
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-based complexes with antiplatelet properties: antagonists of the platelet-activating factor receptor (PAFR) and other aggregating agents
Athanassios I. Philippopoulos
 *a and
Constantinos A. Demopoulosb
*a and
Constantinos A. Demopoulosb
aLaboratory of Inorganic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis Zografou 15771, Athens, Greece. E-mail: atphilip@chem.uoa.gr
bLaboratory of Biochemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis Zografou 15771, Athens, Greece
First published on 24th February 2025
Abstract
Metal complexes displaying anti-inflammatory and antithrombotic properties are a promising research area. Development of new and effective anti-inflammatory and antithrombotic agents is necessary to prevent inflammatory-assisted diseases, thromboembolic disease and oxidative stress. In this frontier article, we report on coordination and organometallic compounds displaying anti-inflammatory and/or antithrombotic potencies, particularly through the inhibition of platelet aggregation. Non -classic targets, such as the platelet-activating factor (PAF), a phospholipid signaling molecule of the immune system and the most potent lipid mediator of inflammation, and its receptor (PAFR), along with collagen, serve as the target molecules in addition to thrombin and ADP (adenosine diphosphate). This article elucidates the progress in this area over the last 15 years, focusing on the great potential of transition metal complexes as possible therapeutic agents to treat inflammatory-assisted diseases, thromboembolic disease and oxidative stress. Metal-based inhibitors of inflammatory mediators could potentially constitute an interesting class of compounds as alternatives to the organic analogues currently in use. Results of this study show that this class of compounds merit further research towards the preparation of new metal-based complexes with improved pharmacological profiles.
Associate Professor Athanassios I. Philippopoulos received his B.S. degree in chemistry from the University of Loannina, Greece, in 1992. In 1997, Athanassios completed his Ph.D. in organometallic chemistry at the same university under the supervision of Prof. N. Hadjiliadis. During his Ph.D. thesis, he spent more than two years in the Laboratoire de Chimie de Coordination du CNRS in Toulouse, France, working with Prof. R. Poilblanc in the field of organometallic chemistry (sandwich PhD thesis). He then undertook postdoctoral research at the Humboldt Universität zu Berlin, Institute of Chemistry, with Prof. Dr A. C. Filippou, working in the fascinating area of transition metal complexes that display triple bonds to a heavier analogue of the 14th group (i.e. Ge, Sn and Pb). In 2003, he joined NCSR Demokritos Greece, as a Research Associate, working with Dr P. Falaras in the field of photovoltaic solar cells. In 2006, he was appointed as a Lecturer at the National and Kapodistian University of Athens, Department of Chemistry. His research interests cover a wide area of inorganic chemistry (synthetic coordination-organometallic chemistry), focusing on the design, synthesis, and characterization of new molecular materials with specific properties and applications in (i) energy conversion (third generation photovoltaic solar cells and dye-sensitized solar cells); (ii) homogeneous catalysis (transfer hydrogenation, α- and β-alkylation reactions, catalyst recycling, etc.) and (iii) bio-inorganic chemistry (metal-based inflammatory mediators and metal-based drugs). He serves as a member of the Editorial Board of “Compounds” (MDPI, 2024–to date). He is the inventor of one patent from the Industrial Property Organization of Greece, a reviewer in more than 32 refereed journals and an evaluator of research proposals from the State Institute of Scholarship, academic ebook repository Kallipos and other organizations. He has supervised six PhD and twelve MSc theses, and he is the Scientist-in-charge of four research projects. Part of his work has been published in international journals (more than 60 publications, number of citations >1800, h index: 20) and in scientific conferences (>70). He is a member of the Association of Greek Chemists. Selected awards during his post-doctoral studies include a Fellowship from the Institute of Physical Chemistry, NCSR “Demokritos, Greece, 2006; IKYDA fellowship, 2003–2005 Joint Greek-Germany collaboration program; and a Fellowship, within the Graduiertenkolleg program entitled “Synthetische, Mechanistische und Reactionstechnische Aspekte von Metallkatalysatoren”, TUB/HUB/FU, 1999–2000. |
Introduction
The serendipitous discovery of the inhibition of cell division in E. coli by cis-diamminedichloridoplatinum(II) (cis-Pt(NH3)2Cl2), which is also known as cisplatin, initiated intense research efforts towards the development and application of inorganic complexes as therapeutic agents against numerous cancer malignancies.1,2 This was the first example of a coordination compound, i.e. a Pt(II) metal ion surrounded by simple non-carbon molecules and ions (NH3 and Cl), acting as a ligand, which was successfully used for combating cancer. To overcome the side effects of the inherent acute toxicity of cisplatin and platinum-containing analogues, a series of new metal-based drugs with other metal ions have been developed.3 To this end, it must be noted that cancer remains responsible for the increased number of deaths worldwide.4 The terms “metal-based drugs” and “medicinal inorganic chemistry” were developed to highlight the high impact of coordination compounds and, more specifically, of the transition metal complexes against a variety of diseases.5Despite the extended research in this field, which resulted in the synthesis and evaluation of a plethora of coordination compounds, interest remains constant towards the rational design of new and more potent anticancer drugs.6
However, relatively less attention has been focused on the investigation of the effects of metal ions and coordination compounds in other bioactivities, such as cardiovascular diseases and thrombosis, which are the leading causes of death worldwide.7 Targeting platelets, thrombin and other aggregating agents is a realistic means to prevent acute thromboembolic artery occlusions in cardiovascular diseases and treat chronic inflammatory diseases.8,9
Thus far, a great majority of compounds with anti-inflammatory and antithrombotic activities are of organic origin (organic small molecules). In this respect, several advantages of metal complexes over organic analogues have been well documented, highlighting their prospects in the development of new therapeutic agents.10 Transition metal complexes display different coordination geometries, ranging from square planar, tetrahedral, square pyramidal and octahedral, in contrast to the typical tetrahedral or trigonal planar geometry of organic congeners. Additional characteristics include their ability to tune thermodynamic and kinetic ligand substitution to obtain different oxidation states and undergo redox reactions.11–13 Finally, it must be stated that several metal ions play a vital role in life and are involved in many natural biological processes14–16 and some diseases.17
This frontier article reports selected examples of coordination and organometallic compounds that have been developed as potent antiplatelet agents, particularly via the inhibition of platelet aggregation.
