
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An overloaded pure silica zeolite ISV synthesized using a phosphonium cation†
Huajian
Yu
,
Salvador R. G.
Balestra‡
 *,
Zihao Rei
Gao§
and
Miguel A.
Camblor
*
*,
Zihao Rei
Gao§
and
Miguel A.
Camblor
*
Instituto de Ciencia de Materiales de Madrid (ICMM), CSIC, Madrid, Spain. E-mail: macamblor@icmm.csic.es; srodriguez9@us.es
First published on 20th January 2025
Abstract
A pure silica zeolite with ISV structure was synthesized using the tricyclopentylmethylphosphonium (Cp3MP+) cation under fluoride media. Analysis of the organic content in the zeolite suggested the presence of more than four cations per unit cell, while fluoride anions were exclusively located in the four double four-membered rings of the structure. 29Si solid-state NMR demonstrated a significant concentration of Q3 Si species, i.e., ![[S with combining low line]](https://www.rsc.org/images/entities/char_0053_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) (OSi)3(OH), in the structure, which afforded charge balance but contrasted with the most common observation of defect-free pure silica zeolites prepared in fluoride media at near-neutral pH. Calculations showed that improved stability was obtained in an overloaded (i.e., containing more Cp3MP+ than F− and three-channel crossings) defective zeolite. The calculated stabilization of ISV was larger with Cp3MP+ than with the cyclohexyl analog used to produce ZEO-1, a zeolite with an extra-large 3D pore system. Substitution of small amounts of Cp3MP+ by tetraethylammonium stopped crystallization instead of producing ISV/BEC intergrowths, despite calculations on the bare zeolites suggesting improved stability of the intergrowths. This rendered the observed difficulty in obtaining such intergrowths puzzling and likely dependent on a disruption of the host–guest assembly. Compared with prior ISV materials, this zeolite displayed a minimum amount of disorder, which, however, was sufficient to impede proper Rietveld refinement.
(OSi)3(OH), in the structure, which afforded charge balance but contrasted with the most common observation of defect-free pure silica zeolites prepared in fluoride media at near-neutral pH. Calculations showed that improved stability was obtained in an overloaded (i.e., containing more Cp3MP+ than F− and three-channel crossings) defective zeolite. The calculated stabilization of ISV was larger with Cp3MP+ than with the cyclohexyl analog used to produce ZEO-1, a zeolite with an extra-large 3D pore system. Substitution of small amounts of Cp3MP+ by tetraethylammonium stopped crystallization instead of producing ISV/BEC intergrowths, despite calculations on the bare zeolites suggesting improved stability of the intergrowths. This rendered the observed difficulty in obtaining such intergrowths puzzling and likely dependent on a disruption of the host–guest assembly. Compared with prior ISV materials, this zeolite displayed a minimum amount of disorder, which, however, was sufficient to impede proper Rietveld refinement.
Introduction
The recent report of the discovery of zeolite ZEO-1 (with zeolite framework type1 code JZO),2 a stable 3D extra-large pore (3D ELP) high-silica zeolite, deserves, in our opinion, careful consideration. The fascinating structures, properties and industrial applications of zeolites have fostered extensive synthetic efforts worldwide, in both academia and industry. However, after over eight decades of tenacious efforts, ZEO-1, reported at the end of 2021, is the first stable zeolite containing a 3D system of extra-large pores, interspersed with an additional 3D system of large pores, with the two systems being fully connected. The diffusion-limiting window in a large pore is made up of 11 or 12 tetrahedra (11-ring or 12-ring, respectively), while in extra-large pores, they contain more than 12 tetrahedra. Prior to ZEO-1, 3D extra-large pore “zeolites” were, in fact, interrupted frameworks (i.e., zeolite-like materials in which there is a structural, as opposed to defective, lack of full connectivity of the tetrahedral atoms), which additionally had a composition that compromised their stability (germanosilicates or gallium phosphates).3–5 After ZEO-1, two even more porous, stable extra-large pore zeolites were reported, namely, ZEO-3 (JZT)6 and ZEO-5 (HZF),7 but they were not synthesized by conventional hydrothermal synthesis. Instead, they were produced from a chain silicate through a 1D-to-3D topotactic condensation in the case of ZEO-3 or by an interchain expanded reaction, followed by a topotactic 3D–3D condensation of the same starting chain in the case of ZEO-5. Another stable zeolite with an extra-large pore size in only one dimension has also been recently reported.8 Thus, ZEO-1 remains the only stable zeolite with a 3D system of extra-large pores obtained by conventional hydrothermal synthesis. Then, it is important to investigate the main factors leading to the discovery of ZEO-1. ZEO-1 was afforded3 by a combination of a particular phosphonium organic structure-directing agent (OSDA), methyltricyclohexylphosphonium (Cy3MP+ ), and a relatively high crystallization temperature (190 °C) during the hydrothermal reaction.Herein, we report our studies of the synthesis of zeolites using the slightly smaller methyltricyclopentylphosphonium (Cp3MP+), which leads to a 3D large pore pure SiO2 zeolite ITQ-7 (ISV) instead of ZEO-1. Not many reports exist on the synthesis of pure silica zeolite ISV with different OSDAs. This zeolite was first discovered using a spiro cation, 1,3,3-trimethyl-6-azoniumtricyclo[3.2.1.46,6]dodecane (TMATCD), synthesized from a commercial bicycloamine,9,10 and later on from a slightly different spiro cation, 1,3,3-trimethyl-6-azoniumtricyclo[3.2.1.56,6]tridecane (TMATCT), derived from the same amine.11 The commercial supply of this bicycloamine was discontinued a few years later; thus, research on this zeolite was hampered. Even though the compound is now on the market again, its current price is considerably expensive.12 Only a few new OSDAs were reported later on to produce ISV and only one, N-butyl-N-cyclohexyl-pyrrolidinium (BCHP), could produce the pure silica version but only with the help of seeds.13 All the OSDAs mentioned up to this point are depicted in Scheme 1. Other reported OSDAs require the presence of Ge to produce ISV.14,15 Thus, the discovery of a new OSDA for pure silica ISV, beyond the spiro cations mentioned above, is significant. In trying to understand structure–direction issues, herein, we have combined experimental results with a detailed theoretical study.
