
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbon–carbon bond formation and cleavage at redox active bis(pyridylimino)isoindole (BPI) germylene compounds†
Antonio I.
Nicasio
 ,
Rosie J.
Somerville
,
Pablo
Sahagún
,
Enrique
Soto
,
Joaquín
López-Serrano
and
Jesús
Campos
*
,
Rosie J.
Somerville
,
Pablo
Sahagún
,
Enrique
Soto
,
Joaquín
López-Serrano
and
Jesús
Campos
*
Instituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica, Facultad de Química, and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Consejo Superior de Investigaciones Científicas (CSIC) and Universidad de Sevilla, 41092 Sevilla, Spain. E-mail: jesus.campos@iiq.csic.es
First published on 3rd January 2025
Abstract
Redox-active ligands provide alternative reaction pathways by facilitating redox events. Among these, tridentate bis(piridylimino)isoindole (BPI) fragments offer great potential, though their redox-active behaviour remains largely underdeveloped. We describe herein a family of BPI germanium(II) complexes and the study of their redox properties. Amido complexes (RBPI)Ge[N(SiMe3)2] 1-R (R = H, Me, Et) showing a κ2-Npy,Niso coordination to the germanium(II) centre were prepared. In contrast, chloride derivatives (RBPI)GeCl, 2-R, display dynamic κ3-Npy,Niso,Npy coordination to the metal centre. The addition of silver bis(trifluoromethane)sulfonimide to compound 1-H generates a dinuclear complex, 3, where the silver atoms are bound to the germanium and one of the imine nitrogen atoms of another BPI fragment. The reduction of 2-R with KC8 generates dinuclear complexes (4-R) characterized by the formation of new C–C bonds between the isoindoline five-membered rings of two different BPI ligands via a radical mechanism, a transformation that does not take place in the absence of germanium. Interestingly, computational and spectroscopic studies support that the reduction takes place exclusively over the RBPI ligand. Strikingly, the newly formed C–C bond is also readily cleaved. Thus, subsequent reduction of 4-R (R = H, Me) using additional KC8 affords dinuclear species 5-R, with polymeric structures between potassium atoms and the corresponding dinuclear Ge2(RBPI2) fragments.
Introduction
The ambiphilic nature of low-valent germanium compounds, with germylenes as prototypical motifs, has attracted significant attention over the last decades.1,2 Undoubtedly, the recognition that these compounds have the potential to mimic some of the reactivity of transition metals has sparked continuous and growing interest in recent times.3 This is exemplified by oxidative addition reactions4 or, for instance, the hydroboration of carbonyl compounds.5 However, the difficulty involved in achieving reductive regeneration of low-valent germanium after oxidative processes has hampered the design of redox cycles based on germanium. To overcome this, a developing strategy entails the incorporation of a transition metal that unlocks reductive processes by cooperation with the germanium site,6 a research avenue that we have pursued over the last years.7–9Alternatively, an underdeveloped approach to enable challenging redox processes in main group elements is the use of redox-active ligands, which can offer alternative redox pathways.10 Among the wide variety of redox-active ligands, a recent report by Breher and co-workers demonstrates the potential of widespread bis(pyridylimino)isoindolide (BPI) ligands as redox-active fragments. In the aforesaid study, the authors were capable of isolating and characterizing up to three different redox states for the same motif.11 Despite these results and their known phosphorescent and electronic properties,12–18 the use of BPI metal compounds that exploit the redox features of the ligand remains underdeveloped. Nonetheless, they have been used as ancillary ligands with f-block metals19 and transition metal complexes having applications in redox flow batteries,20,21 water-splitting,22,23 homogeneous catalysis,24–28 and polymerase mimics.29,30 However, BPI complexes of main group metals have been scarcely developed, and as far as we know only a few examples containing aluminium,31–33 boron, gallium and indium have been described.32
On these grounds, we questioned whether we could use non-innocent BPI ligands to stabilise low-valent germanium compounds with the aim of opening the gates to a new series of main group complexes able to perform reversible redox chemistry. Therefore, in the present work we report the synthesis, characterization and computational studies of a series of BPI germanium complexes and their redox reactivity.
Results and discussion
Ge(II) mononuclear BPI complexes
We started our investigation by synthesizing the 1,3-bis(pyridylimino)isoindoline derivatives RBPI (R = H, Me, Et). We hypothesized that modifying the 6-position of the lateral pyridine rings from hydrogen atoms to alkyl groups could offer a way of modifying the binding modes to low-valent germanium complexes. The ligands were prepared using a modification of Siegl's method starting from phthalonitrile and the corresponding aminopyridine.34 For the ethyl substituent, the synthesis of EtBPI from 6-ethyl(2-aminopyridine) requires increasing the temperature and the use of n-hexanol as solvent.35 Next, we treated the BPI derivatives with Lappert's bis-amido germylene Ge[N(SiMe3)2]2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 as the germanium source, affording the germanium amido derivatives (RBPI)Ge[N(SiMe3)2] 1-R as dark-orange solids in good yields (67–83%) (Scheme 1). The basicity of the silylamido ligands in the Ge(II) precursor avoids the need for external base to deprotonate the isoindoline nitrogen atom of the BPI ligand.
