | 1 | 35 | 1.8:0:1 |
| 4 | 4 | 1 | 26 | 7.1:0:1 |
| 5 | 3 | 16 | >99 | 0:0:1 |
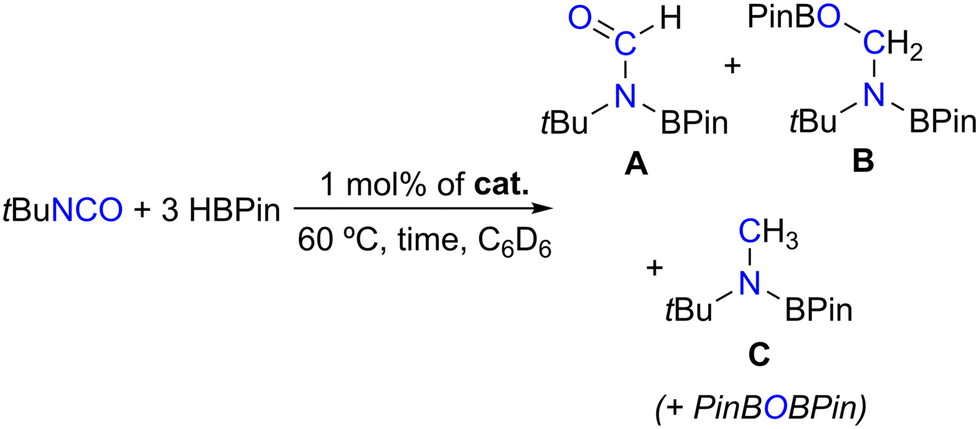
Table 2 shows that complexes 1–4 achieved HBPin conversions of 9–35% after 1 h of reaction, increasing in the order 1 < 2 < 4 < 3 (Table 2, entries 1–4). This again indicates that the borane functionalities present in complexes 3 and 4 are beneficial for their catalytic activity. The catalytic reactions performed in the presence of complexes 3 and 4 were accompanied by the observation of hydroboration products resulting from different degrees of tert-butyl isocyanate reduction: formamide (A), methanamide (B) and methylamide (C) levels, the latter also concomitantly forming with PinBOBPin, in A:B:C ratios equal to 1.8–7.1:0:1. The formation of the hydroboration products of tert-butyl isocyanate was detected by 1H NMR spectroscopy, matching the data found in the literature,12 and their ratio was calculated by relative integration of the respective resonances. When performing the hydroboration of tert-butyl isocyanate with HBPin using 1 mol% of complex 3 for 16 h at 60 °C, the reaction proceeded to full conversion with exclusive selectivity toward the methylated product C, along with PinBOBPin (Table 2, entry 5). Performing the same reactions using 1 or 2 equivalents of HBPin with respect to tert-butyl isocyanate did not change the selectivity in the products of catalysis. Under the same reaction conditions, N,N′-dicyclohexylcarbodiimide remained unreactive.
To further understand the reactivity profile of the hydroboration of tert-butyl isocyanate with HBPin catalyzed by 1 mol% of complex 3, the reaction conversion and yields were monitored over time at 60 °C (Fig. 2).
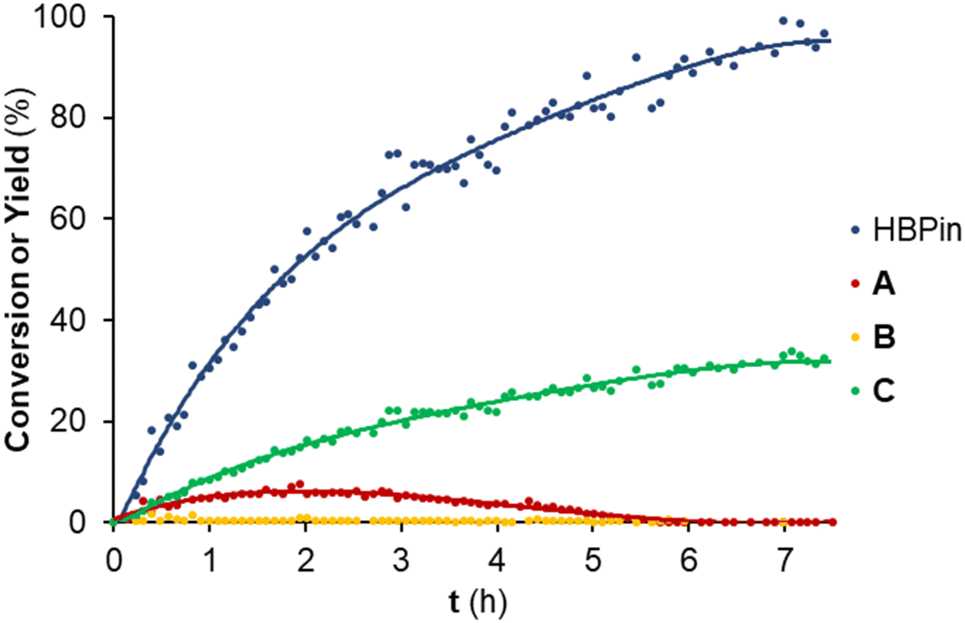 | ||
| Fig. 2 Conversion and yield monitoring of the hydroboration of tert-butyl isocyanate with HBPin catalyzed by 3 over time at 60 °C: evolution of the conversion of HBPin (blue data) and the yields of the boramidate products resulting from the mono (red data), double (yellow data) and triple (green data) reduction of tert-butyl isocyanate, that is, products A, B and C in Table 3, respectively. The colored lines are a visual guide. Conditions: 1 mol% of 3; 0.5 mmol HBPin; 0.5 mmol tert-butyl isocyanate; solvent: C6D6 (0.5 mL). The conversions and yields were determined by 1H NMR spectroscopy by relative integration of the resonances of the HBPin and boramidate products. | ||
The reaction conversion and yield monitoring of the hydroboration of tert-butyl isocyanate with HBPin at 60 °C catalyzed by 1 mol% of complex 3 over time led to the detection of compounds A, B and C (and PinBOBPin) at early reaction times (<30 minutes), with compound B being only observed in trace amounts. Compound A reached a maximum yield of 8% after 2 h and gradually disappeared, thus converting to compound C. After 6 h of the reaction, compound C (along with PinBOBPin) is the only observable product and achieved the maximum yield after 7.5 h. The maximum TOF of this reaction, measured at the highest rate of HBPin conversion, is equal to 33 h−1. The conversion profile of the hydroboration of tert-butyl isocyanate catalyzed by complex 3 is somewhat analogous to that observed for the hydroboration of CO2 catalyzed by complex [(L3)ZnCl2]/2KHBEt3, meaning that the methylamide product was sequentially formed from the formamide and methanamide ones. The system [(L3)ZnCl2]/2KHBEt3 also catalyzed the hydroboration of tert-butyl isocyanate, selectively forming the corresponding methyl boramidate product in quantitative yield. This latter result is though misleading, considering that KHBEt3 itself is an efficient isocyanate hydroboration catalyst26 and using complex [(L3)ZnCl2] on its own did not achieve any recordable conversion after 16 h at 60 °C.