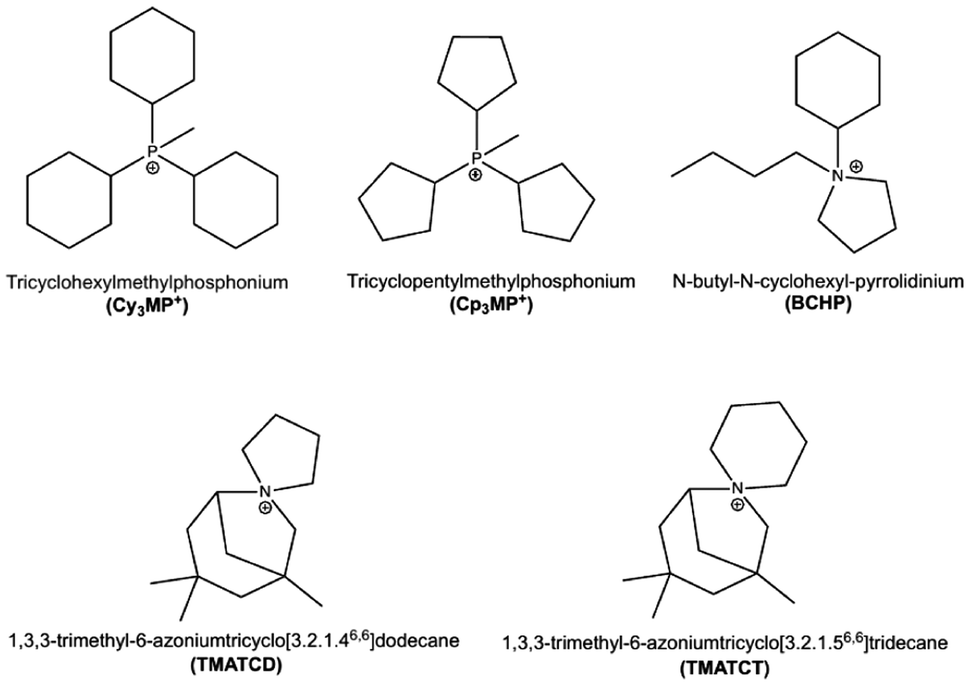 | ||
| Scheme 1 OSDA cations relevant to this work. Only Cy3MP+ is able to produce JZO, while the rest can direct to pure silica ISV (BCHP requires the additional assistance of ISV seeds). | ||
Experimental
Synthesis of OSDA
For the synthesis of tricyclopentylmethylphosphonium iodide (Cp3MP+I−), 50 g tricyclopentylphosphine (10 wt% in hexane solution, STREM, 20.98 mmol) was dissolved in 100 mL hexane and placed in an ice bath. Then, 17.88 g methyl iodide (99%, Aldrich, 126 mmol) was added dropwise under magnetic stirring (CAUTION: phosphines and methyl iodide are harmful; work under a hood and use proper personal protective equipment). After 3 days of reaction, the precipitated solid was recovered by filtration, washed with hexane (100 mL) and diethyl ether (100 mL) and dried at 50 °C overnight. Tricyclopentylmethylphosphonium iodide salt (Cp3MP+I−) was dissolved in CDCl3 for liquid NMR (1H, 13C and 31P), which revealed that except for a small impurity (about 5% based on P), the solid was the expected phosphonium (see Fig. S1–S3 in the ESI†).Anion exchange
14.9975 g (39.43 mmol) Cp3MP+I− and 100 mL of wet resin (Aldrich Dowex Monosphere 550A resin in hydroxide form with an exchange capacity of 1.2 mol L−1 of wet resin) were added in a flask and stirred overnight for anion exchange. The iodide salt is not very soluble in water; thus, initially, a white powder was clearly distinguished together with the resin. However, as the exchange reaction proceeded, the amount of white powder diminished and finally disappeared, indicating a much higher solubility of the hydroxide. Subsequently, the solution was filtered to remove the resin. After extensively washing the resin with water, the pH of the filtered water was almost neutral. The solution was then collected and concentrated using a rotary evaporator under vacuum at 72 °C to get a concentrated OSDA hydroxide solution that was subsequently titrated with dilute HCl to determine the hydroxide concentration.Zeolite synthesis
Pure SiO2ISV was synthesized from a gel with a composition of 0.5 OSDAOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 HF:1.0 SiO2:5 H2O at 175 °C or 190 °C within Teflon-lined tumbling autoclaves. In a typical synthesis, 17.8844 g (4.999 mmol, [OH−] = 0.2795 mmol g−1) OSDAOH and 2.0922 g TEOS (tetraethylorthosilicate, 98%, Aldrich 10.042 mmol) were added in a weighted beaker. After stirring overnight for hydrolysis, 181 μL hydrofluoric acid (HF 48%, Aldrich, 4.996 mmol) was added. After concentrating in an oven at 86 °C until the water amount reached its target, as monitored by the weight, the final gel was mixed by hand as homogeneously as possible and placed into different autoclaves. After heating for selected times, the product was collected by filtration and washed with water and acetone twice. The solid products were dried at room temperature and, when it was found to be pure ISV, it amounted to 21–27 g per 100 g of gel, implying about 80–100% yield of calcined zeolite based on silica. This high yields contrast with recent claims that zeolite yields by the fluoride route are low16 but are in full agreement with our abundantly reported previous observations.17 Other synthesis conditions are described in Table 1.
:0.5 HF:1.0 SiO2:5 H2O at 175 °C or 190 °C within Teflon-lined tumbling autoclaves. In a typical synthesis, 17.8844 g (4.999 mmol, [OH−] = 0.2795 mmol g−1) OSDAOH and 2.0922 g TEOS (tetraethylorthosilicate, 98%, Aldrich 10.042 mmol) were added in a weighted beaker. After stirring overnight for hydrolysis, 181 μL hydrofluoric acid (HF 48%, Aldrich, 4.996 mmol) was added. After concentrating in an oven at 86 °C until the water amount reached its target, as monitored by the weight, the final gel was mixed by hand as homogeneously as possible and placed into different autoclaves. After heating for selected times, the product was collected by filtration and washed with water and acetone twice. The solid products were dried at room temperature and, when it was found to be pure ISV, it amounted to 21–27 g per 100 g of gel, implying about 80–100% yield of calcined zeolite based on silica. This high yields contrast with recent claims that zeolite yields by the fluoride route are low16 but are in full agreement with our abundantly reported previous observations.17 Other synthesis conditions are described in Table 1.
| Alfb | OSDA/T | H2O/T | OSDA/F | T °C | Time | Yieldc | XRD |
|---|---|---|---|---|---|---|---|
| a Am. amorphous, IC-ill crystallized, UU unknown unstable. b Al molar fraction (Al/Al + Si). c g per 100 g of gel. d TEA+ added as 1% OSDA. e TEA+ added as 5% OSDA. | |||||||
| 0.02 | 0.5 | 10 | No HF | 190 | 5 d | 16.71 | Am. |
| 12 d | 20.50 | Am. +IWV | |||||
| 24 d | 27.94 | Am. (+IWV) | |||||
| 0.02 | 0.5 | 10 | No HF | 175 | 15 d | 38.84 | IC |
| 40 d | 42.39 | IC | |||||
| 0.0 | 0.5 | 10 | No HF | 190 | 5 d | 14.76 | UU |
| 12 d | 20.55 | Am. | |||||
| 24 d | 19.75 | Am. (+ISV) | |||||
| 0.0 | 0.5 | 15 | No HF | 190 | 5 d | 0 | No solid |
| 15 d | 13.18 | Am. | |||||
| 30 d | 14.15 | UU | |||||
| 0.0 | 0.5 | 10 | No HF | 175 | 15 d | 40.97 | UU |
| 40 d | 31.55 | Am. | |||||
| 0.0 | 0.5 | 5 | 1 | 190 | 12 d | 26.93 | ISV |
| 0.0 | 0.5 | 5 | 1 | 175 | 12 d | 39.81 | Am.(+ISV) |
| 24 d | 21.11 | ISV | |||||
| 26 d | 21.43 | ISV | |||||
| 0.0 | 0.5 | 5 | 1 | 190 | 12 dd | 27.67 | Am. +ISV |
| 12 de | 42.44 | IC | |||||
Characterization techniques
1H and 13C liquid nuclear magnetic resonance (NMR) spectra of the OSDA were measured on a Bruker Avance III-HD Nanobay 300 MHz. Multinuclear (13C, 19F, 29Si and 31P) solid-state magic angle spinning (MAS) NMR spectra were recorded on a Bruker AV-400-WB equipment and the conditions, with the exception of 31P measurements, have been reported elsewhere.18 The 31P spectra of the as-made zeolite were acquired at a resonance frequency of 161.97 MHz in a 4 mm triple channel probe with a π/2 pulse at 60 kHz, spectral width of 100 kHz, relaxation delay of 40 s, 10 kHz spinning speed and 64 scans accumulated. In addition to direct irradiation spectra, {1H} 31P cross polarization (CP) spectra were also acquired with an 1H excitation pulse of 3 μs, 3 ms contact time, 100 kHz spectral width, tppm15 decoupling at 80 kHz and 256 scans. The relaxation delay was 5 s. In both cases, (NH4)H2PO4 was used as a secondary reference (0.81 ppm from 85% H3PO4 solution as a primary reference). Laboratory powder X-ray diffraction (PXRD, Bruker D8 Advance diffractometer) was used to identify the products with Cu Kα radiation, λ = 1.5418 Å. Synchrotron PXRD data patterns were collected at the Spanish synchrotron light source (ALBA, Cerdanyola del Vallès, Barcelona) using the MAD26 multicrystal detector on ALBA's beamline 04 (MSPD) with a wavelength of 0.61928 Å in capillary mode (0.7 and 0.9 mm diameter for the as-made and calcined, respectively). The crystal size and morphology were examined by field-emission scanning electron microscopy (FE-SEM) imaging on a FEI Nova NanoSEM 230 microscope, which was also equipped with a Genesis XM2i detector for microanalysis by energy dispersive spectroscopy (EDS). CHN elemental analysis of the zeolite was done on a LECO CHNS-932 analyzer. The Fourier transform infrared (FT-IR) spectra of the as-made and calcined samples were obtained in the transmission mode after dilution in KBr on a Bruker Vertex 70 V spectrophotometer at a resolution of 2 cm−1 and performing 100 scans per sample. For thermogravimetric analysis (TG), an SDT Q600 TA instrument under air flow (100 mL min−1) was used to analyze the weight loss of the as-prepared zeolite from 25 °C to 1000 °C at 10 °C min−1.Computational methods
The initial zeolite structure was sourced from the IZA database.1 Fluoride anions were positioned at the centre of d4r (as supported by Experimental data, see below). The templates were loaded in the pore of the zeolite following the procedure explained in the ESI.† To the best of our knowledge, there is no specific parametrization for the interatomic potentials among the P-bearing cation types under consideration (Cy3MP+ and Cp3MP+ cations), the zeolitic framework (SiO2), and F− anions. Therefore, instead of using a standard Monte Carlo procedure with generic potentials (such as UFF or Dreiding potential), the organic cations were loaded into the ISV structure using the CEGA workflow, which was developed by Balestra19 and extended later in the works by Gao et al.20 and Núñez-Rico et al.21 This workflow allows us to study the insertion of molecules in a porous system automatically at reasonable accuracy. Some details about the followed procedure (like the insertion of templates and the construction of crystalline phases) may be relevant to the discussion in this work and are included in the ESI.†We have also used reference materials for the calculation of enthalpies of formation at standard conditions. We used quartz for SiO2 composition and built hypothetical structures for OSDA salts/hydroxide, including Cy3MP+ F−, Cp3MP+ F−, and Cp3MP+ OH−. We have described the generation and optimization of these salts/hydroxide in the ESI.† Once we had constructed an initial spatial configuration of the (defect-free) crystalline materials, we performed fully flexible structural optimizations (i.e., optimizing all atom coordinates and lattice parameters, without considering symmetry) using the recalibrated r2SCAN + rVV10 theoretical level with the VASP program,22 as explained in the ESI.†
In this work, following the experimental characterization, we have also considered ISV zeolites with defects. For structures with negative charge-bearing defects, we have to insert an extra cation per defect. To achieve the defect densities seen in the Results section of this work, we had to go to cells with large numbers of atoms, making energy optimization with the r2SCAN + rVV10 method simply too computationally expensive. For this reason, we used TB calculations using the GFN2-xTB theoretical level (as explained in the ESI†),23 which offer reasonable reliability in terms of the computational cost.