36 as the germanium source, affording the germanium amido derivatives (RBPI)Ge[N(SiMe3)2] 1-R as dark-orange solids in good yields (67–83%) (Scheme 1). The basicity of the silylamido ligands in the Ge(II) precursor avoids the need for external base to deprotonate the isoindoline nitrogen atom of the BPI ligand.
 | ||
| Scheme 1 Formation of complexes 1-R. | ||
Compounds 1-R are stable in dried and degassed solutions of benzene for several days allowing their spectroscopic characterization. The 1H-NMR spectrum in benzene-d6 of 1-Me exhibits two singlets, at 2.70 and 2.38 ppm, corresponding to two non-equivalent methyl groups and 10 signals in the aromatic region from 8.05 to 6.09, corresponding to the isoindole and pyridine protons (see ESI† for details). The spectroscopic data indicate a non-symmetric structure of the molecule, which is consistent with a κ2-Niso,Npy-coordination of the BPI ligand, as reported by Gade for (BPI)iridium(I) complexes.37,38 The κ2-coordination was confirmed by X-ray diffraction analysis of single crystals of complex 1-Me (Fig. 1).
 | ||
| Fig. 1 ORTEP of 1-Me with 50% probability ellipsoids. Hydrogen atoms were omitted and some groups were represented in wireframe for the sake of clarity. | ||
The germanium(II) centre shows a pyramidal geometry with the nitrogen atoms of the isoindoline (Ge–N(3) = 1.965(2) Å) and the coordinated pyridine (Ge–N(1) = 2.156(2) Å) in the same plane and the nitrogen atom of the bis(trimethylsilyl) amido group exhibiting angles of 98.88(9)° (N(1)–Ge–N(6)) and 98.19(9)° (N(3)–Ge–N(6)). The arrangement of the uncoordinated pyridyl unit is similar to that reported by Gade in iridium(I) complexes where the 1,3-bis(2-(4-tert-butylpyridylimino)-5,6-dimethylisoindoline) ligand coordinated in a κ2-Npy,Niso fashion.37 Since only one pyridyl group is coordinated, the other can freely rotate. In fact, both groups undergo chemical exchange in which only one of the two is bound to the germanium atom at any given time. We carried out 2D-exchange spectroscopy (EXSY) experiments to determine the conformational exchange rates in the temperature interval 278 to 303 K. The corresponding Eyring plot (Fig. S40†) yielded activation parameters of ΔH‡ = 17 ± 2 kcal mol−1, ΔS‡ = 5 ± 8 cal mol−1 K−1, and ΔG‡300 K = 16 ± 1 kcal mol−1, consistent with the process taking place at room temperature.
In order to test whether a pincer κ3-N,N,N-geometry would be possible, we decided to use GeCl2 as germanium source, hoping that the smaller chloride substituents would allow for the weak interaction of the second pyridyl group. The desired geometry was indeed obtained in a two-step synthesis adding potassium bis(trimethylsilyl)amide as a base and, subsequently, germanium(II) chloride dioxane adduct to afford the mononuclear derivatives (RBPI)GeCl, 2-R (Scheme 2) as orange powders in 60–70% yield.
 | ||
| Scheme 2 Formation of 2-R. | ||
As expected, the 1H NMR spectrum of 2-H shows half the number of signals than that of 1-H, due to the symmetry of the new pincer-type compound. The κ3-Npy,Niso,Npy coordination of the germanium centre to the HBPI ligand was further confirmed by X-ray diffraction analysis of single crystals obtained from saturated THF solutions of compounds 2-R at −30 °C (Fig. 2). Interestingly, the 1H-NMR spectra of derivatives 2-Me and 2-Et contain two sets of resonances, pinpointing to the existence of an equilibrium in solution between the minor compound bearing the κ2-Niso,Npy-coordination, related to compounds 1, and the major species showing a κ3-Npy,Niso,Npy coordination of the RBPI ligand to the germanium centre. 2D-EXSY experiments confirm the presence of this equilibrium between both compounds (see Fig. S41†). Not surprisingly, the introduction of alkyl groups in the 6-position of the pyridine rings affects the equilibrium, with the proportion of κ2:κ3 coordination increasing from a ratio of 1:2 to 1:1 from 2-Me to 2-Et.
