
A bi-functional S-scheme cobalt-porphyrin conjugated polymer/C3N4 heterojunction for cooperative CO2 reduction and tetracycline degradation†
Chao
Xu
a,
Shien
Guo
 *b,
Jiaxin
Wang
a,
Yiqing
Jiang
a,
Xiaomin
Wu
a,
Dandan
Lin
a and
Yuting
Xiao
*a
*b,
Jiaxin
Wang
a,
Yiqing
Jiang
a,
Xiaomin
Wu
a,
Dandan
Lin
a and
Yuting
Xiao
*a
aKey Laboratory of Jiangxi Province for Persistent Pollutants Prevention Control and Resources Reuse, Nanchang Hangkong University, Nanchang 330063, Republic of China. E-mail: yutingxiao9@sina.com
bInstitute of Advanced Materials (IAM), College of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang 330022, China. E-mail: guoshien@jxnu.edu.cn
First published on 3rd March 2025
Abstract
The design of bifunctional photocatalysts for the removal of contaminants and the reduction of CO2 is of significant practical importance in addressing pollution and energy challenges. However, the photocatalytic efficiency is limited by the inadequate redox ability, high carrier recombination rate, and insufficient reactive sites of existing photocatalysts. Herein, a 2D/2D S-scheme heterojunction composed of cobalt-porphyrin conjugated polymer nanoflakes and C3N4 nanosheets (CoPor-DBE/CN) was rationally synthesized, exhibiting matched redox ability and favorable CO2 adsorption properties. The layered structure and functional groups of CoPor-DBE/CN provide numerous active sites, thereby enhancing the separation and transfer of charge carriers as well as the adsorption of reactants. Under visible light illumination, the optimized 50CoPor-DBE/CN hybrid achieved a CO production rate of 16.7 μmol g−1 h−1 and a tetracycline removal rate of 93.8%, which are significantly higher than those of the individual CN material. By employing X-ray photoelectron spectroscopy (XPS), ultraviolet photoelectron spectroscopy (UPS), and photo-irradiated Kelvin probe force microscopy (KPFM), we demonstrate that the transfer of charge carriers within the CoPor-DBE/CN system follows the S-scheme heterojunction mechanism. This work offers a promising blueprint for the design of multifunctional S-scheme photocatalysts aimed at the simultaneous efficient reduction of CO2 and degradation of organic pollutants.
Introduction
Environmental pollution and energy shortages have significantly impacted people's lives, highlighting the necessity to explore affordable technologies that address environmental issues while generating energy without causing secondary pollution.1–3 Recently, photocatalysis technology has emerged as a promising and sustainable solution to meet current demands.4–6 In particular, photocatalytic CO2 reduction serves not only to produce solar fuels but also to alleviate the greenhouse effect, positioning it as an effective strategy for tackling the pressing environmental and energy dilemmas.7–10 At present, a majority of researchers concentrate chiefly on the reductive half-reaction of CO2 photoreduction, often overlooking its oxidative counterpart. While the innovative coupling of photo-redox organic oxidation synthesis with CO2 reduction has been acknowledged as an attractive remedy for such challenges,11–13 it can potentially generate secondary pollution due to its by-products and remaining organic solvents. In recent years, wastewater containing antibiotics, particularly tetracycline (TC), has posed significant risks to human health and the ecosystem.14,15 Recently, researchers have developed various techniques, including adsorption,16 microwave catalysis,17 and photocatalytic degradation,18 to eliminate antibiotics from contaminated water. Among these methods, the photocatalytic degradation of TC has emerged as a promising solution, attributed to its high efficiency, economic feasibility, and simplicity in operation. By altering the oxidative half-reaction to organic pollutant decomposition, an ideal integration of CO2 photoreduction and pollutant degradation has been proposed.19,20 This strategy aims to achieve a mutually beneficial outcome for both energy production and environmental sustainability, while also ensuring the complete utilization of photoelectrons and holes.To achieve this synergistic coupled reaction system, it is crucial to design effective photocatalysts that exhibit strong redox capabilities and high efficiency in charge separation. Conjugated porous organic polymers represent an attractive platform for photocatalysis due to their broad light absorption, good structural regularity, and design possibilities.21–23 In particular, metalloporphyrin-based conjugated polymers have great potential in CO2 photocatalytic reduction due to their advantages in CO2 adsorption and activation.24–26 Fang et al.27 synthesized three porphyrin-based covalent organic frameworks by polymerizing triptycene and porphyrin; JUC-640-Co exhibits an excellent CO production rate and selectivity. However, such photocatalytic systems often require the addition of sacrificial reagents, including ascorbic acid, triethylamine, isopropanol, and triethanolamine. The underlying reasons for this requirement are primarily related to the shallow valence band (VB) and the inadequate oxidation capacity.
One of the most effective approaches for addressing the aforementioned photocatalytic challenges is to construct an appropriate step-scheme (S-scheme) heterojunction system.28–30 The S-scheme heterojunction primarily comprises a reduction photocatalyst (RP) and an oxidation photocatalyst (OP).31 In an S-scheme system, the photoinduced electrons from the OP, which has a limited reduction capability, can recombine with the photoinduced holes from the RP, which has a limited oxidation capability, at the interface. Consequently, the S-scheme heterojunction facilitates not only more efficient separation and transfer of photoinduced charge carriers but also preserves their maximum redox capability, thereby thermodynamically benefiting photocatalytic reactions.32,33 Recently, several novel S-scheme heterojunctions have been developed, including ZnIn2S4@CdS,34 Bi2MoO6−x/MoS2,35 and Bi2WO6/NiO,36 which exhibit exceptional photoactivity for CO2 reduction coupled with the oxidation of organic pollutants. Metalloporphyrin-based conjugated polymers are considered as promising RPs due to their potential in CO2 conversion, which can form S-scheme heterojunctions with a suitable OP. Carbon nitride (C3N4) is an intriguing 2D visible-light-active photocatalyst known for its outstanding electronic properties, low cost, and thermodynamic stability.37–39 It has been extensively explored in various photocatalytic applications and is considered an ideal candidate as an OP. Motivated by the remarkable properties of metalloporphyrin-based conjugated polymers and C3N4, we aim to construct an S-scheme heterostructure photocatalyst by integrating 2D lamellar C3N4 with metalloporphyrin-based conjugated polymer nanosheets. The resulting composite is expected to harness the advantages of S-scheme and 2D/2D heterojunctions, leading to a synergistic effect that optimizes photocatalytic performance for CO2 reduction coupled with contaminant degradation.
Herein, a unique 2D/2D S-scheme heterojunction composed of a cobalt-porphyrin conjugated polymer and C3N4 (CoPor-DBE/CN) was designed, in which the CoPor-DBE nanoflakes serve as the CO2 reduction centre, while the CN nanosheets synergistically function as a TC oxidation booster. The well-structured interface between the CoPor-DBE and CN nanosheets facilitated interfacial charge transfer, while the S-scheme heterojunction maintained a strong redox capability. This combination endowed the CoPor-DBE/CN heterojunction with significantly enhanced photocatalytic performance. Under visible light illumination, the optimized 50CoPor-DBE/CN heterojunction achieved a TC removal rate of 93.8%, along with a CO production rate of 16.7 μmol g−1 h−1. Based on the experimental results and characterization, the mechanism of the cooperative effect was studied in detail.