Regarding the estimation of zeolite stabilities, we calculated the formation enthalpy δHi of the zeolites using as reference the formation enthalpy of quartz per Si atom and the enthalpies of OSDA+F− salts and OSDA+ OH−. We calculated all formation enthalpies using the same theoretical level. Thus, ΔHx = H(NO,F x, NO,F F, NO,d) − NSih(SiO2)quartz − NO,FH(xF)salt − NO,dH(xOH)salt for x = Cy3MP+ and Cp3MP+, where H(NO,F x, NO,F F, NO,d) is the enthalpy of the zeolite system, NO,F is the number of pairs of x-type OSDA cations and F− anions, NO,d is (in the presence of negative bearing defects) the number of x-type OSDA and defects (–SiOH, and –Si–O− pairs), NSi is the number of Si-atoms in the simulation box, and H(xF)salt and H(xOH)salt are the enthalpy of the corresponding fluoride and hydroxide. The enthalpy per T-atom is Δhx = N−1Si ΔHx. This formation enthalpy allows us to compare the stability between different compositions.
Results and discussion
Zeolite synthesis
Table 1 shows the synthesis experiments using Cp3MP+OH− while varying the H2O/T ratio, temperature and presence or absence of HF. We first attempted experiments without HF at 175 and 190 °C. With Cy3MP+, the preferred conditions to get ZEO-1 were high temperature with Al,2 while at 175 °C in pure SiO2, the chain silicate ZEO-2 was obtained.6 With the slightly smaller Cp3MP+, the corresponding experiments produced amorphous, unstable or ill-crystallized solids, although some traces of IWV (when using Al) or ISV (without Al) were also detected. Since the synthesis of pure silica d4r-containing zeolites without fluoride has never been achieved by a direct hydrothermal synthesis,4 we suspected the traces of ISV that we obtained might be due to small amounts of fluoride coming from the Teflon liners, where it might be adsorbed from previous synthesis and/or during their washing. This has been corroborated by the presence of a small fluoride Kα peak in the EDS spectra of certain crystal-like particles observed in that solid. By contrast, IWV, which contains a 2D system of large 12-ring pores with 14-ring stretches, was synthesized as an aluminosilicate by a fluoride-free hydroxide route using diquats formed by linking two 1,3,4,5-tetramethylimidazole molecules with chains of four, five or six methylenes.24 The initial discovery of IWV was made, however, in fluoride media using a phosphonium cation.25 Our results suggests that Cp3MP+ could possibly be a good OSDA for aluminosilicate IWV in hydroxide medium by increasing the Al content in the gel (here, it is smaller than in the original report). The PXRD patterns and SEM images of several products of nominally HF-free synthesis are collected in Fig. S4 and S5,† respectively.When fluoride is intentionally added to the pure SiO2 synthesis mixture, ISV is the only crystalline phase obtained under the conditions used at both temperatures tried (175 and 190 °C). The addition of small fractions of tetraethylammonium (TEA+) to replace equimolar amounts of Cp3MP+ in the synthesis gel in an attempt to force an intergrowth of ISV with BEC resulted in a very small amount of pure ISV (for a 1% mol OSDA replacement) and an ill-crystallized phase (for a 5% mol OSDA replacement). This very strong disrupting effect of small TEA+ additions may seem intriguing, given the highly similar structures of BEC and ISV, with layer sequences AAAA and ABAB, respectively, and the possibility to synthesize BEC with TEA under some circumstances (presence of Ge).26 Actually, we have calculated the energies of ISV/BEC-ordered structures with ABBA and ABBBA layer sequences (Fig. S7†), as well as of BEC and ISV (ABAB), using the Sanders et al. potential,27 and the GFN2-xTB method; the results (Table S1†) show that all the structures have similar stabilities and the order of decreasing stability is BEC, BAAAB, BAAB and ISV (see structures in Fig. S7†). Given that the calculations refer to SiO2 without guests, we can attribute the observations to a highly disruptive effect of TEA in the assembly of cations and SiO2 structures.