 | ||
| Fig. 2 ORTEPs of 2-H (left), 2-Me (middle), 2-Et (right) with 50% probability ellipsoids. Hydrogen atoms were omitted for clarity. | ||
These spectroscopic observations align well with the crystal structures of 2-R. Although we could only determine the structure of a single isomer for compounds 2-Me and 2-Et, their structures reflect the increasing difficulties in accessing the κ3-Npy,Niso,Npy coordination of the RBPI ligand, showing a significant torsion of the pyridyl units upon increasing the size of the substituents at the 6-position (Fig. 3). The bond distance between the germanium and the isoindole nitrogen atom is similar in the three species (2-H, Ge–N(3) = 1.989(9) Å; 2-Me, Ge(1)–N(3) = 1.991(9) Å, Ge(2)–N(8) = 1.964(8) Å (two molecules in the asymmetric unit); 2-Et, Ge–N(3) = 1.966(2) Å). In contrast, the germanium-pyridine distances of 2-Me are between 0.02–0.14 Å longer than the observed values of 2-H, and the distances of 2-Et are between 0.06–0.15 Å longer than the values of 2-H. As stated above, these observations correlate well with the additional peaks present in the NMR spectra discussed previously.
 | ||
| Fig. 3 ORTEPs of 2-H (above), 2-Me (middle), 2-Et (below) with 50% probability ellipsoids. Hydrogen atoms were omitted for clarity. | ||
The capacity to coordinate two pyridyl groups prompted us to investigate whether we could access a cationic germylene [(BPI)Ge+] fragment stabilized by three N-based donors in a pincer environment much alike its transition metal counterparts. To do so, we treated chloride derivatives 2-R with common halide abstractors (i.e. silver salts, GaCl3 and NaBArF); however, all attempts were unsuccessful, leading to highly insoluble materials or affording mostly intractable mixtures. However, when the amido derivative 1-H was reacted with 1 equivalent of silver bis(trifluoromethane)sulfonimide the dimeric species [{(HBPI)Ge(N(SiMe3)2)}(μ-Ag)2]2(NTf2)2 (3) (NTf2 = [N(SO2CF3)2]−) was formed (Scheme 3). Compound 3 was characterized spectroscopically by 1H and 13C{1H} NMR. The 1H NMR spectrum exhibits a broad signal centred at 0.06 ppm in chloroform-d1 at room temperature, which corresponds to the eighteen protons of the fluxional SiMe3 groups on the amido ligands. Upon lowering the temperature of the solution this resonance resolves into two singlets, one for each pair of inequivalent SiMe3 groups (Fig. S26†).
 | ||
| Scheme 3 Synthesis of complex 3. | ||
Again, the asymmetry of this species is evident in the aromatic region of the 1H NMR spectrum, showing 12 different signals between 8.55 and 6.63 ppm corresponding to the aromatic protons of the BPI ligand. The κ2-Npy,Niso coordination of the BPI ligand to the germanium atom was again supported by X-ray diffraction analysis (Fig. 4). The geometry around the germanium centre is tetrahedral, surrounded by the bridging silver atom and the trimethylsilyl amido ligand. The Ge–Ag distances of 2.4041(11) Å and 2.4020(12) Å are similar to those found in the literature.39–41 The Ge–N(py) distances of 2.029(8) Å and 2.171(8) Å lie in the reported range of 1.9–2.3 Å for Ge–N(py) bonds.42–44
 | ||
| Fig. 4 ORTEP of 3 with 50% probability ellipsoids. Hydrogen atoms and NTf2− anions were omitted for clarity. | ||
Reduction of germanium-BPI complexes
Cyclic voltammetry (CV) studies were performed on the germanium(II)-BPI mononuclear complexes 1-H and 2-H in THF to test their redox behaviour (Fig. 5). The voltammogram of the amido derivative 1-H shows a quasi-reversible reduction event at Epc2 = −2.00 V (vs. Fc/Fc+) associated with an oxidation process at Epa3 = −1.64 V (Table 1) and a second quasi-reversible reduction process (Epc3 = −2.41 V and Epa3 = −1.97 V). These observations are in agreement with the findings of Breher for the reduced 1,3-bis-(4′-methylpyridylimino) isoindoline complexes .11 In addition, a third oxidation event whose nature we are not certain about can be observed at Epc1 = −1.18 V. In contrast, the chloride derivative 2-H shows two irreversible reduction events at Epc1 = −2.01 and Epc2 = −2.68 V, not surprising considering the unavailability of chloride ligands to coordinate back to germanium ([NBu4][PF6] used as electrolyte). In turn, the oxidation processes were barely detectable.
.11 In addition, a third oxidation event whose nature we are not certain about can be observed at Epc1 = −1.18 V. In contrast, the chloride derivative 2-H shows two irreversible reduction events at Epc1 = −2.01 and Epc2 = −2.68 V, not surprising considering the unavailability of chloride ligands to coordinate back to germanium ([NBu4][PF6] used as electrolyte). In turn, the oxidation processes were barely detectable.