Experimental section
Preparation of C3N4 (CN) nanosheets
1 g of melamine and 1.2 g of phosphoric acid were dissolved in 100 mL of deionized water. The resulting solution was placed in a water bath at a constant temperature of 80 °C and stirred rigorously for one hour. Afterward, the solution was transferred to a polytetrafluoroethylene autoclave and heated to 180 °C for 10 hours. At the end of this period, the mixture was subjected to centrifugation, washed with deionized water, and dried at 60 °C to obtain the precursor. Subsequently, 0.8 grams of the precursor were combined with a mixed solution of 5 mL of glycerol and 15 mL of ethanol, and the mixture was refluxed at 90 °C for three hours. After this reflux process, the precipitate was washed three times with ethanol and dried at 60 °C. Lastly, the precipitate was heated in a muffle furnace at 520 °C for 4 hours with the temperature raised at a rate of 2 °C min−1.Preparation of the cobalt-porphyrin conjugated polymer (CoPor-DBE)
A total of 134 mg of trans-4,4′-dibromostilbene, 152 mg of Co(II)-5,10,15,20-tetrakis(4-ethynylphenyl)porphyrin, 3 mL of diisopropylamine, and 30 mL of chloroform were placed together in a 100 mL round-bottom flask. The resulting mixture was subjected to argon replacement, followed by 10-minute sonication and degassing via three freeze–thaw cycles. After allowing it to stir at room temperature for 12 hours, 9.2 mg of tetrakis (triphenylphosphine) palladium and 2.3 mg of copper iodide were incorporated into the flask and the mixture was heated at 80 °C under an argon atmosphere for 36 hours. The crude product was subsequently washed in sequence with toluene, 1,4-dioxane, dichloromethane, and methanol, and then extracted using a Soxhlet apparatus with dichloromethane over the course of three days. Finally, the product was dried in a vacuum oven at 60 °C, producing CoPor-DBE.Preparation of the CoPor-DBE/CN heterojunction
In a typical procedure, 100 mg of CN was mixed with an appropriate weight ratio of CoPor-CP in 150 mL of ethanol. The resulting suspension was sonicated for one hour and subsequently heated to 80 °C with stirring until complete evaporation of the solvent occurred. The amount of CoPor-DBE added was adjusted to 30, 40, 50, and 60 mg in combination with 100 mg of CN, and the resulting products are denoted as 30CoPor-DBE/CN, 40CoPor-DBE/CN, 50CoPor-DBE/CN, and 60CoPor-DBE/CN.Photocatalytic performance test
The coupling of CO2 conversion with the decomposition of tetracycline via photoredox processes was examined using a CO2 reaction setup (PerfectLight Labsolar 6A). A 300 W xenon lamp (PLS-SME300) fitted with a 400 nm UV cut filter served as the light source. The power density of the light source was determined to be 65.2 mW cm−2. Generally, 20 mg of the sample was added to 100 mL of tetracycline solution (20 mg L−1) and subjected to sonication for 10 minutes. The reactor and associated reaction apparatus were then assembled under vacuum conditions. Ultra-pure CO2 was subsequently introduced into the reaction solution as the reactant gas. The reaction was maintained at a temperature of 15 °C by circulating condensate water. Gas products were analyzed utilizing a FuLi GC9790II gas chromatograph. Following this, the liquid phase was extracted and centrifuged to yield the supernatant, which was analyzed using a Hitachi U-3310 UV-visible spectrophotometer.Results and discussion
The CoPor-DBE/CN heterojunction was fabricated by a facile self-assembly method, as shown in Fig. 1a. The morphologies and detailed microstructures of the as-prepared samples were elucidated using transmission electron microscopy (TEM) and atomic force microscopy (AFM). The TEM image of CN reveals a smooth lamellar morphology embedded with numerous nanoscale pores (Fig. S1†). CoPor-DBE exhibits a stacked nanoflake structure with lateral dimensions of 40–60 nm (Fig. S2†). The TEM image of 50CoPor-DBE/CN (Fig. 1b) reveals that dense CoPor-DBE flakes cover the surface of the layered CN. Furthermore, the magnified TEM image (Fig. 1c) clearly shows distinct interfaces between the two components. This close interface is anticipated to enhance charge separation during the photocatalytic process. Additionally, the AFM image of 50CoPor-DBE/CN illustrates a well-defined sheet-stacked structure (Fig. S3†), where CoPor-DBE and CN can be differentiated by their respective thicknesses in the height cutaway view. The thicknesses of CoPor-DBE and CN are approximately 9.2 nm and 2.4 nm, respectively. Additionally, energy-dispersive X-ray analysis (EDX) elemental mapping images indicate that C, N, and Co are uniformly distributed throughout the CoPor-DBE/CN hybrid (Fig. 1d), confirming the successful synthesis of the heterojunction. | ||
| Fig. 1 (a) Schematic diagram of the 2D/2D CoPor-DBE/CN heterojunction prepared via a self-assembly process. (b) TEM image of 50CoPor-DBE/CN and (c) enlarged TEM image of the selected area in (b). (d) EDX elemental mapping images of the 50CoPor-DBE/CN composite. | ||
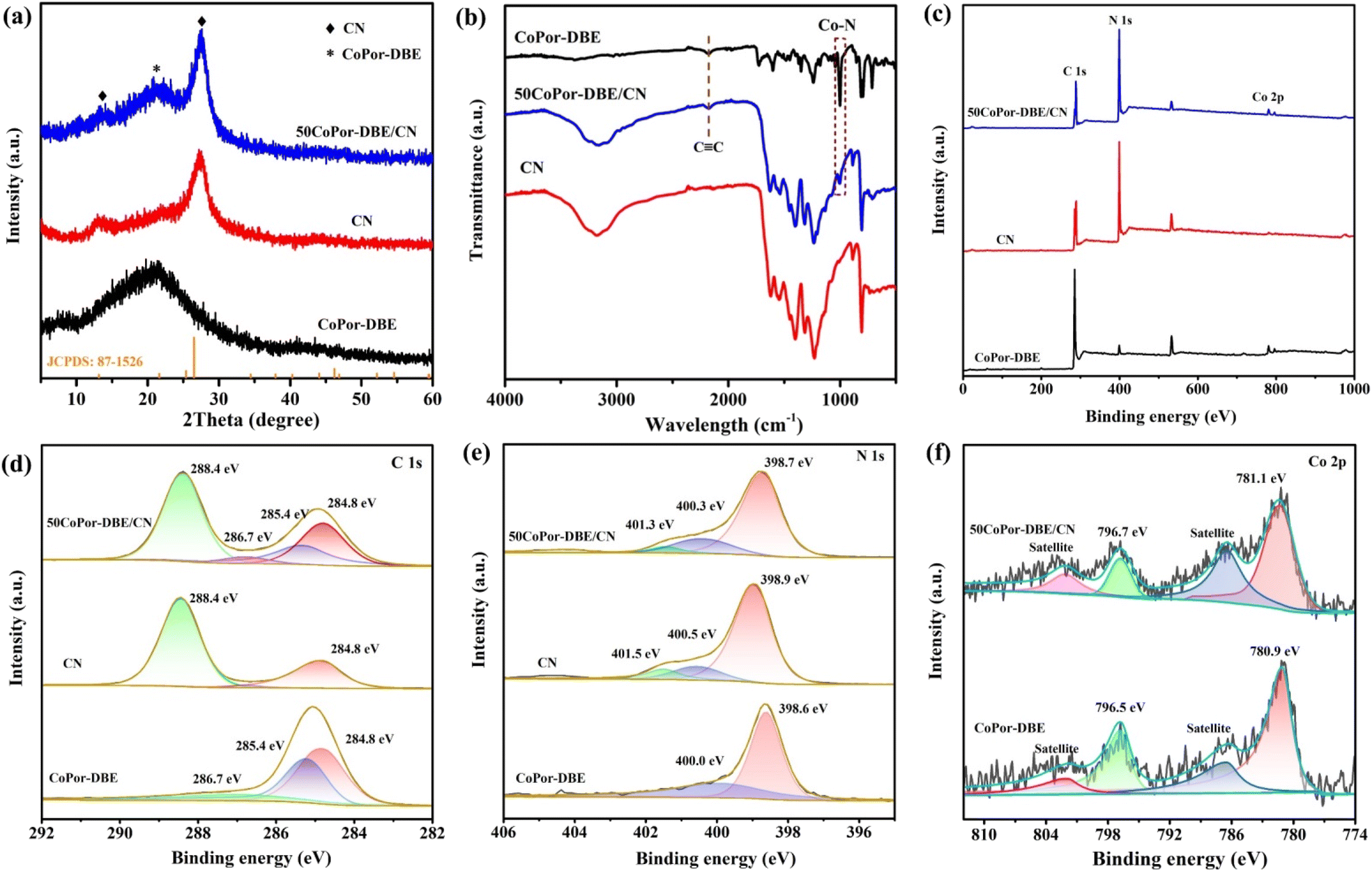
The structures of the CoPor-DBE/CN composites were clearly confirmed by X-ray diffraction (XRD) patterns and Fourier transform infrared (FT-IR) spectra. The XRD pattern of CoPor-DBE exhibits a broad diffraction peak at approximately 20.8°, indicating its amorphous characteristics alongside a partially stacked structure (Fig. 2a).40 CN displays two distinct diffraction peaks located at 13.1° and 27.4°, which correspond to the (100) crystal lattices originating from interlayer stacking and the (002) crystal lattices attributed to the in-plane tri-s-triazine units, respectively. The diffraction peaks of the 50CoPor-DBE/CN composites are well matched with those of individual CoPor-DBE and CN components. As shown in the FT-IR spectra (Fig. 2b), CoPor-DBE exhibited characteristic signals corresponding to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching vibration bands at 2192 cm−1.41 Furthermore, the N–H stretching bands in the region of 3100–3400 cm−1 are significantly attenuated, while a new characteristic peak for Co–N vibration at 1008 cm−1 was observed.42 This indicates that Co ion was successfully coordinated to the porphyrin ring. The chemical structure of CoPor-DBE was further validated by 13C NMR spectroscopy (Fig. S4†). The representative signals for alkynyl (–CC–) groups are observed at 80.2 ppm and 83.2 ppm, while the signal for vinyl (–C