Despite the similarity of the calculated energies of the different structures, ISV/BEC intergrowths have not been clearly realized so far.28 The only claim of such an intergrowth that we are aware of is not very strongly supported by the experimental evidence14 since when comparing the DIFFaX simulations with the experimental patterns, the latter can be assigned to either ordered ISV or ordered BEC. There is a large interest in controlling the adsorption and catalytic properties of zeolites by controlling their degree of intergrowth.29 Our work with these and other OSDAs (pending publication) indicates that the ISV/BEC intergrowth is surprisingly difficult and elusive despite the obvious structural relationship and apparent ease of connectivity between both phases.9,30
The synthesis of pure silica ISV with Cp3MP+ contrasts with previous reports of zeolite synthesis using the same OSDA, which only obtained this phase in the presence of high Ge fractions (Gef = Ge/(Ge + Si) = 0.25 or 0.5).31 We attribute their failure to the significantly lower temperature used (150 °C). As we already discussed for materials prepared with Cy3MP+, the use of more stable phosphonium cations may afford the synthesis of materials with higher apparent activation energies not accessible at lower crystallization temperatures.3
Zeolite characterization
PXRD reveals a high crystallinity of pure silica ISV synthesized with Cp3MP+ (Fig. 1). However, attempts to refine the structure by the Rietveld method against synchrotron PXRD data under different space groups failed, and this was also the case for Le Bail full profile fittings (Fig. S6†). This is attributed to disorder (see below). Disorder in our previously reported ISV was not related to stacking faults, which was determined by high-resolution electron microscopy and was proposed to be associated with lattice strain, likely related to missing or incompletely built d4r.30 Comparison of the PXRD patterns of the as-made and calcined ISV in this work with those we reported earlier suggests that the new ISV does have disorder although to a minimum degree (denoted as Dmin in ref. 30).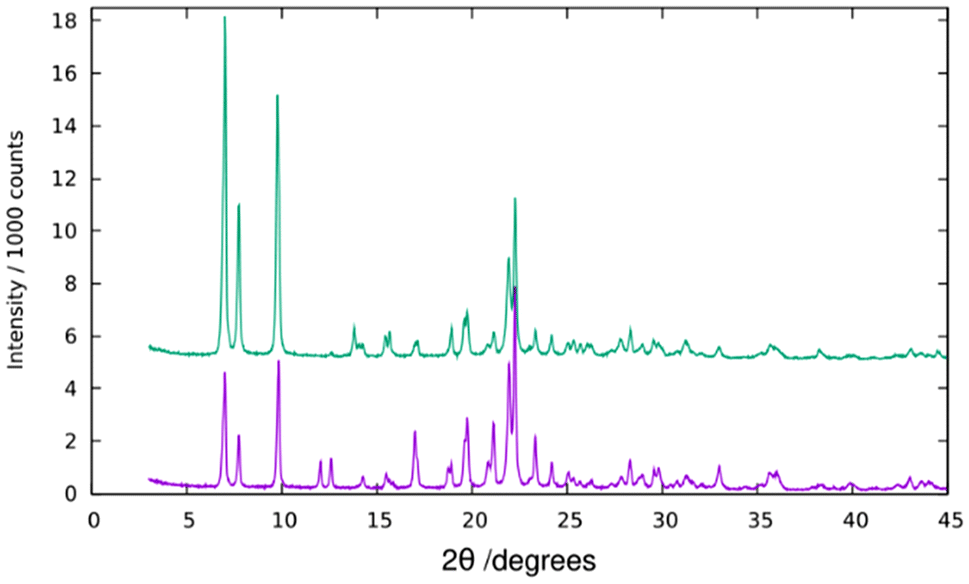 | ||
| Fig. 1 PXRD of the as-made (bottom) and calcined Cp3MP+-SiO2-ISV. | ||
TGA (Fig. 2) mainly shows a single step starting at about 400 °C and a total weight loss of ca. 21.7%. This is slightly smaller than expected if all the OSDA and F were removed (22.07 for the ideal content of 4 cations per unit cell) to yield SiO2, which suggests that a portion of P remains in the zeolite as phosphorus oxides. This has been confirmed by EDS, which showed the presence of P after calcination, although in amounts much smaller (0.6–1 P uc−1) than those in the as-prepared materials (ideally 4 P uc−1). This amount of P2O5 retained in the final solid would amount to a decreased weight loss of about 0.8 to 1.4 wt% in the TGA curve. The relatively low retention of P contrasts with a much larger retention of P in the aluminosilicate ZEO-1 despite its larger pores, suggesting that framework Al might play a role in phosphorus retention in that case (P/Al is close to 1 by chemical analysis in calcined ZEO-1). Extensively washing a calcined zeolite with water effectively removes P to below the detection limits of EDS.
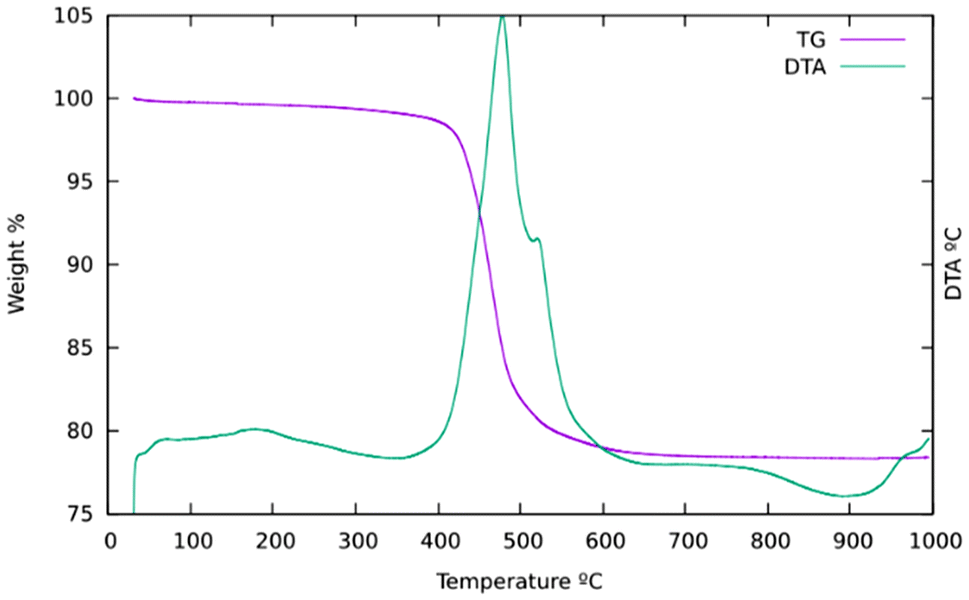 | ||
| Fig. 2 TG (purple) and DTA (green) curves of Cp3MP+-SiO2-ISV. | ||
C and H analysis reveals an H/C ratio of 2.02 well above the value in Cp3MP+ (1.87). C analysis indicates an empirical formula of [C16H30PF]4.31[SiO2]64, assuming that the cation is counterbalanced by fluoride (theoretical 16.5% C, 2.6% H; found 16.5% C, 2.8% H). The excess with regard to an ideal [C16H30PF]4[SiO2]64 (theoretical 15.6% C, 2.45% H) cannot be attributed to an amorphous phase because the FE-SEM images only show the roughly squared crystals typical of ISV zeolites (Fig. 3) without evidence of amorphous particles.9 Since all fluoride atoms are occluded in d4r according to 19F MAS NMR (see below), we find likely that the excess organic is balanced by Q3 observed by 29Si MAS NMR (see below). The question of the chemical composition of ISV will be discussed later, after considering the loading optimization calculations (see below). We describe this cation loading as “overloading” not only because the amount of cations exceeds the amount of fluoride anions but also because it exceeds the number of three-channel crossings (straight/straight/sinusoidal at (0, 0.5, 0.75) where the cations presumably sit (see below)).
 | ||
| Fig. 3 FESEM image of the Cp3MP+-SiO2-ISV zeolite. | ||
31P (single resonance at 40 ppm) and 13C MAS NMR (unresolved resonances at about −0.9, 1.5, 27.6 and 31 ppm) spectra of the as-made zeolite prove the OSDA is occluded intact in its pores (Fig. S8 and S9, respectively†). The 31P {1H} CP spectrum confirms no additional signals (Fig. S10†). The 19F spectrum shows a single and highly asymmetric resonance at about −40 ppm (Fig. 4), a chemical shift highly characteristic of fluoride occluded in pure silica d4r cages in zeolites.32 The absence of any additional 19F resonances demonstrates that there is no fluoride outside the d4r cages, which limits the maximum possible amount of fluoride anions to that of d4r, i.e., 4 F uc−1. A deconvolution of the spectrum shows that there are actually two signals with almost equal intensities (areas 0.98:1) but large differences in widths (FWHM of about 2 and 1.6 ppm, respectively) centered at −39.3 and −40.2 ppm, respectively, confirming that both types of crystallographically distinct d4r cages present in ISV are occupied by fluoride, as was also the case in our first reports of ITQ-7 synthesized with TMTACD.9,30
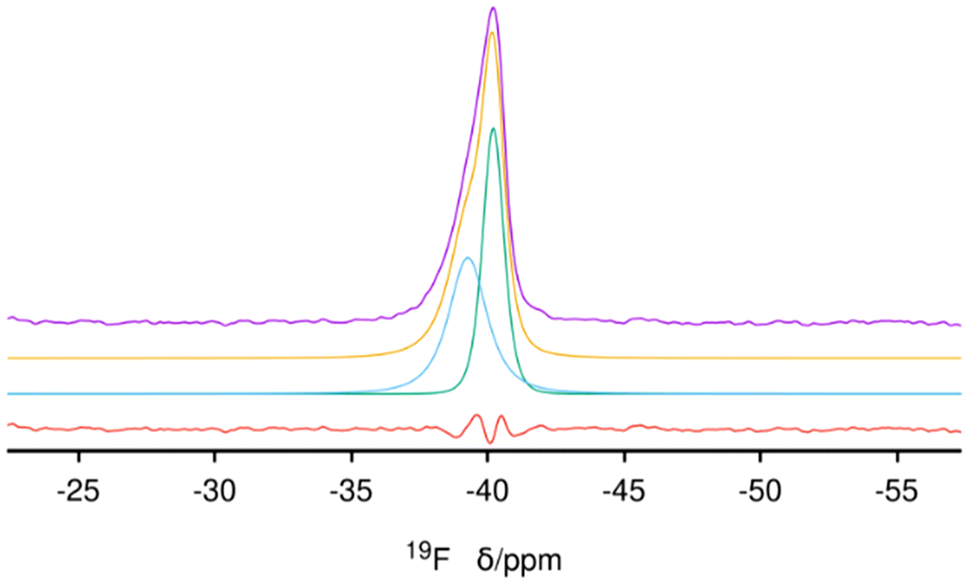 | ||
| Fig. 4 19F MAS NMR spectrum of Cp3MP+-SiO2-ISV zeolite. From top: experimental (purple), deconvoluted (yellow), individual components (blue and green) and difference experimental-deconvoluted (red) curves. | ||
29Si MAS NMR of the as-made ISV needs a long relaxation delay (240 s) due to the lack of interaction with paramagnetic oxygen, as is typical for as-prepared zeolites.33 A small, broad and unresolved resonance in the −95 to −102 ppm range is assigned to Q3 connectivity defects related to the presence of excess Cp3MP+ (Fig. 5 bottom). After calcination and extensive washing with water to remove P, the Q3 species remain (Fig. 5 top), and a noticeable change of the whole spectrum to high field occurs. The resolution of the spectrum of the calcined material is poorer than that observed in the most disordered calcined material prepared with TMATCD (Dmax-ITQ-7),30 which we attribute to the presence of defects in the zeolite framework,34 in addition to framework disorder (see below).