 | ||
| Fig. 5 [−3.5 V; −0.7 V] region of cyclic voltammogram of 3.0 mM THF solutions of complex 1-H (blue) and 2-H (red) recorded in 0.1 M [NBu4][PF6] under N2 at 25 °C, at a scan rate of 100 mV s−1, referenced against Fc+/Fc. | ||
To better understand the aforediscussed redox features, we conducted a computational study of the redox properties of the germanium-BPI complexes using DFT calculations (SMD-B3LYP-D3BJ/6-311+g(2d,p)//SMD-B3LYP-D3BJ/6-31+g(d,p); see the ESI† for details).45 Our calculations reproduced the experimental solid-state geometry of 2-H, where the BPI ligand coordinates to the germanium atom in a κ3-Npy,Niso,Npy fashion. Localized orbital analysis (NBO)46 of the bonding in this species is consistent with a germanium(II) formulation. Specifically, the germanium atom has one s lone pair and three predominantly p vacant orbitals: one along the Npy–Ge–Npy axis accepts electron density from both pyridyl fragments, and the other two located on the perpendicular plane receive electron density from the chlorine and Niso atoms (see Fig. S04†). Considering the canonical MOs, the HOMO involves a σ* combination of one sp-hybrid orbital from the germanium and orbitals of the Npy, Niso, and chlorine atoms. The LUMO is part of the π-system of the BPI, with no significant germanium orbital contribution. Analysing the frontier orbitals of the reduced form, 2-H−, reveals that the SOMO spatially aligns with the LUMO of 2-H. In addition, the spin density is localized on the BPI ligand (Fig. 6). Similar results were obtained for 1-H and 1-H−. For these species two conformers were considered, differing in the coordination mode of the BPI ligand: κ3-Npy,Niso,Npy, and κ2-Npy,Niso. Calculations indicate that both are nearly isoenergetic, with the latter being slightly more stable by approximately ΔG° ∼−1.5 kcal mol−1 (Fig. S06†).
 | ||
| Fig. 6 Spin density (left, 0.004 a.u. isovalue) and SOMO (right, 0.06 a.u. isovalue) of the reduced form of 2-H, 2-H−. | ||
We also calculated the redox potentials of the pairs, 1-H−/1-H, 2-H−/2-H, and BPIGe+/BPIGe relative to the first reduction of the BPI ligand (see the ESI† for details). For the first two pairs, the calculated potentials are −2.01 V, and −1.90 V, respectively. However, the calculated redox potential for the BPIGe+/BPIGe pair is −1.23 V. These results support that the first reduction events seen in the CV experiments for 1-H and 2-H correspond to one-electron reduction processes taking place at the BPI ligand.
With all this information in hand, we next moved to investigate the actual synthesis of reduced forms of BPI-germanium species. Therefore, complexes 2-R (R = H, Me, Et) were each treated with 1 equivalent of potassium graphite KC8 as the reducing agent (E[K+/K] = −2.93 V). Intriguingly, these reactions afforded the exotic dinuclear species 4-R in around 60% isolated yields, characterized by the formation of a new C–C bond across two BPI ligands from two different germylene units (Scheme 4). Despite the vast coordination chemistry of BPI ligands, a process alike has not been observed, though it has reminiscence to other C–C coupling events observed in other pyridine-based ligands.47–52 Importantly, when the free BPI ligands were treated with KC8 leading to their reduced form,11 and then treated with germanium dichloride, no trace of compounds 4 was detected. Therefore, the germylene centres are pivotal in providing the template that enables the selective C–C coupling event that is further discussed below.
 | ||
| Scheme 4 Synthesis of 4-R and 5-R. | ||
Compounds 4 were characterized spectroscopically by 1H and 13C{1H} NMR. The former clearly evidences that the RBPI ligand has undergone a transformation, as the chemical shifts of the pyridyl protons have shifted compared to both 1-R and 2-R. Interestingly, the 13C{1H} NMR spectra of 4-R species each show a singlet at 94.3–94.4 ppm (see ESI† for details). These signals correspond to new quaternary carbon centres that are formed via C–C coupling during the reduction of the RBPI ligand. The structure of these complexes was confirmed by X-ray diffraction analysis of single-crystals obtained by slow diffusion of pentane into saturated toluene solutions of complexes 4-R. The unusual geometry of compound 4-H (Fig. 7) exhibits a new carbon–carbon bond (1.63(2) Å) between both HBPI ligands forming a new five-membered heterometalacycle with the germanium centre in one of the apexes of the pentagon. The germanium atom remains bound to the nitrogen atoms of the pyridyl ring and the isoindoline ring; however, a new bond between it and the nitrogen atom of the imine fragment of the other HBPI ligand is formed. As stated above, the formation of this unusual carbon–carbon bond promoted by germanium has to our knowledge no precedent, neither in the BPI nor in the germanium chemistry. Comparison of the crystal structures of the dinuclear species 4-H and the mono-germanium precursor 2-H reveals that whereas the C–N bond distances of the iminopyridine are similar, the C–N bond distance of the imino-isoindoline unit has been elongated by 0.14 Å, from 1.303(13) (C(6)–N(2)) Å in 2-H to 1.441(14) (C(1)–N(2)) Å in 4-H. This indicates that the C(1)–N(2) double bond character in 4-H is lost due to the reduction of the HBPI ligand. Similar structural features were observed for structures 4-Me (Fig. S01†) and 4-Et (Fig. S02†).
