
Synthesis of a dilithiobutadiene bearing extremely bulky silyl substituents and its reactivity toward functionalized silanes†‡
Katharina
Münster
 a,
Shunsuke
Kudo
a,
Takuya
Kuwabara
a,
Eriko
Shimamura
a,
Shunsuke
Furukawa
a,
Yusuke
Yoshida
b,
Shintaro
Ishida
b,
Takeaki
Iwamoto
b,
Kazuki
Tanifuji
c,
Yasuhiro
Ohki
c,
Mao
Minoura
d and
Masaichi
Saito
*a
a,
Shunsuke
Kudo
a,
Takuya
Kuwabara
a,
Eriko
Shimamura
a,
Shunsuke
Furukawa
a,
Yusuke
Yoshida
b,
Shintaro
Ishida
b,
Takeaki
Iwamoto
b,
Kazuki
Tanifuji
c,
Yasuhiro
Ohki
c,
Mao
Minoura
d and
Masaichi
Saito
*a
aDepartment of Chemistry, Graduate School of Science and Engineering, Saitama University, Shimo-okubo, Sakura-ku, Saitama-city, Saitama, 338-8570, Japan. E-mail: masaichi@chem.saitama-u.ac.jp
bDepartment of Chemistry, Graduate School of Science, Tohoku University, Sendai, Miyagi 980-8578, Japan
cInstitute of Chemical Research, Kyoto University, Uji, Kyoto, 611-0011, Japan
dDepartment of Chemistry, College of Science, Rikkyo University, Toshima-ku, Tokyo, 171-8501, Japan
First published on 4th February 2025
Abstract
The synthesis and full characterization of 1,4-dilithio-1,4-bis(triisopropylsilyl)-2,3-diphenylbuta-1,3-diene (1b) are reported. This molecule featuring extremely bulky silyl groups at the 1- and 4-positions serves as a precursor for the synthesis of 2,5-bis(triisopropylsilyl)-3,4-diphenyl-1-silacyclopenta-1,3-dienes (siloles) bearing various substituents at the silicon atom (SiR2 = SiH2 (4), SiH(OMe) (5), SiF2 (6), SiBr2 (7), SiBr(OMe) (8)). Importantly, compounds 6 and 7 reacted with lithium to afford 2,5-bis(triisopropylsilyl)-3,4-diphenyldilithiosilole (9). The solid-state molecular structure and solution NMR spectra reveal the formation of an aromatic ring system, as opposed to the precursors 6 and 7, with two Li cations coordinated by the silacycle in η5-fashions. The sterically bulky dilithiosilole 9 can be applied as an important starting material in the pursuit of low-valent silicon species without donor stabilization.
Introduction
Silylenes, divalent silicon species, have long received considerable attention in terms of their electronic characteristics and reactivity,1–12 and many types of isolable silylenes stabilized by sterically demanding groups and/or by donor coordination have been reported.12–16 Among such intensively studied silylenes, a cyclic diaminosilylene reported by West is worthy of attention because its divalent silicon center is incorporated into a 6π-electron aromatic ring.17–19 On the other hand, a 1-silacyclopentadienylidene, where the divalent silicon center is situated within a butadiene skeleton, is of considerable interest because of its predicted planar structure featuring a potential 4π-electron system.20,21 Although a variety of 1-silacyclopenta-1,3-dienes (siloles) and derivatives thereof were reported,22,23 none of them emerged as a suitable precursor to obtain a donor-free silylene.16 Bulky substituents on the 1,4-positions should be necessary to sterically protect the reactive silylene center and avoid the necessity of stabilization by a Lewis base.16 To prepare such a silacyclopentadienylidene, 1,4-dilithio-1,3-butadienes24 are reasonable precursors and they can be simply prepared by the reduction of phenylacetylenes with lithium.25,26 We have previously reported on the reactivity of 1,4-substituted dilithiobutadienes obtained by such reduction of phenylacetylenes bearing bulky silyl substituents SiR3 (SiR3 = tBuMe2, SitBu3, SiiPr3) (Scheme 1).27,28 The reduction of phenyl(tert-butyldimethylsilyl)acetylene with lithium exclusively afforded dilithiobutadiene 1a, whereas reduction of phenyl(tri-tert-butylsilyl)acetylene bearing the bulkiest silyl group among these compounds yielded only tri-tert-butylsilylacetylene (3c). In the reduction of phenyl(triisopropylsilyl)acetylene, highly crystalline dilithiodibenzopentalene 2b was isolated, even though dilithiobutadiene 1b formed as the main product as evidenced by NMR spectroscopy. We now report the successful isolation and full characterization of dilithiobutadiene 1b, which possesses thus far the bulkiest silyl groups on the 1,4-positions. Furthermore, we demonstrate the reaction of 1b with functionalized silanes yielding siloles that serve as starting materials for the synthesis of a new dilithiosilole (9). This compound would act as a promising precursor in the pursuit of a silacyclopentadienylidene.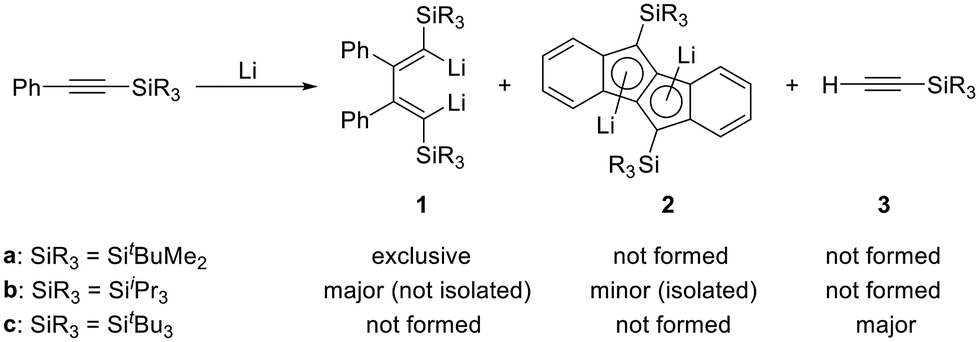 | ||
| Scheme 1 Reduction of phenyl(SiR3)acetylenes with lithium. | ||
Results and discussion
Synthesis and characterization of 1,4-dilithio-1,4-bis(triisopropylsilyl)-2,3-diphenylbuta-1,3-diene (1b)