The new complexes 1–4 also catalyzed the hydroboration of ethyl benzoate, giving rise to the respective benzyl and ethyl boronic esters, as indicated in Table 3.
a
|
|||
|---|---|---|---|
| Entry | Cat. | Time (h) | Conversionb (%) |
| a Conditions: 1 mol% cat.; 0.5 mmol of HBPin; 0.5 mmol of ethyl benzoate.b Conversions were determined by 1H NMR spectroscopy by relative integration of the resonances of the HBPin and products and were based on HBPin. | |||
| 1 | 1 | 16 | 7 |
| 2 | 2 | 16 | 11 |
| 3 | 3 | 16 | 52 |
| 4 | 4 | 16 | 29 |
| 5 | 3 | 1 | 15 |
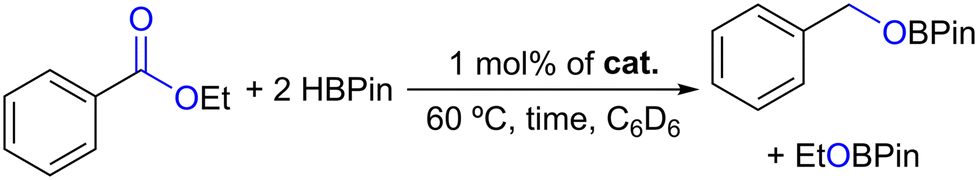
By looking at Table 3, the hydroboration of ethyl benzoate catalyzed by 1 mol% of complexes 1–4 for 16 h at 60 °C led to conversions of 7–52%, increasing in the following order, 1 < 2 < 4 < 3 (Table 3, entries 1–4), again indicating the relevance of the borane functionality to the catalytic activity. In addition, when using complex 3 as a catalyst, the HBPin conversion decreased to 15% when performing the reaction for 1 h (Table 3, entry 5).
The last substrate to be preliminarily screened for hydroboration using complexes 1–4 as catalysts was diphenylacetonitrile and the respective catalytic results are presented in Table 4.
a
|
|||
|---|---|---|---|
| Entry | Cat. | Time (h) | Conversionb (%) |
| a Conditions: 1 mol% cat.; 0.5 mmol of HBPin; 0.5 mmol of diphenylacetonitrile.b Conversions were determined by 1H NMR spectroscopy by relative integration of the resonances of the HBPin and products and were based on HBPin. | |||
| 1 | 1 | 1 | 7 |
| 2 | 2 | 1 | 15 |
| 3 | 3 | 1 | 75 |
| 4 | 4 | 1 | 71 |
| 5 | 3 | 16 | >99 |
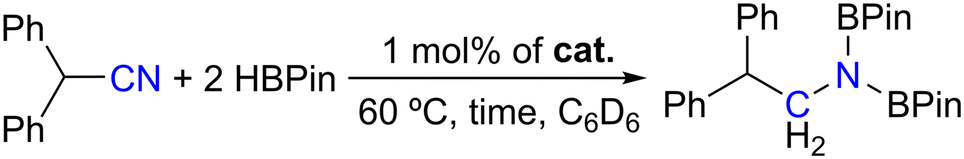
Table 4 indicates that only complexes 3 and 4 functioned as good catalysts for the dihydroboration of diphenylacetonitrile, leading to the formation of the corresponding boramidate, achieving 75% conversion after 1 h at 60 °C (Table 4, entries 3 and 4), while complexes 1 and 2 did not lead to conversions higher than 15% (Table 4, entries 1 and 2). This follows the trend already observed for the substrates considered above. Full conversion of diphenylacetonitrile was achieved using complex 3 as a catalyst by extending the reaction time to 16 h (Table 4, entry 5). The formation of the imine PinBNCH(CHPh2), resulting from the monohydroboration of diphenylacetylene with HBPin was never observed throughout all hydroboration runs, even when employing stoichiometric amounts of HBPin.
As disclosed so far, 1 mol% of complex 3 was able to catalyze the hydroboration of CO2, tert-butyl isocyanate, ethyl benzoate and diphenylacetonitrile. Taking this into account, the substrate scope of this catalyst system was explored under the optimized conditions. In effect, 1 mol% of complex 3 successfully catalyzed the hydroboration of a variety of isocyanates and esters and the dihydroboration of nitriles, as summarized in Scheme 2.
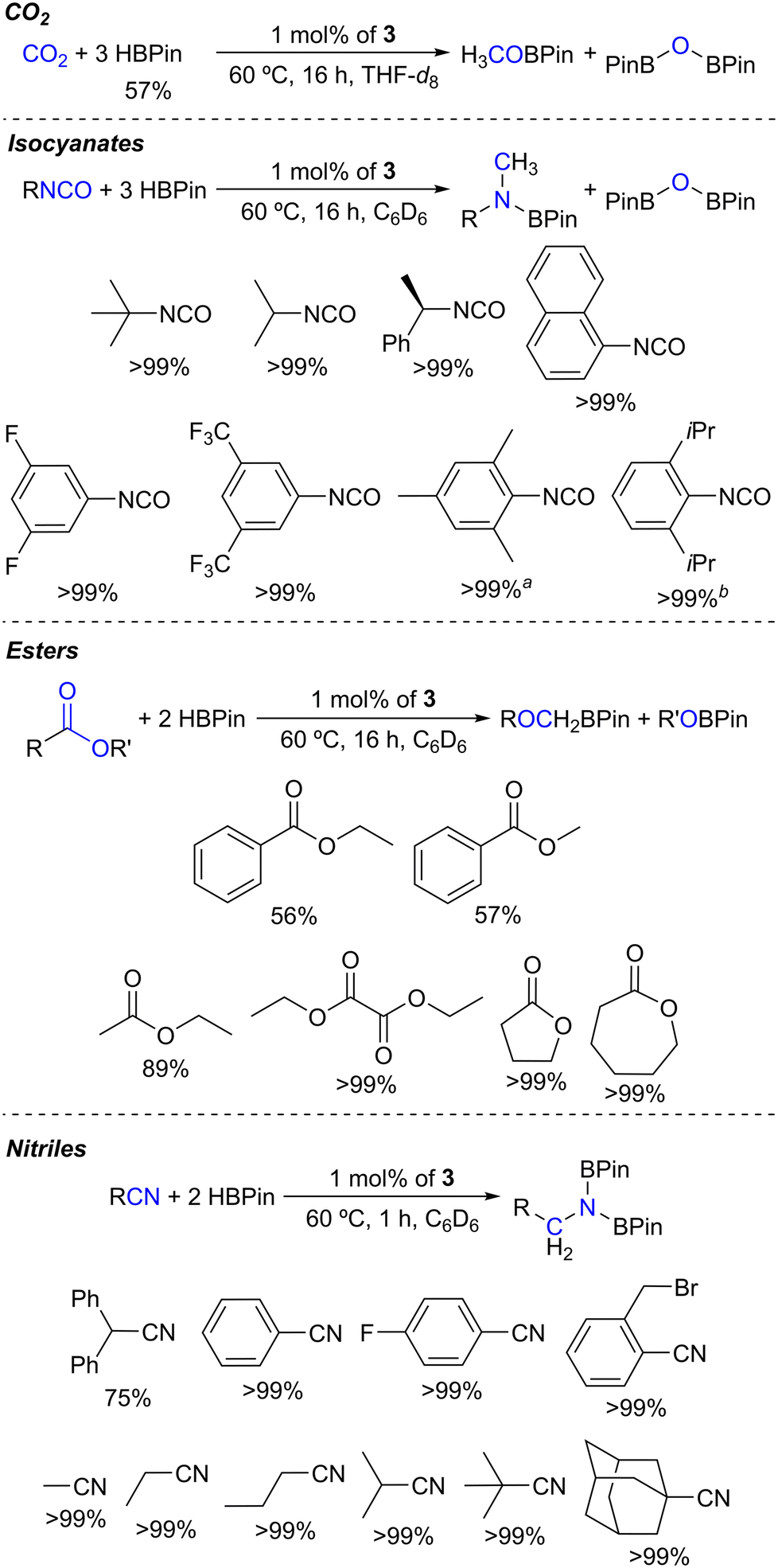 | ||
| Scheme 2 Substrate scope of the hydroboration reactions catalyzed by complex 3. The percentages correspond to the substrate conversion based on HBPin. For the indicated isocyanate substrates, a mixture of the methylene and methyl boramidate products was observed in a a1:3.6 and b1:2.1 ratio. | ||
Complex 3 selectively catalyzed the hydroboration of a set of isocyanates. Several aliphatic and electronically differentiated aromatic isocyanates, including 1-isocyanatonaphthalene, were successfully reduced to the respective methyl boramidate products along with the formation of PinBOBPin (Fig. S19–S26 in the ESI†). The formation of the products was confirmed by comparing the 1H NMR resonances with those reported in the literature.12,29 When using isocyanate substrates bearing mesityl and diisopropylphenyl substituents, the formation of methylene boramidate products was also observed in methylene:methyl ratios of 1:3.6 and 1:2.1, respectively. This observation is likely due to the very high stereochemical hindrance imparted by these substrates.