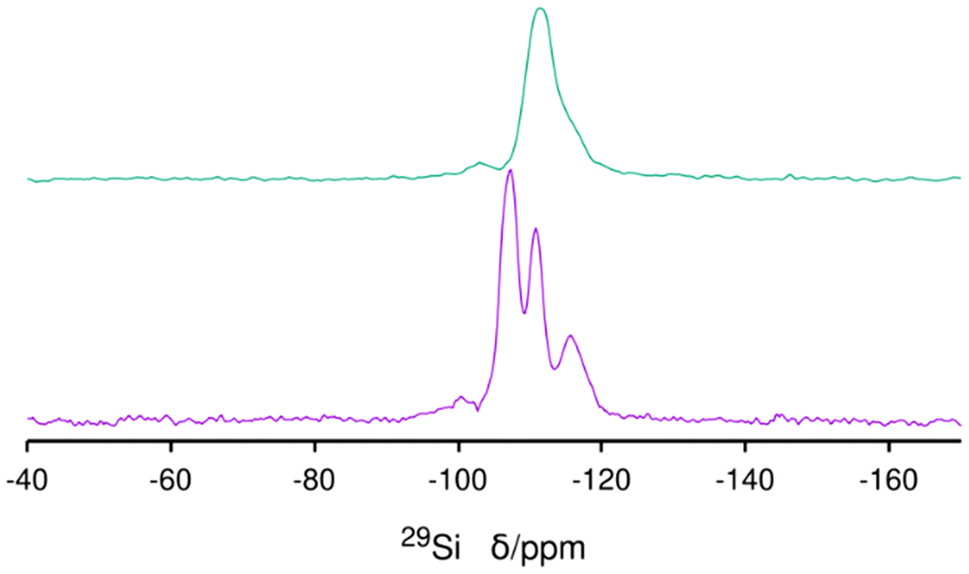 | ||
| Fig. 5 29Si MAS NMR spectra of the as-made (bottom) and calcined ISV prepared with Cp3MP+. | ||
Infrared bands at 2965, 2919 and 2876 cm−1 assigned to the C–H stretching vibration of the OSDA in the as-prepared zeolite and at 1456 cm−1, assigned to C–H bending, disappear by calcination at 600 °C (Fig. S11†). The integrity of the ISV structure upon calcination is demonstrated by PXRD (Fig. 1 top) and affords the presence of sharp bands in the region of framework vibrations below 820 cm−1 (Fig. 6 top). Vibration bands at 504 cm−1 and 484 cm−1 in the as-prepared and calcined zeolites are assigned to the d4r unit with or without F−, respectively.35 On the other hand, there is a noticeable blue shift of the strong symmetric and asymmetric Si–O stretching bands in the 750–850 and 1000–1300 cm−1 regions, respectively, as usually observed when as-prepared high silica zeolites are calcined.36
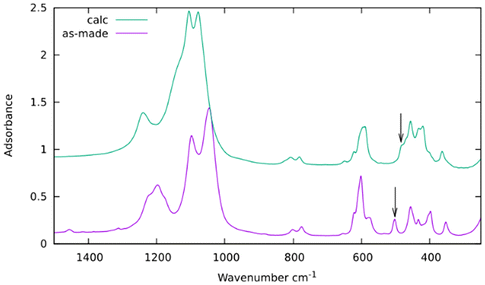 | ||
| Fig. 6 Infrared spectra in KBr of as-made (purple) and calcined ISV (green). The arrows mark the position of the bands assigned to d4r cages with (bottom) or without (top) occluded fluoride.35 | ||
Disorder in SiO2-ISV
According to ref. 30, disorder in ISV materials prepared with TMATCD resulted in differences in the PXRD pattern and in the 19F and 29Si MAS NMR spectra. Certain PXRD peaks became broader and changed positions while others remained sharp and invariant. For instance, the peak at about 22.8° is always sharp and invariant, while that at 21.8° is sometimes broader and shifted to lower angles with increasing degree of disorder. Comparison of the PXRD patterns of calcined Cp3MP-ISV and the ones in the reference mentioned above strongly suggests that Cp3MP-SiO2-ISV has some disorder, very similar to the one characterized as Dmin-ITQ-7. This can be observed in Fig. S12,† showing that the patterns of both materials are almost superimposable. Given the very similar crystal size and morphology (Fig. 3 here and Fig. S4† in ref. 30), preferred orientation effects are not likely to distort the comparison. When the 19F MAS NMR is considered, a decreased resolution of the resonances corresponding to fluoride in each crystallographically distinct d4r also suggest some disorder.30 However, we cannot rule out that interaction with a different cation might contribute to the decreased resolution. In fact, the chemical shifts in our material (−40.2 and −39.3 ppm) are slightly different from those we previously reported (−39.3 and −38.5 ppm), although in both cases, the separation of the two resonances are almost identical. With regard to 29Si MAS NMR, the very low resolution (Fig. 5) can be assigned to the combination of disorder and a significant concentration of defects. Thus, for the material reported here, disorder is mainly supported by PXRD (including the impossibility to perform a Rietveld refinement or even a LeBail profile fitting, in addition to the comparison shown in Fig. S12†), although it is also compatible with the NMR results.Computational results
The relative enthalpy of ISV above BEC zeolites (calcined SiO2 zeolites) using the r2SCAN + rVV10 method is 0.64 kJ per mol per T-atom (BEC more stable). This agrees with the same calculation performed using the SLC potential and the GFN2-xTB theoretical level, with 0.52 and 0.15 kJ per mol per T-atom (see Table S1† for other measures of stability of pure SiO2 zeolites). The cell parameters of the optimized ISV structures using the r2SCAN + rVV10 method, containing different OSDA cations are a = 12.77 Å, b = 12.81 Å, c = 25.14 Å, and a = 12.73 Å, b = 12.64 Å, and c = 24.86 Å, for Cy3MP+ and Cp3MP+ cations, respectively, both with α, β, and γ ∼90°. The corresponding difference of formation enthalpies considering the same theoretical level between the two structures is Δδh(Cy3MP+ → Cp3MP+) = −3.05 kJ per mol per T-atom. Thus, the synthesized structure using the Cp3MP+ cation is expected to be more stable; see Fig. 7 for the optimized structures. The calculated molecular volumes of both cations inside the optimized zeolite systems are much different, viz., 409.3 Å3 (Cp3MP+) and 500.6 Å3 (Cy3MP+), and their conformations can be seen in Fig. 8. The difference between the formation energies is also reflected in the volume of the structures relative to the volume of the optimized pure silica ISV structure, V/V0, through the molecular pressure on the zeolite pore. The V(Cy3MP+)/V0 = 1.01 (i.e., a 1.37% increase), while V(Cp3MP+)/V0 = 0.98 (i.e., a 1.41% decrease), suggesting that the larger cation “pushes” the zeolite walls outwards, while the smallest cation, with also a larger stabilization energy, “pulls” them inwards.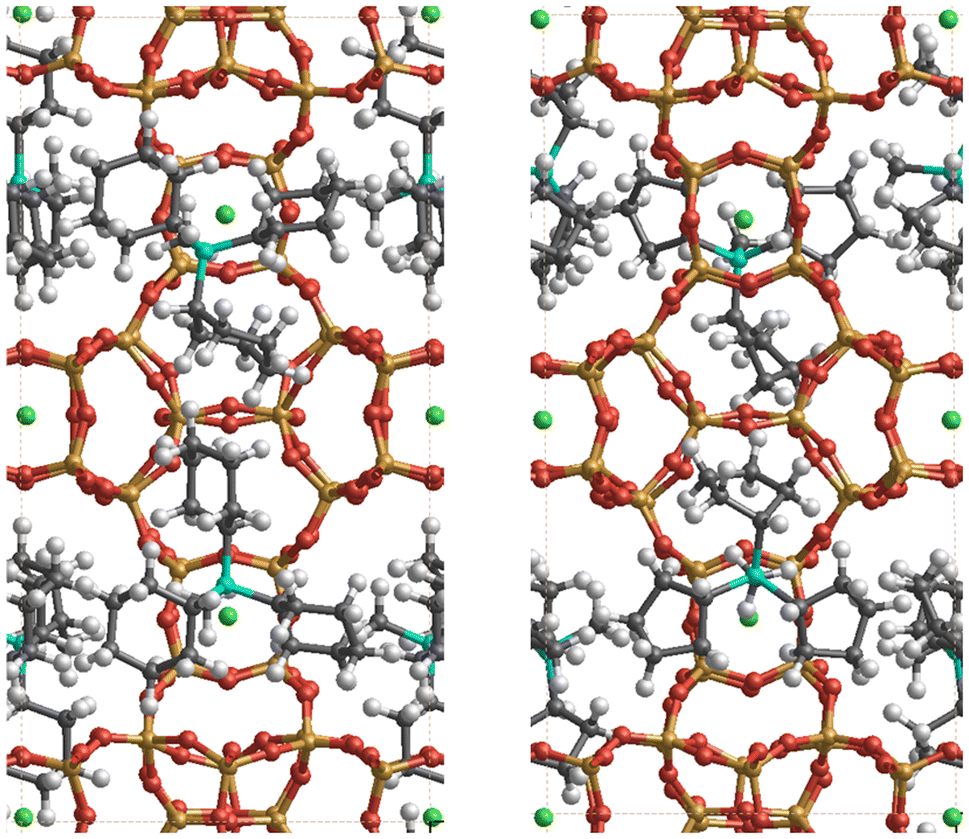 | ||