We focus our study on metal-based complexes, highlighting their great potential for possible use in antiplatelet therapy, including the therapy of inflammatory-assisted diseases, thromboembolic disease and oxidative stress. Finally, restrictions on the application of these metal-based compounds are covered, along with their prospects for the future and the need for additional studies to improve their efficacy and safety.
Current antiplatelet therapies
Platelets are anucleate small circulating blood cells that play a vital role in haemostatic processes because they prevent excessive blood loss upon vascular damage through blood clothing.8,18 Inappropriate activation of platelets leads to thrombosis, reducing the blood supply to the heart and brain, leading to heart attacks and strokes.19 Pharmacologic platelet aggregation inhibitors constitute a class of diverse agents that act in a different way towards platelet adhesion–activation–aggregation progression. In general, the primary mechanisms of action include the interruption of platelet intracellular signalling pathways and/or the blockade of ligand receptors on the platelet membrane.9,20Currently, there are several antiplatelet drugs in clinical practice (Fig. 1), which can be divided into five main categories: (i) cyclooxygenase 1 (COX 1) inhibitors, with aspirin as the typical example; (ii) inhibitors of the adenosine diphosphate (ADP) P2Y12 receptor (clopidogrel, cangrelor, and prasugrel); (iii) PAR1 (proteinase-activated receptor) antagonists of the thrombin-related pathways (vorapaxar); (iv) glycoprotein GPIIb/IIIa receptor inhibitors (Abciximab, eptifibatide) of the collagen-related pathways; and (v) inhibitors of the platelet-activating factor receptor (PAFR), with rupatadine (Rupafin) the only existing drug (Fig. 2).21,22
 | ||
| Fig. 1 Antiplatelet drugs used in clinical practice. | ||
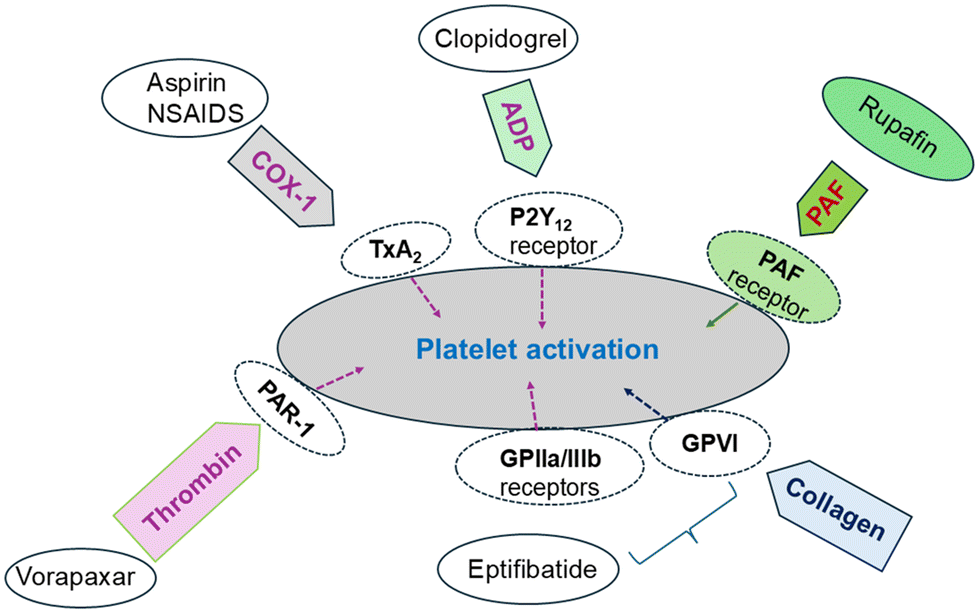 | ||
| Fig. 2 Targets of antiplatelet therapies. | ||
However, these agents, which are the cornerstone of therapy for acute coronary syndromes, exert undesirable side effects, such as bleeding risk. This is also associated with adverse cardiovascular outcomes and mortality. Consequently, several new antiplatelet drugs, focusing either on new drug targets and/or not associated with bleeding, have been developed.18
Antiplatelet metal-based complexes
Metal-based complexes as inhibitors of PAF-induced aggregation
To explore the action of metal ions and their coordination compounds towards molecules of biological interest (biological probes), our recent research focused on the platelet-activating factor (PAF), which is the most potent lipid mediator of inflammation and is also involved in thrombosis and oxidative stress. The systematic name of PAF is 1-O-alkyl-2-acetyl-sn-glycero-3-phosphochloline and its structure is depicted in Fig. 3.23 This phospholipid, which was reported almost 45 years ago, is one member of a family of structurally related phospholipid signaling molecules and exerts its biological activity by binding to its receptor (PAFR), a G-protein-coupled receptor with a seven-transmembrane topology that has been a therapeutic target for many years.24 Various natural and synthetic organic compounds have demonstrated an inhibitory effect on the PAF/PAF receptor acting either through direct antagonistic/competitive effects by binding to the PAFR or through indirect mechanisms.25 In our approach, metal-based complexes have been examined in addition to the classic organic molecules that act as inhibitors of PAF action. Evaluation of the antiplatelet activity expressed by these compounds may be performed by applying an internationally tested method, which is the inhibition of the PAF-induced aggregation in washed rabbit (WRPs) and human platelets (hWPs) via their interaction with PAFR and subsequent blocking of the PAFR activation.26,27 Accordingly, because PAF is the most potent lipid mediator of inflammation, the anti-PAF activity exerted by a new substance can be potentially considered proof of its anti-inflammatory activity. This is of interest considering that organic compounds represent well-known antagonists in this area, while metal-based inhibitors have been totally ignored (Fig. 3).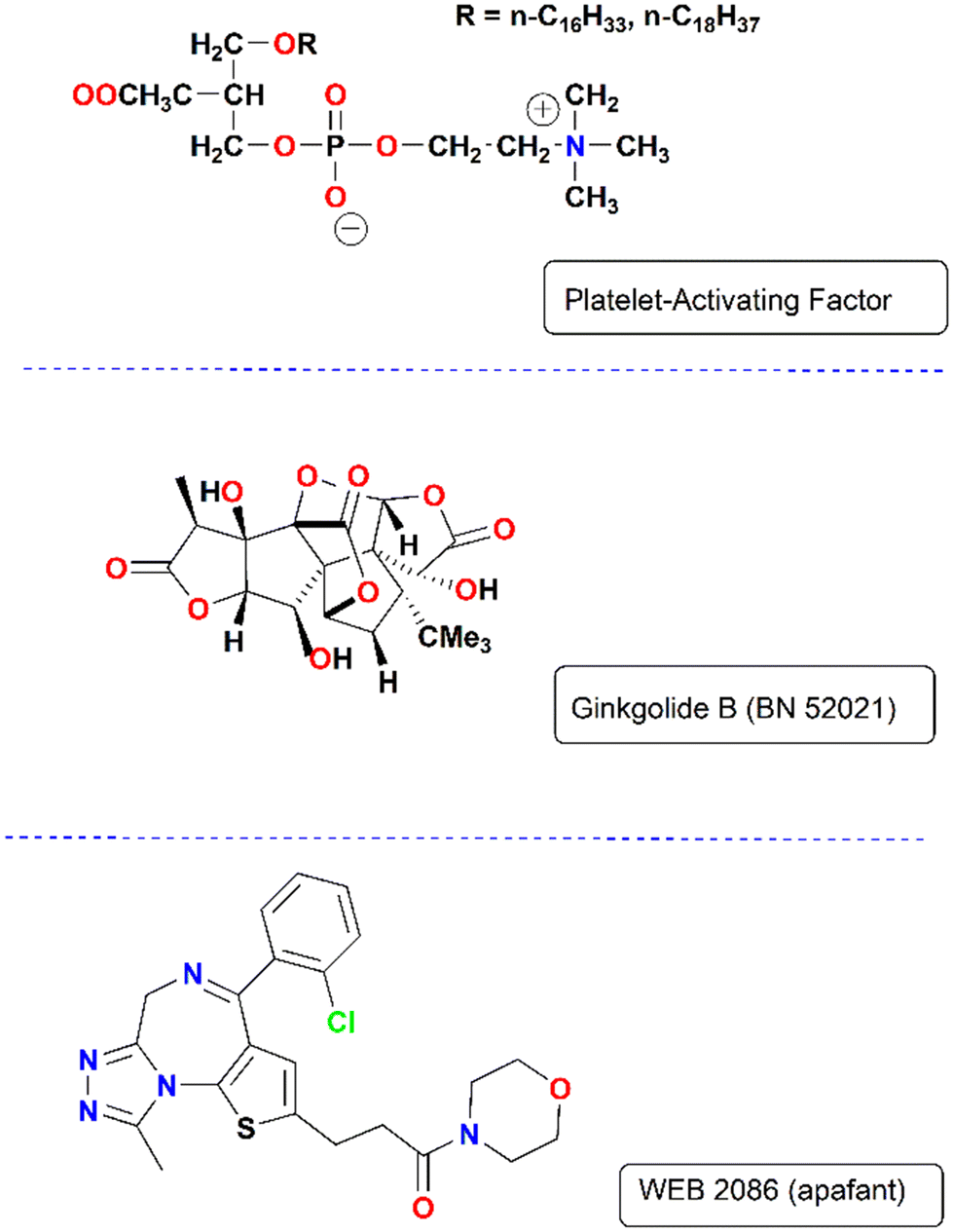 | ||
| Fig. 3 Structures of PAF and organic PAFR antagonists. | ||
Early attempts towards this goal trace back to 2009, i.e. when we started a systematic approach for the development of a library of coordination and organometallic compounds with anti-inflammatory and antithrombotic activities.28,29
Since then, the idea of preparing metal complexes as scaffolds for the design of PAF inhibitors has become more realistic, and this approach can be considered a therapeutic opportunity using PAF-receptor pharmacological antagonists.30 Very recently, following this strategy, the mononuclear Cr(III)-pqx (IC50 = 4.5 μM), Co(II)-pqx (IC50 = 4.1 μM), Cu(II)-pqx (IC50 = 10.6 μM), Zn(II)-pqx (IC50 = 3.3 μM) (1–4) and dinuclear complexes Mn(II)-pqx (IC50 = 39 μM), Fe(II)-pqx (IC50 = 1.79 μM) and Ni(II)-pqx (IC50 = 6.83 μM) (5–7), containing the 2-(2′-pyridyl)quinoxaline ligand (pqx, IC50 = 32 μM), were synthesized and biologically tested as inhibitors of the PAF and thrombin-induced aggregation in WRPs (Fig. 4A).31 The coordination geometries in these complexes range from tetrahedral to trigonal pyramidal and octahedral. The Fe(II) complex 6 was the most active against PAF-induced aggregation, whereas the bulkiest manganese complex 5 was identified as the least active. Considering that washed rabbit platelets exhibit physiological responses to the understudied compounds—free from interference by other blood components—on PAF and other aggregating agent receptors during platelet aggregation, this model is widely regarded as one of the most internationally accepted experimental approaches for replicating findings in human platelets.32 The antithrombotic activities of these metal complexes have been examined, revealing that the Fe-pqx complex (6) is the most potent inhibitor, which exerts similar affinities for both the PAFR and PAR receptors.31 Cytotoxicity studies (in vitro) have been performed for all relevant complexes in HEK 293T (human embryonic kidney cells) and HeLa cells (cervical cancer cells) via the MTT assay. In the HEK 293T cell line, the Cu-pqx analogue (3) was quite toxic with potencies higher than that of cisplatin, which served as a reference compound (Table 1). Previously, a series of Cu(II)(O–O)-PNP (IC50 = ∼1.0 μM, 8), Co(II)(O–Se)-PNP (IC50 = 0.018 ± 0.005 μM) (9), Ni(II)(O–O)-PNP (X = Cl−, IC50–16 μM, 10a; X = Br−, IC50–30 μM, 10a) and Zn(II)(O–O)-PNP (IC50 = 0.54 μM) (11) complexes, bearing chalcogenated imidodiphosphinato ligands (PNP), have been reported (Fig. 4B).33,34 The anti-PAF activities of the PNP-based series were in the sub-micromolar region, showing that the different ligand sphere around the metal centre has a dramatic effect on the biological activity expressed. However, these complexes could be cautiously considered PAFR antagonists because there are no indications of binding to the PAF receptor. In addition, cytotoxicity measurements have not been recorded, which is a drawback for further investigation. Moreover, both square planar complexes 10a and 10b proved unstable in DMSO, rendering them unsuitable for inhibitory action under the experimental conditions reported. Consequently, the aggregation of thrombin-induced WRP by these complexes was not investigated. In this respect, docking theoretical calculations could be very helpful (vide infra in the case of Rh(I)/Rh(III) complexes).