Regarding ester substrates (Fig. S27–S32 in the ESI†), 1 mol% of complex 3 catalyzed the hydroboration of ethyl and methyl benzoate, forming PhCH2OBPin (along with H3CH2COBPin for ethyl benzoate and H3COBPin for methyl benzoate) in conversions of 57% and 56%, respectively. The hydroborations of the ester substrates ethyl acetate and diethyl oxalate were smoothly catalyzed by complex 3, achieving 89 and >99% conversions, respectively. Cyclic esters, γ-butyrolactone and ε-caprolactone, were also quantitatively hydroborated in the present study. The formation of products was confirmed by comparing the 1H NMR resonances with those reported in the literature.17a The catalyst system comprising 1 mol% of complex 3 also promoted the hydroborative deoxygenation of N,N-diethylbenzamide to N-benzyl-N-ethylethanamine in a near-quantitative conversion at 60 °C in C6D6 after 16 h (Fig. S33 in the ESI†). The formation of this product, along with PinBOBPin, was confirmed by comparing its 1H NMR spectrum with that reported in the literature.50 This latter result is significant, since deoxygenation of tertiary amides via hydroboration is a rare occurrence in the literature.50
Complex 3 also catalyzed the dihydroboration of nitrile substrates (Fig. S34–S43 in the ESI†). With this system, a number of aliphatic and electronically differentiated aromatic substrates were successfully dihydroborated. Also, complex 3 catalyzed the chemoselective dihydroboration of 2-(bromomethyl)benzonitrile. The formation of the products was confirmed by comparing the 1H NMR resonances with those reported in the literature.25b
No catalytic activity was ever observed when performing the reactions at 25 °C or in the absence of catalyst for all preliminarily tested substrates (Fig. S44 in the ESI†). Complexes 1–4 did not catalyze the hydroboration of the heterocyclic substrate pyridine.
In order to evaluate the relative reactivity of the different substrate families when exposed to the present hydroboration conditions, selected competitive experiments were performed (Scheme 3A). When performing the competitive hydroboration with 1-isocyanatonaphthalene or benzonitrile with ethyl benzoate, only the products resulting from the conversion of isocyanate or nitrile substrates were observed. On the other hand, when performing the competitive hydroboration between 1-isocyanatonaphthalene and benzonitrile, a mixture of products was observed, in which the dihydroboration product of benzonitrile corresponds to 36% of the mixture, while 64% resulted from the conversion of 1-isocyanatonaphthalene, forming the formyl and methyl boramidate (along with PinBOBPin) products in a 0.8:1 ratio. The spectra relevant to the competition experiments are presented in Fig. S45–S47 in the ESI.† These results show that the present catalyst system is chemoselective toward nitrile or isocyanate substrates in comparison with ester ones.
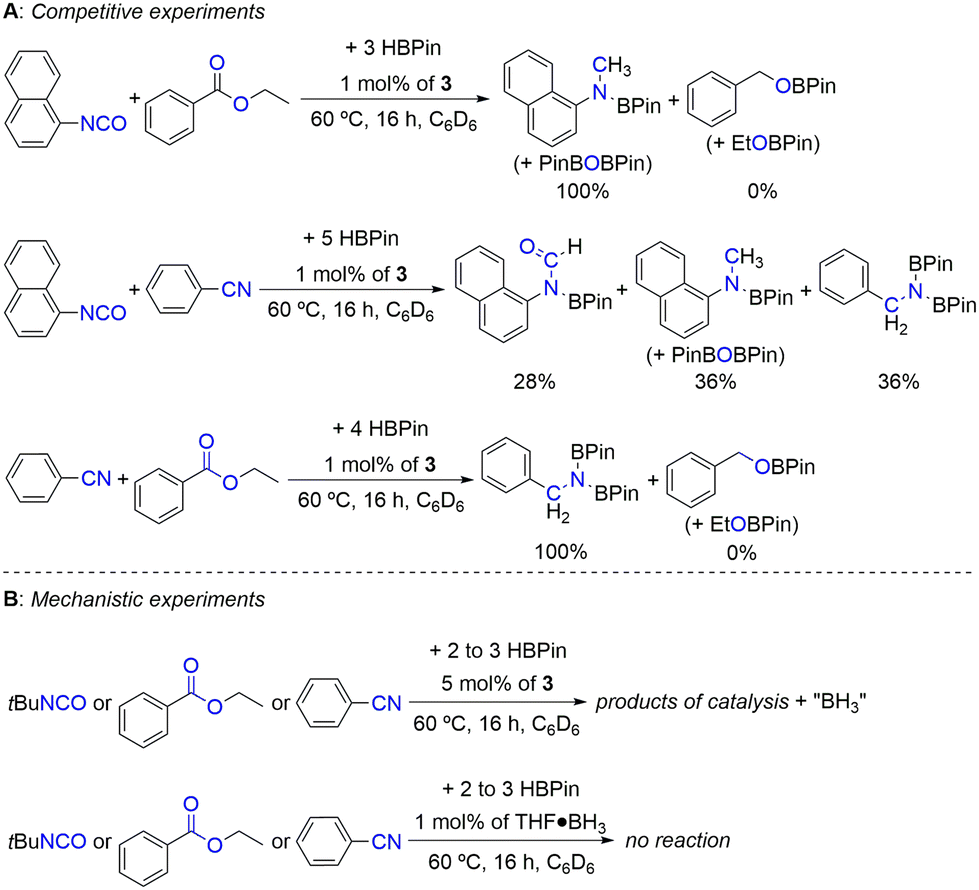 | ||
| Scheme 3 Competitive (A) and mechanistic (B) experiments. The percentages correspond to the products’ ratio. | ||
The catalytic hydroboration reactions of tert-butyl isocyanate, ethyl benzoate and diphenylacetonitrile using 5 mol% of complex 3 were also performed (Scheme 3B), with the corresponding magnified versions of the 11B NMR spectra being presented in Fig. S48 in the ESI.† The 11B NMR spectra acquired from these reactions revealed the appearance of quartet resonances centered at −12 ppm (1JBH = 99 Hz), indicative of the presence of BH3.51 The observation of BH3 in the reaction mixtures arises from the dissociation of the borohydride co-ligands in complex 3, readily forming the respective zinc dihydride intermediate [(L3)ZnH2]. The formation and structure of this intermediate have already been discussed in a previous publication.41 It may not be ruled out that the BH3 formed during the catalytic reactions is trapped by the coordinative pressure of the substrates, leading to the formation of substrate·BH3 Lewis pairs. In fact, for the tert-butyl isocyanate substrate, two further triplets (1JBH = 119 Hz) appeared in the 11B NMR spectrum, indicating that “BH2” species are present, likely originating from stoichiometric hydroboration reactions with BH3 formed during the catalytic initiation process.