As described in the previous article,27 the reduction of phenyl(triisopropylsilyl)acetylene leads to the simultaneous formation of dilithiobutadiene 1b and dilithiodibenzopentalene 2b (Scheme 2). Although 1b was shown to be the major product in this reaction evidenced by NMR spectroscopy, the isolated yields of 1b and 2b are comparable because the isolation of 1b is hampered by the facile crystallization of 2b in the shape of orange crystals. Therefore, to isolate pure compound 1b, repeated recrystallization from n-hexane or n-hexane/Et2O is necessary (Experimental section). By this method, 1b contaminated by <2% of 2b can be obtained in ca. 13% yield as a dark-red crystalline solid usually containing residual Et2O and/or n-hexane. The compound is highly air- and moisture-sensitive, but can be stored over a prolonged period of time under inert conditions at ambient temperature without decomposition. In the solid state, decomposition is observed at a temperature of about 80 °C. In order to promote the crystallization of pure 1b, other solvents for the recrystallization were attempted. However, although 1b can be crystallized as a THF- or N,N,N′,N′-tetramethylethane-1,2-diamine-coordinated (TMEDA) compound by using the respective coordinating solvent in combination with n-hexane at −30 °C, this method does not prevent 2b from simultaneous crystallization. Furthermore, the higher polarity of THF and TMEDA in comparison to Et2O makes it difficult to find a suitable concentration of the crystallization solution leading to the formation of isolable solid materials. As a result, in our experience the crystallization from Et2O and n-hexane is the most convenient method to obtain 1b. In this context, we would also like to note that coordinated Et2O from the reaction is replaced by THF or TMEDA, respectively; but that the replacement or removal of the latter solvents is very difficult. This should be taken into consideration for the following reactions where compound 1b is planned to be used.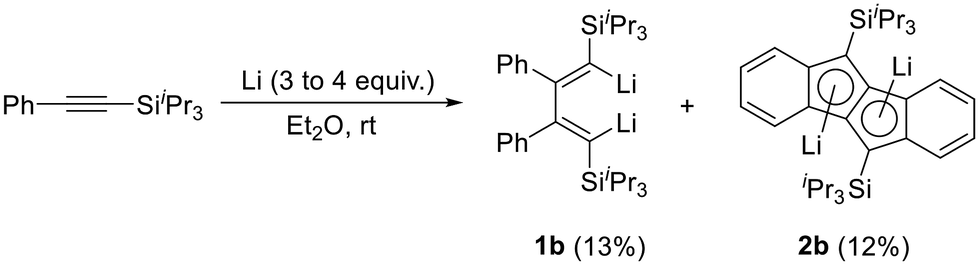 | ||
| Scheme 2 Synthesis of dilithiobutadiene 1b and dilithiodibenzopentalene 2b. | ||
A characteristic feature of compound 1b is its 7Li NMR chemical shift of δ = 1.16 ppm (C6D6), which lies in the range of resonances of vinyllithiums.29–33 Notably, 7Li NMR spectra of 1b sometimes exhibited additional resonances in the region between δ = 1–2 ppm, which disappeared upon addition of Et2O to the NMR sample (refer to ESI‡ for details). Organolithium compounds are known to form aggregates in solution34,35 and therefore, we attribute these additional resonances not to impurities, but to structurally differing molecules of 1b caused by the absence of sufficient coordinating solvent (Et2O). In contrast to 1b, the 7Li NMR resonance of 2b can be observed in the high-field region at δ ≈ −8 ppm,27 characteristic of contact ion pairs in which the Li ion is coordinated to an aromatic ring system.32,36 Moreover, the 13C NMR spectrum of dilithiobutadiene 1b features a resonance at δ = 205.81 ppm indicative of a lithiated vinyl carbon atom.29 The solid-state structure of 1b was determined by X-ray diffraction analysis of a single crystal obtained from a solution of 1b in Et2O and n-hexane at −30 °C (Fig. 1). The molecular structure reveals coordination of the Li atoms to the terminal carbon atoms of the butadiene framework. The Li–C bond distances lying in the range of 2.10–2.20 Å agree with those observed in analogous 1,4-disilyldilithiobutadienes (SiR3 = SiMe3, SitBuMe2).27,29 Similar to the literature-reported 1,4-bis(trimethylsilyl)-substituted species containing two molecules of TMEDA,29 in compound 1b every Li atom is coordinated by one Et2O molecule. In contrast, the SitBuMe2-functionalized version lacks a coordinating solvent leading to a dimeric structure with a Li4 tetrahedron.27 The C4 chain in 1b exhibits the expected alternation of bond lengths and a torsion angle of ca. 20°, which can be traced to the steric repulsion between the Ph and the silyl substituents. The latter effect is far less pronounced in the SiMe3 derivative (2.4°) in line with the smaller steric hindrance of the substituents.29
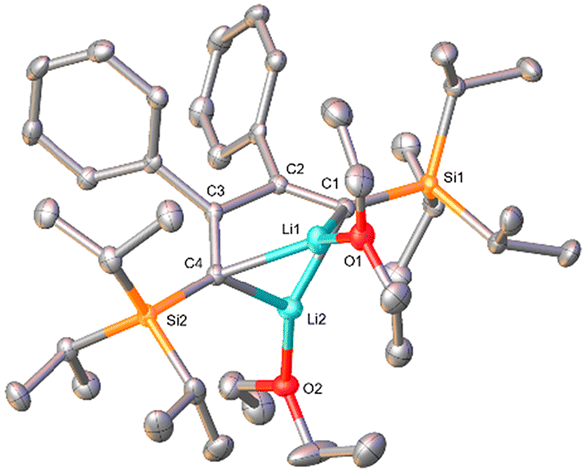 | ||
| Fig. 1 The solid-state molecular structure of 1b. The H atoms are omitted for clarity. Ellipsoids are depicted at the 50% probability level. Space group P21/n (monoclinic). Selected bond distances (Å) and (torsion) angles (°): C1–C2 1.3604(19), C2–C3 1.5614(18), C3–C4 1.3552(19), Li1–C1 2.111(3), Li1–C4 2.184(3), Li2–C1 2.160(3), Li2–C4 2.109(3), C1–Si1 1.8635(13), C4–Si2 1.8647(13), Li1–O1 1.975(3), Li2–O2 1.948(3), Li1··Li2 2.579(4), Li1–C1–C2 86.67(11), Li1–C4–C3 80.15(10), Li2–C1–C2 85.34(11), Li2–C4–C3 91.85(11), C1–C2–C3–C4 2.4. | ||