C stretching vibration bands at 2192 cm−1.41 Furthermore, the N–H stretching bands in the region of 3100–3400 cm−1 are significantly attenuated, while a new characteristic peak for Co–N vibration at 1008 cm−1 was observed.42 This indicates that Co ion was successfully coordinated to the porphyrin ring. The chemical structure of CoPor-DBE was further validated by 13C NMR spectroscopy (Fig. S4†). The representative signals for alkynyl (–CC–) groups are observed at 80.2 ppm and 83.2 ppm, while the signal for vinyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–) appears at 125.6 ppm.43 In the FT-IR spectrum of CN, the prominent absorption peak at 810 cm−1 is attributed to the breathing vibration mode of the heptazine ring. In addition, a series of absorption peaks between 1100 and 1700 cm−1 correspond to the typical stretching mode of aromatic heterocyclic skeletons. The broadband absorption between 3000 and 3500 cm−1 is attributed to the stretching vibrations of the amine group and the hydroxyl group. As for the 50CoPor-DBE/CN composite, the characteristic vibrational bands of both CoPor-DBE and CN can be observed, indicating that the hybrid photocatalyst has been successfully prepared.
C–) appears at 125.6 ppm.43 In the FT-IR spectrum of CN, the prominent absorption peak at 810 cm−1 is attributed to the breathing vibration mode of the heptazine ring. In addition, a series of absorption peaks between 1100 and 1700 cm−1 correspond to the typical stretching mode of aromatic heterocyclic skeletons. The broadband absorption between 3000 and 3500 cm−1 is attributed to the stretching vibrations of the amine group and the hydroxyl group. As for the 50CoPor-DBE/CN composite, the characteristic vibrational bands of both CoPor-DBE and CN can be observed, indicating that the hybrid photocatalyst has been successfully prepared.
 | ||
| Fig. 2 (a) XRD profiles, (b) FTIR spectra and (c) XPS survey spectra of CoPor-DBE, CN and the 50CoPor-DBE/CN composite. (c) High-resolution C 1s spectra of CoPor-DBE, CN and 50CoPor-DBE/CN, (d) N 1s spectra of CN and 50CoPor-DBE/CN, and (e) Co 2p spectra of CoPor-DBE and 50CoPor-DBE/CN. | ||
The surface chemical states of the samples were analyzed using X-ray photoelectron spectroscopy (XPS). The XPS survey spectrum of 50CoPor-DBE/CN confirms the co-existence of C, N, and Co elements (Fig. 2c), which is consistent with the results from EDX elemental mapping. As shown in Fig. 2d, the C 1s region of CN displays two peaks at 284.8 eV and 288.4 eV, corresponding to the adventitious hydrocarbon and N–CN coordination, respectively. In the case of 50CoPor-DBE/CN, the deconvolution of the high-resolution C 1s spectrum reveals four peaks at 284.8 eV, 285.4 eV, 286.7 eV, and 288.4 eV, which are assignable to the bonding of CC, CC, C–N, and N–CN, respectively.44,45 This confirms the successful preparation of the CoPor-DBE/CN hybrid. The N 1s spectra of CoPor-DBE show the peaks of pyrrole nitrogen at 398.6 eV and Co–N coordination at 400.0 eV, respectively. The high-resolution N 1s spectrum of CN can be deconvoluted into three peaks at 398.9, 400.5, and 401.5 eV, which correspond to nitrogen species in the C–NC, N–(C)3, and NHx groups of melon (Fig. 2e). In comparison with pure CN, the N 1s binding energies of the 50CoPor-DBE/CN composite are shifted by 0.2 eV to lower energies, suggesting the migration of electrons from CoPor-DBE to CN.46,47 As shown in Fig. 2f, the binding energy signal peaks for Co 2p1/2 and Co 2p3/2 were observed at 796.5 eV and 780.9 eV, respectively, in CoPor-DBE, accompanied by two satellite peaks at 786.7 eV and 802.1 eV, indicating the +2 state of Co species in CoPor-DBE.48 In the case of 50CoPor-DBE/CN, the binding energy of Co 2p increased by 0.2 eV compared to that in CoPor-DBE, suggesting a reduction in the electron density of the Co atoms. These shifts in the peaks demonstrate that a strong chemical interaction and intimate contact have formed between CoPor-DBE and CN within the 2D/2D heterostructure.
The specific surface areas and porosities of CN, CoPor-DBE, and 50CoPor-DBE/CN were investigated using N2 sorption measurements at 77 K, with the results summarized in Table S1.† As illustrated in Fig. S5,† CoPor-DBE exhibits a type IV adsorption–desorption isotherm, while CN displays a type II hysteresis loop, indicating the presence of microporous and mesoporous structures,49,50 respectively. The Brunauer–Emmett–Teller surface areas of CoPor-DBE, CN, and the 50CoPor-DBE/CN composite were calculated to be 442.34, 51.49, and 105.83 m2 g−1, respectively. In comparison with CoPor-DBE, the decrease in the specific surface area of the 50CoPor-DBE/CN composite can be attributed to the stacking of CoPor-DBE plates and CN nanosheets (Table S1†), which further illustrates the strong interaction between the two components.51 The pore size was also determined, and two pore sizes were observed in the 50CoPor-DBE/CN composite with pore diameters of approximately 1.2 nm and 2.5 nm, respectively. This porous structure is not only beneficial for the mass transfer of reactants and product molecules in the 2D/2D CoPor-DBE/CN heterojunction, but also provides more surface reaction sites, thereby improving photocatalytic redox reactions.