The spontaneous liberation of BH3 from complex 3 was never observed, not even after prolonged heating at 60 °C. To discard the possibility of substrate·BH3 adducts operating as catalysts themselves, the catalytic hydroborations of CO2, tert-butyl isocyanate, ethyl benzoate and diphenylacetonitrile with HBPin in the presence of 1 mol% of THF·BH3 were performed (Scheme 3B). In our hands, no catalytic activity was ever observed. In fact, even though BH3 adducts have been known to catalyze the hydroboration of nitriles with HBPin, they did so in very low yields.52 Control reactions performed with 1 mol% of THF·BH3 in the presence of L3 also did not lead to any catalytic activity. In addition, reactions of complex 3 with 1 bar of CO2 or 5 equivalents of 1,3-difluoro-5-isocyanatobenzene or 4-fluorobenzonitrile were performed, with the respective 1H NMR spectra being presented in Fig. S49 in the ESI.† Even though such reactions led to complex mixtures of products, the formation of new complexes was evident and the appearance of deshielded singlet resonances at 7.81–9.16 ppm was observed, attributed to zinc formate (for CO2), formamide (for 1,3-difluoro-5-isocyanatobenzene) or azavinylidene (for 4-fluorobenzonitrile) proton resonances. This indicates the occurrence of substrate insertion reactions in the putative zinc dihydride intermediate [(L3)ZnH2], likely formed upon liberation of BH3 from complex 3. Such deshielded singlet resonances have been detected in zinc formate (8.33 ppm),41 formamide (7.93 ppm)29 and azavinylidene (9.59 ppm)30e complexes.
In order to gain further insight into the activation of the catalysts, the spontaneous liberation of BH3 from complex 3 and the reactions of complex 3 with the different families of substrates, giving rise to the putative dihydride complex [(L3)ZnH2] and corresponding substrate·BH3 adducts, were studied by DFT calculations53 and the respective Gibbs energy balances were established (Scheme 4).
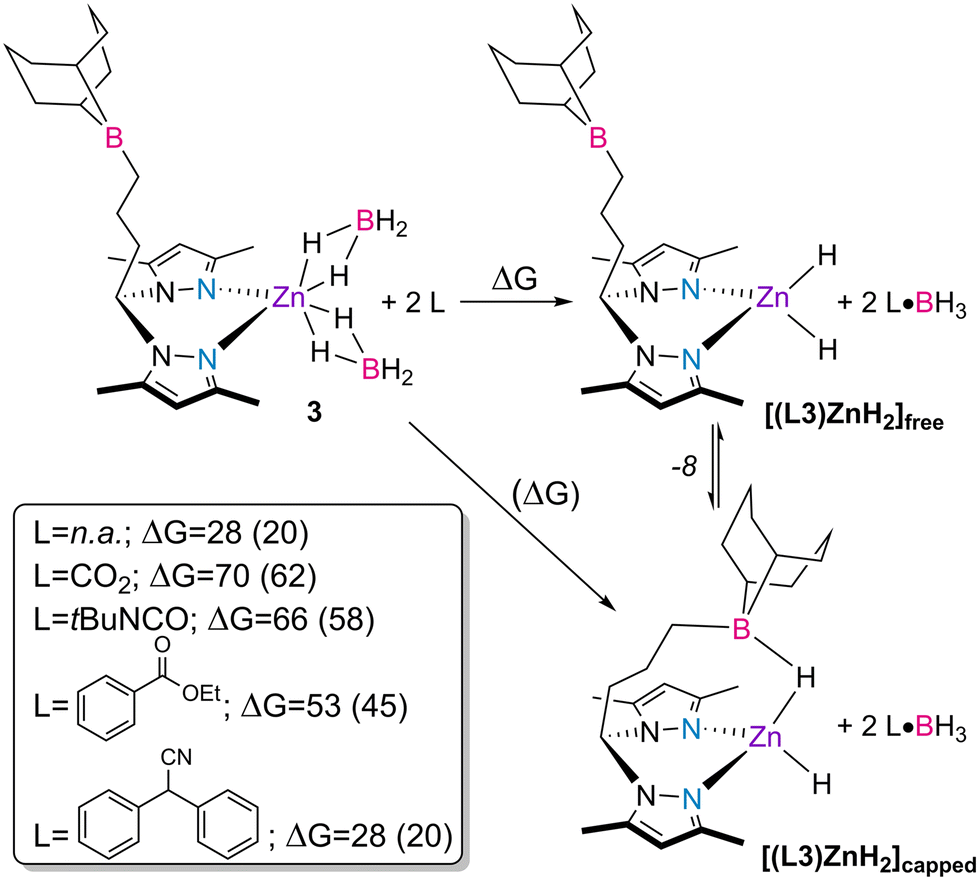 | ||
| Scheme 4 Gibbs energy balances (in kcal mol−1) for the reactions involving the liberation of BH3 from the bis(κ2-borohydride) complex 3, yielding complex [(L3)ZnH2]free or [(L3)ZnH2]capped (numbers in parentheses) determined by DFT calculations. | ||
By looking at the energy balances determined by DFT calculations, the spontaneous liberation of BH3 from complex 3 yielding complex [(L3)ZnH2]free is less unfavorable than that induced by the different substrate molecules with a free energy balance of ΔG = 28 kcal mol−1. This value is further reduced to 20 kcal mol−1 when considering that the spontaneous liberation of BH3 from complex 3 yields complex [(L3)ZnH2]capped, in which the pending borane intramolecularly stabilizes one of the hydride ligands. The structure of complex [(L3)ZnH2]capped has been discussed in a previous publication.41 For all other substrate families, except nitriles, the liberation and subsequent trapping processes of BH3 using CO2, tert-butyl isocyanate and ethyl benzoate are highly unfavored, resulting in Gibbs energies in the range of 45–62 kcal mol−1. The presented energy balances are, thus, in line with the systematic observation of BH3 over the course of the catalytic reactions, regardless of the type of substrate used. In addition, they further corroborate the increased catalytic activity observed in the borane-substituted catalysts 3 and 4 in comparison with the unfunctionalized complexes 1 and 2, giving the favorable formation of the capped form of the complex.
Given the experimental and theoretical information described above, as well as the conclusions taken from our previous work,41 a mechanistic discussion may be proposed (Scheme 5).