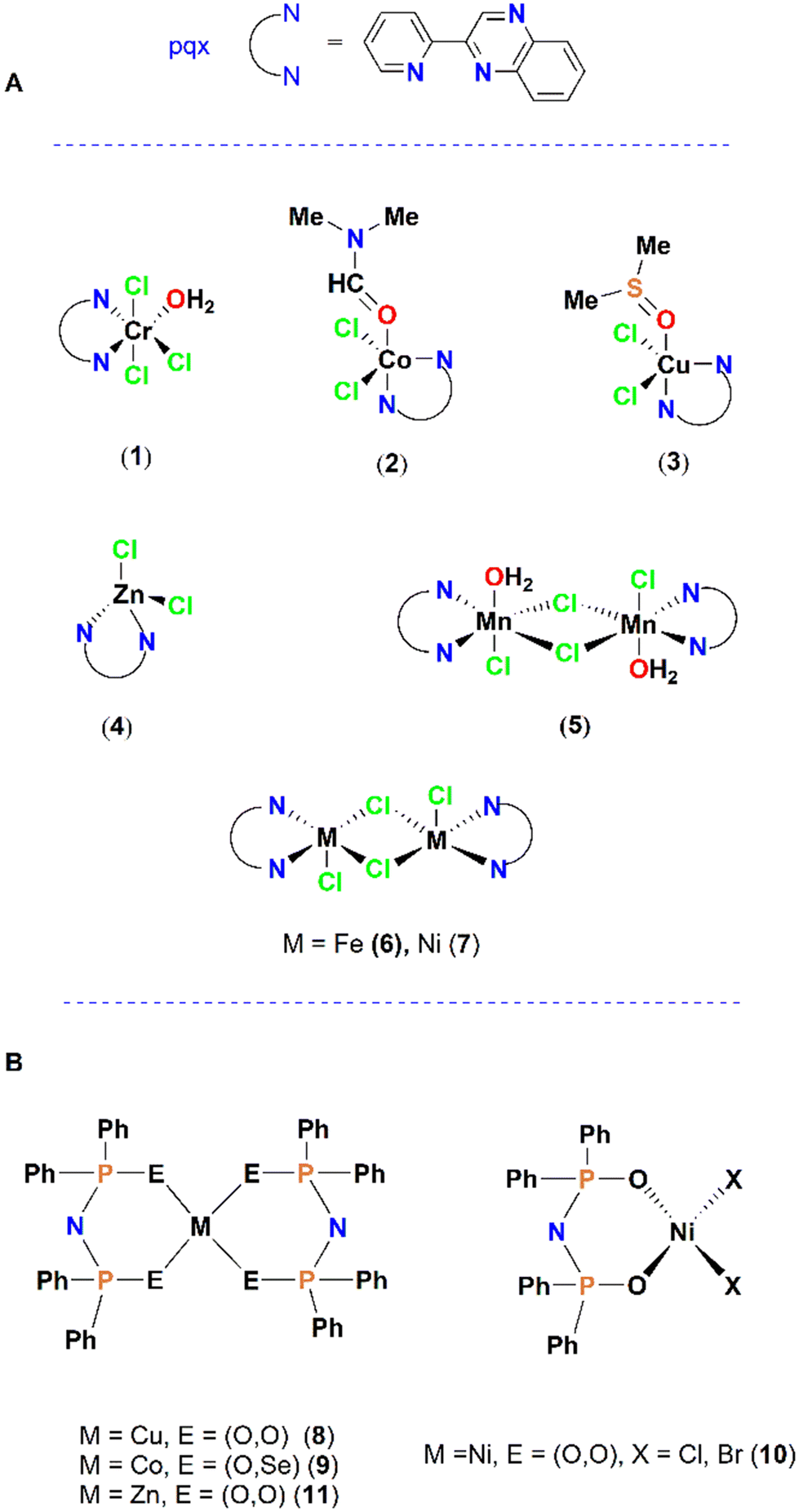 | ||
| Fig. 4 (A) Structures of pqx containing metal-based PAF-inhibitors; (B) structures of PNP-based PAF-inhibitors. | ||
| Complexes | IC50 (PAF in WRPs, μM) target PAFR | In vitro cytotoxicity (HEK293T) | IC50 (thrombin in WRPs, μM) Target PAR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Symbols * and ** represent statistically significant differences with p < 0.05 and p < 0.005, respectively, when these metal complexes are compared with all the other ones.a Towards PAF in hPRPs.b MCF-7 cancer cell line.c ADP-aggregation in hPRPs.d Collagen-induced aggregation in hPRP. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cr-pqx (1) | 4.5 ± 0.9* | 99.05 ± 6.16 | 54.6 ± 10.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Co-pqx (2) | 4.1 ± 0.6* | 67.03 ± 12.58 | 8.9 ± 1.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cu-pqx (3) | 10.6 ± 1.4 | 2.10 ± 0.52 | 3.1 ± 0.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Zn-pqx (4) | 3.3 ± 0.3** | 94.92 ± 23.16 | 3.5 ± 0.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mn-pqx (5) | 39 ± 6 | 28.83 ± 6.16 | 14.0 ± 2.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Fe-pqx (6) | 1.79 ± 0.12** | 65.81 ± 13.17 | 0.46 ± 0.03 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ni-pqx (7) | 6.83 ± 0.42 | 25.53 ± 8.61 | 5.60 ± 0.34 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Pqx | 32 ± 15 | 576 | 54.6 ± 10.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cisplatin | 0.55 ± 0.22 | 4.44 ± 1.29 | 56 ± 16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cu(O–O)-PNP (8) | ∼1.0 | — | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Co(O–Se)-PNP (9) | 0.018 ± 0.005 | — | 7.86 ± 4.77 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ni(O–O)-PNP, X = Cl− (10a) | ∼16 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ni(O–O)-PNP, X = Br− (10b) | ∼30 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Zn(O–O)-PNP (11) | 0.54 | — | 12.80 ± 5.62 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Rh(cod)(pqx)]Cl (12) | 0.016 ± 0.015 | — | 4.5 ± 2.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Rh(cod)(pqx)](NO3) (13) | 0.015 ± 0.015 | — | 37.9 ± 6.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cis-[Rh(pqx)2Cl2]Cl (14) | 0.12 ± 0.11 | 54% | 37.0 ± 8.4 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 77%b viability | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cis-[Rh(cpq)2Cl2]Cl (15) | 0.51 ± 0.23 | — | 0.195 ± 0.098 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cis-[Rh(dcbpyH2)2Cl2]Cl (16) | 0.35 ± 0.20 | — | 0.34 ± 0.11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mer-[Rh(pqx)Cl3(MeOH)] (17) | 2.6 ± 2.0 | — | 35 ± 12 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mer-[Rh(Br-Qpy)Cl3(MeOH)] (18) | 1.0 ± 0.6 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mer-[Rh(OH-Ph-Qpy)Cl3(MeOH)] (19) | 3.9 ± 0.2 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Rh(cod)Cl(tpc)] (20) | 1.91 ± 0.50 | ∼30% viability | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Rh(cod)Cl(tpc)] (20) | 122.6 ± 5.6a | 195 ± 8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 353 ± 64c | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 83.7 ± 11.3d | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Metal-based inhibitors of PAF and thrombin, including second and third row metal ions (heavier analogues)
Several rhodium complexes were synthesized and subsequently evaluated as PAFR and thrombin antagonists (anti-inflammatory and antithrombotic action) (Fig. 5). Notably, the organometallic square planar Rh(I) complexes [Rh(cod)(pqx)]X (cod = cis-1,5-cyclooctadiene), X = Cl− (IC50 = 0.016 ± 0.015 μM, 12), and NO3− (IC50 = 0.015 ± 0.014 μM, 13) displayed very strong antiplatelet activity in the nanomolar scale against PAF. Theoretical docking calculations suggested that these molecules could be accommodated within the ligand-binding site of PAFR (Fig. 6). The bulkier octahedral complexes cis-[Rh(L)2Cl2]Cl (L = pqx, (IC50 = 0.51 ± 0.23 μM, 14); cpq = 4-carboxy-2-(2′-pyridyl)quinoline, (IC50 = 0.35 ± 0.20 μM, 15); and dcbpyH2 = 2,2′-bipyridine-4,4′-dicarboxylic acid (IC50 = 0.12 ± 0.11 μM, 16)) were less potent and could not fit within the PAFR binding site.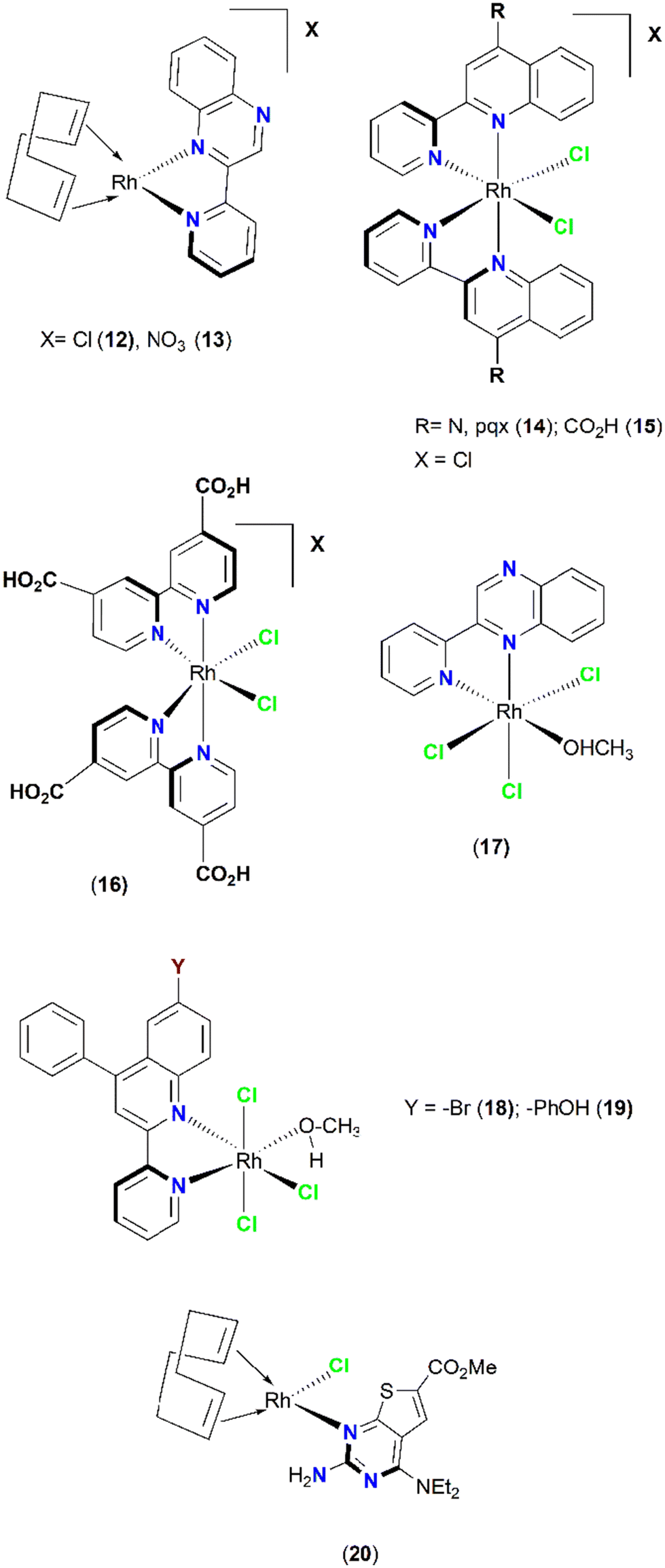 | ||
| Fig. 5 Rhodium-based PAF inhibitors. | ||
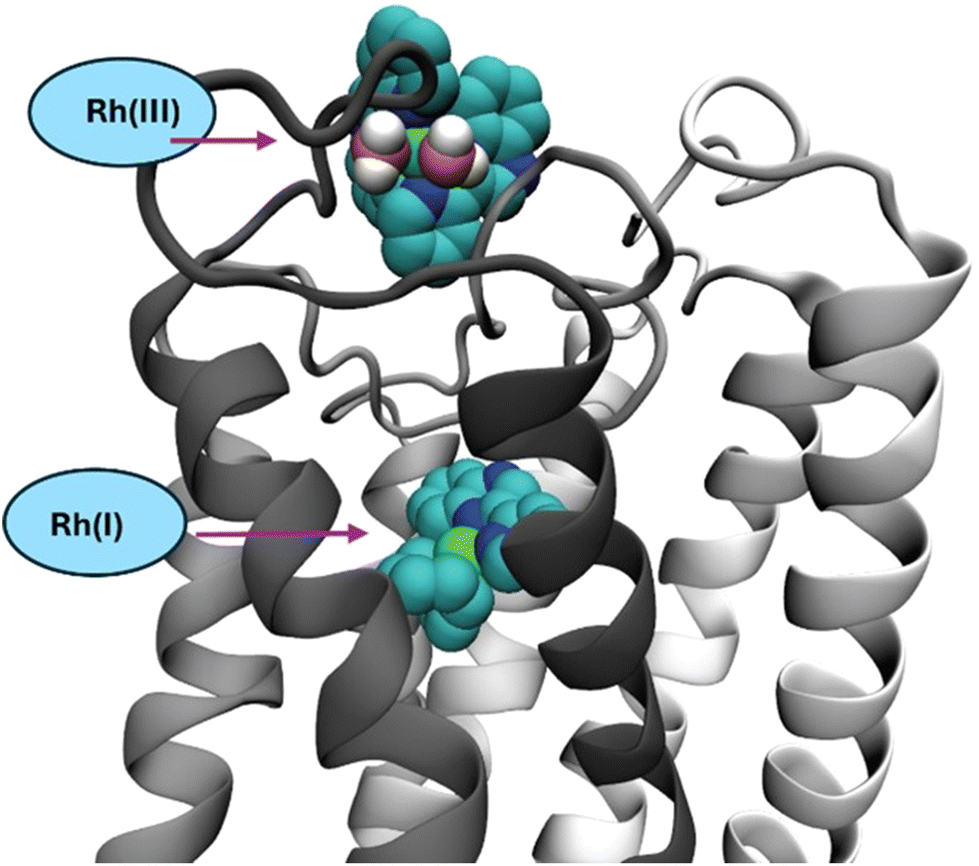 | ||
| Fig. 6 Schematic representation of the molecular model of the PAFR with two predicted binding sites of square planar Rh(I) complexes 12 and 13 and the octahedral Rh(III) complex 14. | ||