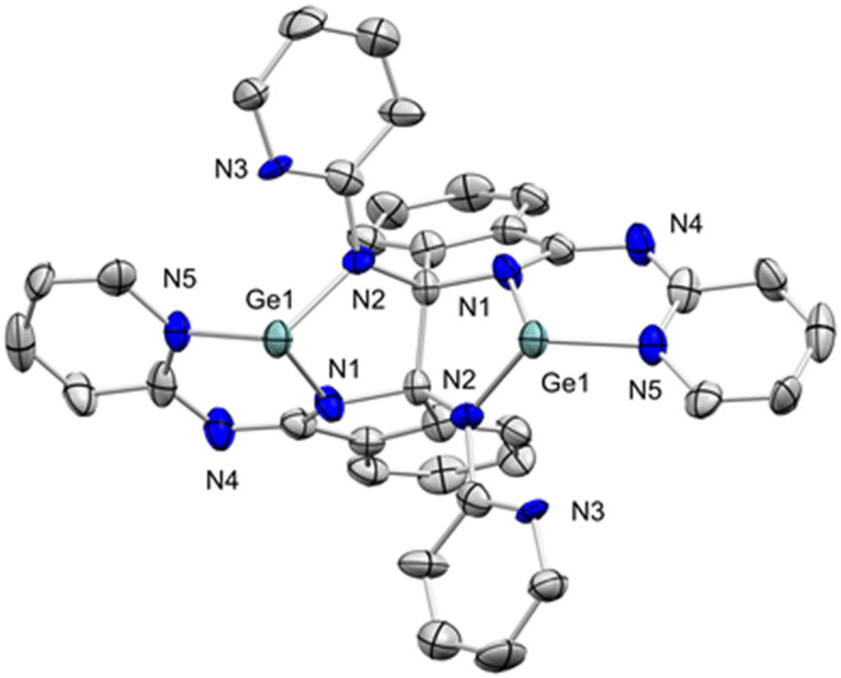 | ||
| Fig. 7 ORTEP of 4-H with 50% probability ellipsoids. Hydrogen atoms were omitted for clarity. | ||
In line with our electrochemical studies, the reduced products 4-R (R = H, Me) can be further reduced using 1 additional equivalent of KC8 per germanium in THF at room temperature. This leads to the formation of new deep purple dinuclear germanium(II) species, 5-R. Despite the low solubility of these compounds, 1H NMR spectroscopic characterization of 5-Me in THF-d8 reveals the existence of coordinated tetrahydrofuran molecules and the presence of two different methyl signals at 2.69 and 2.27 ppm. X-ray diffraction analyses of 5-H (Fig. 8) and 5-Me (Fig. S03†) confirm that the coordination mode of the BPI ligand is similar to 4-R, with a κ2-Npy,Niso coordination of the ligand to one of the germanium centres and a κ1-Nimine coordination to the other germanium atom. Strikingly, the incorporation of two additional electrons into these dimers triggers the cleavage of the previously formed C–C bond between the two RBPI ligands. The coordination of potassium atoms bridging the reduced Ge(RBPI) units completes the structure and results in the appearance of coordination polymers. A related BPI coordination polymer has been observed in the reduced potassium species reported by Breher, where the reduced 1,3-bis(4′-methyl-pyridylimino)isoindole ligand crystallises as a coordination copolymer of 4′-MeBPI and 18-crown-6 moieties between potassium ions.11 Analysis of the bond lengths in 2-H and 5-H reveals that the double bond character between the imine nitrogen atoms and the five-membered ring isoindoline carbon atoms in 2-H has been formally reduced with two electrons. The C(6)–N(2) bond distance has been lengthened by 0.07–0.09 Å, and the C(13)–N(4) bond distance has been increased by 0.13–0.15 Å.
 | ||
| Fig. 8 ORTEP of 5-H with 50% probability ellipsoids. Hydrogen atoms and tetrahydrofuran were omitted for clarity. | ||
The structure of 5-H can be better understood by analysing the resonance forms of the [BPI]3− anion. The first one (Scheme 5, left) contains one formal charge on a bridging nitrogen that has amide character. That could be the reason why the isoindolinic and pyridinic nitrogen atoms coordinate to one of the germanium centres, whereas the amidic nitrogen atom coordinates to the second germanium atom. The resonance forms are consistent with the bond distances found in the X-ray structure of 5-H. For example, the bond length between the iminic nitrogen and the pyridine carbon, C(5)–N(2), of 1.314 Å, is in agreement with the double bond character of the resonance form. On the other hand, this result contrasts with the reduced BPI-potassium structure that Breher reported,10 which could be explained by the second resonance form (Scheme 5, right). In this line, our reported structure contains two pyridine nitrogen anions coordinated to the potassium cations. The formation of the dimeric species 5-H could explain the irreversibility of the redox process observed on the cyclic voltammetry of 1-H and 2-H derivatives. Unfortunately, CV of the reduced products could not be analysed due to decomposition of the complexes under the redox conditions. Similarly, attempts to reoxidize compounds 5 towards 2 or 4 lead to complex mixtures where hydrolysis products were formed in considerable amounts.