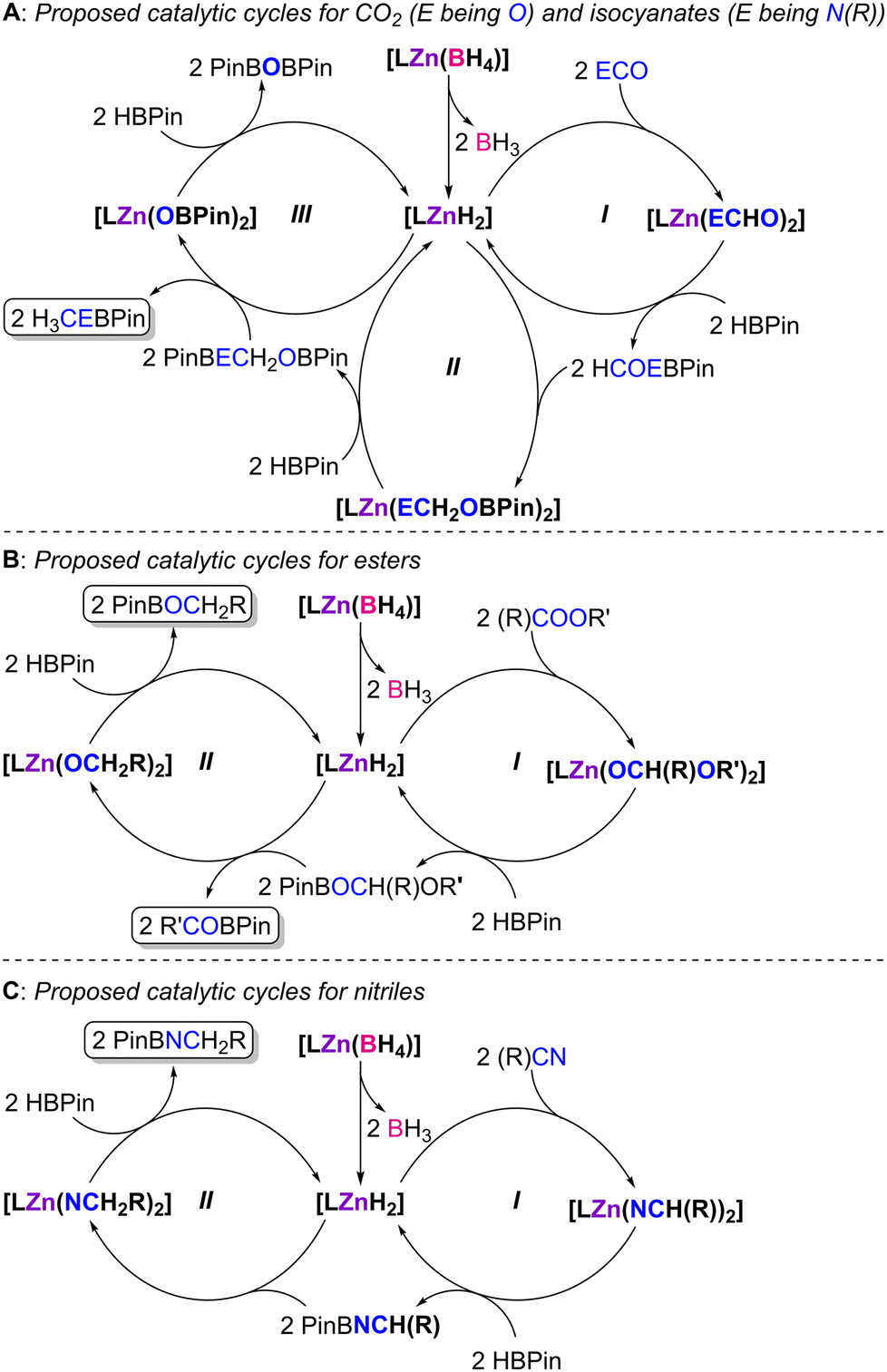 | ||
| Scheme 5 Mechanistic proposal for the hydroboration of CO2 and isocyanates (A), esters (B) and nitriles (C) with HBPin catalyzed by complexes 1–4, where L denotes ligands L1–4. | ||
The activation step in the mechanistic hypothesis is common to all substrate families: elimination of BH3 from the κ2-borohydride ligands in the complexes, giving rise to the formation of [LZnH2] intermediates. The [LZnH2] intermediates bearing the borane-functionalized ligands L3 and L4 may either exhibit the borane in a tri-coordinated, free conformation or where the borane is inter- (via dimerization) or intramolecularly stabilizing one of the hydride ligands.41 Subsequently, the substrates undergo insertion into the [LZnH2] intermediates, forming the respective formate ([LZn(OCHO)2], for CO2), formamide ([LZn(N(R)CHO)2], for isocyanates), methanolate ([LZn(OCH2(R)OR′)2], for esters) and azavinylidene ([LZn(NCH(R))2], for nitriles) intermediates.
In the case of the hydroboration of CO2 and isocyanates (Scheme 5A), the formate or formamide intermediates, respectively, resulting from their insertion into the [LZnH2] intermediates, give rise, upon reaction with an excess amount HBPin, to the generation of the formyl or formamide compounds HCOEBPin (E being O or N(R)), regenerating [LZnH2] (Scheme 5A, cycle I). Compounds HCOEBPin undergo further insertion reactions into the Zn–H bonds of the [LZnH2] intermediates, generating complexes of type [LZn(OCH2EBPin)2] (Scheme 5A, cycle II). Intermediate complexes [LZn(OCH2EBPin)2] may further react with HBPin, leading to the formation of H3CEBPin along with intermediates [LZn(OBPin)2]. Complexes [LZn(OBPin)2] give rise to PinBOBPin by further reaction with HBPin, again regenerating [LZnH2] (Scheme 5A, cycle III). This mechanism was originally proposed by Trovitch7 and was analogously reported by us in our previous study concerned with the hydroboration of CO2 catalyzed by complexes [(L1–4)ZnCl2]/KHBEt3,41 and is supported by the fact that the present catalyst systems give rise to methyl-level products.
With respect to the hydroboration of esters (Scheme 5B), further reaction of the methanolate intermediates [LZn(OCH2(R)OR′)2] with HBPin would lead to the formation of compounds PinBOCH2(R)OR′ and regeneration of intermediates [LZnH2] (Scheme 5B, cycle I). Subsequent reaction of PinBOCH2(R)OR′ and [LZnH2] would likely induce an ether cleavage reaction, generating intermediate [LZn(OCH2R)2] and compounds R′OBPin, the latter products being systematically observed in all catalytic reactions. The reaction of intermediate complex [LZn(OCH2R)2] with HBPin would also give rise to compounds PinBOCH2R and regenerate the dihydride intermediates [LZnH2] (Scheme 5B, cycle II). This ester hydroboration mechanism has been reported by Nembenna and co-workers.17
Lastly, the hydroboration of nitriles (Scheme 5C) and the reaction of the azavinylidene intermediates [LZn(NCH(R))2] with HBPin would give rise to the imine PinBNCH(R) and regeneration of the [LZnH2] intermediates (Scheme 5C, cycle I). The imine PinBNCH(R) likely undergoes a facile insertion reaction into the Zn–H bonds of the [LZnH2] intermediates, generating the intermediate amido complexes [LZn(NCH2R)2]. Such amido complexes may react with HBPin and give rise to products PinBNCH2R and the regeneration of the [LZnH2] active species (Scheme 5C, cycle II).