The UV–vis absorption spectra were recorded to examine the light absorption capabilities of the materials. As shown in Fig. 3a, the absorption edges for CN and CoPor-DBE are approximately 456 nm and 838 nm, respectively, indicating their excellent ability to harvest visible light. The 50CoPor-DBE/CN heterojunction samples exhibited intermediate absorption behavior, which may have resulted from the strong electronic coupling between CoPor-DBE and CN. Furthermore, the band gap (Eg) of the samples was determined using the Tauc diagram and calculated with the equation (αhν)2 = A(hν − Eg),52 where α, hν, A, and Eg represent the absorption coefficient, the photon energy, a constant, and the band gap, respectively. Consequently, the Eg values for CN and the CoPor-DBE nanosheets were estimated to be 2.72 eV and 1.48 eV, respectively. In order to elucidate the energy band structures of CN and CoPor-DBE, the flat band potentials of the two materials were analyzed using the Mott–Schottky diagram. As shown in Fig. 3b and c, the Mott–Schottky slopes of both CN and CoPor-DBE are positive, confirming that both samples are n-type semiconductors. The flat band potential values for CoPor-DBE and CN are −0.86 V and −0.52 V relative to the normal hydrogen electrode (NHE), respectively. For n-type semiconductors, the conduction band (CB) edge is approximately equal to the flat band potential.53 Thus, the CB potentials (ECB) for CoPor-DBE and CN were determined to be −0.86 V and −0.52 V vs. NHE, respectively. Additionally, based on the equation EVB = ECB + Eg, the valence band edge (EVB) for CoPor-DBE and CN were calculated to be 0.62 eV and 2.20 eV, respectively. As illustrated in Fig. 3d, the energy band structures of CoPor-DBE and CN exhibit a staggered band diagram, which is essential for the construction of an S-scheme heterojunction.
 | ||
| Fig. 3 (a) UV–vis–NIR differential reflectance spectra and (inset: Tauc plot) of CoPor-DBE, CN and 50CoPor-DBE/CN. Mott–Schottky plots of (b) CoPor-DBE and (c) CN. (d) Energy band structures of CoPor-DBE and CN. | ||
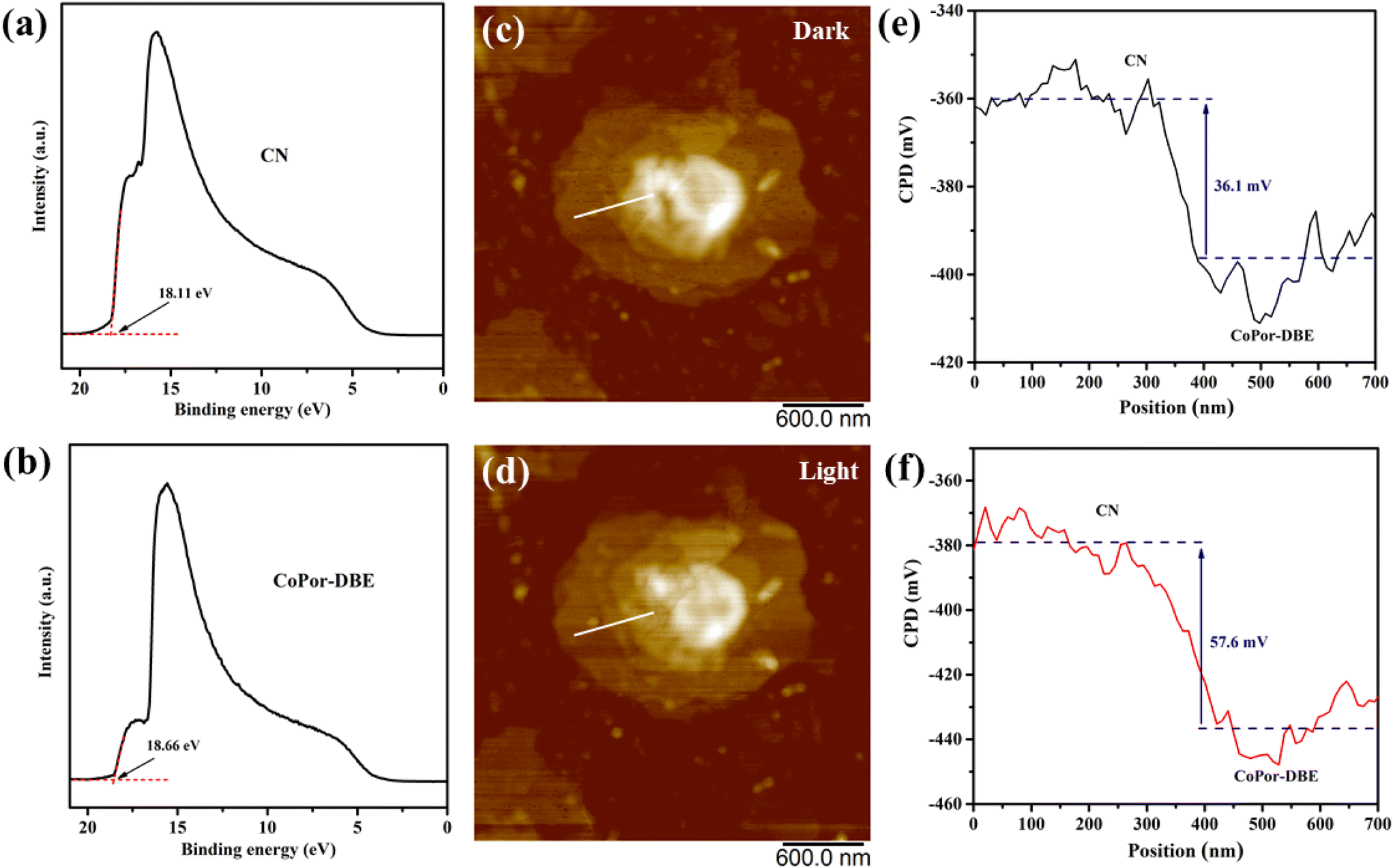
To investigate the electron-transfer pathway in the CoPor-DBE/CN heterojunction, the work functions (Φ) of both the CoPor-DBE and CN materials were analyzed using ultraviolet electron spectroscopy (UPS). As shown in Fig. 4a and b, the secondary electron cutoff edge (Ecutoff) for CN and CoPor-DBE was determined to be 18.11 eV and 18.66 eV, respectively. The corresponding work function can be calculated using the formula Φ = hv − Ecutoff, where hv represents the He I photoelectron energy, with a value of 21.22 eV. As a result, the calculated Φ values are 3.11 eV for CN and 2.56 eV for CoPor-DBE. The higher Φ value of CN indicates that it possesses lower Fermi levels compared to CoPor-DBE. Consequently, when CoPor-DBE and CN are brought into contact to form the CoPor-DBE/CN heterojunction, electrons spontaneously transfer from CoPor-DBE through the interface to CN until their Fermi levels reach equilibrium (Fig. S6†). This process establishes an internal electric field directed from CoPor-DBE to CN in the interface region. As the charge density of CoPor-DBE decreases and the charge density of CN increases, the energy band edges of CoPor-DBE and CN bend upward and downward, respectively. Under illumination, the photogenerated electrons in the CB of CN rapidly recombine with the holes in the VB of CoPor-DBE driven by the built-in electric field, energy band bending, and electrostatic interactions. This recombination eliminates electrons and holes with low redox abilities while retaining those with high redox abilities. This unique S-scheme charge transfer mechanism not only enhances the charge separation of the CoPor-DBE/CN heterojunction but also preserves its exceptional redox capacity for subsequent photocatalytic reactions.