 | ||
| Scheme 5 Resonance forms for the [BPI]3− trianion. | ||
To further characterize the reported structures and their nuclearity we performed diffusion ordered spectroscopy (DOSY) experiments on THF solutions of the methyl derivatives 2-Me, 4-Me and 5-Me (Fig. S42–44 in the ESI†). The diffusion coefficient of the potassium-bridged species 5-Me (6.46 × 10−10 m2 s−1) is similar to that of the carbon–carbon bound dimer 4-Me (7.58 × 10−10 m2 s−1). These data indicate that, although the potassium atoms bridge Ge(RBPI) units in a polymeric fashion in the solid state, the structure is dimeric in solution and solvated by THF molecules. The possibility of using other non-coordinating solvents to maintain the polymeric structure was prevented due to high insolubility. In addition, the value of the diffusion coefficient of the monomeric species 2-Me (1.02 × 10−9 m2 s−1) is consistent with its smaller size in comparison with the dinuclear species 4-Me and 5-Me. The estimated hydrodynamic radius of 2-Me calculated using the Stokes–Einstein equation (4.69 Å) is also consistent with the estimated value for the dimeric species 4-Me of 6.31 Å and that of the dinuclear potassium-bridged complex 5-Me (7.41 Å).
The formation of 5-Me from 4-Me using 1 equivalent of KC8 was monitored by 1H-NMR spectroscopy using hexamethylbenzene as internal standard, with spectroscopic yields of 60% (Fig. S37†). Treatment of the chloride derivative 2-R (R = H, Me) with 2 equivalents of KC8 allows the potassium-bridged dimer 5-R to be obtained in a single step, in similar yields, without the need to isolate the 4-R intermediate. Treatment of the amido derivative 1-R with 1 equivalent of the reductant does not afford a clean reaction; however, the addition of 2 equivalents of KC8 does afford the synthesis of 5-R, albeit in lower yields than from the chloride derivative.
Our joint electrochemical and computational studies suggest that the reduction takes place at the BPI ligand, without direct participation of germanium. In the same vein, the C–C coupling event leading to compounds 4 further pinpoints the formation of two C-based radicals that are quenched in solution. With the aim of detecting these ligand-based radical intermediates, we conducted EPR spectroscopic experiments. Gratifyingly, these investigations allowed us to detect the radical intermediate that forms during the reaction between 2-H and a reductant (Fig. 9). We first attempted to monitor by EPR the reaction between 2-H and KC8. However, with such a strong reducing agent the reaction is extremely rapid even at low temperature and we could not observe any signal. In contrast, the use of a solution of Jones’ quasi-universal reductant Mg(DippNacNac)53 afforded control over the reduction to 4-H at low temperature and detection of the radical, showing a resonance with a g value of 2.0065 (THF, 77 K, Fig. 9). This value is in agreement with the observations of Breher for the reduced 4′-MeBPI potassium complex mentioned above.11 Due to the low concentration of the intermediate, a modulation amplitude of 1 G had to be chosen in order to observe the signal. Due to this, the putative hyperfine coupling could not be observed. The temperature of the sample was increased to RT to let the reaction finish, and subsequent measurement of the refrozen sample by EPR reveals the virtually complete disappearance of the paramagnetic intermediate (due to evolution into diamagnetic species 4-H).
 | ||
| Fig. 9 EPR spectra of the reaction upon combining 2-H and Mg(NacNacDipp) in THF (10−3 M, modulation amplitude: 1 G, 9.635588 GHz, 77 K, 32 scans, g = 2.0065). | ||
Conclusions
In summary, we have synthesized and structurally characterized a family of germanium(II) complexes containing bis(pyridylimino)isoindoline (RBPI) ligands with different alkyl groups at the 6-position of the pyridyl unit. Using precursor Ge[N(SiMe3)2]2 leads to BPI-complexes coordinating in a bidentate fashion, whereas precursor GeCl2 yields variable ratios of bidentate vs. pincer-type coordination. Access to cationic Ge(II) species was hampered by solubility, but a dimeric species containing silver bridges between both (HBPI)Ge units could be isolated and characterized. Electrochemical and computational studies support two feasible reductions strictly centred at the BPI ligands, as corroborated by reactivity studies and EPR spectroscopy of a transient intermediate. However, the presence of germanium imparts a unique chemistry that does not take place in its absence, acting as a template for the selective formation of new C–C bonds between two BPI units upon reduction with KC8. Moreover, subsequent addition of a second equivalent of KC8 allows a second reduction of the BPI ligand that triggers the cleavage of the previously formed C–C bond yielding a coordination polymer. Although this rather unusual formation and cleavage of C–C bonds occurs relatively far from the germanium centres (>3 Å), their presence is pivotal, offering potential pathways to temper the radical chemistry of redox-active ligands by using main group elements as templating motifs.Author contributions
A. I. N., R. J. S. and P. S. carried out the experimental work: synthesis and characterization of new complexes and reactivity studies. E. S. performed EPR studies. J. L. S. carried out computational investigations. J. C. supervised the overall work. A. I. N. and R. J. S. wrote the manuscript with participation of all authors.Data availability
Data relating to the synthesis and characterization of new compounds, general methods, NMR spectra, crystal structure determination and computational studies are available in the ESI.†Crystallographic data for 1-H, 2-H, 2-Me, 2-Et, 3, 4-H, 4-Me, 4-Et, 5-H and 5-Me have been deposited at the CCDC under 2403134–2403143†.