| Fig. 7 Snapshots of the r2SCAN + rVV10 optimized structures of the ISV zeolite with Cy3MP+ (left panel) and Cp3MP+ (right panel) cations (four cations per unit cell). The colour code is red, orange, grey, green, cyan, and white for O, Si, C, F, P, and H atoms, respectively. F atoms are always (by construction) occluded in the d4r. The P atoms have a similar distribution in the pore for both compositions. Thus, the two systems mainly differ in the conformation of the cyclic groups in each organic cation, as well as in some structural distortions of the ISV framework (see text and Fig. 8). | ||
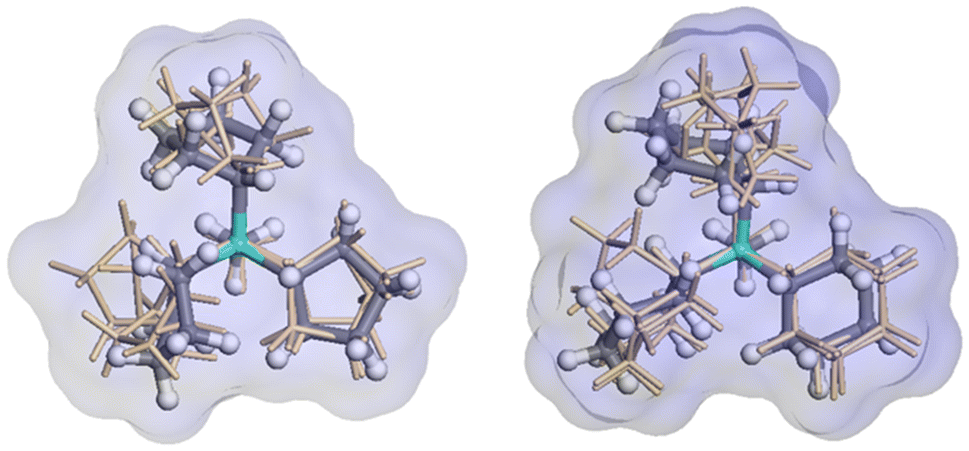 | ||
| Fig. 8 Molecular conformation of Cp3MP+ (left) and Cy3MP+ (right) inside the ISV pores of the corresponding r2SCAN + rVV10 optimized structures. The ball-and-stick model represents the conformer with the minimum energy in vacuum (green P, grey C, white H). The superposed golden stick structures represent the conformers in the pore of the ISV structure, close in energy to the best, and the depicted surface corresponds to the complete ensemble of conformers. | ||
The structures obtained from the energy relaxations using the r2SCAN + rVV10 functional help us understand the two zeolite systems at an atomic level. Fig. 7 shows that the cations distort the ISV framework. These distortions can be studied using the histogram of geometric measurements, such as T–O distances or T–O–T angles, with respect to the pure silica ISV framework. We have shown these in Fig. 9. Both Cy3MP+ and Cp3MP+ cations make the average T–O distances slightly longer than in the calcined material. However, Cp3MP+ cations increase the number of smaller TOT angles (the distortions in the ISV structure in Fig. 7), explaining how the final zeolite volume is smaller than that of the calcined zeolite with this cation (longer T–O distances but more acute TOT angles). For Cy3MP+, similar or larger T–O distances with larger TOT angles result in an expansion of the cell volume. These distortions are reflected in the estimated 29Si NMR chemical shift (Fig. 10).
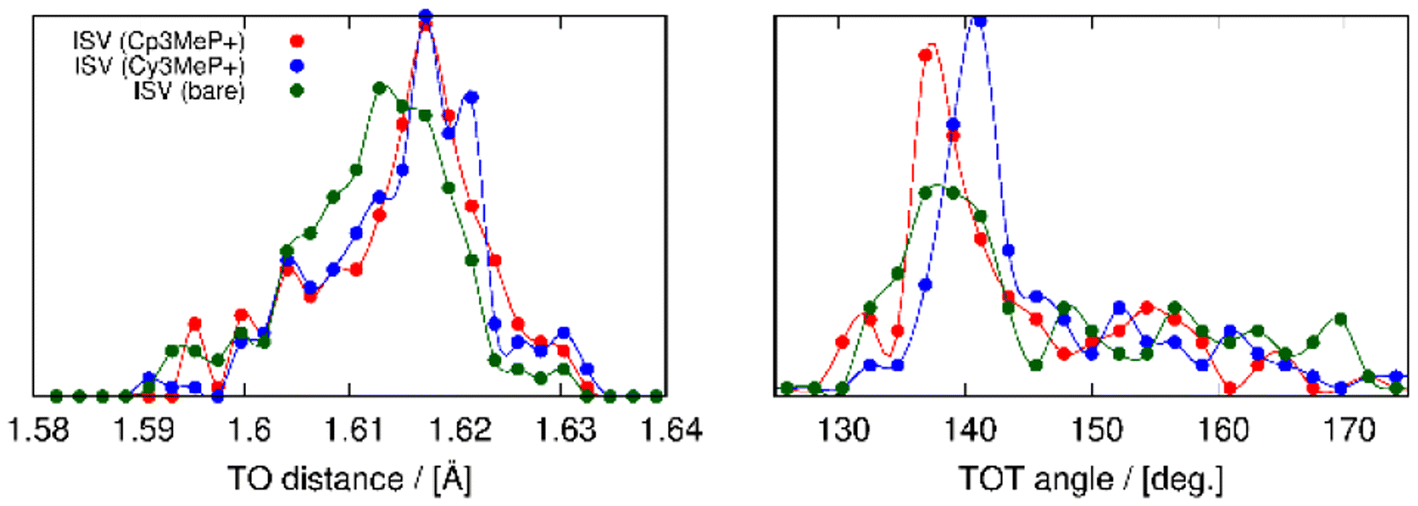 | ||
| Fig. 9 Distribution of TO distances (left panel) and TOT angles (right panel). In red, blue, and green are the compositions for ISV with Cp3MP+, Cy3MP+ and without guests, respectively. | ||
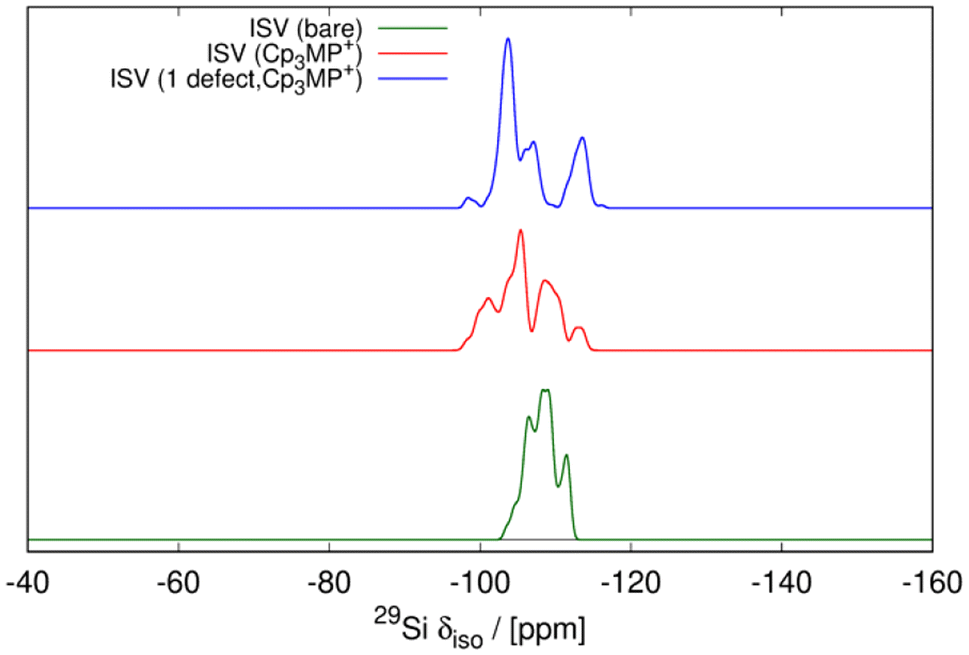 | ||
| Fig. 10 Simulated 29Si NMR spectra for empty ISV (green), ISV with 4 F− and Cp3MP+ ion pairs per unit cell (red), and ISV with 4 F− anions, 4.25 Cp3MP+ cations per unit cell, and 1/4 negatively charged structural defect (SiOH, SiO− pair). | ||
In addition, the influence of structural defects has been considered, consistent with experimental findings (29Si MAS NMR and C, H, N analysis). To model the defect concentration found experimentally, we used a 2 × 2 × 1 supercell containing one defect. The defect consists of a silanol–silanolate pair, introducing a local lattice disruption, and a cation, yielding 17 cations in the supercell, equivalent to 4.25 cations per unit cell. Fig. 11 shows the defective overloaded ISV structure, highlighting the defect and the extra cation. The defective structure exhibits slightly enhanced stability compared to the structure without defects, with a calculated energy of 12.43 kJ per mol per T-atom versus 13.17 kJ per mol per T-atom, calculated using the GFN2-xTB theoretical level (see Table S2†). The defective structure's volume decreases by 0.91% relative to the calcined ISV zeolite but remains slightly larger than that of the defect-free structure. In the initial optimization with 4 cations per unit cell, the cations are located very close to the three channels crossing at (0, 0.5, 0.75), a site we here call S1. The extra cation is positioned at a different site, S2. To accommodate the cation at S2, the four neighboring cations at S1 are displaced by circa 1–2 Å from their equilibrium positions. This arrangement is depicted in Fig. 11, where the cation at S2 is marked with large spheres. In the 2 × 2 × 1 supercell, there are four S2 sites (one per unit cell). However, due to the required displacement of cations in S1 sites, introducing more than one cation at S2 becomes energetically unfavorable as adding a second cation increases the enthalpy to 22.2 kJ per mol per T-atom, still accessible at room temperature, but exceeding the enthalpy of the defect-free structure (13.7 kJ per mol per T-atom) at the same theoretical level (see Table S2†). The presence of defects also alters the ISV structure, as evidenced by the shift in the 29Si NMR chemical shift (see the blue line in Fig. 10). This shift indicates a modification in the local silicon environment due to the introduced defects.