The role of the appended boranes in complexes 3 and 4 is that of kinetically stabilizing intermediates in all catalytic cycles by means of establishing Lewis pairs between hydrogen/oxygen/nitrogen-based ligands and the respective borane functions via Zn–H–B/Zn–O–B/Zn–N–B interactions. These interactions may facilitate insertion and σ-bond metathesis reactions by weakening the respective Zn–H/Zn–O/Zn–N bonds.
Conclusions
In this work, new bis(κ2-borohydride) zinc complexes of borane-functionalized heteroscorpionate ligands served as catalysts for the hydroboration of a broad scope of substrates.Four new bis(κ2-borohydride) zinc complexes 1–4 were synthesized. Complexes 1 and 2 contained unfunctionalized and allyl-functionalized bis(3,5-dimethylpyrazolyl)methane ligands, while complexes 3 and 4 contained borane functionalities on these scaffolds. Complexes 1–4 were characterized by NMR spectroscopy, FTIR spectroscopy and elemental analysis while complex 3 was additionally characterized by single-crystal X-ray diffraction. All complexes displayed the κ2-coodination mode of borohydride, as confirmed by the spectroscopic techniques used.
The new complexes were used as catalysts in the hydroboration of a series of unsaturated substrates containing heteroatoms, namely CO2 at 1 bar, tert-butyl isocyanate, and ethyl benzoate and the dihydroboration of diphenylacetonitrile at 60 °C with 1 mol% of catalyst loading. The borane-functionalized complexes 3 and 4 achieved conversions of 29–99% and, as a common feature, exhibited an average 5-fold increase in their catalytic activities in comparison with the unfunctionalized complexes 1 and 2. In the case of CO2 and isocyanates, the methylated products (along with PinBOBPin) were preferred after 16 h. In addition, 1 mol% of complex 3 catalyzed the hydroboration of several isocyanates, esters and nitriles with yields in the range of 56–99%.
The catalytic competence of the new complexes is dependent on the release of BH3 from the borohydride ligands, as confirmed by a set of experimental and theoretical observations. This generates dihydride zinc complexes, through which catalytic turnover takes place. The borane functionalities in complexes 3 and 4 provide a catalytic advantage because they are able to establish Lewis pairs with oxygen- and nitrogen-containing catalytic intermediates.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Centro de Química Estrutural is financed by the Fundação para a Ciência e a Tecnologia, I.P./MCTES through national funds PIDDAC – CQE UIDB/00100/2020 and UIDP/00100/2020 (https://doi.org/10.54499/UIDP/00100/2020) and the Institute of Molecular Sciences Associated Laboratory is funded by Project LA/P/0056/2020 (https://doi.org/10.54499/LA/P/0056/2020).References
- (a) V. P. Ananikov and M. Tanaka, Hydrofunctionalization, Springer, 2013, vol. 43 CrossRef; (b) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- (a) C. Erken, A. Kaithal, S. Sen, T. Weyhermüller, M. Hölscher, C. Werlé and W. Leitner, Nat. Commun., 2018, 9, 4521–4529 CrossRef PubMed; (b) S. Kostera, M. Peruzzini, K. Kirchner and L. Gonsalvi, ChemCatChem, 2020, 12, 4625–4631 CrossRef CAS; (c) P. Ghosh and A. J. von Wangelin, Angew. Chem., Int. Ed., 2021, 60, 16035–16043 CrossRef CAS PubMed.
- (a) G. Jin, C. G. Werncke, Y. Escudié, S. Sabo-Etienne and S. Bontemps, J. Am. Chem. Soc., 2015, 137, 9563–9566 CrossRef CAS PubMed; (b) S. Lau, C. B. Provis-Evans, A. P. James and R. L. Webster, Dalton Trans., 2021, 50, 10696–10700 RSC; (c) H. Gao, J. Jia, C.-H. Tung and I. Wang, Organometallics, 2023, 42, 944–951 CrossRef CAS.
- S. R. Tamang and M. Findlater, Dalton Trans., 2018, 47, 8199–8203 RSC.
- H.-W. Suh, L. M. Guard and N. Hazari, Polyhedron, 2014, 84, 37–43 CrossRef CAS.
- R. Shintani and K. Nozaki, Organometallics, 2013, 32, 2459–2462 CrossRef CAS.
- R. Pal, T. L. Groy and R. J. Trovitch, Inorg. Chem., 2015, 54, 7506–7515 CrossRef CAS PubMed.
- (a) S. Bontemps, L. Vendier and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 1671–1674 CrossRef CAS PubMed; (b) M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343–11345 CrossRef CAS PubMed; (c) S. Bontemps and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2013, 52, 10253–10255 CrossRef CAS PubMed; (d) S. Bontemps, L. Vendier and S. Sabo-Etienne, J. Am. Chem. Soc., 2014, 136, 4419–4425 CrossRef CAS PubMed; (e) C. K. Ng, J. Wu, T. S. A. Hor and H.-K. Luo, Chem. Commun., 2016, 52, 11842–11845 RSC; (f) L. Li, H. Zhu, L. Liu, D. Song and M. Lei, Inorg. Chem., 2018, 57, 3054–3060 CrossRef CAS PubMed.
- H.-W. Suh, L. M. Guard and N. A. Hazari, Chem. Sci., 2014, 5, 3859–3872 RSC.
- (a) M. D. Anker, M. Arrowsmith, P. Bellham, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot, M. F. Mahon and C. Weetman, Chem. Sci., 2014, 5, 2826–2830 RSC; (b) J. A. B. Abdalla, I. M. Riddlestone, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 5098–5102 CrossRef CAS PubMed; (c) Z. Lu, H. Hausmann, S. Becker and H. A. Wegner, J. Am. Chem. Soc., 2015, 137, 5332–5335 CrossRef CAS PubMed; (d) T. J. Hadlington, C. E. Kefalidis, L. Maron and C. Jones, ACS Catal., 2017, 7, 1853–1859 CrossRef CAS; (e) X. Cao, W. Wang, K. Lu, W. Yao, F. Xue and M. Ma, Dalton Trans., 2020, 49, 2776–2780 RSC; (f) C.-C. Chia, W.-C. Teo, N. Cham, S. Y.-F. Ho, Z.-H. Ng, H.-M. Toh, N. Mézailles and C.-W. So, Inorg. Chem., 2021, 60, 4569–4577 CrossRef CAS PubMed; (g) J. Fan, J.-Q. Mah, M.-C. Yang, M.-D. Su and C.-W. So, J. Am. Chem. Soc., 2021, 143, 4993–5002 CrossRef PubMed; (h) B. Yan, S. Dutta, X. Ma, C. Ni, D. Koley, Z. Yang and H. W. Roesky, Dalton Trans., 2022, 51, 6756–6765 RSC.
- M. R. Espinosa, D. J. Charboneau, A. G. de Oliveira and N. Hazari, ACS Catal., 2019, 9, 301–314 CrossRef CAS.
- K. A. Gudun, S. Tussupbayev, A. Slamovac and A. Y. Khalimon, Org. Biomol. Chem., 2022, 20, 6821–6830 RSC.
- L. E. English, T. M. H. Downie, C. L. Lyall, M. F. Mahon, C. L. McMullin, S. E. Neale, C. M. Saunders and D. J. Liptrot, Chem. Commun., 2023, 59, 1074–1077 RSC.
- V. K. Pandey, S. Sahoo and A. Rit, Chem. Commun., 2022, 58, 5514–5517 RSC.