It has been suggested that they could bind to the extracellular domain of the receptor and, therefore, antagonize the substrate's entrance to PAFR.26 In the case of 14, the previously reported findings from theoretical calculations corroborate the specific binding results of 3H-labelled platelet-activating factor ([3H]-PAF) to WRPs and its inhibition exerted by this rhodium(III) complex. In this case, its inhibitory effect is attributed only in part to the inhibition of PAF binding within the PAF receptor. It has been proposed that at low concentrations, 14 affects PAFR more effectively and probably inhibits PAF action in another pathway. Notably, complex 12–14 demonstrates the strongest inhibitory effect on the PAF-induced aggregation of WRPs, affecting the thrombin-related pathway of WRP aggregation less. This implies that these complexes may be considered more selective inhibitors of the PAF-related signalling pathways than thrombin. Interestingly, the most potent PAFR inhibitors (12–13) exhibited moderate cytotoxicity against the HEK 293 cell lines, which is in accord with the increased antiplatelet (anti-inflammatory) activity observed (Table 1). Cytotoxicity measurements of 15–19 are currently underway, and the results will be described elsewhere. The mononuclear methanol adducts mer-[(Br-Qpy)Cl3(MeOH)] (IC50 = 1.0 ± 0.6 μM, 18) and mer-[Rh(OH-Ph-Qpy)Cl3(MeOH)] (IC50 = 3.9 ± 0.2 μM, 19) (Br-Qpy) = 6-bromo-4-phenyl-2-pyridin-2-ylquinoline (IC50 = 3.4 ± 0.2 μM) and OH-Ph-Qpy = 4-(4-phenyl-2-(pyridin-2-yl)quinolin-6-yl)phenol (IC50 = 4.4 ± 0.2 μM) are also evaluated and potentially added to the library of metal-based complexes displaying antiplatelet effects in the micromolar range.26,35 Remarkably, these metal-based inhibitors display comparable biological activity (IC50 values) with those of known natural PAF antagonists from the series of Gingolides B (Fig. 3).36 This research has also been extended to a series of substituted thieno-[2,3-d]-pyrimidines and their Rh(I) analogues based on the structural similarity of thieno-[2,3-d]-pyrimidines with thienopyridines.37 Generally, the latter constitute the main organic scaffold for inhibitors of the adenosine diphosphate (ADP) binding irreversibly to the P2Y12 receptor, while ticlopidine, clopidogrel, and prasugrel are the most representative prodrugs. In this respect, the neutral Rh(I) organometallic complex of the formula [Rh(cod)Cl(tpc)] (IC50 = 1.91 μM, 20) (cod = cis-1,5-cyclooctadiene; tpc = methyl 2-amino-4-(diethylamino)-thieno-[2,3-d]-pyrimidine-6-carboxylate, IC50 = 8.12 μM) showed antiplatelet activity in vitro, in both human and washed rabbit platelets (Fig. 5).38 As expected in human platelets and in conditions close to the in vivo ones, the inhibitory effect is dramatically reduced owing to the presence of plasma proteins along with other biomolecules.38,39 Based on molecular docking calculations performed, it has been proposed that the rhodium(I) complex 20 fit within the ligand-binding site of PAF receptor and block the activity of PAF (Fig. 7). Furthermore, the activity of this complex has been studied (in human platelet-rich plasmas) against other inflammatory and thrombotic agents, such as thrombin (IC50 = 195 ± 8 μM), along with platelet agonists, such as ADP (IC50 = 353 ± 64 μM) and collagen (IC50 = 83.7 ± 11.3 μM). This implies a higher affinity of 20 for the collagen-induced pathway. This is an interesting feature, rendering complex 20 a very promising antiplatelet agent. Cytotoxicity studies by MTT assay for 48 h of drug activity, in HEK 293 cells, reveal a dose-dependent behavior, reaching ∼30% viability at the concentration of 100 μM (∼2% cell viability, for cisplatin).38
 | ||
| Fig. 7 (A) Ribbon representation of the PAF-receptor (PAFR) in complex with the antagonist SR27417 from the X-ray structure with PDB ID 5ZKP. The antagonist bound within the PAF-binding site is shown with green C spheres, while the seven helices and the N- and C-termini of the receptor, are also indicated. (B) Close-up view of the PAF-binding site from the extracellular (top) side of the PAFR, illustrating the docked pose of SR27417 (SR-D) in comparison with the crystallographic structure (SR-X). The antagonist is shown with green C sticks (X-ray) or orange C (docked), while N atoms are colored blue, O red, and S yellow. (C) Top-ranked conformation of the PAF bound to the PAFR, with the P atom colored brown. (D and E) Two most populated clusters of docked poses for complex 20 (20-(a) and 20-(b)). Rhodium(I) center is shown as a light mauve sphere, and the coordinated chloride as a green sphere, while cod and tpc ligands are coded with the same color, as that of SR-D and PAF. Selected, top-ranked conformations of the bound ligand tpc. | ||
Previous studies have focused on several Ru(II) and Ru(III) heterocyclic complexes incorporating bidentate (N^N) and tridentate ligands (N^N^N) (21–26), which possess potent antiplatelet and anti-PAF activities (Fig. 8).25,40 Interestingly, the anti-PAF activity of cis-[Ru(dcbpyH2)2(pqx)](NO3)2 (IC50 = 0.18 ± 0.01 μM, 23) and cis-[Ru(dcbpyH2)2(cpq)](NO3)2 (IC50 = 0.24 ± 0.24 μM, 24), which contain carboxylic acid groups in the ligand periphery, is comparable to that of rupatadine fumarate, a potent PAF receptor antagonist that is currently in clinical use with the brand name Rupafin (Fig. 1).41 To this end, selected cytotoxicity studies in HEK-293 cells are performed only for these two ruthenium(II) complexes which exert strong anti-PAF activities. Interestingly, the results demonstrated that PAF inhibitors 23 and 24 are less cytotoxic compared to cisplatin, with IC50 values of 60.2 ± 1.1 and 71.0 ± 2.8 μM, respectively (Table 1). The anti-PAF potencies for all other ruthenium(II) complexes are in the micromolar range (25a, IC50 = 3.1 ± 0.3 μM; 26a, IC50 = 11.8 ± 0.1 μM; 26b, IC50 = 6.4 ± 1.1 μM).