 | ||
| Fig. 11 c-View of the ISV structure (2 × 2 × 1) supercell with 17 Cp3MP+ cations, 16 F− anions, and 1 structural defect. The cations are located in sites S1 and S2. The cation in the S1 site is shown by the ball-and-stick model, and the cation in the S2 site, as well as the –SiOH defect, by the CPK (large spheres) model. The displacement of nearby cations at site S1 upon accommodation of an additional cation at site S2 is shown in Fig. S14.† | ||
The simulation of 29Si MAS NMR spectrum of the bare zeolite agrees reasonably well with the spectrum in our first report of ISV in ref. 9 but not with the current one in Fig. 5, which we attribute to structural disorder, as commented above, as well as to the presence of Q3 in the current material. On the other hand, the simulation of ISV with the cation inside (red curve in Fig. 10) does not agree with the one of the as-prepared sample in Fig. 5. However, the simulation of the overloaded defective zeolite (blue curve) is in reasonable agreement with the experimental one in Fig. 5, confirming the overloading and validating the computational approach.
Conclusions
The new synthesis of the 3D large pore zeolite ISV without the addition of Ge or seeds has been accomplished. The synthesis makes use of Cp3MP+ as the OSDA. This cation is slightly smaller than the Cy3MP+ used to synthesize the 3D extra-large pore zeolite ZEO-1. Calculations indicate a larger flexibility and entropy of Cp3MP+ and a better fit in ISV and larger stabilization energy than those calculated for Cy3MP+, explaining the observations. Experimental results suggest that the zeolite is overloaded with cations and contain defects to balance the extra charges. Calculations demonstrate that this overloaded configuration is possible and energetically favorable by introducing one additional cation every four unit cells. The obtained zeolite contains disorder that, albeit small, prevents a successful Rietveld refinement. The replacement of small fractions of Cp3MP+ by TEA+ stops the crystallization instead of promoting ISV/BEC intergrowths despite our calculations, which suggested that such intergrowths may be more stable than ISV itself. We attribute this to a disrupting effect of TEA+ on the packing of Cp3MP+ inside the zeolite.Data availability
Data for this article, including PXRD, Le Bail fitting, multinuclear liquid and MAS NMR, calculated bond lengths and angles and infrared data, are available at DIGITAL.CSIC at https://hdl.handle.net/10261/374491.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science Innovation (PID2022-137889OB-I00, TED2021-131223B-I00, MCIN/AEI/10.13039/501100011033) is gratefully acknowledged. Synchrotron powder X-ray diffraction data experiments were performed at the Materials Science Powder Diffraction beamline bl04 at the ALBA Spanish Synchrotron with the collaboration of the ALBA staff. H. Y. is grateful to the China Scholarship Council for a PhD grant. S. R. G. B. acknowledges grant RYC2022-036070-I funded by MICIU/AEI/10.13039/501100011033 and by “ESF+”. Authors would like to thank the computing time provided by the Servicio de Supercomputación de la Universidad de Granada.References
- C. Baerlocher and L. B. McCusker, Database of Zeolite Structures, https://www.iza-structure.org/databases/, (accessed December 3rd, 2024).
- Q.-F. Lin, Z. R. Gao, C. Lin, S. Zhang, J. Chen, Z. Li, X. Liu, W. Fan, J. Li, X. Chen, M. A. Camblor and F.-J. Chen, Science, 2021, 374, 1605–1608 CrossRef CAS PubMed.
- H. Yu, L. A. Villaescusa, Z. R. Gao and M. A. Camblor, Angew. Chem., Int. Ed., 2024, 63, e202412170 CrossRef CAS PubMed.
- F.-J. Chen, Z.-H. Gao, L.-L. Liang, J. Zhang and H.-B. Du, CrystEngComm, 2016, 18, 2735–2741 RSC.
- R. Fricke, H.-L. Zubowa, M. Richter and H. Kosslick, C. R. Chim., 2005, 8, 549–559 CrossRef CAS; A. Corma, M. J. Díaz-Cabañas, J. Jiang, M. Afeworki, D. L. Dorset, S. L. Soled and K. G. Strohmaier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13997–14002 CrossRef PubMed.