- (a) Y. Yang, M. D. Anker, J. Fang, M. F. Mahon, L. Maron, C. Weetman and M. S. Hill, Chem. Sci., 2017, 8, 3529–3537 RSC; (b) N. Sarkar, R. K. Sahoo and S. Nembenna, Eur. J. Org. Chem., 2022, e202200941 CrossRef CAS; (c) C. Ni, X. Ma, Z. Yang and H. W. Roesky, ChemistrySelect, 2022, 7, e202202878 CrossRef CAS; (d) J. Shi, M. Luo, X. Zhang, T. Yuan, X. Chen and M. Ma, Org. Biomol. Chem., 2023, 21, 3628–3635 RSC.
- K. Makarov, I. Ritacco, N. Fridman, L. Caporaso and M. S. Eisen, ACS Catal., 2023, 13, 11798–11814 CrossRef CAS.
- (a) N. Sarkar, R. K. Sahoo and S. Nembenna, Chem. – Eur. J., 2023, 29, e202203023 CrossRef CAS PubMed; (b) Y. Zheng, X. Zhu, X. Xu, S. Zhou, W. Lua and M. Xue, New J. Chem., 2023, 47, 19367–19371 RSC.
- (a) T. T. Nguyen, J.-H. Kim, S. Kim, C. Oh, M. Flores, T. L. Groy, M.-H. Baik and R. J. Trovitch, Chem. Commun., 2020, 56, 3959–3962 RSC; (b) M. R. Elsby, C. Oh, M. Son, S. Y. H. Kim, M.-H. Baik and R. T. Baker, Chem. Sci., 2022, 13, 12550–12559 RSC.
- (a) A. R. Bazkiaei, M. Wiseman and M. Findlater, RSC Adv., 2021, 11, 15284–15289 RSC; (b) T. Komuro, K. Hayasaka, K. Takahashi, N. Ishiwata, K. Yamauchi, H. Tobita and H. Hashimoto, Dalton Trans., 2024, 53, 4041–4047 RSC.
- (a) A. D. Ibrahim, S. W. Entsminger and A. R. Fout, ACS Catal., 2017, 7, 3730–3734 CrossRef CAS; (b) H. Ben-Daat, C. L. Rock, M. Flores, T. L. Groy, A. C. Bowman and R. J. Trovitch, Chem. Commun., 2017, 53, 7333–7336 RSC; (c) C. Ghosh, S. Kim, M. R. Mena, J.-H. Kim, R. Pal, C. L. Rock, T. L. Groy, M.-H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2019, 141, 15327–15337 CrossRef CAS PubMed; (d) J. Pecak, W. Eder, B. Stöger, S. Realista, P. N. Martinho, M. J. Calhorda, W. Linert and K. Kirchner, Organometallics, 2020, 39, 2594–2601 CrossRef CAS PubMed; (e) K. A. Gudun, A. Slamova, D. Hayrapetyan and A. Y. Khalimon, Chem. – Eur. J., 2020, 26, 4963–4968 CrossRef CAS PubMed; (f) C. Li, S. Song, Y. Li, C. Xu, Q. Luo, Y. Guo and X. Wang, Nat. Commun., 2021, 12, 3813–3820 CrossRef CAS PubMed.
- (a) S. Ataie and R. T. Baker, Inorg. Chem., 2022, 61, 19998–20007 CrossRef CAS PubMed; (b) M. Afandiyeva, X. Wu, W. W. Brennessel, A. A. Kadam and C. R. Kennedy, Chem. Commun., 2023, 59, 13450–13453 RSC.
- (a) J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed; (b) A. Kaithal, B. Chatterjee and C. Gunanathan, J. Org. Chem., 2016, 81, 11153–11161 CrossRef CAS PubMed; (c) T. Kitano, T. Komuro and H. Tobita, Organometallics, 2019, 38, 1417–1420 CrossRef CAS.
- V. K. Pandey, C. S. Tiwari and A. Rit, Org. Lett., 2021, 23, 1681–1686 CrossRef CAS PubMed.
- (a) C. Weetman, M. D. Anker, M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. Liptrot and M. F. Mahon, Chem. Sci., 2016, 7, 628–641 RSC; (b) A. Harinath, J. Bhattacharjee and T. K. Panda, Adv. Synth. Catal., 2019, 361, 850–857 CrossRef CAS; (c) W. Liu, Y. Ding, D. Jin, Q. Shen, B. Yan, X. Ma and Z. Yang, Green Chem., 2019, 21, 3812–3815 RSC; (d) Y. Ding, X. Ma, Y. Liu, W. Liu, Z. Yang and H. W. Roesky, Organometallics, 2019, 38, 3092–3097 CrossRef CAS; (e) N. Sarkar, S. Bera and S. Nembenna, J. Org. Chem., 2020, 85, 4999–5009 CrossRef CAS PubMed; (f) P. Ghosh and A. J. von Wangelin, Org. Chem. Front., 2020, 7, 960–966 RSC; (g) D. Bedi, A. Brar and M. Findlater, Green Chem., 2020, 22, 1125–1128 RSC; (h) B. Yan, X. He, C. Ni, Z. Yang and X. Ma, ChemCatChem, 2021, 13, 851–854 CrossRef CAS; (i) J. Peng, Y. Song, Y. Wang, Z. Liu and X. Chen, Org. Chem. Front., 2022, 9, 1536–1540 RSC; (j) J. E. Seok, H. T. Kim, J. Kim, J. H. Lee, A. K. Jaladi, H. Hwang and D. K. An, Asian J. Org. Chem., 2022, 11, e202200405 CrossRef CAS.
- (a) Z. Huang, S. Wang, X. Zhu, Q. Yuan, Y. Wei, S. Zhou and X. Mu, Inorg. Chem., 2018, 57, 15069–15078 CrossRef CAS PubMed; (b) S. Saha and M. S. Eisen, ACS Catal., 2019, 9, 5947–5956 CrossRef CAS.
- Z. Du, B. Behera, A. Kumar and Y. Ding, J. Organomet. Chem., 2021, 950, 121982 CrossRef CAS.
- (a) J. Bhattacharjee, D. Bockfeld and M. Tamm, J. Org. Chem., 2022, 87, 1098–1109 CrossRef CAS PubMed; (b) R. Narvariya, S. Das, S. Mandal, A. Jain and T. K. Panda, Eur. J. Inorg. Chem., 2023, 26, e202300247 CrossRef CAS; (c) R. Kumar, K. Bano, M. Choudhary, J. Sharma, K. Pal, S. K. Singh and T. K. Panda, Organometallics, 2023, 42, 2216–2227 CrossRef CAS; (d) H. J. Han, S. Y. Park, S. E. Jeon, J. S. Kwak, J. H. Lee, A. K. Jaladi, H. Hwang and D. K. An, Molecules, 2023, 28, 7090–7111 CrossRef CAS PubMed.
- (a) D. Mukherjee, A.-K. Wiegand, T. P. Spaniol and J. Okuda, Dalton Trans., 2017, 46, 6183–6186 RSC; (b) X. Wang, K. Chang and X. Xu, Dalton Trans., 2020, 49, 7324–7327 RSC; (c) R. Chambenahalli, R. M. Bhargav, K. N. McCabe, A. P. Andrews, F. Ritter, J. Okuda, L. Maron and A. Venugopal, Chem. – Eur. J., 2021, 27, 7391–7401 CrossRef CAS PubMed; (d) D. G. Shlian, E. Amemiya and G. Parkin, Chem. Commun., 2022, 58, 4188–4191 RSC.