 | ||
| Fig. 8 Selected Ru-, Re- and Au-based inhibitors of the PAF. | ||
Within the series of these ruthenium complexes, the ionic ones were found to be more potent compared to the neutral analogues. These were the first examples of Ru(II)/Ru(III) complexes with anti-PAF activities reported.40 This sounds interesting, considering that in the field of medicinal chemistry, ruthenium complexes constitute an important class of compounds with various potential medicinal and pharmaceutical applications, such as antidiabetic, anti-HIV, anti-Alzheimer's and anti-cancer agents.42,43 Next, the fac-[Re(phendione)(CO)3Cl] complex (27) (phendione = 1,10-phenanthroline-5,6-dione) has been evaluated as another example of a heavier 3d metal complex (Fig. 8). The metal precursor [Re(CO)5Cl] showed an increased inhibitory effect (IC50 = 0.17 ± 0.09 μM) compared to phendione (IC50 = 0.92 ± 0.13 μM) and the Re(I) complex (IC50 = 0.86 ± 0.15 μM), respectively.44 This contrasts with the synergistic effect, which is a general trend that emerged upon thorough investigation of several metal-based PAF inhibitors. This trend showcases the positive effect of ligand coordination on a metal center that generally leads to an increase in the antiplatelet activity exerted.25 In the absence of theoretical calculation studies or specific [3H]-PAF labelled experiments, we cannot suggest if this Re(I) complex fits into the binding site of PAFR, as reported for other similar complexes. Complex 27 showed moderate cytotoxicity ∼70% viability against the breast cancer cell line (MCF-7).
Various aurate(I) salts consisting of the typical [AuCl2]− anion and the [NHC·H]+ counter cation (NHC = N-heterocyclic carbene, IC50 = 0.98 ± 0.15 μM, 28) have been investigated against the PAF-induced aggregation in human platelets in vitro (Fig. 8).45 It seems that the anti-PAF activity observed cannot be clearly attributed to the presence of the [AuCl2]− counter anion because all complexes studied display very similar anti-PAF activities; additionally, the potency of the free ligand (NHC·HCl, IC50 = 3.52 ± 1.39 μM) is within the same range.46 It must be pointed out, however, that further studies with thrombin or other well-established platelet agonists (i.e. ADP or collagen) to maintain the proposed antithrombotic activity have not been performed. Nonetheless, these aurates are promising PAF inhibitors and could be evaluated further because this is the first time in which the antiplatelet properties of N-heterocyclic carbene complexes toward PAF-induced aggregation in human platelets at concentrations comparable to those of similar metal-based PAF inhibitors is reported. For further consideration, cytotoxicity assays are required.
Despite the large pool of information collected from transition metal ions coordinated with different organic ligands, the main group of elements has been studied less. The amphiphilic oxygen tripodal Kläui ligands [(η5-C5R5)Co{P(OEt)2O}3]−, {R = H, (NaLOEt, IC50 = 0.88 ± 0.15 μM); Me (NaL*OEt, IC50 = 0.95 ± 0.21 μM)}, served as scaffolds for the coordination of Sn(II) and Sn(IV) ions. The LOEtSnCl (IC50 = 10.3 ± 1.1 μM 29a), L*OEtSnCl (IC50 = 0.5 ± 0.1 μM, 29b) and LOEtSnPh3 (IC50 = 0.5 ± 0.1 μM, 30a), L*OEtSnPh3 (IC50 = 4.4 ± 0.6 μM, 30b) complexes were found to be potent inhibitors of the PAF and thrombin-induced aggregation in WPRPs and in rabbit platelet rich plasma (rPRPs) aggregation assays in the micromolar range (Fig. 9).47 For the case of thrombin, the six-coordinate organotin analogues 30a and 30b displayed a rather strong antithrombotic effect as expressed by the IC50 values of 0.6 ± 0.1 μM and 0.23 ± 0.02 μM, respectively. In particular, for 30b, this further demonstrates a higher affinity for the PAR receptors of thrombin.
 | ||
| Fig. 9 Sn(II), Sn(IV) and Ga(III) inhibitors of the PAF. | ||
Within this series, complex 29b was the most potent PAFR inhibitor, inducing platelet aggregation in WRPs at higher concentrations. Cross-desensitization tests performed reveal that at high concentration levels, this complex could have a rather weak agonistic activity through the PAF/PAF-R related pathway of platelet aggregation, but, at lower levels, its inhibitory activities against PAF prevail. Cross-desensitization tests performed reveal that this complex could have the agonistic activity through the PAF-R-related pathway of platelet aggregation. Tests conducted with varying concentrations of complex 29b added to WRPs indicate that at high concentrations, the complex exhibits weak agonistic activity through the PAF/PAF-R-related pathway of platelet aggregation. However, at lower concentrations, its inhibitory effects against PAF predominate. Furthermore, these tests suggest that 29b could act as an agonist of the PAF/PAF-receptor pathway, in higher EC50 (concentration effective to produce 50% of the maximum response) concentrations than its IC50 value (inhibitory concentration that reduces the activity/binding of an inducer to its receptor and thus the associated pathway of platelet aggregation). Apparently, 29b can substantially reduce the activation–aggregation of platelets induced by PAF. Using molecular docking calculations, it has been suggested that all organotin complexes that bear the sterically demanding Kläui oxygen tripodal ligand cannot fit inside the PAF-binding site of the receptor. However, they can interact with the extracellular domain of the receptor and block the entrance of the PAF inside the receptor. The in vitro cytotoxic activities of the organotin complexes 29–30 were tested against the Jurkat T lymphoblastic tumour cell line. Within the series, 29a showed rather moderate activity (∼50% viability) in the range of 1–20 μM (Table 2). The homoleptic Ga(III)(O–O)-PNP (IC50 = 0.062 μM ± 0.045 μM, 31) consisting of three bidentate PNP ligands with O,O as the donor atoms displayed an antiplatelet activity in the nanomolar range against the PAF-induced WRP aggregation (Fig. 9).34 It must be noted that this complex did not inhibit the thrombin-induced aggregation of WRPs, even at high doses, which indicates that probably it antagonizes the platelet aggregation through the selective inhibition of the PAF-receptor pathway. Molecular modeling studies would be very helpful to assist this hypothesis.26
| Complex | IC50 (PAF in WRPs, μM) Target-PAFR | In vitro cytotoxicity (HEK293T) | IC50 (thrombin in WRPs, μM) Target-PAR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a MCF-7 cancer cell line.b Jurkat T lymphoblastic tumour cell line. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cis-[Ru(bpy)2Cl2] (21) | 7.0 ± 0.7 | — | 56 ± 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cis-[Ru(dcbpyH2)2Cl2] (22) | 4.5 ± 0.5 | — | 40 ± 5 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cis-[Ru(dcbpyH2)2(pqx)](NO3)2 (23) | 0.18 ± 0.01 | 60.2 ± 1.1 (66.5 ± 1.2)a | No inhibition | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cis-[Ru(dcbpyH2)2(cpq)](NO3)2 (24) | 0.24 ± 0.24 | 71.0 ± 2.8 (81.0 ± 2.1)a | 6.1 ± 0.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Ru(bpp)(dcbpyH)Cl] (25a) | 3.1 ± 0.3 | — | 25 ± 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Ru(bpp)(dcbpyH2)Cl]Cl (26a) | 11.8 ± 0.1 | — | No inhibition | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Ru(bdmpp)(dcbpyH2)Cl](PF6) (26b) | 6.4 ± 1.1 | — | 2.1 ± 0.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| fac-[Re(phendione)(CO)3Cl] (27) | 0.86 ± 0.15 | ∼70% viabilitya | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| [Ph-NHC·H][AuCl2] | 0.98 ± 0.24 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ph-NHC·HCl | 3.52 ± 1.39 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LOEtSnCl (29a) | 10.3 ± 1.1 | ∼50% (range 1–20 μM)b | 10.3 ± 1.3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L*OEtSnCl (29b) | 0.5 ± 0.1 | Cytotoxic at >10 μMb | 1.8 ± 0.7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LOEtSnPh3 (30a) | 0.5 ± 0.1 | Cytotoxic at >5 μMb | 0.6 ± 0.1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| L*OEtSnPh3 (30b) | 4.4 ± 0.6 | Cytotoxic at >5 μMb | 0.23 ± 0.02 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ga(III)(O–O)-PNP (31) | 0.062 ± 0.045 | — | — | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Structure–activity relationship studies
Since the first report on this topic, the number of metal-based complexes with anti-PAF and/or antiplatelet activities against other aggregating agents that have been evaluated as PAF and/or thrombin-induced aggregation inhibitors has significantly increased. From the library of ∼60 compounds created, and upon a thorough study, some preliminary structure–activity relationships have been established based on simple coordination chemistry principles.25,48Diverse parameters are considered, such as the coordination geometry of the metal complex (square planar vs. octahedral), the nature of the ligands (bidentate, tridentate), the effect of the counter anions and the total charge and size of the complex. As previously reported, the bulkier octahedral Rh(III) analogues (14–19) were less potent than the square-planar Rh(I) complexes (12–13).25,26 This further demonstrates the effect of different coordination geometry on the biological effect exerted. Within the various Ru(II) and Ru(III) complexes synthesized as possible PAF and thrombin inhibitors, an increased potency was determined upon exchanging the chloride for a hexafluorophosphate counter anion. This behaviour could be attributed to the higher inhibitory effect towards PAF of fluoride containing substances, such as the trifluoroacetyl analogues, compared to the trichloroacetyl ones.25 Moreover, the activity of ionic complexes was higher than that of the neutral congeners.