Synthesis and characterization of 2,5-bis(triisopropylsilyl)-3,4-diphenylsiloles (SiR2 = SiH2 (4), SiH(OMe) (5), SiF2 (6), SiBr2 (7) and SiBr(OMe) (8))
Scheme 3 shows an overview over the synthetic route toward siloles 4–8 starting from dilithiobutadiene 1b. Upon reaction with 2 equivalents of trimethoxysilane (HSi(OMe)3), compounds 4 and 5 are formed simultaneously. In contrast to our expectation, methoxy-substituted silole 5 is obtained as a minor product (4%), whereas dihydrosilole 4 emerges as the main species (22%). The compounds are air- and moisture-stable and can be separated by column chromatography with subsequent recrystallization. Although the mechanism of this reaction was not investigated in more detail, we attribute this outcome to the ability of basic alkaline compounds such as 1b to catalyze the disproportionation of alkoxysilanes yielding SiH4 among other species.37 Similar behaviour was also observed upon synthesis of a silacyclopentane from a 1,4-dilithio-1,1,4,4-tetraarylbutane treated with an excess amount of HSi(OMe)3.38 Variation of the reaction conditions (stoichiometry, solvent and temperature) did not alter the result shown here. In order to increase the efficiency in synthesizing 4 and 5, and since pure 1b was obtained in a low yield, a one-pot reaction starting from phenyl(triisopropylsilyl)acetylene (Scheme 2) was developed, in which the intermediately formed dilithiobutadiene was used for further reactions without prior isolation (for more details, refer to the Experimental section). Although a variety of compounds inherently formed in this reaction (e.g. hydrolyzed 2b), purification by column chromatography afforded compounds 4 and 5. In contrast, the synthesis of difluorosilole 6 was conducted using pure 1b, since a column-chromatographic purification was expected to cause decomposition of the target compound. Instead, 6 was obtained after recrystallization under inert conditions in 34% yield. To expand the library of halide-functionalized siloles, compounds 4 and 5 were treated with N-bromosuccinimide to afford the corresponding bromosiloles 7 and 8 in moderate to good yields. The siloles 4–8 are thermally stable compounds, which melt reversibly between 90 and 150 °C. Compounds 4 and 5 are insensitive to moisture and air, while compounds 6–8 do not decompose upon short exposure to atmospheric conditions; however, they should be handled under inert conditions to avoid hydrolysis. The purity of the compounds was confirmed by elemental and NMR spectroscopic analyses.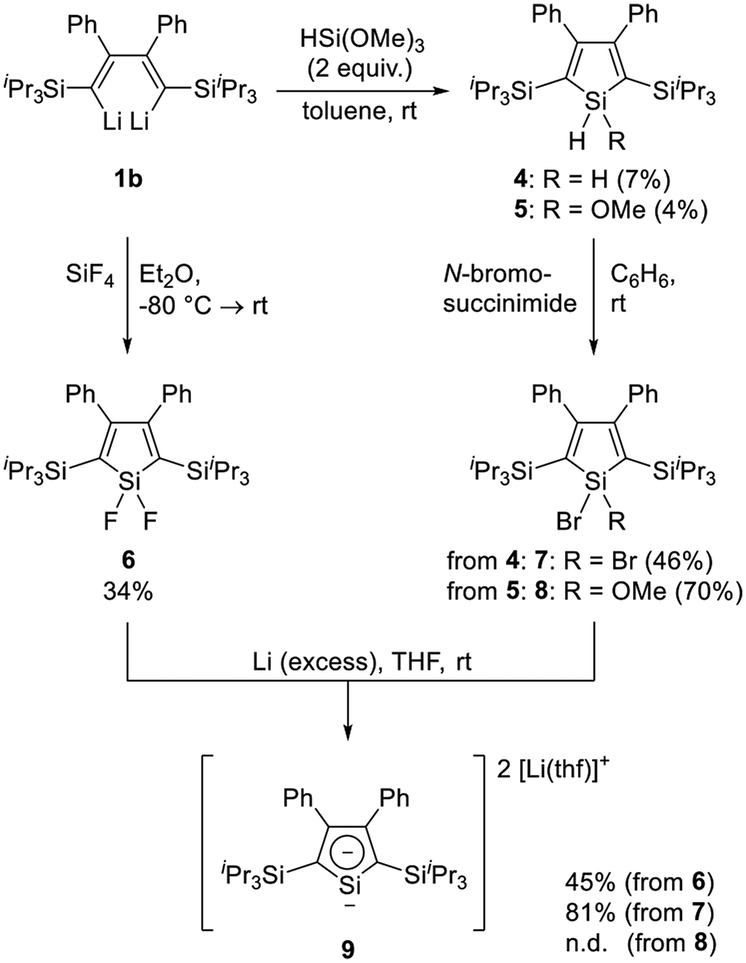 | ||
| Scheme 3 Synthesis of siloles 4–8 and dilithiosilole 9. | ||
| Compound | Siring | SiiPr | Cα | Cβ |
|---|---|---|---|---|
| a (CDCl3, ppm) of compound 4 b (C6D6, ppm) of compounds 5–9. | ||||
| 4 | −18.4 | 0.8 | 135.85 | 172.71 |
| 1 J Si,H = 196 Hz | ||||
| 5 | 9.3 | 0.7 | 135.04 | 171.80 |
| 1 J Si,H = 212 Hz | ||||
| 6 | −11.0 | 1.4 | n. o. | 173.85 |
| 1 J Si,F = 332 Hz | 3 J C,F = 7.2 Hz | |||
| 7 | 5.8 | 2.2 | 136.39 | 169.35 |
| 8 | 0.4 | 1.0 | 134.71 | 169.81 |
| 9 | 130.7 | −0.1 | 139.72 | 142.66 |
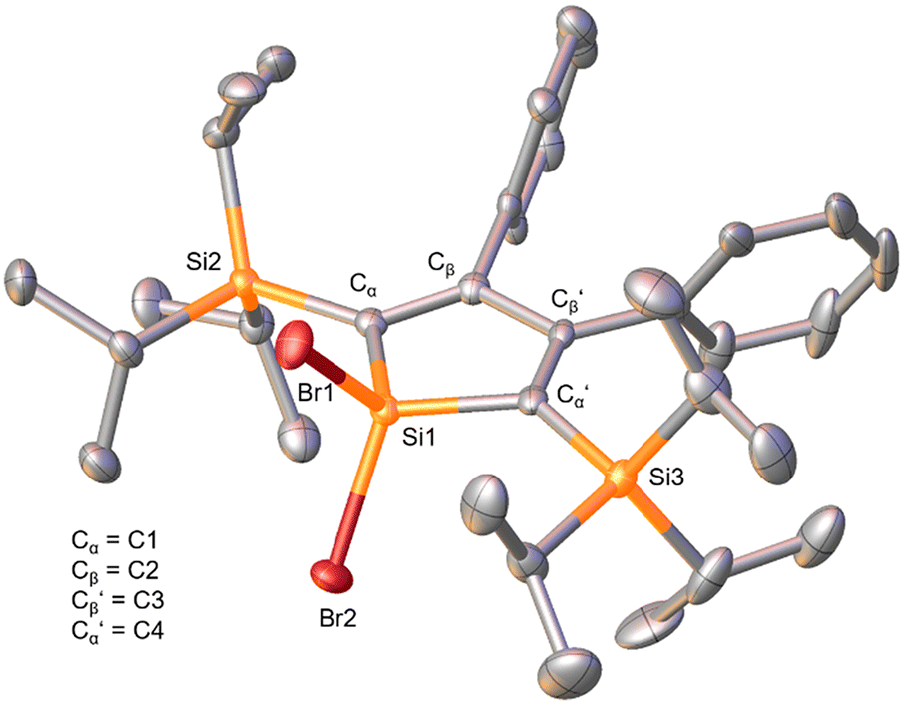 | ||
| Fig. 2 The solid-state molecular structure of 7. One of the two independent molecules in the unit cell is shown. The H atoms and the second molecule in the asymmetric unit are omitted for clarity. Ellipsoids are depicted at the 50% probability level. Space group I121 (monoclinic). Selected bond distances (Å) and (torsion) angles (°): Cα–Cβ 1.351(6), Cβ–Cβ′ 1.537(6), Cβ′–Cα′ 1.350(6), Si1–Cα 1.857(4), Si1–Cα′ 1.848(4), Si1–Br1 2.208(2), Si1–Br2 2.212(2), Cα–Si1–Cα′ 99.3(2), Si1–Cα–Cβ 101.8(3), Cα–Cβ–Cβ′ 118.3(4), Cβ′–Cα′–Si1 102.0(3), Cα–Cβ–Cβ′–Cα′ 5.7(6), Si2–Cα–Cα′–Si3 41.6, angle between planes (Cα–Cβ–Cβ′–Cα′)–(Cα–Si1–Cα′) 2.0. | ||
The X-ray diffraction analysis revealed the expected structure of a silacyclopentadiene with two bromide substituents at the 1-position. The bonding parameters are in good agreement with those of previously reported siloles whose solid-state molecular structures were established by X-ray diffraction analysis.41,42,45,46 The five-membered silacycle possesses almost ideal planarity with the angle between the planes formed by the atoms (Cα–Cβ–Cβ′–Cα′) and (Cα–Si1–Cα′), respectively, being 2.0°. In addition, the three ring C–C distances alternate accounting for the presence of two double and one single bond, showing its diene character. The Si–C bond lengths within the silole ring were determined as 1.857(4) Å and 1.848(4) Å, respectively, being in the range of literature-reported values (1.85–1.88 Å).41,45,46 The silole silicon atom (Si1) features a distorted tetrahedral bonding sphere (angle Cα–Si1–Cα′ 99.3(2)°).