- R. K. Sahoo, N. Sarkar and S. Nembenna, Angew. Chem., Int. Ed., 2021, 60, 11991–12000 CrossRef CAS PubMed.
- (a) S. Das, J. Bhattacharjee and T. K. Panda, New J. Chem., 2019, 43, 16812–16818 RSC; (b) X. Wang and X. Xu, RSC Adv., 2021, 11, 1128–1133 RSC; (c) S. Ataie, J. S. Ovens and R. T. Baker, Chem. Commun., 2022, 58, 8266–8269 RSC; (d) S. Mondal, T. Singh, S. Baguli, S. Ghosh and D. Mukherjee, Chem. – Eur. J., 2023, 29, e202300508 CrossRef CAS PubMed; (e) R. K. Sahoo, S. Rajput, S. Dutta, K. Sahu and S. Nembenna, Organometallics, 2023, 42, 2293–2303 CrossRef CAS; (f) S. Ataie and R. T. Baker, ChemCatChem, 2023, 15, e202300649 CrossRef CAS.
- H. Karmakar, G. S. Kumar, K. Pal, V. Chandrasekhar and T. K. Panda, Dalton Trans., 2024, 53, 10592–10602 RSC.
- M. H. Drover, Chem. Soc. Rev., 2022, 51, 1861–1880 RSC.
- D. Specklin, M.-C. Boegli, A. Coffinet, L. Escomel, L. Vendier, M. Grellier and A. Simonneau, Chem. Sci., 2023, 14, 14262–14270 RSC.
- (a) A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2008, 130, 11874–11875 CrossRef CAS PubMed; (b) T. G. Ostapowicz, C. Merkens, M. Hölscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2013, 135, 2104–2107 CrossRef CAS PubMed.
- (a) M. L. Clapson and M. W. Drover, Nat. Synth., 2022, 1, 267–268 CrossRef CAS; (b) M. L. Clapson, H. Sharma, J. A. Zurakowski and M. W. Drover, Chem. – Eur. J., 2023, 29, e202203763 CrossRef CAS PubMed; (c) J. A. Zurakowski, K. R. Brown and M. W. Drover, Inorg. Chem., 2023, 62, 7053–7060 CrossRef CAS PubMed.
- B. J. H. Austen, M. L. Clapson and M. W. Drover, RSC Adv., 2023, 13, 19158–19163 RSC.
- (a) J. J. Kiernicki, M. Zeller and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 18194–18197 CrossRef CAS PubMed; (b) J. J. Kiernicki, J. P. Shanahan, M. Zeller and N. K. Szymczak, Chem. Sci., 2019, 10, 5539–5545 RSC; (c) J. J. Kiernicki, E. E. Norwine, M. Zeller and N. K. Szymczak, Chem. Commun., 2019, 55, 11896–11899 RSC; (d) J. J. Kiernicki, M. Zeller and N. K. Szymczak, Organometallics, 2021, 40, 2658–2665 CrossRef CAS.
- (a) E. E. Norwine, J. J. Kiernicki, M. Zeller and N. K. Szymczak, J. Am. Chem. Soc., 2022, 144, 15038–15046 CrossRef CAS PubMed; (b) E. E. Norwine, J. J. Kiernicki, M. Zeller and N. K. Szymczak, Inorg. Chem., 2024, 63, 18519–18523 CrossRef CAS PubMed.
- H. Song and N. K. Szymczak, Angew. Chem., Int. Ed., 2024, e202411099 CAS.
- (a) V. Chugh, B. Chatterjee, W.-C. Chang, H. H. Cramer, C. Hindemith, H. Randel, T. Weyhermüller, C. Farès and C. Werlé, Angew. Chem., Int. Ed., 2022, 61, e202205515 CrossRef CAS PubMed; (b) S. Jena, L. Frenzen, V. Chugh, J. Wu, T. Weyhermüller, A. A. Auer and C. A. Werlé, J. Am. Chem. Soc., 2023, 145, 27922–27932 CrossRef CAS PubMed.
- T. F. C. Cruz, V. Loupy and L. F. Veiros, Inorg. Chem., 2024, 63, 8244–8256 CrossRef CAS PubMed.
- J. Reedijk and J. Verbiest, Transition Met. Chem., 1979, 4, 239–243 CrossRef CAS.
- S. Hermanek, Chem. Rev., 1992, 92, 325–362 CrossRef CAS.
- G. W. Kramer and H. C. Brown, J. Organomet. Chem., 1974, 73, 1–15 CrossRef CAS.
- T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS; (b) R. Puerta-Oteo, M. V. Jiménez, F. J. Lahoz, F. J. Modrego, V. Passarelli and J. J. Pérez-Torrente, Inorg. Chem., 2018, 57, 5526–5543 CrossRef CAS PubMed.
- (a) Q. Gu, L. Gao, Y. Guo, Y. Tan, Z. Tang, K. S. Wallwork, F. Zhangc and X. Yu, Energy Environ. Sci., 2012, 5, 7590–7600 RSC; (b) S. M. I. Al-Rafia, P. A. Lummis, A. K. Swarnakar, K. C. Deutsch, M. J. Ferguson, R. McDonald and E. Rivard, Aust. J. Chem., 2013, 66, 1235–1245 CrossRef; (c) K. Kadota, N. T. Duong, Y. Nishiyama, E. Sivaniah, S. Kitagawad and S. Horike, Chem. Sci., 2019, 10, 6193–6198 RSC.
- S. R. Tamang and M. Findlater, Dalton Trans., 2018, 47, 8199–8203 RSC.
- (a) P. Ye, Y. Shao, X. Ye, F. Zhang, R. Li, J. Sun, B. Xu and J. Chen, Org. Lett., 2020, 22, 1306–1310 CrossRef CAS PubMed; (b) G. S. Kumar, J. Bhattacharjee, K. Kumari, S. Moorthy, A. Bandyopadhyay, S. K. Singh and T. K. Panda, Polyhedron, 2022, 219, 115784 CrossRef; (c) A. Adilkhanova, V. F. Frolova, A. Yessengazin, Ö. Öztopçu, K. A. Gudun, M. Segizbayev, N. A. Matsokin, A. Dmitrienko, M. Pilkington and A. Y. Khalimon, Dalton Trans., 2023, 52, 2872–2886 RSC.
- (a) A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed; (b) B. Yan, X. Ma, Z. Pang and Z. Yang, New J. Chem., 2023, 47, 3202–3206 RSC.
- (a) F. Meger, A. C. W. Kwok, F. Gilch, D. R. Willcox, A. J. Hendy, K. Nicholson, A. D. Bage, T. Langer, T. A. Hunt and S. P. Thomas, Beilstein J. Org. Chem., 2022, 18, 1332–1337 CrossRef CAS PubMed; (b) R. Kumar, R. K. Meher, J. Sharma, A. Sal and T. K. Panda, Org. Lett., 2023, 25, 7923–7927 CrossRef CAS PubMed.
- (a) G. R. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed; (b) Calculations performed at the PBE0-D3/6-311++G*(d,p)//PBE0/(SDD*,6-31G(d,p)) level using the GAUSSIAN 09 package. Solvent effects (benzene) were considered using the PCM/SMD model. A full account of the computational details and a complete list of references are provided in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2401462. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03363a |
| This journal is © The Royal Society of Chemistry 2025 |