 | ||
| Fig. 4 UPS spectra of (a) CoPor-DBE and (b) CN. KPFM spectra of the 50CoPor-DBE/CN heterojunction under (c) darkness and (d) light irradiation; (e) and (f) the corresponding line-scanning surface potentials. | ||
In situ Kelvin probe force microscopy (KPFM) was utilized to investigate the surface potential distribution of the sample, thereby elucidating the charge transfer route in the CoPor-DBE/CN heterojunction. Fig. 4c and d present the topographic images of the 50CoPor-DBE/CN heterojunction in darkness and under light, respectively, while the corresponding line scan diagram illustrating the surface potential difference between CN and CoPor-DBE is shown in Fig. 4e and f. Prior to illumination, the surface potential of CoPor-DBE is approximately 36.1 mV lower than that of CN. Under light irradiation, the potential difference between CoPor-DBE and CN significantly increases to 57.6 mV. This observation indicates that photoinduced electrons and holes are predominantly concentrated on the surfaces of CoPor-DBE and CN, respectively. This phenomenon arises from the built-in electric field in the S-scheme heterojunction, which extracts photoinduced holes from CoPor-DBE and subsequently quenches them with photoinduced electrons from CN within the depletion layer.54 This result demonstrated an S-scheme charge transfer mechanism in the CoPor-DBE/CN heterojunction.
The S-scheme charge transfer pathway in the CoPor-DBE/CN heterojunction was further validated through ESR spin-trapping experiments. As shown in Fig. S7a,† distinct characteristic peaks for DMPO-˙O2− were observed in all samples, clearly indicating the sufficient reduction ability of the photogenerated electrons to convert O2 into ˙O2− species. Notably, the 50CoPor-DBE/CN heterojunction exhibited the most pronounced signals, contrasting sharply with the significantly weaker signals observed in the individual CoPor-DBE and CN samples. These results strongly suggest a notable accumulation of photogenerated electrons within the 50CoPor-DBE/CN heterojunction. The ESR spectra of DMPO-˙OH (Fig. S7b†) revealed that both CN and 50CoPor-DBE/CN presented signals associated with the DMPO-˙OH species, while CoPor-DBE did not exhibit any detectable signal. This absence of the DMPO-˙OH signal in CoPor-DBE can be attributed to the low oxidation potential of the photoexcited holes. Significantly, the signals for DMPO-˙O2− and DMPO-˙OH in the 50CoPor-DBE/CN heterojunction were notably greater than those from their individual counterparts, further emphasizing the enhanced accumulation of both electrons and holes in this heterojunction. Thus, the results conclusively confirm that the photoexcited electrons transferred from the CB of CN to the VB of CoPor-DBE in the CoPor-DBE/CN heterojunction following an S-scheme charge-transfer mechanism.
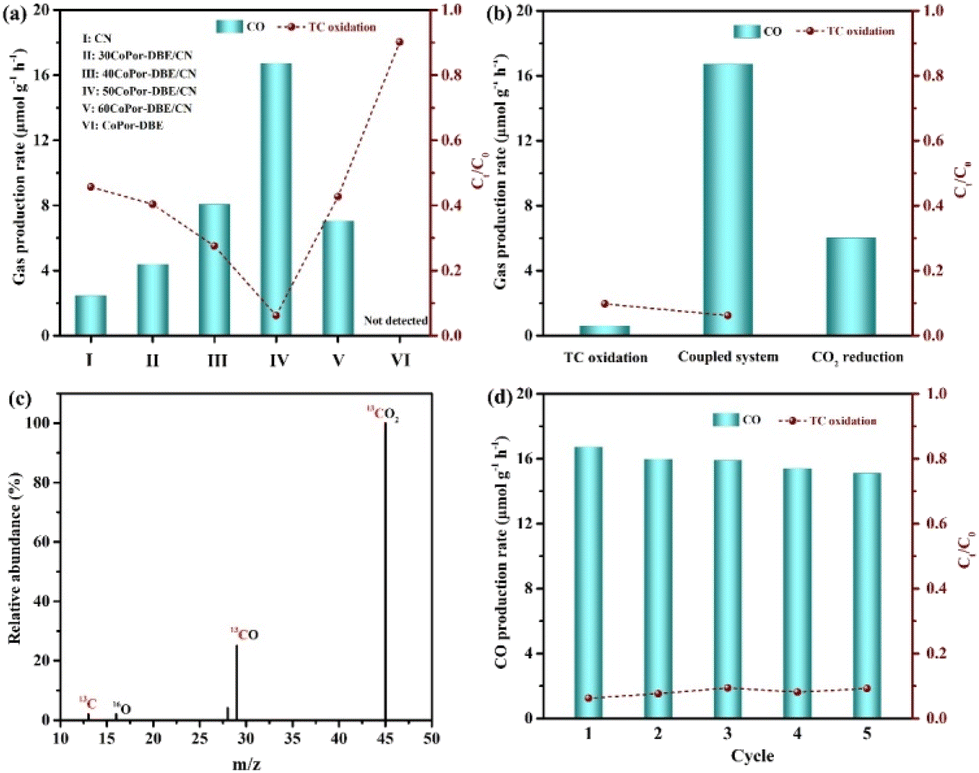
To investigate the photocatalytic performance of the synthesized materials, we conducted CO2 photoreduction coupled with TC degradation in a cooperative catalytic system under visible light irradiation (Fig. S8†). For all evaluated photocatalysts, CO was detected as the primary CO2 reduction product during the photocatalytic reaction. As illustrated in Fig. 5a, pristine CoPor-DBE exhibited low photocatalytic activity toward TC degradation, achieving a removal efficiency of only 10.5%, attributed to its insufficient oxidation capacity. CN exhibited moderate CO2 photoreduction activity with a CO evolution rate of 2.3 μmol g−1 h−1 and a TC removal efficiency of 54.6%. The formation of the 2D/2D CoPor-DBE/CN heterojunction significantly enhanced the visible light-driven photoredox performance. The photocatalytic activity of CoPor-DBE/CN progressively improved with increasing contents of CoPor-DBE. Notably, the optimal sample (50CoPor-DBE/CN) exhibited the highest performance, yielding 16.7 μmol g−1 h−1 of CO and achieving a TC degradation efficiency of 93.8%, which is comparable to that of any heterojunction catalysts reported (Table S2†). However, further increasing the mass percentage of CoPor-DBE was found to be detrimental to photocatalytic performance. This decline is attributed to the excessive CoPor-DBE, which hinders light absorption and the generation of photoexcited carriers in CN.55,56 Additionally, the relatively low photocatalytic performance may also stem from the excess CoPor-DBE acting as a charge recombination center. Subsequently, the 50CoPor-DBE/CN heterojunction was used as a model catalyst in the following experiments for further discussions.
 | ||
| Fig. 5 (a) CO yield rate and TC degradation efficiency of the as-obtained samples, (b) photoredox performance of 50CoPor-DBE/CN in diverse reaction systems, (c) mass spectrum of the produced CO over 50CoPor-DBE/CN using 13CO2 as a substrate, and (d) cycling runs of 50CoPor-DBE/CN for photo-redox coupling CO2 conversion with TC degradation. | ||
Given that the coupled reaction system has the potential to enhance the full utilization of carriers, it thereby affects the photocatalytic redox processes. Consequently, we explored the impacts of various reaction systems on 50CoPor-DBE/CN. As expected, in comparison with the separate half-reactions of CO2 photoreduction and independent TC oxidation under an Ar atmosphere (Fig. 5b), the proposed coupled reaction system demonstrated markedly improved catalytic efficiency compared to the two half-reactions. To ascertain the origin of the gaseous products, 13C isotope labeling experiments were conducted. When the CO2 feedstock was replaced with 13CO2, a strong signal with an m/z value of 29 was detected, robustly confirming that the generated CO originates from CO2 molecules (Fig. 5c). The cycling stability of the CoPor-DBE/CN heterojunction was assessed through photocatalytic CO2 reduction cycle experiments, with the results presented in Fig. 5d. These cycling experiments demonstrate that the CoPor-DBE/CN heterojunction retains its photocatalytic activity after four consecutive cycles. Furthermore, significant changes were seen in the XRD patterns, FT-IR spectra, and XPS spectra (Fig. S9 and S10†) before and after the cyclic reaction. These findings indicate that the CoPor-DBE/CN heterojunction exhibits good stability in the coupled reaction system.