Synthesis and characterization of 2,5-bis(triisopropylsilyl)-3,4-diphenyldilithiosilole (9)
Literature contains a number of studies on dianionic silacyclopentadienes with different substitution patterns and counter cations.42–44,47–55 Generally, the synthesis is accomplished by reaction of a 1,1-dihalide-substituted silole with an alkaline metal (Li, Na, K), and in one case even a 1-methyl-1-hydrosilole was used as the starting material.53 Similarly, we obtained 2,5-disilyl-substituted dilithiosilole 9 by the reaction of either difluorosilole 6 or dibromosilole 7 with an excess amount of Li in THF at ambient temperature in moderate (45%) and good yield (81%), respectively. As evidenced by NMR spectroscopy, the methoxy-functionalized bromosilole 8 can also serve as the starting material indicated by a high-field resonance in the 7Li NMR spectrum; however, multiple other unidentified species were observed by 7Li NMR spectroscopy and the target compound could not be isolated in pure form (for details refer to ESI‡). It is expected that the cleavage of the methoxy group is slow compared to halide cleavage, resulting in a longer reaction time,53 accompanied by side-reactions, e.g. leading to the formation of 1-substituted monoanionic compounds or dimeric species.42,44,48,49Compound 9 was obtained as a dark-red crystalline solid after recrystallization from Et2O and n-hexane. In the solid state, decomposition was observed above 150 °C. The purity was confirmed by NMR spectroscopy (for selected NMR chemical shifts, refer to Table 1), since the high sensitivity against air and moisture prevented the collection of satisfying results in the elemental analysis. The 1H NMR spectrum shows well-resolved signals for the aryl protons between δ = 6.95 and 7.12 ppm, slightly low-field shifted compared to those of dihalosiloles 6 and 7 (δ = 6.87–6.73 and 6.84–6.67 ppm, for 6 and 7, respectively). The presence of two coordinated THF molecules is observed as broadened resonances at δ = 1.31 and 3.58 ppm, differing from the chemical shifts of uncoordinated THF (δ = 1.40, 3.57 ppm in C6D6).56 In the 13C NMR spectrum, all resonances appear at chemical shifts smaller than δ = 150 ppm, which could indicate the introduction of two negative charges into the silole framework compared to the neutral precursor.44 The latter exhibit characteristic 13C NMR resonances at δ ≈ 170 and 135 ppm for the β- and α-C atoms, respectively, of the silacycle. Upon transformation to the dilithiosilole, the β-C resonance is shifted to higher field (δ ≈ 142.7 ppm; assigned by 2D NMR, see ESI‡). Similar observations were made by the groups of Kovács and Müller upon investigation of 2,5-bis(trialkylsilyl)-substituted siloles,43,44 and can also be observed in alkyl- and aryl-substituted derivatives.42,52 In contrast, the resonance of the α-C atom shifts to lower field (δ = 139.72 ppm), which is also observed in 2,3,4,5-tetraphenyldilithiosilole (δ = 129.71 ppm).48 The characteristic 29Si NMR chemical shifts at δ = 130.7 ppm for the ring Si atom and δ = −0.1 ppm for the triisopropylsilyl group are in line with the formation of a dianionic silacycle and again agree with the reports cited before.43,44 In particular, the presence of silyl substituents in the 2,5-positions induces the pronounced low-field shift, probably due to the increase in a silylene character in 9, as it was observed in 2,5-silylated dilithiostannoles.28,44 On the other hand, the previous investigations on 2,5-alkyl- and 2,5-aryl-substituted silole derivatives found relatively high-field-shifted 29Si NMR resonances,42 as for example 2,3,4,5-tetraphenyldilithiosilole (δ = 68.5 ppm)47,48 and 2,3,4,5-tetramethyldilithiosilole (δ = 29.8 ppm).55 Moreover, the 7Li NMR shift of δ = −6.16 ppm indicates the presence of two (on the NMR time scale) chemically equivalent Li atoms coordinated by an aromatic ring system.32,36 On the other hand, a previous report discussed the solution structure of tetraphenyldilithiosilole, which exhibits a 7Li NMR resonance at δ = 0.23 ppm, contrasting our findings for compound 9.47
The solid-state molecular structure (Fig. 3) confirmed the results of the NMR analysis. Single-crystals for the X-ray diffraction analysis were obtained from a solution of 9 in Et2O, C6H6 and n-hexane at −30 °C. As indicated by the 7Li NMR chemical shift, two Li atoms are found coordinated by the five-membered aromatic ring in η5 fashions with an average distance of 2.23 Å between a carbon and a lithium atom, and each is additionally coordinated by one THF molecule. Similar to the dibromosilole discussed in the previous section, the silacycle is close to ideal planarity, as also observed for other derivatives.42,44,47 A pronounced difference compared to the silole 7 is found in the C–C bond distances within the five-membered ring. While in the neutral species values representing two double-bonds and one single-bond were found, the Cα–Cβ and Cβ–Cβ bond distances are now equal within the measurement uncertainty (ca. 1.45 Å). This suggests substantial delocalization of electrons within the ring and is diagnostic of the aromatic nature of this system.44 The Si1–Cα bond distance is slightly elongated relative to those of the precursors 6 and 7 (ca. 0.03 Å), which is a result of elongation by silylene character and shortening by electronic delocalization to increase its double-bond character.57 Upon comparison to the dipotassio-2,5-bis(trimethylsilyl)-3,4-diphenylsilole reported by Müller et al., which forms a coordination polymer through the η5-coordination of K+ ions to the aromatic ring and the ring Si atom (κ-Si),44,58 we conclude that in the present case of compound 9 the steric bulk of the triisopropylsilyl groups in 2,5-positions is sufficient to prevent the Si atom from such coordination. This is a promising structural feature with regard to the synthesis of a base-free silylene from this molecule.