All other datasets supporting this article have been uploaded as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Spanish Ministry of Science and Innovation (PID2022-139782NB-I00 and TED2021-132225B-I00) and Junta de Andalucia (P18-FR-4688). JLS acknowledges the use of computational resources of the CSIRC of the Universidad de Granada – cluster Albaicín. The use of computational resources of the Galicia Supercomputing Centre is also acknowledged. Dr Nadir Jori is gratefully acknowledged for his contributions for the electrochemical studies.References
- F. E. Hahn, Chem. Rev., 2018, 118, 9455–9456 CrossRef CAS PubMed
.
-
V. Y. Lee and A. Sekiguchi, Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds, John Wiley & Sons, 2011, pp. 139–197 Search PubMed
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed
- T. J. Hadlington, M. Hermann, G. Frenking and C. Jones, J. Am. Chem. Soc., 2014, 136, 3028–3031 CrossRef CAS PubMed
- R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 2021, 3488–3498 CrossRef CAS PubMed
- S. Bajo, C. A. Theulier and J. Campos, ChemCatChem, 2022, 14, e202200157 CrossRef CAS PubMed
- M. Fernández-Buenestado, R. J. Somerville, J. López-Serrano and J. Campos, Chem. Commun., 2023, 59, 8826–8829 RSC
- S. Bajo, E. Soto, M. Fernández-Buenestado, J. López-Serrano and J. Campos, Nat. Commun., 2024, 15, 9656, DOI:10.1038/s41467-024-53940-9
- S. Biswas, N. Patel, R. Deb and M. Majundar, Chem. Rec., 2022, 22, e202200003 CrossRef CAS PubMed
- J. O. Wenzel, I. Fernández and F. Breher, Eur. J. Inorg. Chem., 2023, 26, e202300315 CrossRef CAS
- K. Hanson, L. Roskop, P. I. Djurovich, F. Zahariev, M. S. Gordon and M. E. Thompson, J. Am. Chem. Soc., 2010, 132, 16247–16255 CrossRef CAS PubMed
- H. M. Wen, Y. H. Wu, Y. Fan, L. Y. Zhang, C. N. Chen and Z. N. Chen, Inorg. Chem., 2010, 49, 2210–2221 CrossRef CAS PubMed
- H. M. Wen, Y. H. Wu, L. J. Xu, L. Y. Zhang, C. N. Chen and Z. N. Chen, Dalton Trans., 2011, 40, 6929–6938 RSC
- K. Hanson, L. Roskop, N. Patel, L. Griffe, P. I. Djurovich, M. S. Gordon and M. E. Thompson, Dalton Trans., 2012, 41, 8648–8659 RSC
- H. M. Wen, J. Y. Wang, B. Li, L. Y. Zhang, C. N. Chen and Z. N. Chen, Eur. J. Inorg. Chem., 2013, 4789–4798 CrossRef CAS
- A. Kaşıkçı, R. Katırcı, S. Özdemir, M. S. Yalçın and M. Özçeşmeci, Dalton Trans., 2023, 52, 9993–10004 RSC
- E. N. Payce, R. C. Knighton, J. A. Platts, P. N. Horton, S. J. Coles and S. J. A. Pope, Inorg. Chem., 2024, 63, 8273–8285 Search PubMed
- P. Zhang, H. Liao, H. Wang, X. Li, F. Yang and S. Zhang, Organometallics, 2017, 36, 2446–2451 CrossRef CAS
- C. S. Sevov, S. L. Fisher, L. T. Thompson and M. S. Sanford, J. Am. Chem. Soc., 2016, 138, 15378–15384 CrossRef CAS PubMed
- S. Gümrükçü, M. Özçeşmeci, N. Koçyiğit, K. Kaya, A. Gül, Y. Şahin and İ. Özçeşmeci, Dalton Trans., 2023, 52, 5265–5276 RSC
- S. Saha, S. T. Sahil, M. M. R. Mazumder, A. M. Stephens, B. Cronin, E. C. Duin, J. W. Jurss and B. H. Farnum, Dalton Trans., 2021, 50, 926–935 RSC
- T. Benkó, D. Lukács, K. Frey, M. Németh, M. M. Móricz, D. Liu, É. Kováts, N. V. May, L. Vayssieres, M. Li and J. S. Pap, Catal. Sci. Technol., 2021, 11, 6411–6424 RSC
- R. R. Gagné and D. N. Marks, Inorg. Chem., 1984, 23, 65–74 CrossRef
- B. Siggelkow, M. B. Meder, C. H. Galka and L. H. Gade, Eur. J. Inorg. Chem., 2004, 3424–3435 CrossRef CAS
- K. N. T. Tseng, A. M. Rizzi and N. K. Szymczak, J. Am. Chem. Soc., 2013, 135, 16352–16355 CrossRef CAS PubMed