- J. Li, Z. R. Gao, Q.-F. Lin, C. Liu, F. Gao, C. Lin, S. Zhang, H. Deng, A. Mayoral, W. Fan, S. Luo, X. Chen, H. He, M. A. Camblor, F.-J. Chen and J. Yu, Science, 2023, 379, 283–287 CrossRef CAS PubMed.
- Z. R. Gao, H. Yu, F.-J. Chen, A. Mayoral, Z. Niu, Z. Niu, X. Li, H. Deng, C. Márquez-Álvarez, H. He, S. Xu, Y. Zhou, J. Xu, H. Xu, W. Fan, S. R. G. Balestra, C. Ma, J. Hao, J. Li, P. Wu, J. Yu and M. A. Camblor, Nature, 2024, 628, 99–103 CrossRef CAS PubMed.
- P. Lu, J. Xu, Y. Sun, R. Guillet-Nicolas, T. Willhammar, M. Fahda, E. Dib, B. Wang, Z. Qin, H. Xu, J. Cho, Z. Liu, H. Yu, X. Yang, Q. Lang, S. Mintova, X. Zou and V. Valtchev, Nature, 2024, 636, 368–373 CrossRef CAS PubMed.
- L. A. Villaescusa, P. A. Barrett and M. A. Camblor, Angew. Chem., Int. Ed., 1999, 38, 1997–2000 CrossRef PubMed.
- L. A. Villaescusa, PhD Thesis, Universidad Politécnica de Valencia, 1999.
- J.-Q. Song, B. Marler and H. Gies, C. R. Chim., 2005, 8, 341–352 CrossRef CAS.
- 1,3,3-trimethyl-6-azabicyclo[3.2.1]octane, https://www.sigmaaldrich.com/ES/es/product/enamine/ena947386605?context=bbe, (accessed December 3rd 2024).
- S. Leiva, M. J. Sabater, S. Valencia, G. Sastre, V. Fornés, F. Rey and A. Corma, C. R. Chim., 2005, 8, 369–378 CrossRef CAS.
- J. Jiang, Y. Xu, P. Cheng, Q. Sun, J. Yu, A. Corma and R. Xu, Chem. Mater., 2011, 23, 4709–4715 CrossRef CAS.
- S. I. Zones, S.-J. Hwang, K. Chaudhuri, C.-Y. Chen and J. Pascual, Microporous Mesoporous Mater., 2023, 358, 112399 CrossRef CAS.
- L. Deng, Y. Ma, T. Zai, X. Yi, Y. Tong, Y. Hui, K. Fan, Q. Wu, Y. Ma, X. Liu, W. Liu, N. Sheng, H. Wang, A. Zheng, L. Wang and F.-S. Xiao, J. Am. Chem. Soc., 2024, 146, 29115–29122 CrossRef CAS PubMed.
- M. A. Camblor, A. Corma and S. Valencia, J. Mater. Chem., 1998, 8, 2137–2145 RSC; M. A. Camblor, A. Corma, P. Lightfoot, L. A. Villaescusa and P. A. Wright, Angew. Chem., Int. Ed. Engl., 1997, 36, 2659–2661 CrossRef CAS; M. A. Camblor, M.-J. Díaz-Cabañas, J. Perez-Pariente, S. J. Teat, W. Clegg, I. J. Shannon, P. Lightfoot, P. A. Wright and R. E. Morris, Angew. Chem., Int. Ed., 1998, 37, 2122–2126 CrossRef PubMed; T. Blasco, M. A. Camblor, A. Corma, P. Esteve, A. Martínez, C. Prieto and S. Valencia, Chem. Commun., 1996, 2367–2368 RSC; M. A. Camblor, A. Corma and L. A. Villaescusa, Chem. Commun., 1997, 749–750 RSC; M. A. Camblor, A. Corma and S. Valencia, Chem. Commun., 1996, 2365–2366 RSC; M. J. Díaz-Cabañas, M. A. Camblor, Z. Liu, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 2002, 12, 249–257 Search PubMed; L. A. Villaescusa and M. A. Camblor, Recent Res. Dev. Chem., 2003, 93–141 Search PubMed.
- A. Rojas, E. Martínez-Morales, C. M. Zicovich-Wilson and M. A. Camblor, J. Am. Chem. Soc., 2012, 134, 2255–2263 CrossRef CAS PubMed.
- S. R. G. Balestra, Conformer Ensemble Generation for Adsorption (v. 1.0), Zenodo, 2018 Search PubMed.
- Z. R. Gao, S. R. G. Balestra, J. Li and M. A. Camblor, Chem. – Eur. J., 2021, 27, 18109–18117 CrossRef CAS PubMed.
- J. L. Núñez-Rico, J. Cabezas-Giménez, V. Lillo, S. R. G. Balestra, J. R. Galán-Mascarós, S. Calero and A. Vidal-Ferran, ACS Appl. Mater. Interfaces, 2023, 15, 33594–39605 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed; G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef; J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22, 022201 CrossRef PubMed; J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- J. E. Schmidt, C.-Y. Chen, S. K. Brand, S. I. Zones and M. E. Davis, Chem. – Eur. J., 2016, 22, 4022–4029 CrossRef CAS PubMed.
- D. L. Dorset, G. J. Kennedy, K. G. Strohmaier, M. J. Diaz-Cabañas, F. Rey and A. Corma, J. Am. Chem. Soc., 2006, 128, 8862–8867 CrossRef CAS PubMed.
- A. Corma, M. T. Navarro, F. Rey, J. Rius and S. Valencia, Angew. Chem., Int. Ed., 2001, 40, 2277–2280 CrossRef CAS PubMed.
- M. J. Sanders, M. Leslie and C. R. A. Catlow, J. Chem. Soc., Chem. Commun., 1984, 1271–1273 RSC; K.-P. Schröder, J. Sauer, M. Leslie, C. Richard, A. Catlow and J. M. Thomas, Chem. Phys. Lett., 1992, 188, 320–325 CrossRef CAS; J. D. Gale and N. J. Henson, J. Chem. Soc., Faraday Trans., 1994, 90, 3175–3179 RSC.
- Y. Wang, C. Tong, Q. Liu, R. Han and C. Liu, Chem. Rev., 2023, 123, 11664–11721 CrossRef CAS PubMed.
- G. R. Millward, S. Ramdas and J. M. Thomas, Proc. R. Soc. London, Ser. A, 1985, 399, 57–71 Search PubMed; N. Y. Chen, J. L. Schlenker, W. E. Garwood and G. T. Kokotailo, J. Catal., 1984, 86, 24–31 CrossRef CAS.
- L. A. Villaescusa, I. Díaz, P. A. Barrett, S. Nair, J. M. LLoris-Cormano, R. Martínez-Mañez, M. Tsapatsis, Z. Liu, O. Terasaki and M. A. Camblor, Chem. Mater., 2007, 19, 1601–1612 CrossRef CAS.
- A. Sala, PhD Thesis, Universitat Politècnica de València, 2022.
- M. A. Camblor, P. A. Barrett, M. A.-J. Díaz-Cabañas, L. A. Villaescusa, M. Puche, T. Boix, E. Pérez and H. Koller, Microporous Mesoporous Mater., 2001, 48, 11–22 CrossRef CAS.
- J. Klinowski, T. A. Carpenter and J. M. Thomas, J. Chem. Soc., Chem. Commun., 1986, 956–958 RSC; D. J. Cookson and B. E. Smith, J. Magn. Reson., 1985, 63, 217–218 Search PubMed.
- J. M. Chezeau, L. Delmotte, J. L. Guth and Z. Gabelica, Zeolites, 1991, 11, 598–606 CrossRef CAS; M. A. Camblor, L. A. Villaescusa and M. J. Díaz-Cabañas, Top. Catal., 1999, 9, 59–76 CrossRef.
- L. A. Villaescusa, F. M. Márquez, C. M. Zicovich-Wilson and M. A. Camblor, J. Phys. Chem. B, 2002, 106, 2796–2800 CrossRef CAS.
- M. A. Camblor, PhD Thesis, Universidad Autónoma de Madrid, 1992.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03454f |
| ‡ Current Address: Departamento de Física Atómica, Molecular y Nuclear, Área de Física Teórica, Universidad de Sevilla, Av. Reina Mercedes s/n, 41012 Seville, Spain. |
| § Current address: Department of Chemical and Biomolecular Engineering and Institute for NanoBioTechnology, Johns Hopkins University, Baltimore, MD, USA. |
| This journal is © The Royal Society of Chemistry 2025 |