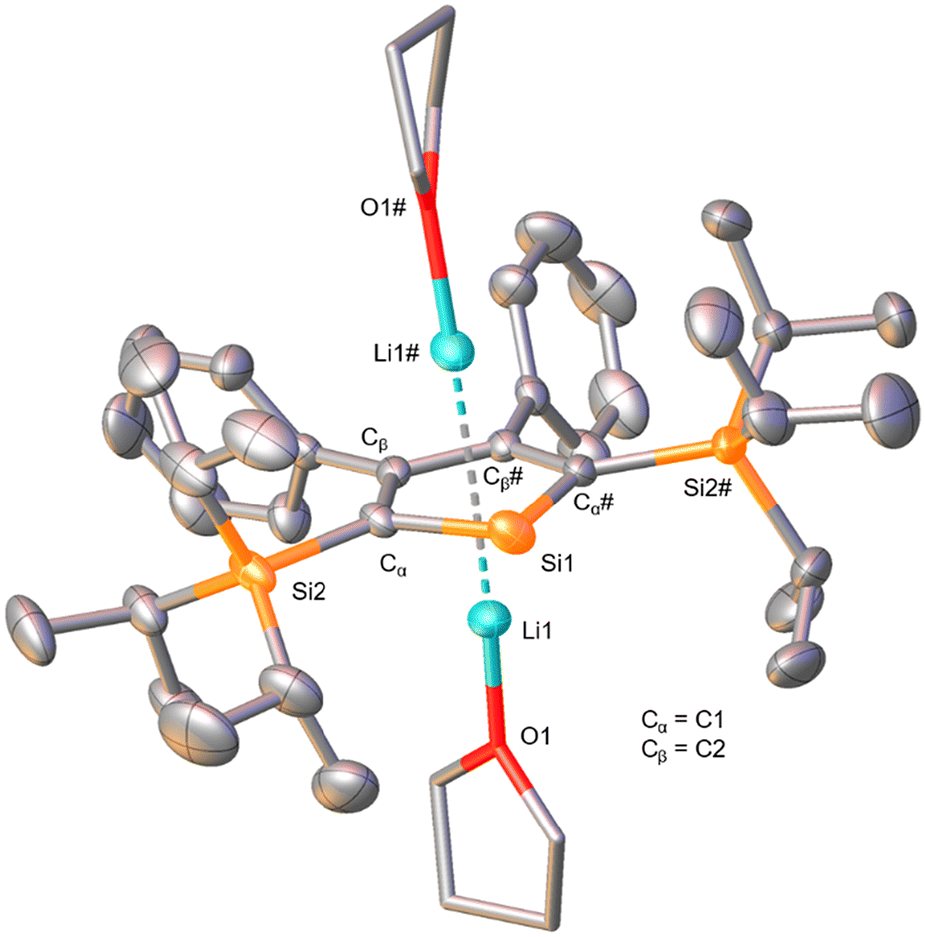 | ||
| Fig. 3 The solid-state molecular structure of 9. The H atoms and the minor part of the two-fold disordered THF molecule are omitted for clarity. Ellipsoids are depicted at the 50% probability level. The coordinated THF molecules are shown as sticks. Space group C2 (monoclinic). Selected bond distances (Å) and (torsion) angles (°): Cα–Cβ 1.446(5), Cβ–Cβ# 1.438(6), Si1–Cα 1.881(4), Si2–Cα 1.875(3), Li1–silole plane 1.829, Li1–O1 1.875(6), Cα–Si1–Cα# 88.3(2), Si1–Cα–Cβ 111.7(2), Cα–Cβ–Cβ# 114.13(18), Cα–Cβ–Cβ#–Cα# 1.0, Si2–Cα–Cα#–Si2# 11.8, angle between planes (Cα–Cβ–Cβ#–Cα#)–(Cα–Si1–Cα#) 0.1. | ||
Conclusions
We succeeded in the synthesis and full characterization of 1,4-dilithio-1,3-butadiene 1b bearing extremely bulky triisopropylsilyl groups at its 1- and 4-positions. The triisopropylsilyl substituents in 1- and 4-positions feature the highest steric demanding among the 1,4-dilithio-1,4-disilyl-1,3-butadienes reported to date. We successfully employed this compound in the synthesis of various functionalized siloles bearing hydro, methoxy and halide substituents at the silole silicon atom. As a representative example, the solid-state molecular structure of dibromosilole 7 was successfully determined by X-ray diffraction analysis, showing its planar five-membered ring with a diene character. In the next step, the dihalide-functionalized siloles 6 and 7 were converted into dilithiosilole (9), which was fully characterized by NMR spectroscopic analysis and X-ray diffraction analysis. The high-fielded 7Li NMR resonance indicates the coordination of Li ions by an aromatic ring system. This was confirmed by the solid-state molecular structure revealing a monomeric species containing a planar silacycle with delocalized bonds and two Li cations coordinated above and below the aromatic silole ring. We strongly believe the dilithiosilole bearing extremely bulky silyl groups will receive considerable attention as a promising precursor for an unconquered stable silacyclopentadienylidene.Experimental
Materials and methods
If not otherwise stated, experiments were performed under an argon atmosphere in a glovebox or using standard Schlenk techniques. Hexane, benzene, diethyl ether, THF and deuterated benzene were purified by potassium mirror before use. Toluene and deuterated chloroform were stored over activated molecular sieves (3 Å). Phenyl(triisopropylsilyl)acetylene was synthesized according to a literature procedure.59N-Bromosuccinimide (FUJIFILM Wako Pure Chemical Corporation) was recrystallized from water and dried prior to use. Commercially purchased trimethoxysilane (Tokyo Chemical Industry Co., Ltd) was stored under argon and used as received. SiF4 gas (Taiyo Nippon Sanso Corporation) was purchased and used without further purification. Lithium powder was prepared according to methods in the cited references from lithium lump (Kanto Chemical Co., Inc.).60–62 Column chromatography was performed using Kanto Silica Gel 60 (spherical, particle size 100–210 μm) and TLC silicagel 60 F254 plates (Merck). The 1H, 13C, 19F, 7Li and 29Si NMR spectra were recorded on Bruker AVANCE-300, Bruker AVANCE-400 Cryo, AVANCE-500 or AVANCE-500T spectrometers at 27 °C. The 1H and 13C{1H} NMR spectra were referenced against signals of residual protonated solvent.56 The 19F, 7Li, 29Si (non-decoupled) and 29Si{1H} NMR spectra were calibrated externally. The chemical shifts (δ) are given in ppm, accompanied by coupling constants J in Hz and resonances classified as s (singlet), d (doublet), t (triplet), sept (septet), br (broad) and Cq (quaternary C atom). The intensity data for X-ray crystallographic analyses were collected under a cold N2 stream on a Rigaku RA-Micro 7HFMR equipped with a Rigaku HyPix-6000HE detector at −150 °C for compound 7, on a Bruker APEX2 for compound 1b, and on a Bruker D8 QUEST ECO equipped with a Bruker PHOTON III detector at −123 °C for compound 9, using graphite-monochromated MoKα radiation (λ = 0.71073 Å). For compound 7: full data collection and reduction were performed using the CrysAlisPro program. The data sets were corrected for absorption using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm. The structures were solved by direct methods using the SHELXT program and refined by full-matrix least squares using the SHELXL program.63,64 For compounds 1b and 9: full data correction and reduction were performed using the APEX5 program software suite. The collected frames were integrated with the Bruker SAINT software. The absorption correction was performed by multi-scan method implemented in SADABS. The structure was solved and refined with the SHELXTL. Melting points were determined by a Yanaco macro melting point MP-S3 apparatus and were uncorrected. Elemental analysis was carried out at the Microanalytical Laboratory of Molecular Analysis and Life Science Center, Saitama University.Synthetic procedures
1st vial: yellow/orange powder; mainly composed of dilithiodibenzopentalene 2b with a trace amount of dilithiobutadiene 1b.* (*In one case, large dark red crystals of 1b were formed among orange powder of 2b. They can be picked out by tweezers and further recrystallized.)