In situ FT-IR was employed to monitor the reaction intermediates involved in photocatalytic CO2 reduction over the 50CoPor-DBE/CN photocatalyst (Fig. S11†). As the illumination time increased, intermediate products gradually emerged. The adsorption peaks observed at 1320, 1473, and 1506 cm−1 were attributed to m-CO32−.57 Peaks at 1352 and 1653 cm−1 were assigned to b-CO32−, while peaks corresponding to the HCO3− group emerged at 1418, 1435 and 1456 cm−1.58 The presence of these carbonate groups suggests that electron transfer of CO2 occurs on the sample surface during photoreduction. Notably, new peaks at 1558 and 1595 cm−1, ascribed to *COOH, appeared, which is generally regarded as a crucial intermediate in the photocatalytic conversion of CO2 to CO.59 Furthermore, several peaks were identified for the *CO adsorption band (1698 cm−1) and gas CO absorption band (2078 cm−1),60 indicating that abundant CO intermediates were generated on the surface of 50CoPor-DBE/CN, facilitating the evolution of more CO gas.
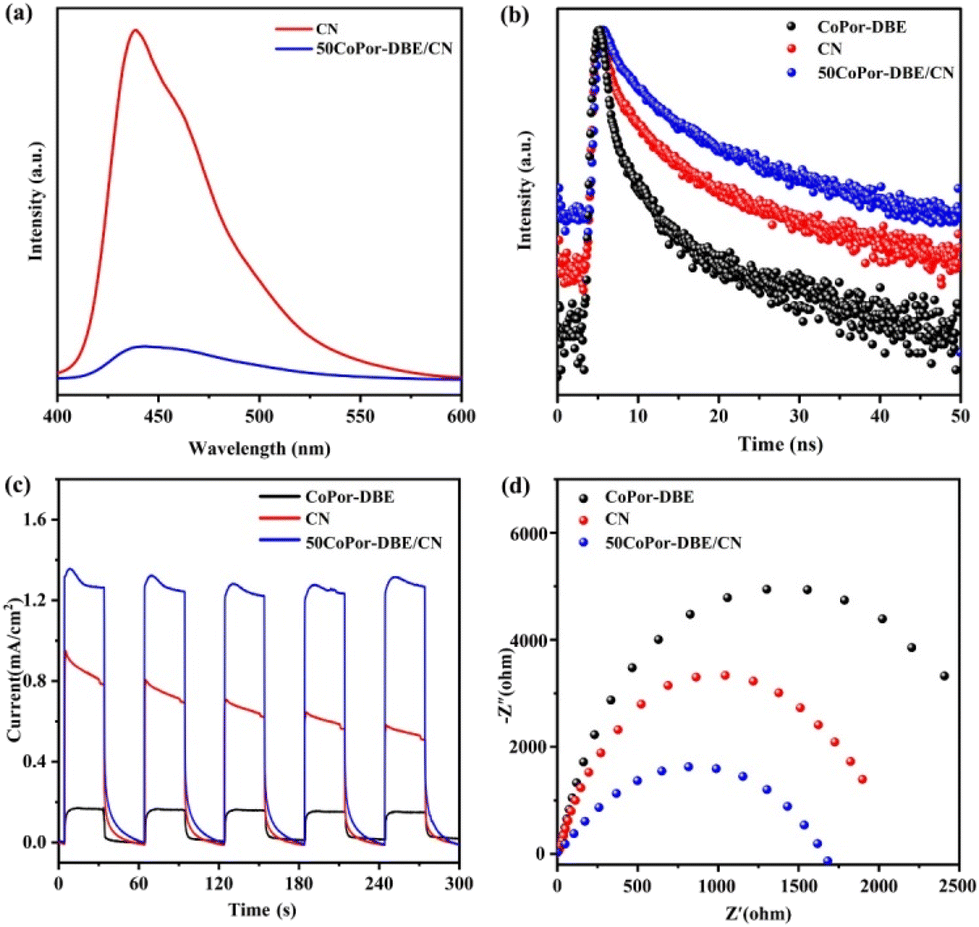
To understand the carrier transfer behavior of the synthesized nanohybrids and their impacts on photocatalytic efficiency, photoluminescence (PL) spectra were recorded at an excitation wavelength of 365 nm. Fig. 6a illustrates that the 50CoPor-DBE/CN composite shows a significantly diminished PL signal intensity relative to bare CN, suggesting effective inhibition of electron and hole recombination. Furthermore, the lifetime and dynamics of photoexcited charge carriers for these specimens were assessed using time-resolved PL (TRPL) spectra. As depicted in Fig. 6b, the TRPL spectrum of 50CoPor-DBE/CN exhibits a decay that is noticeably slower compared to those of CoPor-DBE and CN. A double-exponential decay kinetics model was utilized to analyze the decay profiles and the results from this fitting are shown in Table S3.† The average lifetime can be determined with the following equation: τave = (A1τ12 + A2τ22)/(A1τ1 + A2τ2). The estimated average PL lifetime for 50CoPor-DBE/CN was found to be 7.34 ns, which is 1.5 times and 4.0 times longer than the lifetimes obtained for CN and CoPor-DBE, respectively. The extended charge carrier lifetime in 50CoPor-DBE/CN is likely due to the presence of numerous heterojunction interfaces and a shortened diffusion path for the charge carriers.61 As a result, the longer lifetime implies that the charge carriers have a higher probability of engaging in redox reactions prior to recombination, thereby enhancing the photocatalytic efficiency.
 | ||
| Fig. 6 (a) Steady-state PL spectra, (b) time-resolved fluorescence decay curves, (c) transient photocurrent responses and (d) Nyquist plots of EIS for CoPor-DBE, CN and 50CoPor-DBE/CN. | ||
In addition, the transient photocurrent response of these samples was also investigated to further elucidate their charge separation capability (Fig. 6c). During visible light switching, 50CoPor-DBE/CN was found to have the highest current density, which is approximately three times that of CoPor-DBE, indicating that more photogenerated carriers are generated in the 50CoPor-DBE/CN heterojunction, and the charge separation efficiency is improved. Furthermore, the electrochemical impedance spectra of all the prepared catalysts were recorded to assess the migration abilities of charge carriers. The charge-transfer resistance is positively correlated with the arc radius of the electrochemical impedance spectroscopy plots. As depicted in Fig. 6d, the 50CoPor-DBE/CN heterojunction exhibited a smaller Nyquist semicircular radius compared to those of CoPor-DBE and CN, suggesting a lower charge-transfer resistance of 50CoPor-DBE/CN,62 which aligns with the findings from the photocurrent measurements. The above results confirm that the S-scheme heterojunction formed between CoPor-DBE and CN is beneficial for rapid charge separation and promotes electron transfer, thereby achieving efficient photo-redox reactions.