Over the last few years, the synthesis of metal-based antagonists of PAF and thrombin and their viability as agents of pharmaceutical interest has been the subject of excellent reviews.30,49 It must be noted that in most cases where the antiplatelet activity was studied, control experiments for metal and ligand precursors were also performed. Consequently, the term synergistic effect cannot be attributed to a cumulative effect of these precursor molecules but to the clear synergism of the metal ions and the coordinated organic molecules.46 To support our findings and test our hypothesis, additional theoretical docking calculations would be advisable. A remarkable contribution to this goal may arise from the fact that the X-ray crystal structure of PAFR in complex with SR27417 antagonist has been recently elucidated.50 Based on the outcome of the biological assays performed, the most potent PAFR inhibitors from the small library of compounds, which have been created and probably fit within the binding site of PAFR, must be re-examined. Besides theoretical calculations and in vitro assays, additional studies, including in vivo biological experiments, are required, along with a thorough investigation of the pharmacodynamics and pharmacokinetics of the compounds examined thus far before possibly entering some of them in the next step of clinical trials. This is an interesting issue considering that clinical trials against several disease pathologies with synthetic PAFR antagonists of organic origin were not very effective.24,51
Metal-based complexes with anti-inflammatory and antithrombotic potencies following other pathways
Although the field is still immature, interest in the field of metal-based anti-inflammatory, antithrombotic agents remains constant, as shown by excellent contributions within recent years.30,49 Selected examples by other research groups are included below.Osborn and Vaiyapuri studied the use of a Ru-thio-chrysin complex (32) to modulate the platelet function, haemostasis and thrombosis using chrysin, a natural flavonoid. Under physiological conditions, the inhibitory effect of this complex in human platelet-rich plasma (hPRP) was enhanced in comparison to chrysin.39 The results further showed that the active targets of thio-chrysin and its ruthenium analogue are the same, inhibiting Akt and FAK phosporylation, induced by collagen-related peptide (CPR-XL), as a platelet agonist. In addition, no toxicity issues were mentioned for platelets under the experimental conditions in this study (Fig. 10).
 | ||
| Fig. 10 Ru(II) complexes as inhibitors of collagen-induced aggregation. | ||
Chang et al. synthesized three Ir(III) complexes of the general formula [Ir(Cp*)(1-(2-pyridyl)-3-phenylimidazo[1,5-a]pyridine)Cl][BF4] (Ir-imid, 33), [Ir(Cp*)(1-(2-pyridyl)-3-(3-nitrophenyl)imidazo [1,5-a]pyridine) Cl][BF4] (Ir-imid-NO2, 34) and [Ir(Cp*)(9-[4-(1-pyridin-2-yl-imidazo[1,5-a]pyridin-3-yl)-phenyl]-9H-carbazole) Cl][BF4] (Ir-imid-OCH3, 35) (Cp* = C5Me5), aiming to investigate the in vitro antiplatelet and in vivo antithrombotic activities (Fig. 11). Within the series, complex 33 was the only active because it potentially inhibited adenosine triphosphate (ATP) release, calcium mobilization ([Ca2+]i) and P-selectin expression induced by collagen-induced aggregation, without cytotoxicity.
 | ||
| Fig. 11 Ru(II) complexes of the TQ series with antiplatelet activities. | ||
Additionally, in vivo studies revealed that the iridium(III) complex 33 significantly prolonged the platelet plug formation and reduced the mortality of adenosine diphosphate (ADP)-induced acute pulmonary thromboembolism in mice. The sole activity of this iridium complex was attributed to the absence of substitution on the phenyl group, which may provide a higher rate of hydrolysis.52
Sheu and Chang showed that TQ-5 and TQ-6, two Ru(II)-based complexes of the formula [Ru(2-(pyridin-5-phenyl-2-yl)-quinoxaline)(η6-p-cymene)Cl][BF4] (36) and [Ru((1H-benzoimidazo-2-yl)-quinoline)(η6-p-cymene)Cl][BF4] (37), displayed an inhibitory effect against collagen-induced aggregation in hWPs in a concentration-dependent way.53,54 Their potencies are higher than that of aspirin, a well-established antithrombotic agent (Fig. 1). Moreover, both complexes inhibited thrombin-induced aggregation but not in a concentration-dependent manner.
It has been suggested that the new Ru(II)-p-cymene analogues inhibit platelet aggregation by suppressing [Ca2+]i mobilization and ATP production with no cytotoxicity. These substances have the potential to be used as therapeutic agents for the treatment of thromboembolic disorders. Based on these interesting results, complex 37 was also investigated owing to its neuroprotective action against microglia activation and middle cerebral artery occlusion (MCAO)-induced embolic stroke. At a concentration of 2 μM, the administration of this ruthenium(II) complex severely influenced (diminished) the expression of inflammatory mediators (nitric oxide/inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX-2)), nuclear factor kappa B (NF-κB) p65 phosphorylation, and nuclear translocation, along with hydroxyl radical OH˙ formation in LPS-stimulated microglia. Moreover, the results show an increase in the expression of nuclear factor erythroid 2-related factor 2 (Nrf2) and heme oxygenase-1 (HO-1), which could be a critical therapeutic target for stroke treatment.55
Previously, the same group presented the synthesis of three Ru(II) structurally related complexes namely [Ru((pyridin-2-yl)-quinoline)(η6-p-cymene)Cl][BF4] (TQ-1, 38), [Ru(2-(pyridin-2-yl)-quinoxaline)(η6-p-cymene)Cl][BF4] (TQ-2, 39), [Ru((diphenyl-2(pyridin-2-yl)-quinolin-6-yl)-amine)(η6-p-cymene)Cl][BF4] (TQ-3, 40) (Fig. 11). Interestingly, in vitro studies revealed that among them, only 40 proved to be a very potent inhibitor (Table 3) of the platelet aggregation induced by collagen and thrombin in washed human platelets.56 The inhibitory effect of 40 was attributed to the inhibition of collagen-induced ATP release, calcium mobilization ([Ca2+]i) and P-selectin expression, without cytotoxicity. A limitation of the studies reported may be that stability tests of the relevant complexes in the appropriate media used for the biological studies were not performed. This could be very helpful in gaining further insight into the nature of the active species in the solution. A recent review article has described more examples related to this topic in detail.57 To this end, relevant targets from these studies are included in Table 3.