2nd vial: yellow/orange crystalline solid (+ powder); mainly composed of dilithiodibenzopentalene 2b.
3rd vial: red crystalline solid or powder, sometimes sticky even after drying; mainly composed of 1b (sometimes still contaminated with up to 20% of 2b).
Usually from 4th vial: red crystalline solid, which becomes solid after drying (not sticky); composed of almost pure 1b (<2% of 2b).
The color of the mother liquor is generally very dark. However, after repetitive recrystallizations to remove 2b, the color changes from orange-red (compound 2b) to deep-red (compound 1b).
Dilithiobutadiene 1b. Yield: red solid, 0.962 g (contains (Et2O)2.5, 1.344 mmol of 1b, 13%). Decomposition point:ca. 80 °C. 1H NMR (400 MHz, C6D6):δ = 7.12 (d, 3JH,H = 8.1 Hz, 4 H, o-Ph), 7.05–6.98 (m, 4 H, m-Ph–H), 6.85 (t, 3JH,H = 7.2 Hz, 2 H, p-Ph–H), 3.19 (q, 3JH,H = 7.4 Hz, 10 H, OCH2), 1.25 (d, 3JH,H = 7.3 Hz, 36 H, iPr–CH3 overlapped with 6H of CH(CH3)2), 1.06 (t, 3JH,H = 7.4 Hz, 15 H, OCH2CH3). The signal for the CH protons of the iPr groups was overlapped with the iPr–CH3 signal, confirmed by HSQC measurement (see, ESI‡). 13C{1H} (101 MHz, C6D6):δ = 205.81 (Cq, Cα), 161.75 (Cq, Cβ), 153.17 (Cq, i-Ph), 129.02 (CH), 126.23 (CH), 123.67 (CH), 65.46 (CH2, OCH2), 20.33 (CH3, iPr–CH3), 14.26 (CH3, OCH2CH3), 13.95 (CH, iPr–CH). 7Li NMR (194 MHz, C6D6):δ = 1.16 (s). Because of the severe sensitivity towards air and moisture, satisfying results for the elemental analysis were not obtained.
Dilithiodibenzopentalene 2b. Yield: orange crystals, 0.328 g (0.620 mmol, 12%). Spectroscopic data were reported previously.27
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Compound 4 (Rf ≈ 0.55 in n-hexane) was usually contained in the second fraction, while 5 was obtained from the last fraction (Rf = 0.00 in n-hexane). To obtain analytically pure compounds 4 and 5, recrystallization from hexane was conducted. Other identified compounds obtained from the column-chromatographic purification include the starting material (phenyl(triisopropylsilyl)acetylene in fraction 1) and dibenzopentalene (deeply orange fraction following dihydrosilole 4).
:1. Compound 4 (Rf ≈ 0.55 in n-hexane) was usually contained in the second fraction, while 5 was obtained from the last fraction (Rf = 0.00 in n-hexane). To obtain analytically pure compounds 4 and 5, recrystallization from hexane was conducted. Other identified compounds obtained from the column-chromatographic purification include the starting material (phenyl(triisopropylsilyl)acetylene in fraction 1) and dibenzopentalene (deeply orange fraction following dihydrosilole 4).
Dihydrosilole 4. Yield: light-yellow solid, 421 mg (0.770 mmol, 7%). Melting point: 89–95 °C. Anal. calc. (%) for C34H54Si3 (M = 547.06 g mol−1) C 74.65, H 9.95; found C 74.06, H 10.17. 1H NMR (500 MHz, CDCl3):δ = 7.02–6.94 (m, 6 H, m- and p-Ph–H), 6.91–6.83 (m, 4 H, o-Ph–H), 4.78 (s, 2 H, SiH2), 1.00 (d, 36 H, 3JH,H = 7.4 Hz, iPr–CH3), 0.84 (sept, 6 H, 3JH,H = 7.4 Hz, iPr–CH). 13C{1H} NMR (126 MHz, CDCl3):δ = 172.71 (Cq, Cβ), 143.21 (Cq, Ph–C), 135.85 (Cq, Cα), 129.12 (CH, o-Ph), 126.63 (CH, m-Ph), 126.35 (CH, p-Ph), 19.54 (CH3, iPr–CH3), 13.43 (CH, iPr–CH). 29Si NMR (99 MHz, CDCl3):δ = 0.8 (m, SiiPr3), −18.4 (t, 1JSi,H = 196.0 Hz, ring-Si).
Methoxysilole 5. Yield: yellow solid, 240 mg (0.416 mmol, 4%). Melting point: 112–113 °C. Anal. calc. (%) for C35H56OSi3 (M = 577.09 g mol−1) C 72.85, H 9.78; found C 72.37, H 9.80. 1H NMR (500 MHz, C6D6):δ = 6.98–6.73 (m, 10 H, Ph–CH), 5.63 (s, 1 H, Si–H), 3.57 (s, 3 H, OCH3), 1.16 (d, 3JH,H = 7.3 Hz, 36 H, iPr–CH3), 1.06–0.97 (m, 6 H, iPr–CH). 13C{1H} NMR (126 MHz, C6D6):δ = 171.80 (Cq, Cβ), 143.10 (Cq, i-Ph), 135.04 (Cq, Cα), 129.54 (CH), 126.93 (CH), 126.69 (CH), 52.74 (CH3, OCH3), 19.74 (CH3, iPr–CH3), 13.67 (CH, iPr–CH). 29Si NMR (99 MHz, C6D6):δ = 9.0 (d, 1JSi,H = 210 Hz, ring-Si), 0.7 (m, SiiPr3).