Based on the previously discussed characterization studies and analyses, a plausible mechanism for photocatalytic CO2 reduction coupled with TC oxidation over the CoPor-DBE/CN heterojunction is proposed, as illustrated in Fig. 7. Upon light irradiation, both CoPor-DBE and CN within the CoPor-DBE/CN composites are excited, generating electrons and holes in their CBs and VBs, respectively. Driven by the existing interfacial electric field, the photoexcited electrons in the CB of CN migrate to the VB of CoPor-DBE, where they recombine with the holes via the S-scheme pathway, thereby enhancing charge separation kinetics. Consequently, the electrons accumulated in the CB of CoPor-DBE/CN can effectively transfer to the Co active sites for CO2 reduction. Notably, since CoP-COP possesses a more negative CB potential than CN, the robust reducibility of electrons can be effectively utilized to promote the conversion of CO2 to CO. Meanwhile, the photogenerated holes remaining in the VB of CN provide sufficient driving force to oxidize the adsorbed H2O, resulting in the formation of ˙OH radicals. These ˙OH active species, in combination with the TC molecule, participate in a series of redox reactions, ultimately mineralizing TC into harmless small molecules. Consequently, the CoPor-DBE/CN composite exhibits excellent photocatalytic activity for CO2 reduction coupled with TC degradation.
 | ||
| Fig. 7 Schematic diagram of the mechanism of CO2 reduction coupled TC oxidation for the CoPor-DBE/CN heterojunction. | ||
Conclusions
In summary, we have successfully fabricated a series of well-structured S-scheme CoPor-DBE/CN heterojunctions by assembling CoPor-DBE flakes on the surface of porous CN nanosheets. The resulting samples exhibit dual functionality as photocatalysts, enabling both solar-driven CO2 reduction and TC degradation. Benefiting from the unique 2D/2D morphology and intimate interfacial interactions, the obtained CoPor-DBE/CN heterojunction facilitates rapid migration and separation of photogenerated carriers. The characterization results from XPS, UPS, and KPFM confirm that charge transfer follows the S-scheme transport route, which efficiently achieves spatial separation of charge carriers while maintaining their strong redox capacity. The optimized 50CoPor-DBE/CN heterojunction achieved a CO production rate of 16.7 μmol g−1 h−1, along with a TC removal rate of 93.8%, significantly higher than those of the two individual counterparts. This work provides a feasible strategy for the fabrication of S-scheme heterojunction photocatalysts, successfully achieving a sustainable dual photoredox process for CO2 conversion coupled with environmental remediation.Data availability
The authors confirmed that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (No. 22109064, 22465024, 22105090 and 52270039), the Jiangxi Provincial Natural Science Foundation (No. 20232BAB204100 and 20242BAB25164), and the Key Laboratory of Jiangxi Province for Persistent Pollutants Prevention Control and Resource Reuse (No. 2023SSY02061). We are grateful for the financial support from the projects and research platform support provided by the laboratory.References
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S.-I. In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- H. Wang, X. Li, X. Zhao, C. Li, X. Song, P. Zhang, P. Huo and X. Li, Chin. J. Catal., 2022, 43, 178–214 CrossRef CAS.
- S. Zhuang, D. Chen, W. P. Ng, L. J. Liu, M. Y. Sun, D. Liu, T. Nawaz, Q. Xia, X. Wu and Y. L. Huang, Angew. Chem., Int. Ed., 2023, 62, e202306696 CrossRef CAS PubMed.
- X. Meng, L. Liu, S. Ouyang, H. Xu, D. Wang, N. Zhao and J. Ye, Adv. Mater., 2016, 28, 6781–6803 CrossRef CAS PubMed.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- V. Vinayagam, K. N. Palani, S. Ganesh, S. Rajesh, V. V. Akula, R. Avoodaiappan, O. S. Kushwaha and A. Pugazhendhi, Environ. Res., 2024, 240, 117500 CrossRef CAS PubMed.
- W. Song, K. C. Chong, G. Qi, Y. Xiao, G. Chen, B. Li, Y. Tang, X. Zhang, Y. Yao, Z. Lin, Z. Zou and B. Liu, J. Am. Chem. Soc., 2024, 146, 3303–3314 CrossRef CAS PubMed.
- W. Gao, L. Shi, W. Hou, C. Ding, Q. Liu, R. Long, H. Chi, Y. Zhang, X. Xu, X. Ma, Z. Tang, Y. Yang, X. Wang, Q. Shen, Y. Xiong, J. Wang, Z. Zou and Y. Zhou, Angew. Chem., Int. Ed., 2024, 63, e202317852 CrossRef CAS PubMed.
- W. Huang, Z. Zhang, J. Xu, H. Cui, K. Tang, D. Crawshaw, J. Wu, X. Zhang, L. Tang and N. Liu, JACS Au, 2024 DOI:10.1021/jacsau.4c00998.
- N. Liu, K. Tang, D. Wang, F. Fei, H. Cui, F. Li, J. Lei, D. Crawshaw, X. Zhang and L. Tang, Sep. Purif. Technol., 2024, 332, 125873 CrossRef CAS.
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS.
- Y. Wang, Y. Liu, L. Wang, S. Perumal, H. Wang, H. Ko, C.-L. Dong, P. Zhang, S. Wang, T. T. T. Nga, Y. D. Kim, Y. Ji, S. Zhao, J.-H. Kim, D.-Y. Yee, Y. Hwang, J. Zhang, M. G. Kim and H. Lee, J. Am. Chem. Soc., 2024, 15, 6047 CAS.
- H. Mao, Y. Tan, Q. Chen, M. Cai, C. Xu, T. Yang, Q. Gao, S. Zhang, Y. Fang, S. Liu, F. Peng and S. Yang, ACS Appl. Nano Mater., 2024, 7, 12935–12943 Search PubMed.
- N. Liu, F. Fei, W. Dai, J. Lei, F. Bi, B. Wang, G. Quan, X. Zhang and L. Tang, J. Colloid Interface Sci., 2022, 625, 965–977 CrossRef CAS PubMed.
- N. Liu, J. Wu, F. Fei, J. Lei, W. Shi, G. Quan, S. Zeng, X. Zhang and L. Tang, J. Colloid Interface Sci., 2022, 612, 1–12 CrossRef CAS PubMed.
- Y. Yang, Y. Zhang, X. Gao, Z. Yang, H. Wang and X. Zhang, Water, 2025, 17, 24 CrossRef.
- J. Xu, G. Jiao, G. Sheng, N. Lin, B. Yang, J. Wang, H. Jiang, Y. Wang and X. Zhang, Sep. Purif. Technol., 2025, 354, 129357 CrossRef CAS.
- Y. Wang, W. Xia, G. Jiao, J. Wang, Y. Gong, Q. Yin, H. Jiang and X. Zhang, Colloids Surf., A, 2025, 705, 135706 CrossRef CAS.
- X. Jia, C. Hu, H. Sun, J. Cao, H. Lin, X. Li and S. Chen, Appl. Catal., B, 2023, 324, 122232 CrossRef CAS.
- H. Sun, X. Jia, J. Cao, S. Chen, Y. Chen and H. Lin, J. Colloid Interface Sci., 2024, 661, 150–163 CrossRef CAS PubMed.
- A. Boruah, B. Boro, R. Paul, C.-C. Chang, S. Mandal, A. Shrotri, C.-W. Pao, B. K. Mai and J. Mondal, ACS Appl. Mater. Interfaces, 2024, 16, 34437–34449 CrossRef CAS PubMed.
- M. Barawi, L. Collado, M. Gomez-Mendoza, F. E. Oropeza, M. Liras and V. A. de la Peña O'Shea, Adv. Energy Mater., 2021, 11, 2101530 CrossRef CAS.