- L. V. A. Hale, T. Malakar, K. N. T. Tseng, P. M. Zimmerman, A. Paul and N. K. Szymczak, ACS Catal., 2016, 6, 4799–4813 CrossRef CAS
- T. Ono, S. Qu, C. Gimbert-Surinach, M. A. Johnson, D. J. Marell, J. Benet-Buchholz, C. J. Cramer and A. Llobet, ACS Catal., 2017, 7, 5932–5940 CrossRef CAS
- J. Kaizer, G. Baráth, G. Speier, M. Réglier and M. Giorgi, Inorg. Chem. Commun., 2007, 10, 292–294 CrossRef CAS
- J. Kaizer, T. Csay, P. Kovári, G. Speier and L. Párkányi, J. Mol. Catal. A: Chem., 2008, 280, 203–209 CrossRef CAS
- K. Bakthavachalam and N. D. Reddy, Organometallics, 2013, 32, 3174–3184 CrossRef CAS
- J. D. Dang and T. P. Bender, Inorg. Chem. Commun., 2013, 30, 147–151 CrossRef CAS
- J. O. Wenzel, J. Werner, A. Allgaier, J. van Slageren, I. Fernández, A. N. Unterreiner and F. Breher, Angew. Chem., Int. Ed., 2024, 63, e202402885 Search PubMed
- W. O. Siegl, J. Org. Chem., 1977, 42, 1872–1878 CrossRef CAS
- M. Kruck, D. C. Sauer, M. Enders, H. Wadepohl and L. H. Gade, Dalton Trans., 2011, 40, 10406 RSC
- M. J. S. Gynane, D. H. Harris, M. F. Lappert, P. P. Power, P. Rivière and M. Rivière-Baudet, J. Chem. Soc., Dalton Trans., 1977, 2004–2009 RSC
- J. A. Camerano, C. Sämann, H. Wadepohl and L. H. Gade, Organometallics, 2011, 30, 379–382 CrossRef CAS
- A. L. Müller, T. Bleith, T. Roth, H. Wadepohl and L. H. Gade, Organometallics, 2015, 34, 2326–2342 CrossRef
- J. Li, C. Schenk, F. Winter, H. Scherer, N. Trapp, A. Higelin, S. Keller, R. Pöttgen, I. Krossing and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 9557–9561 CrossRef CAS PubMed
- J. Hlina, H. Arp, M. Walewska, U. Flörke, K. Zangger, C. Marschner and J. Baumgartner, Organometallics, 2014, 33, 7069–7077 CrossRef CAS PubMed
- R. K. Raut and M. Majumdar, Chem. Commun., 2017, 53, 1467–1469 RSC
- J. A. Cissell, T. P. Vaid, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2007, 46, 7713–7715 CrossRef CAS PubMed
- E. Magdzinski, P. Gobbo, M. S. Workentin and P. J. Ragogna, Inorg. Chem., 2013, 52, 11311–11319 CrossRef CAS PubMed
- Y. T. Wey, F. S. Yang, H. C. Yu, T. S. Kuo and Y. C. Tsai, Angew. Chem., Int. Ed., 2017, 56, 15108–15112 CrossRef CAS PubMed
- Computations were performed with the Gaussian 16 software package, Revision C. 01, Gaussian, Inc., Wallingford, CT, 2016; additional computational details can be found in the ESI.†.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed
- M. Gallardo-Villagrán, F. Vidal, P. Palma, E. Álvarez, E. Y.-X. Chen, J. Cámpora and A. Rodríguez-Delgado, Dalton Trans., 2019, 48, 9104–9116 RSC
- R. Arevalo, R. López, L. R. Falvello, L. Riera and J. Perez, Chem. – Eur. J., 2021, 27, 379–289 CrossRef CAS PubMed
- T. W. Myers, N. Kazem, S. Stoll, R. D. Britt, M. Shanmugam and L. A. Berben, J. Am. Chem. Soc., 2011, 133, 8662–8672 CrossRef CAS PubMed
- M. Westerhausen, T. Bollwein, N. Makropoulos, S. Schneiderbauer, M. Suter, H. Nöth, P. Mayer, H. Piotrowski, K. Polborn and A. Pfitzner, Eur. J. Inorg. Chem., 2002, 389–404 CrossRef CAS
- Y. Li, K. C. Mondal, P. Stollberg, H. Zhu, H. W. Roesky, R. Herbst-Irmer, D. Stalke and H. Fliegl, Chem. Commun., 2014, 50, 3356–3358 RSC
- M. Majumdar, R. K. Raut, P. Sahoo and V. Kumar, Chem. Commun., 2018, 54, 10839–10842 RSC
- C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2403134–2403143. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03489a |
| This journal is © The Royal Society of Chemistry 2025 |