Yield: light-yellow solid, 204 mg (0.350 mmol, 34%). Melting point: 99 °C. Anal. calc. (%) for C34H52F2Si3 (M = 583.04 g mol−1) C 70.04, H 8.99; found C 69.96, H 9.17. 1H NMR (400 MHz, C6D6):δ = 6.87–6.73 (m, 10 H, Ph–H), 1.12 (d, 3JH,H = 7.0 Hz, 36 H, iPr–CH3), 1.06–0.95 (m, 6 H, iPr–CH). 13C{1H} NMR (101 MHz, C6D6):δ = 173.85 (Cq, t, 3JC,F = 7.2 Hz, Cβ), 141.88 (Cq, t, 2JC,F = 2.5 Hz, i-Ph), 128.78 (CH, Ph–C), 127.22 (CH, p-Ph–C), 127.06 (CH, Ph–C), 19.41 (CH3, iPr–CH3), 13.38 (CH, iPr–CH). The resonance of the Cα atom was not observed and is probably covered by the signal of C6D6. 19F NMR (282 MHz, C6D6):δ = −141.1 (s). 29Si{1H} NMR (99 MHz, C6D6):δ = 1.4 (s, SiiPr3), −11.0 (t, 1JSi,F = 332 Hz, ring-Si).
Yield: yellow solid, 273 mg (0.387 mmol, 46%). Melting point: 101 °C. Anal. calc. (%) for C34H52Br2Si3 (M = 704.8 g mol−1) C 57.94, H 7.44; found C 55.75 (55.45), H 7.11 (7.23). Despite several attempts, a satisfying result could not be obtained, probably due to incomplete combustion even in the presence of a combustion additive. 1H NMR (400 MHz, C6D6):δ = 6.84–6.67 (m, 10 H, Ph–CH), 1.29–1.13 (m, 42 H, CH, CH3 (iPr)). 13C{1H} NMR (101 MHz, C6D6):δ = 169.35 (Cq, Cβ), 141.00 (Cq, i-Ph), 136.39 (Cq, Cα), 129.20 (CH, Ph–CH), 127.26 (CH, Ph–CH), 127.02 (CH, Ph–CH), 20.18 (CH3, iPr–CH3), 14.04 (CH, iPr–CH). 29Si{1H} NMR (99 MHz, C6D6):δ = 5.8 (s, ring-Si), 2.2 (s, SiiPr3).
Yield: yellow solid, 292 mg (0.445 mmol, 70%). Melting point: 145–148 °C. Anal. calc. (%) for C35H55BrOSi3 (M = 655.98 g mol−1) C 64.08, H 8.45; found C 63.85, H 8.53. 1H NMR (400 MHz, C6D6):δ = 6.92–6.69 (m, 10 H, Ph–CH), 3.63 (s, 3 H, OCH3), 1.26–1.07 (m, 42 H, CH, CH3 (iPr)). 13C{1H} NMR (101 MHz, C6D6):δ = 169.81 (Cq, Cβ), 142.04 (Cq, i-Ph), 134.71 (Cq, Cα), 129.23 (CH, Ph–CH), 129.16 (CH, Ph–CH), 127.02 (CH, Ph–CH), 126.93 (CH, Ph–CH), 126.84 (CH, Ph–CH), 52.76 (CH3, OCH3), 20.10 (CH3, iPr–CH3), 19.73 (CH3, iPr–CH3), 13.99 (CH, iPr–CH). 29Si{1H} NMR (99 MHz, C6D6):δ = 1.0 (s, SiiPr3), 0.4 (s, ring-Si).
Method A. Dibromosilole 7 (125 mg, 0.177 mmol, 1.0 equiv.) was dissolved in THF (1.5 mL) and Li powder (10 mg, 1.44 mmol, 8.1 equiv.) was added. The reaction mixture was stirred at room temperature for ca. 3 days yielding a dark-red solution and unreacted Li. After removal of the solvent in vacuum, the residue was extracted with C6H6 (3 mL) and insoluble components were removed by filtration over Celite®. The filtrate was dried and recrystallized from Et2O and hexane at −30 °C. Yield: dark-red crystalline solid, 67 mg (contains (thf)2, 0.0953 mmol, 81%).
Method B. Difluorosilole 6 (166 mg, 0.285 mmol, 1.0 equiv.) was dissolved in THF (1.5 mL) and Li powder (20 mg, 2.85 mmol, 10.0 equiv.) was added. The reaction mixture was stirred at room temperature for ca. 3 days yielding a dark-red solution and unreacted Li. After removal of the solvent in vacuum, the residue was extracted with C6H6 (3 mL) and insoluble components were removed by filtration over Celite®. The filtrate was dried and recrystallized from Et2O and hexane at −30 °C. Yield: dark-red crystalline solid, 91 mg (contains (thf)2, 0.129 mmol, 45%).
Author contributions
K. M. performed all the synthetic works and prepared the manuscript. S. K. and T. K. performed the preliminary synthetic works. E. S. performed the isolation of the dilithiobutadiene and its spectral analysis. S. F. contributed to preliminary discussion and helped experimental works. Y. Y., S. I. and T. I. contributed to the synthesis of the fluorosilole. K. T. and Y. O. performed the X-ray diffraction analysis of the bromosilole. M. M. performed preliminary X-ray diffraction analysis of siloles and contributed to the refinement of the structure of the dilithiosilole. M. S. conceived and directed the project, performed the X-ray diffraction analysis of the dilithiobutadiene and the dilithiosilole, and prepared the manuscript.Data availability
The crystallographic data were deposited in the Cambridge Crystallographic Data Centre (CCDC): Dilithiobutadiene 1b (2402955), dibromosilole 7 (2402953), dilithiosilole 9 (2402954).‡ ESI‡ include NMR charts of all compounds demonstrated in this work and crystallographic data.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a JSPS post-doctoral fellowship (K. M.), research grants from the Takahashi Industrial and Economic Research Foundation (M. S.) and the White Rock Foundation (M. S.), and the Grants-in-Aid for Scientific Research of 23H01974 for Y. O. and 22K05143 for K. T. from the Ministry of Education, Culture, Sports, Science and Technology, Japan. The authors thank Professor Makoto Yamashita (Institute of Science Tokyo, Japan) for helping us to prepare lithium powder.References
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- N. J. Hill and R. West, J. Organomet. Chem., 2004, 689, 4165–4183 CrossRef CAS.
- P. P. Gaspar and R. West, in The Chemistry of Organic Silicon Compounds, 1998, pp. 2463–2568. DOI:10.1002/0470857250.ch43.
- G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC.
- R. S. Ghadwal, R. Azhakar and H. W. Roesky, Acc. Chem. Res., 2013, 46, 444–456 CrossRef CAS PubMed.
- S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS.
- M. Weidenbruch, Coord. Chem. Rev., 1994, 130, 275–300 CrossRef CAS.
- Y.-P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed.
- S. Raoufmoghaddam, Y.-P. Zhou, Y. Wang and M. Driess, J. Organomet. Chem., 2017, 829, 2–10 CrossRef CAS.
- S. S. Sen, S. Khan, S. Nagendran and H. W. Roesky, Acc. Chem. Res., 2012, 45, 578–587 CrossRef CAS PubMed.
- L. Wang, Y. Li, Z. Li and M. Kira, Coord. Chem. Rev., 2022, 457, 214413 CrossRef CAS.
- B. Gehrhus and M. F. Lappert, J. Organomet. Chem., 2001, 617–618, 209–223 CrossRef CAS.
- M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed.