| Complex | In vitro IC50 (collagen in washed hPRPs, μM) | Target | In vitro IC50 (thrombin in washed hPRPs, μM) | Cytotoxicity | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Collagen-related peptide (CPR-XL).b Plasma-rich platelets (PRP).c Not specific for the GP VI receptor.d In vivo prolonged closure time. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ru-thio-chrysin (32) | Concentration-dependent inhibitiona | Inhibition of Akt, FAK phosphorylation induced by CPR-XL. Inhibited ATP (dense granule secretion), [Ca2+]i | —d | No toxic effects lactate dehydrogenase (LDH) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ir-imid (33) | 11.1% ± 3.7% (32.5% ± 2.6%)b | Inhibition of the Akt/PKC pathways, subsequently suppressing the activation of MAPKs | —d | No toxic effects (LDH) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ir-imid-NO2 (34) | No effects up to 50 μM | No effects on these targets | — | No toxic effects (LDH cytotoxicity assays) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ir-imid-OCH3 (35) | No effects up to 50 μM | No effects on these targets | — | No toxic effects (LDH) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TQ-5 (36) | 1–5 | Suppression of Akt/JNK signalling cascades (phosphorylation), potentially inhibited collagen-induced (ATP) release, calcium mobilization ([Ca2+]i) | >100 μM [suppressed thrombotic plug formation in vivo] | 3–10 μM (LDH) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TQ-6 (37) | ∼0.3 ex vivo | Inhibition of collagen-induced (ATP) release, calcium mobilization ([Ca2+]I), the agonist receptors-mediated inside-out signaling such as the Src-Syk-PLC_2 cascade. Inhibition of the MAPK signalling pathwaysc | ∼60 | 20–100 μM (LDH activity) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TQ-1 (38) | No response even at 250 μM | Main possible targets Akt, MAPKs | Up to 250 μM | Up to 250 μM(LDH) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TQ-2 (39) | No response even at 250 μM | Main targets, SFK/Akt, intracellular calcium mobilization [Ca2+]i and ATP production | Up to 250 μM | Up to 250 μM (LDH) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TQ-3 (40) | 1–5 μM | Suppression of the Syk-Lyn-Fyn cascade and destruction of Akt, JNK and p38 MAPK activation. Reduction of ATP level, surface P-selectin expression and calcium mobilization ([Ca2+]i) | 0.63 | 5 μM | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Metal-based complexes with anti-inflammatory activities
Relatively less attention has been given towards the development of metal complexes with anti-inflammatory activities although many recent studies have suggested they hold great promises.58 Common strategies include the coordination of bioactive ligands, mainly non-steroidal anti-inflammatory drugs (NSAIDs), affording metal complexes with improved anti-inflammatory properties. Most of them inhibit prostaglandin synthesis by inhibiting the cyclo-oxygenase (COX). As a representative example, tetrakis-μ-acetylsalicylato-dicopper(II) was more potent than aspirin in rats or mice.59Independently, platelets that are commonly associated with their involvement in thrombosis and haemostasis also play significant roles in inflammation and immunity.60 Considering that PAF is the most potent lipid mediator of inflammation, which is also involved in thrombosis and oxidative stress, the previously reported metal-based complexes with anti-PAF activities could potentially be examined in the treatment of inflammatory-assisted diseases, thromboembolic disease and oxidative stress.61
Recent advances in the field provide encouraging results, which stem from the anti-inflammatory evaluation (in vitro and in vivo animal models) of novel synthetic ruthenium(II) complexes (namely TQ compounds) consisting of the p-cymene moiety and a series of substituted pyridine-quinoline-based ligands.62 It has been suggested that [Ru((4-phenyl-2-pyridin-2-yl)-quinoline)(η6-p-cymene)Cl][BF4] (TQ-4, 41) (Fig. 11) presents very promising anti-inflammatory activity against mouse liver injury and RAW 264.7 macrophages by down regulating the inflammatory mediators JNK (Jun N-terminal kinases) phosphorylation and NF-κB (nuclear factor-kappa B) signalling pathways.63
In this respect, complex (TQ-6, 37) proved very potent against lipopolysaccharide (LPS)-induced in vitro inflammation in macrophage and in vivo liver injury in mice. Moreover, the results revealed that LPS-induced expression of tumour necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β) were reduced upon treatment of LPS-stimulated RAW 264.7 cells, with this complex. The authors finally suggested that NF-κB could be a promising target for protecting against LPS-induced inflammation and liver injury by administering a ruthenium(II) complex. Accordingly, 37 can be used to treat inflammatory-related diseases.64
A series of ruthenium, osmium and iridium half-sandwich complexes incorporating 1,2-dicarba-closo-dodecarborane-1,2-dithiolato and benzene-1,2-dithiolatoligands were synthesized, and their anti-inflammatory activity was evaluated in RAW 264.7 murine macrophages and MRC-5 fibroblast cell lines, after nitric oxide production and inflammation response, induced by bacterial endotoxin lipopolysaccharide (LPS). Notably, the iridium complex [Ir(η5-pentamethylcyclopentadiene)(benzene-1,2-dithiolato)] (Ir-dithiol, 42) was found to be non-cytotoxic and triggered an anti-inflammatory response against LPS-induced NO production (Fig. 12). The authors suggested that this could be considered a new avenue for the development of non-cytotoxic anti-inflammatory drugs.65
 | ||
| Fig. 12 Ir(III) and Cu(II) complexes with anti-inflammatory activities. | ||
Previous studies have included a series of copper(II) Schiff base compounds with S,N-heterocyclic adducts, which have been assessed for their anti-inflammatory activity. In these studies, the rat carrageenan-induced paw oedema assay was employed as a model for acute inflammation.66 Among them, the [Cu(dienOO)(2a-5 mt)Br2] complex (Cu-5mt, 43), where 2a-5 mt denote 2-amino-5-methyl-thiazole, was the most potent anti-inflammatory agent (Fig. 12). Analogous assays have been surveyed with different Schiff bases coordinated to diverse metal ions, and their roles as anti-inflammatory drugs have been examined.67
The limitations of the aforementioned examples include dose-limiting side effects, resistance to several compounds used in the treatment, and toxicity issues. Apparently, in vivo studies in different animal models may be considered to elucidate the possible mechanisms of action. Additional problems acquired with current treatment strategies include low bioavailability along with non-specific distribution against the biological targets studied.
In this respect, new challenges and perspectives on inflammatory therapy have emerged very recently, in which transition metal-based smart nanosystems (TMSNs) are specifically engineered to block the mechanisms of initiating inflammatory responses. These nanomaterials are also proposed as nanocarriers for delivering anti-inflammatory drugs. Complex synthetic procedures for these nanoparticles, including controllable production and reproducibility issues, constitute severe drawbacks that may limit further application. Moreover, the long-term safety of TMSNs remains an obstacle to clinical use.68
Conclusions and perspectives
The results of this study revealed the high potency of metal-based complexes as anti-inflammatory and antithrombotic agents. The study of the effects of a variety of metal complexes on a biological target, such as the platelet-activating factor and its receptor, has emerged as a versatile approach, towards this goal. The synthesis of a series of metal-based inhibitors of PAF-induced platelet aggregation (PAFR antagonists) along with other aggregating agents (collagen, thrombin) showcases the high impact of coordination chemistry, which remains a powerful tool for inorganic chemists to rational design molecules against various diseases.However, for most of the promising metal-based complexes reported, in vivo studies must be performed to support the aforementioned in vitro results. Additionally, theoretical docking calculations (molecular modelling studies) for the most potent PAFR inhibitors would be very informative considering that the X-ray crystal structure of PAFR in complex with SR27417 antagonist has been reported recently. These calculations will provide the required information to predict the best properties for binding of the appropriate inhibitor and reschedule, if necessary, our synthetic strategy towards the final aim.
Moreover, these activities would enable research on this topic to improve and suggest more potent metal-based PAFR inhibitors in the future. Though the field is immature, the recent examples reported herein, including the TQ ruthenium series and other similar complexes, clearly demonstrate the progress of therapeutic strategies for the treatment of inflammatory-assisted diseases, thromboembolic disease and oxidative stress. This is a cutting-edge research field with a high public health impact due to the increased demands of our society for new compounds that act as potent therapeutic agents.
At this point, it is worth mentioning another critical dimension related to this topic. Recent studies on various diseases have demonstrated a beneficial effect, i.e., an increase in pharmacological action, upon co-administration of PAFR inhibitors along with the specific drugs required to treat each disease. This comes from the fact that elevated levels of PAF can be measured in response to almost every type of pathology where inflammation and cell damage/death are involved.51 Thus, for example, dual administration of PAFR inhibitors and specific anticancer agents towards certain tumor cells leads to an improved pharmacologic profile.69 This is an important issue considering that metal-based complexes constitute important drugs to treat inflammation-related diseases, such as cancer.
Finally, further research on the preparation of new metal-based complexes with improved pharmacological profiles is required to explore them in pre-clinical or clinical trials.
Author contributions
AP: conceptualization; investigation; writing – review & editing; CAD: investigation; writing; conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AIP gratefully acknowledges financial support from “The Special Research Account of the National and Kapodistrian University of Athens (NKUA)” (grant number 21207).References
- B. Rosenberg, E. Renshaw, L. Vancamp, J. Hartwick and J. Drobnik, J. Bacteriol., 1967, 93, 716 CrossRef CAS PubMed.
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- H. Madec, F. Figueiredo, K. Cariou, S. Roland, M. Sollogou and G. Gasser, Chem. Sci., 2023, 14, 409 RSC.
- E. Boros, P. J. Dyson and G. Gasser, Chem, 2020, 9, 641 Search PubMed.
- S. H. Van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089 Search PubMed.
- K. S. Lovejoy and S. J. Lippard, Dalton Trans., 2009, 10651 Search PubMed.
- M. Nagahvi, Lancet, 2015, 385, 117 Search PubMed.
- L. K. Jennings, Am. J. Cardiovasc. Dis., 2009, 103(Supplement), 4A CAS.
- M. M. Shifrin and S. B. Widmar, Nurs. Clin. North Am., 2016, 51, 29 CrossRef PubMed.
- T. W. Hambley, Science, 2007, 318, 1392 Search PubMed.
- C. S. Allardyce, A. Dorcier, C. Scolaro and P. J. Dyson, Appl. Organomet. Chem., 2005, 19, 1 Search PubMed.
- E. Meggers, Chem. Commun., 2009, 1001 Search PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3 Search PubMed.
- N. Farrell, Coord. Chem. Rev., 2002, 232, 1 CrossRef CAS.
- H. Gary, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3563 Search PubMed.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764 Search PubMed.
- S. Lippard and J. M. Berg, Curr. Opin. Chem. Biol., 2000, 4, 137 CrossRef CAS.
- A. Huseynov, J. Reinhardt, L. Chandra, D. Dürschmied and H. F. Langer, Int. J. Mol. Sci., 2023, 24, 6280 CrossRef CAS PubMed.
- B. Furie and B. C. Furie, N. Engl. J. Med., 2008, 359, 938 CrossRef CAS PubMed.
- D. Varga-Szabo, I. Pleines and B. Nieswandt, Arterioscler. Thromb. Vasc. Biol., 2008, 28, 403 CrossRef CAS PubMed.
- J. McFadyen, M. Schaff and K. Peter, Nat. Rev. Cardiol., 2018, 15, 181 CrossRef CAS PubMed.
- R. Hall and C. D. Mazer, Anesth. Analg., 2011, 112, 292 CrossRef CAS PubMed.
- C. Demopoulos, R. Pinckard and D. Hanahan, Biol. Chem., 1979, 254, 9355 Search PubMed.
- R. Lordan, A. Tsoupras, I. Zabetakis and C. A. Demopoulos, Molecules, 2019, 24, 4414 CrossRef CAS PubMed.
- V. D. Papakonstantinou, N. Lagopati, E. C. Tsilibary, C. A. Demopoulos and A. I. Philippopouloos, Bioinorg. Chem. Appl., 2017, 2017, 6947034 Search PubMed.
- A. B. Tsoupras, A. Papakyriakou, C. A. Demopoulos and A. I. Philippopoulos, J. Inorg. Biochem., 2013, 120, 63 CrossRef CAS PubMed.
- R. Lordan, A. Tsoupras and I. Zabetakis, Blood Rev., 2021, 45, 100694 CrossRef CAS PubMed.