- Z. Zhou, Y. Xiao, J. Tian, N. Nan, R. Song and J. Li, J. Mater. Chem. A, 2023, 11, 3245–3261 RSC.
- H. Lv, R. Sa, P. Li, D. Yuan, X. Wang and R. Wang, Sci. China: Chem., 2020, 63, 1289–1294 CrossRef CAS.
- J. H. Lee, Y. Kim, S. Oh and W.-D. Jang, Appl. Phys. Rev., 2024, 11(3), 031319 CAS.
- Z. Jia, Y. Xiao, S. Guo, L. Xiong, P. Yu, T. Lu and R. Song, ACS Appl. Mater. Interfaces, 2023, 15, 47070–47080 CrossRef CAS PubMed.
- J. Ding, X. Guan, J. Lv, X. Chen, Y. Zhang, H. Li, D. Zhang, S. Qiu, H.-L. Jiang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 3248–3254 CrossRef CAS PubMed.
- W. Tao, Q. Tang, J. Hu, Z. Wang, B. Jiang, Y. Xiao, R. Song and S. Guo, J. Mater. Chem. A, 2023, 11, 24999–25007 RSC.
- G. Sun, J. Zhang, B. Cheng, H. Yu, J. Yu and J. Xu, Chem. Eng. J., 2023, 476, 146818 CrossRef CAS.
- P. Sharma, A. Kumar, T. Wang, P. Dhiman, G. Sharma and C. W. Lai, J. Environ. Chem. Eng., 2024, 12, 112851 CrossRef CAS.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- X. Deng, J. Zhang, K. Qi, G. Liang, F. Xu and J. Yu, Nat. Commun., 2024, 15, 4807 CrossRef CAS PubMed.
- B. Zhu, J. Sun, Y. Zhao, L. Zhang and J. Yu, Adv. Mater., 2024, 36, 2310600 CrossRef CAS PubMed.
- Y. Wang, J. Pu, J. An, X. Liang, W. Li, Y. Huang, J. Yang, T. Chen and Y. Yao, Inorg. Chem., 2024, 63, 5269–5280 CrossRef CAS PubMed.
- H. Wei, F. Meng, H. Zhang, W. Yu, J. Li and S. Yao, J. Mater. Sci. Technol., 2024, 185, 107–120 CrossRef CAS.
- Z. Li, Z. Li, J. Liang, W. Fan, Y. Li, Y. Shen, D. Huang, Z. Yu, S. Wang and Y. Hou, Sep. Purif. Technol., 2023, 310, 123197 CrossRef CAS.
- Y. Xiao, G. Tian, W. Li, Y. Xie, B. Jiang, C. Tian, D. Zhao and H. Fu, J. Am. Chem. Soc., 2019, 141, 2508–2515 CrossRef CAS PubMed.
- M. Raaja Rajeshwari, S. Kokilavani and S. Sudheer Khan, Chemosphere, 2022, 291, 132735 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- S. Guo, Z. Yu, C. Xu, Z. Wang, W. Tao, Q. Tang, Z. Liu and Y. Xiao, J. Mater. Chem. A, 2024, 12, 22990–22997 RSC.
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef CAS PubMed.
- Z. Yu, Y. Xiao, S. Guo, F. Min, Q. Sun, R. Song and J. Li, ChemSusChem, 2022, 15, e202200424 CrossRef CAS PubMed.
- H. Yu, F. Zhang, Q. Chen, P.-K. Zhou, W. Xing, S. Wang, G. Zhang, Y. Jiang and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202402297 CrossRef CAS PubMed.
- N. Yang, S. Tian, Y. Feng, Z. Hu, H. Liu, X. Tian, L. Xu, C. Hu and J. Yang, Small, 2023, 19, 2207253 CrossRef CAS PubMed.
- Y. Niu, D. Xie, S. Wei, N. Li, J. Ye and Y. Ma, Sens. Actuators, B, 2024, 409, 135653 CrossRef CAS.
- J. Liu, X. Sun, Y. Fan, Y. Yu, Q. Li, J. Zhou, H. Gu, K. Shi and B. Jiang, Small, 2024, 20, 2306344 CrossRef CAS PubMed.
- T. Li, T. Lu, X. Li, L. Xu, Y. Zhang, Z. Tian, J. Yang, H. Pang, Y. Tang and J. Xue, ACS Nano, 2021, 15, 20032–20041 CrossRef CAS PubMed.
- C. Zhao, C. Yang, X. Lv, S. Wang, C. Hu, G. Zheng and Q. Han, Adv. Mater., 2024, 36, 2401004 CrossRef CAS PubMed.
- F. Bi, J. Wei, Z. Zhou, Y. Zhang, B. Gao, N. Liu, J. Xu, B. Liu, Y. Huang and X. Zhang, JACS Au, 2025, 5, 363–380 CrossRef CAS PubMed.
- J. Huang, X. Feng, F. Bi, G. Huang, R. Rao, R. Qiao and X. Zhang, Sep. Purif. Technol., 2025, 359, 130804 CrossRef CAS.
- J. Yu, H. Zhang, Q. Liu, J. Yu, J. Zhu, Y. Li, R. Li and J. Wang, Sep. Purif. Technol., 2023, 312, 123442 CrossRef CAS.
- N. Liu, F. Fei, W. Dai, J. Lei, F. Bi, B. Wang, G. Quan, X. Zhang and L. Tang, J. Colloid Interface Sci., 2022, 625, 965–977 CrossRef CAS PubMed.
- Q. Tang, W. Tao, J. Hu, T. Gui, Z. Wang, Y. Xiao, R. Song, Y. Jiang and S. Guo, ACS Appl. Nano Mater., 2023, 6, 17130–17139 CrossRef CAS.
- S. Wan, W. Wang, B. Cheng, G. Luo, Q. Shen, J. Yu, J. Zhang, S. Cao and L. Zhang, Nat. Commun., 2024, 15, 9612 CrossRef CAS PubMed.
- J. Wang, Y. Yu, J. Cui, X. Li, Y. Zhang, C. Wang, X. Yu and J. Ye, Appl. Catal., B, 2022, 301, 120814 CrossRef CAS.
- H. Li, D. Wang, C. Miao, F. Xia, Y. Wang, Y. Wang, C. Liu and G. Che, J. Environ. Chem. Eng., 2022, 10, 108201 CrossRef CAS.
- Y. Jiang, L. Xiong, S. Guo, C. Xu, J. Wang, X. Wu, Y. Xiao and R. Song, J. Mater. Chem. A, 2024, 12, 32045–32053 RSC.
- L. Wu, Y. Li, B. Zhou, J. Liu, D. Cheng, S. Guo, K. Xu, C. Yuan, M. Wang, G. J. Hong Melvin, J. Ortiz-Medina, S. Ali, T. Yang, Y. A. Kim and Z. Wang, Carbon, 2023, 207, 36–48 CrossRef CAS.
- Q. Wang, M. He, P. Yang, Y. Zhou, X. Wang, K. Zhang, Z. Jiang, Q. Luo, S. Gao and J. Yang, ACS Nano, 2024, 18, 33576–33586 CrossRef CAS PubMed.
- S. Hu, P. Qiao, X. Yi, Y. Lei, H. Hu, J. Ye and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202304585 CrossRef CAS PubMed.
- Y. Xiao, S. Guo, G. Tian, B. Jiang, Z. Ren, C. Tian, W. Li and H. Fu, Sci. Bull., 2021, 66, 275–283 CrossRef CAS PubMed.
- N. Yang, S. Tian, Y. Feng, Z. Hu, H. Liu, X. Tian, L. Xu, C. Hu and J. Yang, Small, 2023, 19, 2207253 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt03522d |
| This journal is © The Royal Society of Chemistry 2025 |