- R. West and M. Denk, Pure Appl. Chem., 1996, 68, 785–788 CrossRef CAS.
- M. Kira, Chem. Commun., 2010, 46, 2893–2903 RSC.
- Y. Gao, J. Zhang, H. Hu and C. Cui, Organometallics, 2010, 29, 3063–3065 CrossRef CAS.
- L. A. Leites, S. S. Bukalov, A. V. Zabula, I. A. Garbuzova, D. F. Moser and R. West, J. Am. Chem. Soc., 2004, 126, 4114–4115 CrossRef CAS.
- L. A. Leites, S. S. Bukalov, M. Denk, R. West and M. Haaf, J. Mol. Struct., 2000, 550–551, 329–335 CrossRef CAS.
- M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691–2692 CrossRef CAS.
- B. Goldfuss and P. V. R. Schleyer, Organometallics, 1997, 16, 1543–1552 CrossRef CAS.
- E. Vessally, M. Nikoorazm, F. Esmaili and E. Fereyduni, J. Organomet. Chem., 2011, 696, 932–939 CrossRef CAS.
- J. Dubac, A. Laporterie and G. Manuel, Chem. Rev., 1990, 90, 215–263 CrossRef CAS.
- E. Colomer, R. J. P. Corriu and M. Lheureux, Chem. Rev., 1990, 90, 265–282 CrossRef CAS.
- Z. Xi, Eur. J. Org. Chem., 2004, 2773–2781 CrossRef CAS.
- L. I. Smith and H. H. Hoehn, J. Am. Chem. Soc., 1941, 63, 1184–1187 CrossRef CAS.
- A. G. Evans, J. C. Evans, P. J. Emes and T. J. Phelan, J. Chem. Soc. B, 1971, 315–318 RSC.
- M. Saito, M. Nakamura, T. Tajima and M. Yoshioka, Angew. Chem., Int. Ed., 2007, 46, 1504–1507 CrossRef CAS PubMed.
- T. Kuwabara, J.-D. Guo, S. Nagase, M. Minoura, R. H. Herber and M. Saito, Organometallics, 2014, 33, 2910–2913 CrossRef CAS.
- A. J. Ashe III, J. W. Kampf and P. M. Savla, Organometallics, 1993, 12, 3350–3353 CrossRef.
- P. A. Scherr, R. J. Hogan and J. P. Oliver, J. Am. Chem. Soc., 1974, 96, 6055–6059 CrossRef CAS.
- M. D. Rausch and L. P. Klemann, J. Am. Chem. Soc., 1967, 89, 5732–5733 CrossRef CAS.
- D. Schmid, A. Seyboldt, K. Eichele and D. Kunz, Dalton Trans., 2017, 46, 29–32 RSC.
- R. H. Cox and H. W. Terry, J. Magn. Reson., 1974, 14, 317–322 CAS.
- H. Oulyadi, Synthesis, 2018, 3603–3614 CrossRef CAS.
- V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320–3334 CrossRef CAS PubMed.
- D. Johnels, A. Boman and U. Edlund, Magn. Reson. Chem., 1998, 36, S151–S156 CrossRef CAS.
- J. W. Ryan, J. Am. Chem. Soc., 1962, 84, 4730–4734 CrossRef CAS.
- R. Kobayashi, S. Ishida and T. Iwamoto, Angew. Chem., Int. Ed., 2019, 58, 9425–9428 CrossRef CAS PubMed.
- M. Bursch, T. Gasevic, J. B. Stückrath and S. Grimme, Inorg. Chem., 2021, 60, 272–285 CrossRef CAS PubMed.
- K.-I. Kanno and M. Kira, Chem. Lett., 2003, 28, 1127–1128 CrossRef.
- J. Braddock-Wilking, Y. Zhang, J. Y. Corey and N. P. Rath, J. Organomet. Chem., 2008, 693, 1233–1242 CrossRef CAS.
- W. P. Freeman, T. D. Tilley, L. M. Liable-Sands and A. L. Rheingold, J. Am. Chem. Soc., 1996, 118, 10457–10468 CrossRef CAS.
- C. Fekete, L. Nyulászi and I. Kovács, Phosphorus, Sulfur Silicon Relat. Elem., 2014, 189, 1076–1083 CrossRef CAS.
- Z. Dong, C. R. W. Reinhold, M. Schmidtmann and T. Müller, Organometallics, 2018, 37, 4736–4743 CrossRef CAS.
- L. Párkányi, J. Organomet. Chem., 1981, 216, 9–16 CrossRef.
- G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed.
- R. West, H. Sohn, U. Bankwitz, J. Calabrese, Y. Apeloig and T. Mueller, J. Am. Chem. Soc., 1995, 117, 11608–11609 CrossRef CAS.
- J.-H. Hong, P. Boudjouk and S. Castellino, Organometallics, 1994, 13, 3387–3389 CrossRef CAS.
- J. H. Hong and P. Boudjouk, J. Am. Chem. Soc., 1993, 115, 5883–5884 CrossRef CAS.
- J.-H. Hong, Molecules, 2011, 16, 8033–8040 CrossRef CAS PubMed.
- Z. Dong, L. Albers and T. Müller, Acc. Chem. Res., 2020, 53, 532–543 CrossRef CAS PubMed.
- W.-C. Joo, J.-H. Hong, S.-B. Choi, H.-E. Son and C. Hwan Kim, J. Organomet. Chem., 1990, 391, 27–36 CrossRef CAS.
- T. Wakahara and W. Ando, Chem. Lett., 2005, 26, 1179–1180 CrossRef.
- M. S. Gordon, P. Boudjouk and F. Anwari, J. Am. Chem. Soc., 1983, 105, 4972–4976 CrossRef CAS.
- U. Bankwitz, H. Sohn, D. R. Powell and R. West, J. Organomet. Chem., 1995, 499, C7–C9 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- M. Saito, M. Sakaguchi, T. Tajima, K. Ishimura, S. Nagase and M. Hada, Science, 2010, 328, 339–342 CrossRef CAS PubMed.
- M. Saito and M. Yoshioka, Coord. Chem. Rev., 2005, 249, 765–780 CrossRef CAS.
- T. He, Z.-W. Qu, H. F. T. Klare, S. Grimme and M. Oestreich, Angew. Chem., Int. Ed., 2022, 61, e202203347 CrossRef CAS PubMed.
- J. Belzner, U. Bunz, K. Semmler, G. Szeimies, K. Opitz and A.-D. Schlüter, Chem. Ber., 1989, 122, 397–398 CrossRef CAS.
- K. Nishihara, T. Kurogi and H. Yorimitsu, ARKIVOC, 2023, 202312017 Search PubMed.
- M. Yus, P. Martínez and D. Guijarro, Tetrahedron, 2001, 57, 10119–10124 CrossRef CAS.
- G. M. Sheldrick, SHELX, Bruker AXS, 1997 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2402953–2402955. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt03537b |
| ‡ This work is dedicated to Professor Vadapalli Chandrasekhar on the occasion of his 65th birthday. |
| This journal is © The Royal Society of Chemistry 2025 |