- A. I. Philippopoulos, N. Tsantila, C. A. Demopoulos, C. P. Raptopoulou, V. Likodimos and P. Falaras, Polyhedron, 2009, 28, 3310 CrossRef CAS.
- P. Falaras and A. I. Philippopoulos, Platelet Activating Factor (PAF) Inhibitors with Possible Antitumor Activity, Industrial Property Organization of Greece Patent Number 1006959, 2009 Search PubMed.
- I. K. Hyland, R. F. O'Toole, J. A. Smith and A. C. Bissemberg, ChemMedChem, 2018, 13, 1873 CrossRef CAS PubMed.
- A. Margariti, V. D. Papakonstantinou, G. M. Stamatakis, C. A. Demopoulos, C. Machalia, E. Emmanouilidou, G. Schnakenburg, M.-C. Nika, N. S. Thomaidis and A. I. Philippopoulos, Molecules, 2023, 28, 6899 Search PubMed.
- M. Siller-Matula, R. Plasenzotti, A. Spiel, P. Quehenberger and B. Jilma, Thromb. Haemostasis, 2008, 100, 397 CrossRef.
- E. Ferentinos, A. B. Tsoupras, M. Roulia, S. D. Chatziefthimiou, C. A. Demopoulos and P. Kyritsis, Inorg. Chim. Acta, 2011, 378, 102 CrossRef CAS.
- A. B. Tsoupras, M. Roulia, E. Ferentinos, I. Stamatopoulos, C. A. Demopoulos and P. Kyritsis, Bioinorg. Chem. Appl., 2010, 731202 Search PubMed.
- A. Margariti, V. D. Papakonstantinou, G. M. Stamatakis, C. A. Demopoulos, G. Schnakenburg, A. K. Andreopoulou, P. Giannopoulos, J. K. Kallitsis and A. I. Philippopoulos, Polyhedron, 2020, 178, 114336 Search PubMed.
- J. R. Fletcher, A. G. DiSimone and M. A. Earnest, Ann. Surg., 1990, 211, 312 CAS.
- A. Kalampalidis, Synthesis and characterization of rhodium complexes incorporating substituted thienopyrimidine ligands. Study of their potent biological activities, Doctoral Dissertation, National and Kapodistrian University of Athens, Athens, Greece, 2021. Persistent URL: https://pergamos.lib.uoa.gr/uoa/dl/object/2936659.A Search PubMed.
- A. Kalampalidis, A. Peppas, G. Schnakenburg, A. Papakyriakoy, A. Tsoupras, I. Zabetakis and A. I. Philippopoulos, Appl. Organomet. Chem., 2021, 35, e6210 CrossRef CAS.
- D. Ravishankar, M. Salamah, A. Attina, R. Pothi, T. M. Vallance, M. Javed, H. F. Williams, E. M. S. Alzahranil, E. Kabove, R. Vaiyapuri, K. Shankland, J. Gebbins, K. Strohfeldt, F. Greco, H. M. I. Osborn and S. Vaiyapuri, Sci. Rep., 2017, 7, 5738 CrossRef PubMed.
- A. B. Tsoupras, V. Papakonstantinou, G. M. Stamatakis, C. A. Demopoulos, P. Falaras and A. I. Philippopoulos, Sci. Lett. J., 2015, 4, 208 Search PubMed.
- M. Merlos, M. Giral, D. Balsa, R. Ferrando, M. Queralt, A. Puigdemont, J. García-Rafanell and J. Forn, J. Pharmacol. Exp. Ther., 1997, 280, 114 CrossRef CAS PubMed.
- A. Skoczynska, A. Lewinski, M. Pokora, P. Paneth and E. Budzisz, Int. J. Mol. Sci., 2023, 24, 9512 CrossRef CAS PubMed.
- K. Lin, Z. Z. Zhao, H. B. Bo, X. J. Hao and J. Q. Wang, Front. Pharmacol., 2018, 9, 1323 CrossRef CAS PubMed.
- M. Kaplanis, G. Stamatakis, V. D. Papakonstantinou, M. Paravatou-Petsotas, C. A. Demopoulos and C. A. Mitsopoulou, J. Inorg. Biochem., 2014, 135, 1 CrossRef CAS PubMed.
- E. Sioriki, R. Lordan, F. Nahra, K. van Hecke, I. Zabetakis and S. P. Nolan, ChemMedChem, 2018, 13, 2484 Search PubMed.
- A. Tsoupras, S. Pafli, C. Stylianoudakis, K. Ladomenou, C. A. Demopoulos and A. I. Philippopoulos, Compounds, 2024, 4, 376 CrossRef CAS.
- A. Kalampalidis, A. Damati, D. Matthopoulos, A. B. Tsoupras, C. A. Demopoulos, G. Schnakenburg and A. I. Philippopoulos, Molecules, 2023, 28, 1859 CrossRef CAS PubMed.
- A. Peppas, A. Kalabalidis, V. D. Papakonstantinou, C. A. Demopoulos, G. Schnakenburg and A. I. Philippopoulos, Rhodium-based Inhibitors of the Platelet Activating Factor (PAF). A new class of potent anti-inflammatory drugs, in Chemical Elements (Fluorine, Rhodium and Rubidium). Properties, Synthesis and Applications, ed. A. Huff, Nova Science Publishers, New York, 2018, pp. 127–169 Search PubMed.
- M. Sohrabi, M. Saeedi, B. Larijani and M. Mahdavi, Eur. J. Med. Chem., 2021, 216, 113308 CrossRef CAS PubMed.
- C. Cao, Q. Tan, C. Xu, L. He, L. Yang, Y. Zhou, Y. Zhou, A. Qiao, M. Lu, C. Yi, G. W. Han, X. Wang, X. Li, H. Yang, Z. Rao, H. Jiang, Y. Zhao, J. Liu, R. C. Stevens, Q. Zhao, X. C. Zhang and B. Wu, Nat. Struct. Mol. Biol., 2018, 25, 488 CrossRef CAS PubMed.
- J. B. Travers, J. G. Rohan and R. P. Sahu, Front. Endocrinol., 2021, 12, 624132 CrossRef PubMed.
- C. H. Yang, C. W. Hsia, T. Jayakumar, J. R. Sheu Sheu, C. H. Hsia, T. Khamrang, Y. J. Chen, M. Manubolu and Y. Chang, Int. J. Mol. Sci., 2018, 19, 3641 Search PubMed.
- K. C. Hung, C. H. Hsia, C. Y. Hsieh, M. Velusamy, T. Jayakumar and J. R. Sheu, Int. J. Mol. Sci., 2017, 18, 916 Search PubMed.
- C. H. Hsia, M. Velusamy, J. R. Sheu, T. Khamrang, T. Jayakumar, W. J. Lu, K. H. Lin and C. C. Chang, Sci. Rep., 2017, 7, 9556 CrossRef PubMed.
- C. H. Hsia, T. Jayakumar, J. R. Sheu, C. W. Hsia, W. C. Huang, M. Velusamy and L. M. Lien, J. Clin. Med., 2020, 9, 996 CrossRef CAS PubMed.
- C. H. Hsia, T. Jayakumar, J. R. Sheu, S. Y. Tsao, M. Velusamy, C. W. Hsia, D. S. Chou, C. C. Chang, C. L. Chung, T. Khamrang and K. C. Lin, Molecules, 2018, 23, 477 CrossRef PubMed.
- A. Tsoupras, T. Adamantidi, M. A. Finos, A. Philippopoulos, P. Detopoulou, I. Tsopoki, M. Kynatidou and C. A. Demopoulos, Front. Biosci., Landmark Ed., 2024, 29, 345 CrossRef PubMed.
- C. H. Leung, S. Li, H. J. Zhong and D. L. Ma, Chem. Sci., 2015, 6, 871 RSC.
- Z. H. Chohan, M. S. Iqbal, H. S. Iqbal, A. Scozzafava and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2002, 17, 87 CrossRef CAS PubMed.
- R. Ross, N. Engl. J. Med., 1999, 340, 115 CrossRef CAS PubMed.
- A. B. Tsoupras, C. Iatrou, C. Frangia and C. A. Demopoulos, Infect. Disord.:Drug Targets, 2009, 9, 390 CAS.
- T. Jayakumar, J. R. Sheu, C. W. Hsia, P. S. Bhavan and C. C. Chang, Appl. Sci., 2021, 11, 10092 CrossRef CAS.
- T. Jayakumar, H. C. Huang, C. W. Hsia, T. H. Fong, T. Khamrang, M. Velusamy, M. Manubolu, J. R. Sheu and C. H. Hsia, Bioorg. Chem., 2020, 96, 103639 CrossRef CAS PubMed.
- C. H. Hsia, M. Velusamy, T. Jayakumar, Y. J. Chen, C. W. Hsia, J. H. Tsai, R. D. Teng and J. R. Sheu, Cells, 2018, 7, 217 CrossRef CAS PubMed.
- J. Zhang, A. Pitto-Barry, L. Shang and N. P. E. Barry, R. Soc. Open Sci., 2017, 4, 170786 CrossRef PubMed.
- E. Pontiki, D. Hadjipavlou-Litina and A. T. Chaviara, J. Enzyme Inhib. Med. Chem., 2008, 23, 1011 CrossRef CAS PubMed.
- S. Qurat-Ul-Ain, M. Pervaiz, A. Majid, U. Younas, Z. Saeed, A. Ashraf, R. R. M. Khan, S. Ullah, F. Ali and S. Jelani, J. Coord. Chem., 2023, 76, 1094 CrossRef.
- Y. Song, Q. You and X. Chen, Adv. Mater., 2023, 35, 2212102 CrossRef CAS PubMed.
- I. A. da Silva Junior, S. C. Stone, R. M. Rossetti, S. Jancar and A. P. Lepique, J. Immunol. Res., 2017, 2017, 5482768 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |