
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bispidine coordination chemistry
Katharina
Bleher
 *ab,
Patrick A.
Cieslik
*ac and
Peter
Comba
*ad
*ab,
Patrick A.
Cieslik
*ac and
Peter
Comba
*ad
aUniversität Heidelberg, Anorganisch-Chemisches Institut, INF 270, 69120 Heidelberg, Germany. E-mail: katharina.bleher@kit.edu; patrick.cieslik@medma.uni-heidelberg.de; peter.comba@aci.uni-heidelberg.de
bInstitute of Functional Interfaces, Karlsruhe Institute of Technology, Eggenstein-Leopoldshafen 76344, Germany
cMolecular Imaging & Radiochemistry, Institute for Clinical Radiology and Nuclear Medicine, Medical Faculty Mannheim of Heidelberg University, Theodor-Kutzer-Ufer 1-3, 68167 Mannheim, Germany
dUniversität Heidelberg, Interdisziplinäres Zentrum für Wissenschaftliches Rechnen (IWR), INF 205, 69120 Heidelberg, Germany
First published on 30th January 2025
Abstract
Bispidines are extremely rigid ligands, easy to prepare in a large variety, with denticities of four to ten, various donor sets and charges, for mono- and oligonuclear transition metal, main group and rare earth complexes. In the last approx. 20 years significantly more than 50 new bispidine based ligands were prepared and their coordination chemistry studied. Biological probes and medicinal applications is one main area in bispidine coordination chemistry, where fast complex formation, high stability, metal ion selectivity and inertness are of utmost importance. Oxygen activation and oxidation catalysis is another main focus in bispidine coordination chemistry, with catalyst efficiency and stability as well as product selectivity as important requirements. Particularly successful applications in these areas are presented and discussed in detail, in addition to fundamental principles that show the importance of ligand rigidity, cavity size and shape as overarching fundamental properties.
 Katharina Bleher | Katharina Bleher (born in 1991) studied chemistry at the University of Heidelberg and obtained her PhD in 2020 in the research group of Peter Comba. Until 2021, she continued her work in the Comba group on high-valent transition metal chemistry. In 2022, she began a postdoctoral position at KIT under Matthias Franzreb, shifting her research focus to bioprocess engineering and the development of new analytical methods. Since 2024, she has been leading the Analytical Biochemistry group in Matthias Franzreb's department. |
 Patrick A. Cieslik | Patrick A. Cieslik (born in 1994) studied chemistry at Heidelberg University. During his master's thesis and subsequent PhD in the Comba group, he focused on the development of metal-selective chelators for manganese(II), lanthanides(III) and actinium(III), with a particular focus on their therapeutic and diagnostic applications. After completing his PhD in 2021, he continued his scientific career as a postdoctoral researcher in Heidelberg. In September 2022, he joined the research group of Prof. Jason P. Holland at the University of Zurich, where he worked on innovative photoligation methods for the radiolabeling of monoclonal antibodies. Since November 2024, Patrick A. Cieslik is employed as Radiochemist at the University Hospital Mannheim and focuses his research on the further development of radiotheranostic methods. |
 Peter Comba | Peter Comba (born in 1953) was educated at ETH Zürich, obtained a PhD from the Université de Neuchâtel and a habilitation from the Universität Basel. His main teachers were Werner Marty, Alan Sargeson and André Merbach, and he started his own research group in 1986 in Basel before moving to Heidelberg in 1992. He is interested in fundamental features of transition metal and rare earth complexes, in particular in ligand-enforced coordination geometries and their influence on electronics, thermodynamics and reactivities. The methods used involve theoretical and computational chemistry, organic synthesis as well as preparative coordination chemistry, mechanistic work, spectroscopy and magnetism. Current projects include oxidation catalysis, molecular magnetism, biological inorganic and medicinal chemistry. |
Introduction
Bispidines (3,7-diazabicyclo[3.3.1]nonane derivatives)1 were first described in 1930 by Mannich and Mohs.2 The very rigid diazaadamantane derived scaffold is generally prepared in two consecutive double Mannich reactions (see Scheme 1). Stetter and Haller reported in 1957 and 1969 the first transition metal complexes.3,4 This was followed by some studies in the early 1990s,5–7 until a 1997 Dalton paper marked the start of modern bispidine coordination chemistry.8 Bispidines have found applications in pharmaceutical chemistry,9–12 as chiral auxiliaries and organocatalysts,13,14 as building blocks for coordination polymers,15 as ligands for metal-based radiopharmaceuticals16 and medicinally active compounds,17 as chelators for lanthanide-based optical sensors18–20 and manganese-based magnetic resonance imaging (MRI) contrast agents,21–23 as well as for fundamental work in molecular magnetism,24–26 and as ligands for bio-inspired catalysts.27–30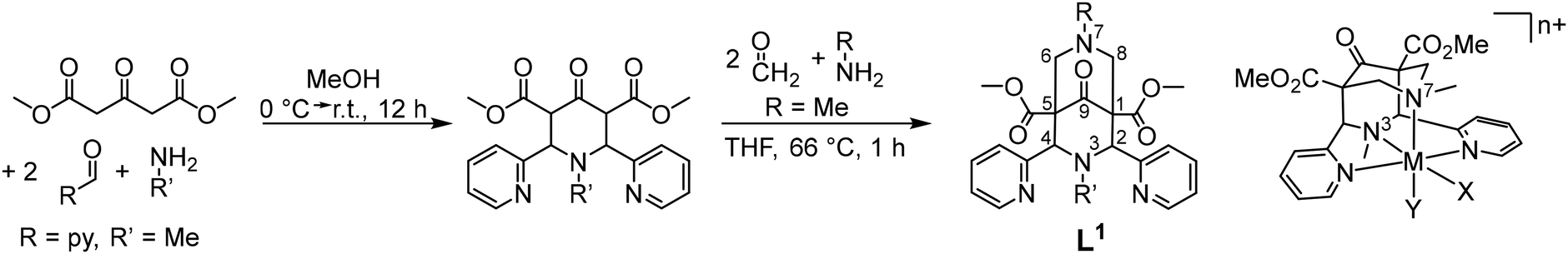 | ||
| Scheme 1 General synthesis of bispidine ligands with atom numbering scheme and a structural plot of an exemplary metal ion complex; this “Mannich bispidine” is L1, the numbering of the other ligands is given in tables and figures throughout, and the specific numbering is generally indicated in relevant tables. | ||
In this essay we describe the properties of bispidine ligands and their metal complexes derived from the original tetradentate “Mannich bispidine” scaffold (L1 = 2,4-bispyridyl-3,7-dimethyl-3,7-diazabicyclo[3.3.1]nonane-9-one-1,5-dimethyl-dicarboxylate, see Scheme 1, which also gives the general atom numbering in bispidines) that enforces square pyramidal or octahedral coordination geometries with one or two co-ligands trans to N3 and N7, i.e. in cis orientation. Ligands with hydrolyzed ester groups at C1 and C5 or a reduced ketone at C9 and/or reduced esters at C1 and C5 are used to vary the stability, charge and donor capacity of the ligands, and derivatives with pendant donors appended to N3 and N7 lead up to decadentate ligands – and this is not the limit in denticity. Not included in the present review are systems without donors at C2 and C4 (often pyridine – ligands with substituted pyridine, histidine, thiazole and quinoline have also been reported),16,31,32 leading to geometries derived from square planar.33,34 Organometallic chemistry is also omitted from this account.35–37 The coordination chemistry of ligands derived from the tetradentate “Mannich bispidine” scaffold has been reviewed.16,30,38,39 A recent publication16 in particular lists all relevant ligands available up to 2018.
The overarching properties of ligands based on the bispidine scaffold are:
(i) Bispidines are highly preorganized. There are two possible orientations for substituents (generally pyridines) at C2 and C4, i.e., exo or endo with respect to N3, and the endo–endo configuration (see Scheme 2) is relevant for coordination to metal ions and generally may be obtained selectively with well-described synthesis protocols.8,38 The two fused six-membered rings with the amine donors N3 (six-ring with the two pyridine donors at C2 and C4) and N7 both may assume chair or boat conformations – the chair–chair conformation is that of relevance for coordination to metal ions and generally is the most stable. Chair–boat conformations (boat at the N7-based ring) of metal-free ligands have been observed, boat–chair conformations (boat at the N3-based ring) are rare (with endo–endo configuration of the bis-pyridyl-bispidines, these would then be forced to axial orientation), and there is only one reported system with boat–boat conformation.38,40 Stabilization of boat conformation occurs with sterically demanding substituents at N7 (or N3), and it may also be stabilized by hydrogen bonding, e.g., with an alcohol at C9 – obviously this then depends on the orientation of the OH substituent, i.e., syn-N3 or syn-N7, and there are protocols for the stereoselective reduction of the C9 keto group to a syn-N7 alcohol.41 Obviously, twist-boat conformations of the two fused six-membered rings are also possible but this has rarely been fully analyzed.42,43 All these stereochemical features have been discussed in detail.8,38,39 The relative energies of chair–chair, chair–boat, boat–chair and boat–boat conformations obviously depends on the substituents at the bispidine scaffold and the level of theory of the computational work but typical energies of the major conformations are 0.0, 50, 120, >150 kJ mol−1 for chair–chair, chair–boat, boat–chair and boat–boat, respectively (see also sections on MRI contrast agents and radiopharmaceutical chemistry below).38,39,42,43 Conformational interconversion is fast if the conformation is not locked by coordination to a metal ion. The conformational equilibria are of importance for the evaluation of formation kinetics and mechanisms, particularly for ligands with multidentate substituents at N3 and N7, and this will be discussed in detail in the sections on MRI contrast agents and radiopharmaceutical chemistry below.42 In general the structure of a fully coordinated bispidine is endo–endo–chair–chair, and usually bispidine ligands are therefore highly preorganized, and this contributes to the often observed high stability of bispidine metal complexes.16,38,39,44
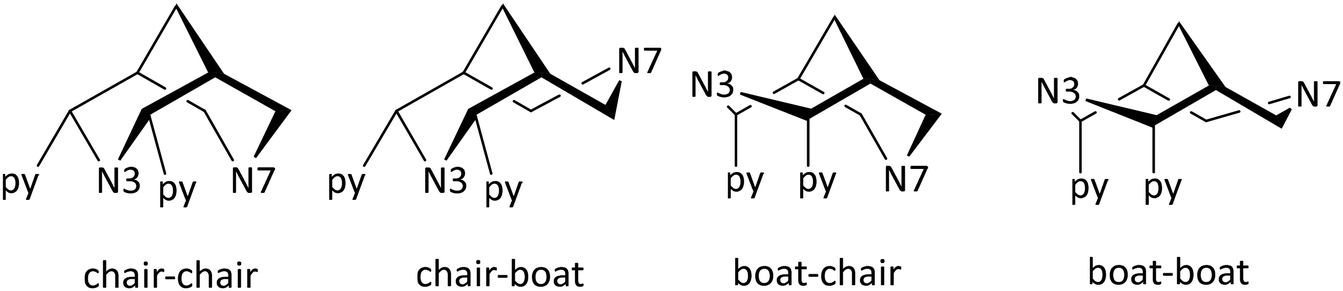 | ||
| Scheme 2 Relevant conformations of bispidines with two pyridine donors at C2 and C4 (i.e., an L1 derivative, equatorial orientation [endo–endo] in the chair–chair and chair–boat conformations). | ||
(ii) Bispidines are very rigid. There are two attributes related to rigidity, i.e., associated with flexibility (conformational flexibility – related to preorganization, see above) and associated with elasticity (variation of bond distances, valence and torsional angles that allow for adaption of the cavity size and shape – related to complementarity).44 In general there is only one isomer of a fully coordinated bispidine (endo–endo–chair–chair), i.e., there is no isomerism in bispidine metal complexes, and the metal-diaza-adamantane structure is rather inelastic, i.e., very rigid.8,16,38,44 However, there is some elasticity with respect to the position of the metal ion in the bispidine cavity, and this has been structurally analyzed.44,45 This may lead to asymmetry and, consequently, to various energy minima with related coordination geometries. In this context, interesting variants of distortional isomerism have been observed with bispidine transition metal complexes, in particular with CuII, where the orientation of the pseudo-Jahn–Teller elongation axis may vary.46–50 Preferences for trigonal vs. tetragonal geometries can also be enforced with substituted bispidines,51 and structural dynamics of CuI systems has been analyzed by X-ray crystallography and NMR spectroscopy,49,52 a property that has been shown to be of relevance for the copper-based activation of dioxygen (see also section on reactivity).27,53,54 The enormous rigidity of the bispidine scaffold with respect to low elasticity of the ligand is shown in Fig. 1a, which is an overlay plot of 183 tetra- to nonadentate ligands coordinated to various metal ions (CSD search in September 2024, all substituents to the tetradentate bispidine scaffold, metal ions and H atoms are omitted). The elasticity of the bispidine coordination sphere is visualized in Fig. 1b, which is Fig. 1a replotted with Bi3+, Cu2+ and Fe2+ coordinated to a nonadentate, a hexadentate and a pentadentate bispidine, respectively (based on the X-ray data; note that, due to the rigidity of the bispidine scaffold, only one conformation is possible for a fully coordinating tetradentate bispidine – obviously this also is true for the backbone of bispidines with higher denticity –, and this also emerges from Fig. 1). Complete encapsulation with high-denticity ligands generally results in very stable metal ion complexes, both thermodynamically and, more importantly, kinetically. As discussed below, this is particularly significant for potential in vivo applications. In the field of radiopharmaceuticals, theranostic pairs must meet specific criteria, such as having the same structure and charge, to ensure that the pharmacokinetic profile of the drug remains unchanged. The rigidity of the bispidine backbone, combined with its inherent conformational stability, makes it an excellent candidate for these applications.
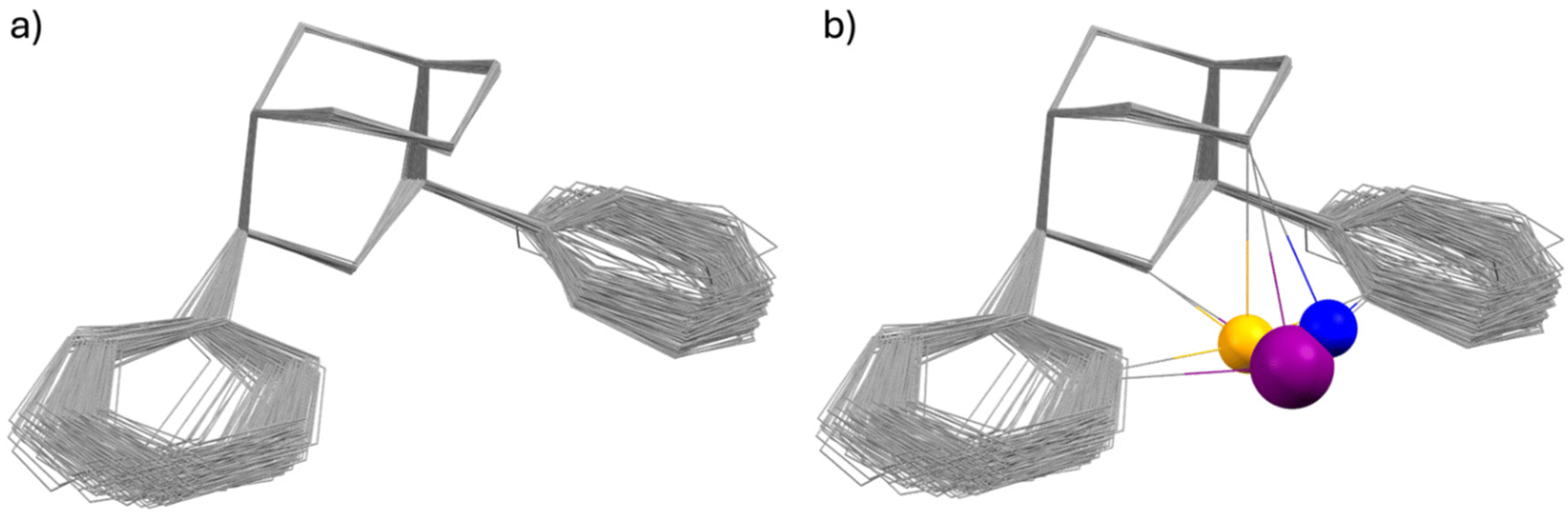 | ||
| Fig. 1 Overlay of the single crystal X-ray structures of 183 bispidine–metal ion complexes depicting (a) the rigidity of the bispidine cavity and (b) the occupation of multiple possible positions of metal ions inside the cavity, with Bi3+ (violet), Cu2+ (blue) and Fe2+ (yellow); note that the position of the metal ion also depends on the pendent donors of the ligand and that sometimes various possible minima are close to degenerate, leading to distortional isomers. | ||
(iii) The bispidine cavity is tetragonal. When coordinated to a metal ion, N3 is part of two very tight 5-membered chelate rings involving the two trans-disposed pyridine donors at C2 and C4: this enforces generally short metal–N3 bonds with very restricted elasticity. N7 on the other hand is part of two more flexible 6-membered chelate rings also involving N3: this also emerges from the overlay plot in Fig. 1. Generally, with CuII (d9, Jahn–Teller active in octahedral symmetry) octahedral geometries with elongated N7–Cu–X axes or square pyramidal geometries with an apical N7 are observed (see Fig. 2). However, the direction of the elongated axis may be varied in dependence of the substituents at N3, N7 and the C2,C4-based pyridine groups as well as possible co-ligands, and square pyramidal geometries with apical N3 as well as octahedral complexes with elongation along N7, N3 or the two pyridine donors have been observed.46–50 This has interesting consequences in terms of complex stabilities (metal ion selectivities), electron transfer properties (stabilization of CuI, CuII, electron self-exchange rates) and reactivities (stabilization of oxygen adducts).27,28,49,54–61 The asymmetry of the bispidine cavity is also of interest in oxidation catalysis with high-valent nonheme iron species because in high-spin configuration of [(L)FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O]2+, one of the four d-electrons is in the dx2–y2 orbital (i.e., part of the degenerate eg set in octahedral symmetry), and stabilization of the dx2–y2 orbital lowers the energy gap between the triplet state (often the electronic ground state) and the quintet state that generally is responsible for the oxidation reactivity (see section on high-valent iron mediated reactions below).62
O]2+, one of the four d-electrons is in the dx2–y2 orbital (i.e., part of the degenerate eg set in octahedral symmetry), and stabilization of the dx2–y2 orbital lowers the energy gap between the triplet state (often the electronic ground state) and the quintet state that generally is responsible for the oxidation reactivity (see section on high-valent iron mediated reactions below).62
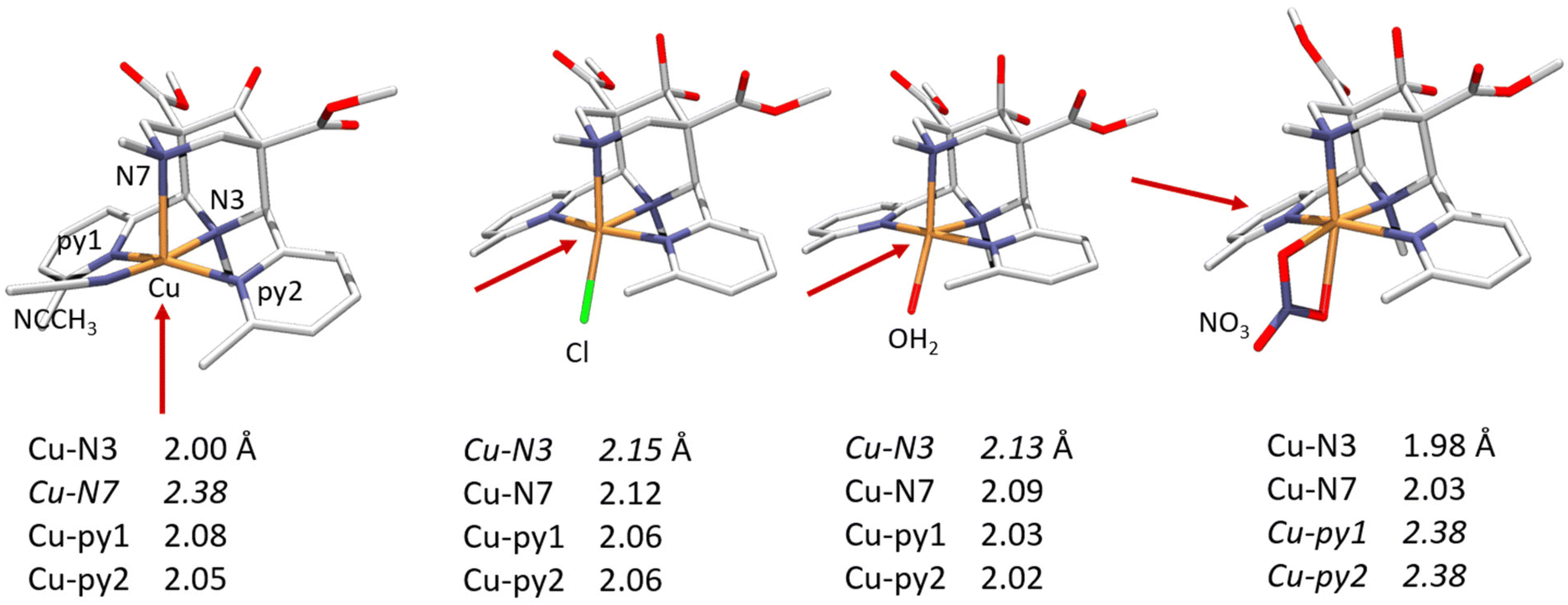 | ||
| Fig. 2 CuII complexes of the tetradentate 2,4-bis-α-methylpyridyl substituted tetradentate bispidine L2, showing how the ligand-enforced tetragonal distortion may be varied by ligand substitution and choice of co-ligands (elongated bonds in italics).50 | ||
(iv) The bispidine cavity is relatively large. As discussed above, the bispidine cavity is very rigid. However, without additional appended donor groups bispidines are not fully encapsulating metal ions, and these may occupy various positions in the cavity (see, e.g., Fig. 1b), i.e., there is asymmetry in terms of energy loss for the coordination of too small and too large metal ions. This has been analyzed by the computation of cavity shape and size curves, generally using molecular mechanics based techniques,63–66 where the loss of steric energy induced to the ligand (variation of ligand-based bond distances, valence and torsional angles and non-bonded interactions) by metal ion coordination is computed by force field calculations, excluding energies based on the metal ions.67 A typical hole-size and shape curve is shown in Fig. 3, confirming that the bispidine cavity is asymmetrical and relatively large.22,23,42,55 This obviously is of importance for metal ion selectivity and high stability, among others for medicinal probes (see sections on MRI contrast agents, radiopharmaceutical probes and optical imaging below). However, it also is of relevance for oxidation catalysis (see section on high-valent iron mediated reactions) since the FeIVO group, e.g., is “too small” for the bispidine cavity and therefore renders the corresponding ferryl oxidants unstable. Since reduction of FeIV to FeIII or FeII is a major decay path and the lower valent iron species are larger, the driving force of bispidine-iron(IV) oxidants is exceptionally large, and the corresponding redox potentials indeed are among the largest ones of nonheme iron(IV) model systems.68–71
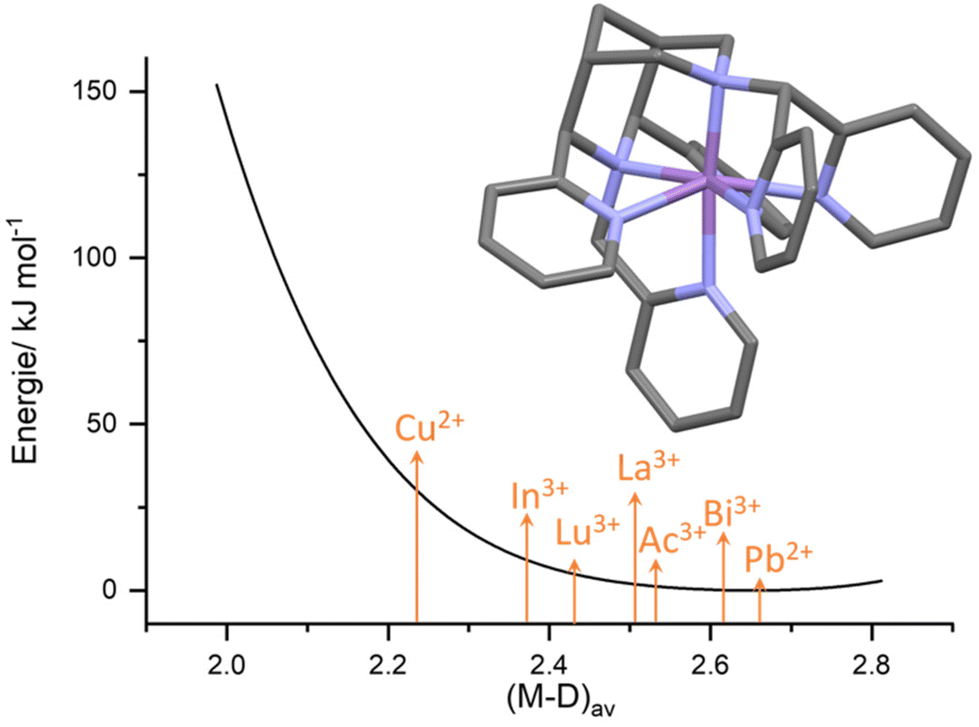 | ||
| Fig. 3 Exemplary hole-size curve calculated for the heptadentate ligand L6 (see Scheme 3 below). Arrows depict the approx. metal ion size (i.e., the average metal–donor distance for the appropriate metal ion – ligand combination) for a range of metal ions relevant for radiopharmaceutical applications.16,22,42,55 | ||
Metal ion selectivity
Thermodynamics and kinetics
For metal-ion-based medicinal probes for radioimaging, radiotherapy, optical imaging and for MRI contrast agents, very high complex stability is a prerequisite. However, inertness (kinetics) is more important than stability (thermodynamics – one reason why thermodynamics is relevant is that medical probes often need to be sterilized, i.e., to ideally withstand thermal sterilization in aqueous solution). In addition to inertness, fast complexation at physiological conditions may be of relevance: physiological conditions are pertinent when sensitive biological vectors such as antibodies are used, and fast complexation is required with radionuclides with relatively short half-lives. The resulting problem is visualized with a potential energy surface plot in Fig. 4: it is obvious that, with a given complex stability and with microscopic reversibility, fast complexation (fast and efficient radiolabeling) and inertness are mutually exclusive. Therefore, the way into and the way out of the ligand cavity need to be enforced to differ, i.e., microscopic reversibility must be prevented. In this respect, bispidines provide two distinct benefits:(i) Complexation of multidentate ligands is a multistep process, also involving isomerization reactions, and less flexible ligands such as macrocycles and cages often lead to slow complexation.72,73 Bispidines are open-chained systems with generally fast complex formation.42,48,74 The “bispidine effect” shown in Fig. 4 originates from the fact that coordination of a metal ion to a ligand is an acid–base reaction, where the coordination of the metal ion competes with protonation of the ligand. The proton at the center of the bispidine cavity is particularly difficult to remove since it is strongly bound to the lone pairs of the two tertiary amines N3 and N7 in chair–chair conformation and further stabilized by the two pyridine donors – the corresponding pKa value generally is very high, i.e., approx. 11.22,75 Once the complex is formed, i.e., when the lone pairs of the tertiary amines N3 and N7 are blocked by a metal ion, reprotonation of the ligand is extremely difficult. That is, proton-assisted decomplexation of bispidine metal complexes is improbable to occur, viz. complexation and decomplexation follow different pathways, i.e., there is no microscopic reversibility,16,22,23,42 and this is an important reason for the generally observed inertness of bispidine complexes. This is well documented with the heptadentate MnII selective bispidines L6 and L7 used as MRI contrast agents (see Scheme 3 and Tables 1, 2 below), where there is no H+ dependent term for transmetallation with Zn2+.
(ii) An additional important feature of bispidines is that the large and rigid cavity only involves two tertiary amines and two pyridines and that the additional donors generally are part of rigid (often planar bi- or tridentate) pendant groups appended to N3 and/or N7 (see the “mousetrap effect” in Fig. 4). While swung out, metal ion coordination to the bispidine cavity is facile and fast, once the pendent groups swing in, the metal ion is trapped – with a favorable design (e.g., a picolinate or terpy pendent arm attached to N3 or N7), protonation of the coordinated multidentate pendant arm does not allow the rigid group to swing out anymore. This is reminiscent of a mouse trap: exit from the trap is not possible once the flap is closed – demetallation does not primarily depend on the thermodynamic stability. A good example to show how efficiently this works is the comparison of complex stabilities (thermodymanics) and serum challenge experiments (kinetics) with In3+ and Lu3+ as the metal ions and the nonadentate bispidine L20 shown in Fig. 4, and compared with DOTA ((1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrayl)tetraacetic acid) and DTPA (diethylenetriamine-pentaacetic acid) as the ligands: the complex stabilities for Lu3+ are similar and high for the three ligands (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 21.0, 23.6 and 22.5 for the bispidine L20, DOTA and DTPA, respectively), for In3+, however, the bispidine stability is much lower (logK = 11.7 vs. 23.9 and 29.0) but the serum stabilities are similar and high for all six systems (see section on radiopharmaceutical chemistry).42,74,75 From the metal ion and bispidine cavity sizes (see above) it was expected that the In3+ bispidine complex has a low thermodynamic stability. However, once the tri- and tetradentate pendent groups are closed in, the metal ion is fully encapsulated and trapped.
K = 21.0, 23.6 and 22.5 for the bispidine L20, DOTA and DTPA, respectively), for In3+, however, the bispidine stability is much lower (logK = 11.7 vs. 23.9 and 29.0) but the serum stabilities are similar and high for all six systems (see section on radiopharmaceutical chemistry).42,74,75 From the metal ion and bispidine cavity sizes (see above) it was expected that the In3+ bispidine complex has a low thermodynamic stability. However, once the tri- and tetradentate pendent groups are closed in, the metal ion is fully encapsulated and trapped.
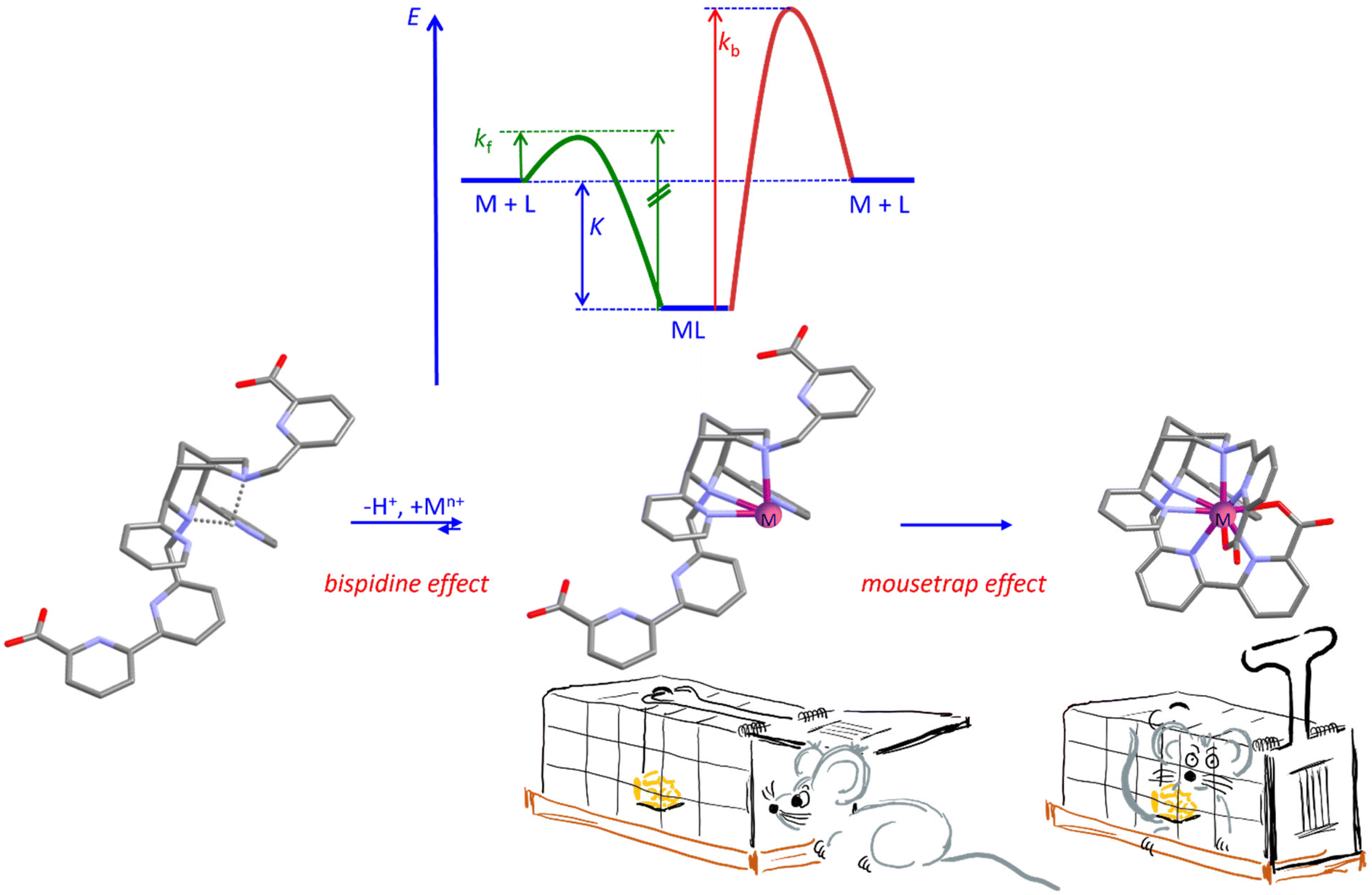 | ||
| Fig. 4 Visualization of the problem of microscopic reversibility, leading to labile coordination with fast complexation (green potential) and prevention of microscopic reversibility with the nonadentate bispidine L20 (see Fig. 6 below), including the “bispidine effect” and the “mousetrap effect” (see text for details). | ||
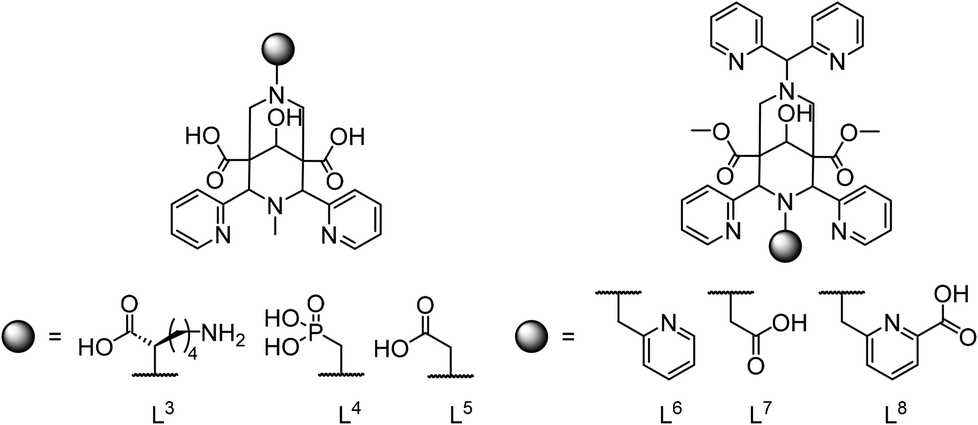 | ||
| Scheme 3 Pentadentate (L3, L4, L5) and hepta- or octadentate bispidines (L6, L7, L8) for the complexation of MnII. | ||
| L3a |
L4a |
L5a |
L6b |
L7b |
L8b |
|
|---|---|---|---|---|---|---|
| a From ref. 82. b From ref. 23. c pM calculated for cM = cL = 10−5 M, pH = 7.4. | ||||||
| logKH1 |
11.44 | 11.15 | 9.54 | 11.05 | 12.26 | 11.90 |
| logKH2 |
10.31 | 7.35 | 5.11 | 6.73 | 6.52 | 5.44 |
| logKH3 |
4.71 | 3.78 | 2.99 | 5.62 | 4.41 | 5.28 |
| logKH4 |
2.76 | 3.02 | — | 5.27 | 2.83 | 1.36 |
| logKH5 |
2.22 | — | — | 2.30 | — | — |
| logKMnL |
12.2 | 13.7 | 11.3 | 24.2 | 19.5 | 24.7 |
| logKZnL |
15.6 | 16.5 | 13.7 | 14.3 | 15.0 | 14.7 |
| pMnc | 6.65 | 7.28 | 7.06 | 12.71 | 9.82 | 12.59 |
| pZnc | 8.40 | 8.76 | 8.29 | 9.50 | 8.73 | 8.58 |
| MnL3a |
MnL4a |
MnL5a |
MnL6b |
MnL7c |
|
|---|---|---|---|---|---|
| a From ref. 82. b Unpublished data. c From ref. 23. | |||||
| CN | 6 | 6 | 6 | 8 | 8 |
| q | 1 | 1 | 1 | 1 | 1 |
| r 1 (mM−1 s−1; 298 K, 20 MHz) | 4.31 | 3.64 | 4.28 | 5.04 | 4.44 |
| k 298ex (107 s−1) | 0.12 | 5.5 | 5.1 | 11.9 | 13.5 |
| ΔH‡ (kJ mol−1) | 25.7 | 14.9 | 10.6 | 31.42 | 34.0 |
| ΔS‡ (Jmol−1 K−1) | −42 | −47 | −62 | — | +25 |
| E rH (kJ mol−1) | 19 | 20 | 22 | 30 | 22 |
| τ r 298H (ps) | 92 | 89 | 100 | 137 | 110 |
MnII selectivity
Manganese complexes have gained increasing importance in medicinal applications, such as MRI contrast agents,76,77 and radiopharmaceutical probes using the positron-emitting isotope Mn-52 are also increasingly attracting interest.78–80 For applications in medicinal chemistry, key factors include inertness, high complex stability and metal ion selectivity, particularly versus ubiquitous first-row transition metal cations, with ZnII being the most abundant and biologically relevant competitor. This highlights the main challenge in developing MnII based drugs, contrast agents, and chelation therapies: MnII complexes are inherently more labile due to the relatively large ionic radius of MnII and the spherical distribution of its d electrons. As a result, MnII coordination compounds tend to have lower stability compared to other first-row transition metal dications, especially ZnII. The Irving–Williams series, established over 75 years ago, describes the stability trend of first-row transition metal dications with any given ligand as MnII < FeII < CoII < NiII < CuII > ZnII.81 The series originally involved mono- and bidentate ligands and coordination geometries derived from octahedral, and it is seen as the basis for the lack of MnII selective ligands.Three possible key design strategies for MnII selectivity, specifically over ZnII, are: (i) preference for MnII over other first-row transition-metal dications due to a large and rigid cavity – depending on the coordination number, MnII is approximately 10% larger than ZnII;22 (ii) using a high-denticity open-chain ligand, where a large cavity may prevent smaller competitors like ZnII from full coordination, thereby reducing the metal–ligand bonding energy; and (iii) ligands with fast MnII complexation – afforded by the fast water exchange rate – that fully encapsulate the metal ion, leading to slow metal ion exchange (see the “mousetrap effect” above, i.e., this is not based on thermodynamics). Bispidine-based ligands fulfill these three prerequisites and have therefore gained prominence in the design of MnII selective ligands. Due to the privileged scaffold, efforts to develop the first MnII selective chelators have been successful.21,43
Pentadentate chelate structures were proposed, featuring a single carboxylate or phosphonate pendent group at N7 or N3.21,43 The corresponding MnII complexes exhibit exceptional kinetic inertness: in challenge experiments involving an excess of ZnII or CuII ions, the MnII complexes demonstrated remarkable kinetic stability, maintaining their integrity even over the span of several months.21,82 However, the thermodynamic stability with the three pentadentate bispidine derivatives L3, L4 and L5 was only modest (see Fig. 5; logKMnL3 = 12.21; logKMnL4 = 11.26; logKMnL5 = 13.72) and significantly lower than for the corresponding ZnII complexes (logKZnL3 = 15.59; logKZnL4 = 13.72; logKZnL5 = 16.54), i.e., these ligands are not MnII selective (see Table 1). Important to note is that the introduction of a phosphonate donor increases the stability of the complexes by approx. 2.5 orders of magnitude but does not change the metal ion selectivity. The rigidity of the ligand skeleton is a critical factor in enhancing the inertness of MnII complexes, and this was described as the “bispidine effect” in Fig. 4 and renders these ligands suitable for imaging applications (see below).
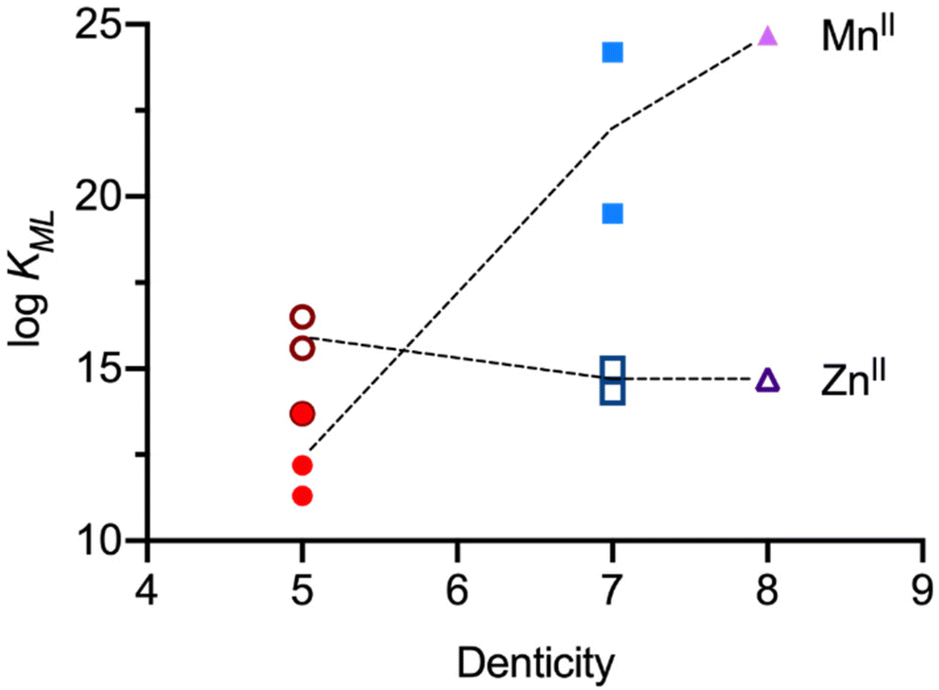 | ||
| Fig. 5 Visualization of the formation constants (logKML) for various MnLn and ZnLn bispidine complexes, categorized by ligand denticity. Filled circles (MnII complexes with pentadentate bispidines), filled squares (MnII complexes with heptadentate bispidines), and filled triangle (MnII complex with an octadentate bispidine); hollow circles (ZnII complexes with pentadentate bispidines), hollow squares (ZnII complexes with heptadentate bispidines), hollow triangle (ZnII complex with octadentate bispidine). The complexes are arranged according to ligand denticity. Dashed lines serve as visual guides, connecting the mean stability constants of the MnII and ZnII complexes for each ligand type. | ||
With the design and synthesis of hepta- and octadentate ligands L6, L7 and L8 (see Scheme 3), the first examples of significant thermodynamic selectivity of MnII over ZnII was reported.22,23 These chelators form octa-coordinate MnII complexes with exceptional stability (logKMnL values that are five orders of magnitude larger than those of the most stable previously known systems), and MnII/ZnII selectivities, with MnII stabilities exceeding those with ZnII by 5–10 orders of magnitude. The complexes achieve pMn values of 12.73, 9.82, and 12.59 for [Mn(L6)(OH2)]2+, [Mn(L7)(OH2)]+ and [Mn(L8)], respectively (see Table 1). These values significantly surpass those of traditional manganese complexes such as [Mn(DOTA)(OH2)], which has a pMn value of only 9.02.83 Note that pM values are conditional parameters (here reported for pH 7.4) and, apart from the very high logK values of the bispidines, the large difference to DOTA is also due to the difference in basicity between the bispidine derivatives and DOTA. To overcome the Irving–Williams series clearly was a challenge and a feat. It can be attributed to the large and rigid diazaadamantane-derived cavity of the bispidine scaffold, which is perfectly suited for MnII coordination, allowing for an eight-coordinate geometry, a rare configuration for MnII complexes. The bispidines efficiently wrap around the MnII ion, facilitating optimal coordination geometry and minimizing steric hindrance. In contrast, competitors like ZnII are too small for this cavity and can only bind 6 of the 7 or 8 potential donor atoms, resulting in both lower binding energy and increased steric strain on the ligand. Interestingly and in stark contrast to MnII, the stabilities of all six ZnII bispidine complexes only differ by max. 2 orders of magnitude (Table 1, and Fig. 5). While the octadentate ligand L8 fully encapsulates MnII, offering the highest known MnII complex stability, the MnII complex with the pentadentate ligands L3–L5 as well as the heptadentate ligands L6 and L7 feature one inner-sphere water molecule in aqueous solution (see Table 2). This makes their MnII complexes promising candidates for use as MRI contrast agents.
MRI contrast agents
MnII bispidine complexes have emerged as promising candidates for MRI contrast agents. The unique electronic and structural characteristics of Mn ions, especially in their divalent state with five unpaired electrons in high-spin configuration and fast H2O exchange rates, make them suitable for enhancing MRI contrast.84 The development of manganese-based contrast agents has been driven by the need for safer alternatives to GdIII based compounds, which have been associated with nephrogenic systemic fibrosis and other adverse effects.85,86 Manganese, being a naturally occurring element in the body, presents a lower risk for toxicity when properly chelated. Recent studies have focused on synthesizing MnII complexes that exhibit high thermodynamic stability and kinetic inertness, which are essential for ensuring that the contrast agents remain effective in vivo without releasing free and therefore toxic MnII.87,88 One of the significant advancements in this field is the design of MnII complexes using bispidine ligands, which enhance the stability and kinetic inertness of these complexes, as discussed above.Table 2 provides a summary of the key characteristics relevant to MRI contrast agents for bispidine complexes of MnII. All complexes have a coordination number of either 6 or 8 with a single water molecule coordinated to the metal center. Notably, the longitudinal relaxivities (r1) for all complexes are high, ranging from 3.64 mM−1 s−1 for [Mn(L4)(OH2)]+ to 5.04 mM−1 s−1 for [Mn(L6)(OH2)]2+.23,82 These values are comparable to [GdIII(DOTA)(OH2)], which has an r1 relaxivity of 4.2 mM−1 s−1 under the same conditions.23,89 The highest relaxivities correspond to the L6 and L7 based octacoordinate complexes, placing them among the highest for mono-hydrated MnII complexes. These octacoordinate MnII complexes exhibit significantly larger water exchange rates than their hexacoordinated counterparts, with rates of 11.9 × 107 s−1 and 13.5 × 107 s−1 for [Mn(L6)(OH2)]2+ and [Mn(L7)(OH2)]+, respectively. This results from the longer and therefore weaker Mn–OH2 bonds, which has steric as well as electronic reasons and is corroborated by the X-ray crystal structures of the [Mn(L6)(OTf)]+ and [Mn(L7)(MeOH)]2+ cations, which show bond lengths to the monodentate ligands of 2.3804(16) Å and 2.182(3) Å, respectively.22,23 Interestingly, the ΔS‡ values of the compounds with pentadentate bispidines (hexacoordinate complexes) are negative, suggesting associative H2O exchange, while that of the octacoordinated complex with the heptadentate L7 is positive, as expected for a dissociative process. All complexes presented in Table 2 exhibit characteristics that make them well-suited for in vivo use as MRI contrast agents. Consequently, [Mn(L4)(OH2)]+ and [Mn(L7)(OH2)]+ were evaluated for their efficiency in initial studies on wild-type mice, showing promising potential as MRI agents.21,23 In both cases, a noticeable enhancement in contrast was observed, with the clearance of the complexes occurring via the renal pathway. Additionally, no accumulation of MnII was detected in various tissues when compared to non-treated mice, indicating the high in vivo stability of these compounds. Nevertheless, further in vivo studies are needed to comprehensively assess their safety and efficiency in clinical settings. Also, it appears that further derivatives may need to be developed to increase water solubility. Note that, while the thermodynamic stability is not of primary importance for in vivo applications – the kinetics, i.e., inertness is the main requirement – stability plays a significant role, e.g., for sterilization and storage.
Radiopharmaceutical chemistry
As mentioned earlier, due to their high thermodynamic stability and kinetic inertness bispidine chelators are ideal for complexing a wide range of metal ions. Importantly, efficient metal ion complexation typically occurs at room temperature, making these systems a promising platform for medicinal applications. Over the past 15 years, bispidine chelators have been extensively studied in radiopharmaceutical research (Fig. 6), especially with the rise of diagnostic tools utilizing radioactive nuclides, such as positron emission tomography (PET) and single photon emission computed tomography (SPECT).16 The growing interest in metal radioisotopes (transition, main group and rare earth metal ions) stems from the fact that radiolabeling with radiometal ions can be achieved in a final step, unlike organic radioactive compounds that often require multiple reaction steps and therefore result in lower radiochemical yields or lower overall activities.90,91 Examples of diagnostic metal radionuclides include Mn-52, Cu-64, Ga-68, Tc-99 m and In-111 for imaging, while therapeutic radionuclides include Cu-67, Tb-161, Lu-177, and Ac-225.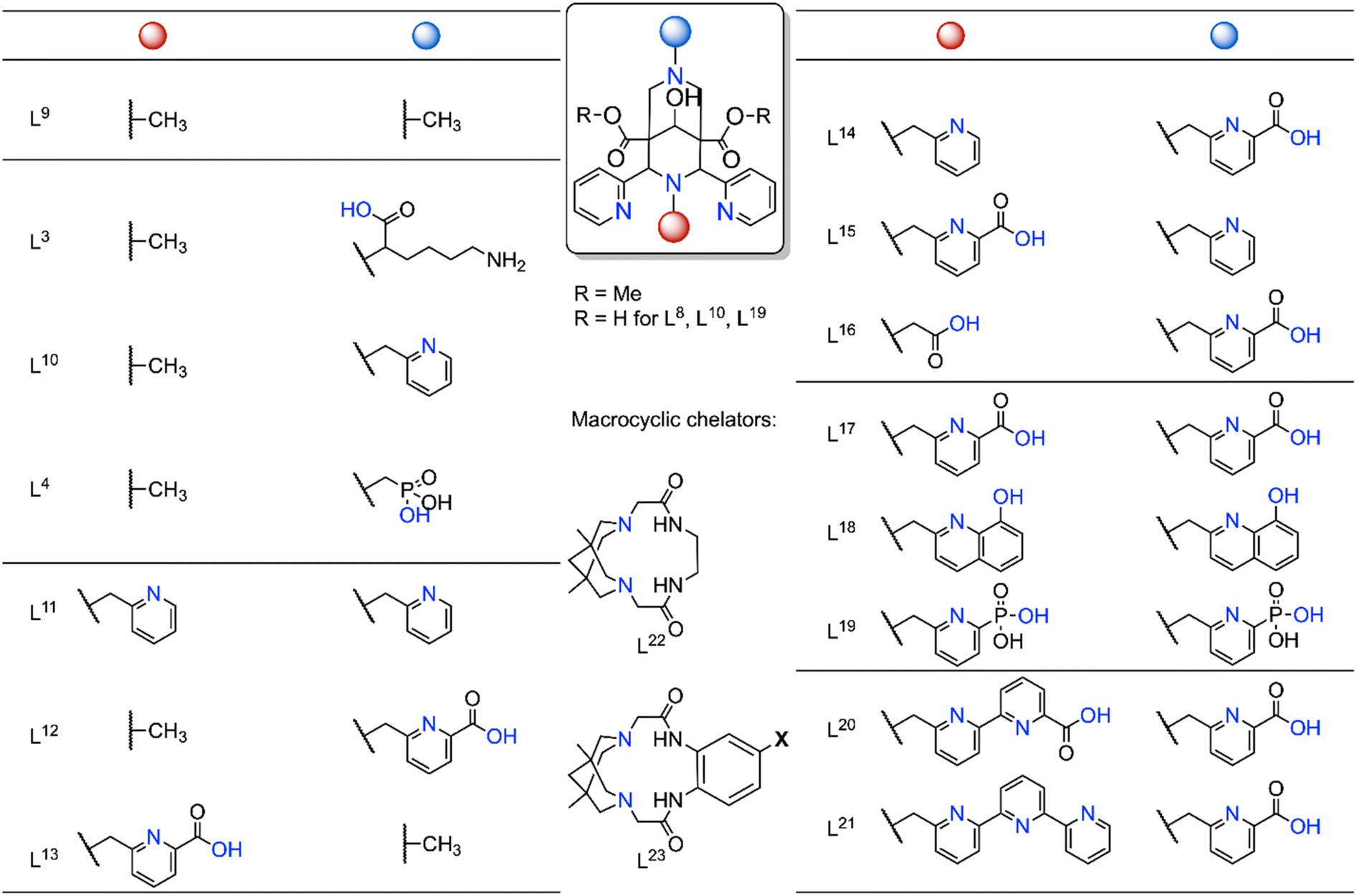 | ||
| Fig. 6 Selected bispidine chelators investigated for radiopharmaceutical applications. Coordinating atoms are marked in blue (L9 and the macrocycles L22 and L23 are tetradentate, L3, L10, L4 are pentadentate, L11–L13 are hexadentate, L14–L16 heptadentate, L17–L19 octadentate, L20, L21 nonadentate, and the latter are suitable for main group and rare earth metal ions and possibly in certain cases also for MnII).16,42,48,55,74,75,92–99 | ||
Copper offers two highly interesting radionuclides for use in radiopharmaceuticals: Cu-64 and Cu-67.100 Cu-64 is a positron-emitting radionuclide with a half-life of 12.7 hours, making it ideal for PET imaging. On the other hand, Cu-67 emits beta particles and has a half-life of 61.7 hours, which makes it suitable for therapeutic purposes. Since both radionuclides belong to the same element, they are considered a true theranostic pair, meaning that copper complexes can be used both for diagnosis and treatment in cancer therapies. In recent decades, significant effort has been invested in developing chelators for stably binding copper ions, particularly CuII, to enable their effective use in medical applications. A major challenge in the development of copper-based radiopharmaceuticals is the in vivo reduction of CuII to CuI, which complicates the task of stably binding copper.101,102 The main reason is that most CuI complexes generally have much lower (and quite similar) thermodynamic stabilities.103,104 This challenge has driven significant research, including efforts to use the bispidine scaffold for CuII chelation. Over the past 15 years, the development of bispidine-type ligands has focused on modifying the structure by varying the substituents at N3 and N7 with different mono-, bi- and tridentate coordinating groups, while keeping the pyridines at C2 and C4 as constant donors (Fig. 6). The bispidine scaffold inherently provides four nitrogen donors (two amines and two pyridines), and two additional donors appended to N3 and/or N7 can be inserted to enhance the CuII binding affinity.92 Additionally, macrocyclic bispidine ligands (L22, L23) have also been developed and tested for their potential in radiopharmaceutical applications.93 While tetradentate chelators were found to lead to fast and unfavorable transchelation,92 hexadentate chelators L11, L12 and L13 lead to complexes of high stability and inertness. Transchelation in superoxide dismutase (SOD) and human serum are negligible for all investigated Cu-64 complexes of the hexadentate chelators L11–L13 (>95% intact complex after 1 h), and are in the same range as DOTA, NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) and diamsar (1,8-diamino-3,6,10,13,16,19-hexaazabicyclo[6,6,6]eicosane) CuII complexes.16,48,55,96,105
Bifunctionalization and the attachment of biological vectors to the bispidine scaffold can be efficiently accomplished through modifications at various positions of the bispidine backbone: functional groups at the C1 or C5 ester positions offer a straightforward route for conjugation, as does the O-alkylation of the C9-hydroxy group on the bispidine backbone. This flexibility allows for the attachment of various bioactive vectors through a range of bioconjugation chemistries, including esterification, amine coupling, amide bond formation, isothiocyanate linkage, maleimide addition, and click chemistry (e.g., alkyne–azide cycloaddition).18,75,92,93,96,105–107 Studies on copper-64 radiolabeled bispidine ligands attached to biological vectors demonstrate that the vector's intrinsic biological activity is maintained and this suggests that the bispidine structure does not interfere with or alter the functionality of the attached vector, underscoring its suitability as a versatile and effective chelating agent. The approval of Ga-68 based radiopharmaceuticals for imaging and the growing interest in Ga-67 for therapeutic purposes108 have spurred efforts to adapt chelators like bispidine for gallium coordination. However, the bispidine scaffold, while promising for other metal ions, seems to face limitations with GaIII.48,109 The need for high concentrations of chelator L3 and elevated temperatures to achieve decent radiochemical yields highlights some inefficiencies. This may stem from the fact that GaIII prefers oxygen donors, whereas bispidine's nitrogen-rich structure may not provide the optimal environment for GaIII binding. However, transchelation with the hexacoordinate ligands has not been studied so far, and we have discussed above that thermodynamic stability might not be of primary importance (see the “mousetrap effect”). Further work could also explore modifying the bispidine framework by introducing oxygen donors or hybrid systems that combine the strengths of bispidines with other ligand types. Alternatively, different scaffolds might offer a more natural fit for gallium coordination.
In the field of theranostic agents, significant progress has been made in developing new chelators for alpha- and beta-particle emitting radionuclides, particularly given the rise of radioimmunotherapy, which leverages antibodies as biological vectors. These vectors are often thermolabile, meaning that chelating agents must facilitate labeling under mild conditions, typically at temperatures ranging from room temperature to 40 °C. Ligands with coordination numbers between six and nine have shown great promise in this regard, and first examples of bispidine–antibody conjugates have been reported.96,110 A major area of interest in chelator development has been the f-block elements, as many therapeutic nuclides, especially those suited for antibody-based delivery, are lanthanide(III) and actinide(III) ions. These tend to form stable complexes with chelators that can provide nonacoordinate environments. Recent efforts in chelator design have focused on improving the selectivity and stability of bispidine chelators for such applications, aiming to ensure that the complexes remain inert under physiological conditions, while chelators are easy to label at low temperatures to preserve the functionality of the antibody vectors.
The exploration of the heptadentate bispidine chelators L14, L15, L16 and the octadentate ligand L17 has yielded initial results in radiolabeling of bispidines with BiIII.95 Due to its potent alpha-emitting properties bismuth-213 is a highly valuable radionuclide for targeted alpha therapy (TAT).111 It is generated as a daughter product of actinium-225. Bi-213 transforms via alpha-decay into Pb-209, which undergoes β− decay to eventually form stable Bi-209. The branching of decay pathways and the emission of high-energy alpha particles make Bi-213 ideal for TAT, where localized delivery of powerful alpha radiation can destroy cancer cells with minimal damage to surrounding healthy tissue. Detailed studies showed that nonadentate bispidines (L20, L21) are not only ideally suited for lanthanides and actinides75 but also for BiIII (as well as PbII).42 In comparison to conventional ligands like DOTA, the bispidine-based chelators exhibit higher labeling efficiency and relatively good stability in challenging conditions, such as competition with DTPA, with the heptadentate and octadentate analogues being less inert. It is to mention that the octadentate ligand L17 demonstrated temperature-dependent complex stability, showing greater resistance to decomposition when radiolabeled at higher temperatures (95 °C compared to rt).95 Temperature-dependent behavior was also noted in studies involving [Mn(L6)(OH2)]2+ (ref. 22) and radiolabeling experiments with L20 and L21,42 suggesting that equilibria involving various isomers may play a significant role in the stability of these complexes.
To gain further insight into the complexation and decomplexation mechanisms, density functional theory (DFT) calculations were employed.42 The preliminary analysis shows multiple coordination pathways, also including chair–boat conformations and out-of-cage complexation, leading to various time regimes in the complexation and possibly less inert species when complexation occurs at low temperature. The bispidine ligands can exist in three forms (see Scheme 3), with the most stable being the chair–chair conformation (Lcc) that is able to fully encapsulates metal ions (see Fig. 7). The chair–boat (Lcb) and boat–chair (Lbc) forms of the metal-free ligand are significantly less stable. Importantly, due to the pyridine substituents at C2 and C4, the boat conformation involving N3 is significantly less stable than that involving N7, i.e., the two relevant conformations are Lcc and Lcb. Complexation of metal ions to multidentate bispidines is a multistep process with initial coordination of the metal ion to the residues attached to N3 or N7 (see Fig. 7). In chair conformation of the corresponding six-ring, the pendent group at N3 may form a tridentate environment for pre-complexation, and the picolinate at N7 in boat conformation of the corresponding six-ring together with N7 also provides tridentate binding. The metal-bound complexes exhibit dynamic isomerization equilibria, such that [MLcc]a, [MLcc]b and [MLcb]a exist in equilibrium with partially protonated ligand forms. Low-energy rotation around C–C single bonds facilitate the transformation of out-of-cage pre-equilibria to the fully encapsulated metal ion. However, formation of the pre-complex [MLcb]a represents a “dead end” since full encapsulation of the metal ion is only possible by cleaving a metal–ligand bond (formation of [MLcb]b), a process that energetically is extremely unfavorable.
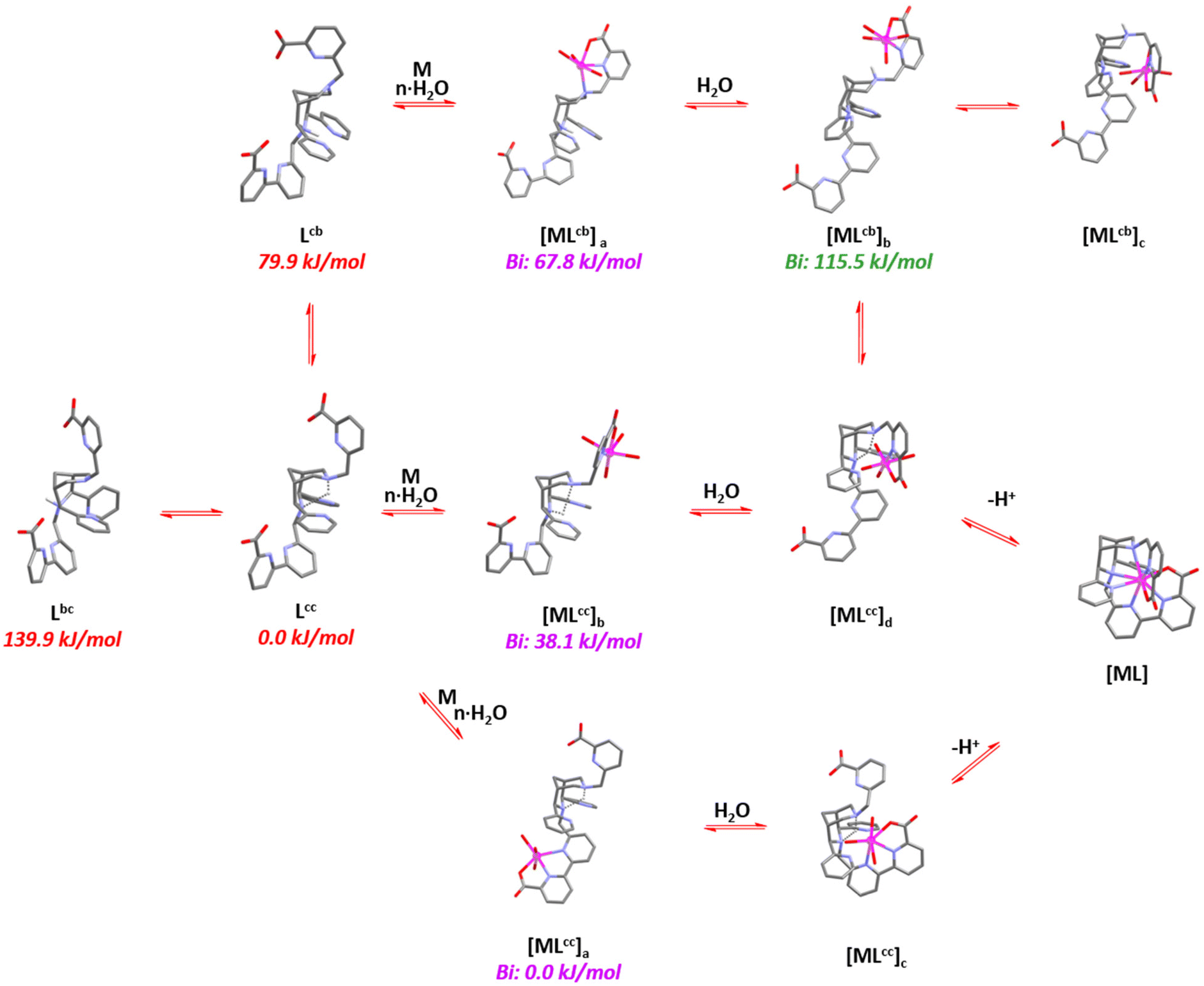 | ||
| Fig. 7 Proposed mechanism of complex formation with the nonadentate bispidine L20 (updated and redrawn from ref. 42). Energies in red (monoprotonated ligand) and blue (“out of cage complex”) are isomers, i.e. the relative red energies as well as the blue onew are comparable. Note that all Bi3+ complexes are computed as 8-coordinate, and this might not be realistic but should not have a major influence. | ||
L17 was further investigated for its ability to complex various metal ions of radiopharmaceutical interest, like In-111, Lu-177 and Ac-225, and the analogue L18 with oxine-donor groups at N3 and N7 was also developed.94,112 Both compounds showed promising labeling efficiency with In-111, a radionuclide valuable for SPECT imaging and discussed as a potential candidate for Auger electron therapy.113 Radio-complexation occurs at room temperature with high molar activities, and the In-111 complexes displayed high stability in human serum, a critical factor for in vivo applications. However, radiolabeling with Lu-177, a β−-emitting radionuclide used in β therapy,114 showed that L17 exhibited inferior labeling efficiency compared to other chelators. In prior studies, structures designed for lanthanide(III) and bismuth(III) ions typically formed complexes with nonacoordinate central ions, with additional mono- or bidentate ligands completing the coordination sphere.94,95,112 As a consequence, two nonadentate chelators, L20 and L21, were developed, which achieved full encapsulation of a variety of lanthanide(III) ions as well as BiIII.42,75 Interestingly, in various examples, very high inertness is obtained despite relatively low thermodynamic stability, as shown with the LuIII complexes of L17 and L18,74 and with the InIII complex of L20.23,42 Obviously, in vivo inertness is the main or only prerequisite for medicinal applications, and the seeming contradiction between kinetics and thermodynamics has been discussed above as the “mousetrap effect” (see Fig. 4).
The investigation of the complexation behavior of the bispidines with various radionuclides, including In-111, La-133, Lu-177, Pb-212, Bi-212/213, and Ac-225, highlights the superior radiolabeling properties of L20 over L17 and L21. The nonadentate L20, featuring two carboxylate donors, achieves quantitative radiolabeling with all radiometal ions at mildly elevated temperatures of 40 °C. The resulting radiocomplexes exhibit remarkable stability in human serum and challenge experiments with competing ligands, like DTPA, comparable to the gold-standard chelator DOTA. Building on its success, L20 was further functionalized with the targeting biovector Tyr3-octreotate,75 and has also been bioconjugated to a monoclonal antibody, targeting the HER2/neu receptor.110 The study involving the peptidic vector, showed similar efficiency in therapeutic studies as the DOTA analogue but the bioconjugated radiocomplex demonstrated significant liver accumulation in tumor bearing mice. However, the primary advantage remains in the mild labeling conditions, allowing for efficient radiolabeling without compromising the integrity of thermolabile biomolecules. Additionally, photoradiolabeling of trastuzumab (a monoclonal antibody) was achieved with a decent radiochemical yield of approximately 14%. The specificity of the radiolabeled antibody towards the HER2-positive tumor was confirmed in ex vivo biodistribution studies, which indicated that the chelator did not significantly alter the biological behavior of the antibody and underscores L20's potential in targeted radiopharmaceuticals, which balances efficient labeling, thermodynamic stability, kinetic inertness and biological compatibility.
Increasing radiochemical yields and reducing nonspecific tumor accumulation remain active goals for optimizing these chelators. Stability studies in human serum have highlighted differences in stability among the radiocomplexes with L20, with the Ac-225 complex maintaining only 85% integrity after seven days, compared to 94% for the Lu-177 analogue, and this suggests a need for continued refinement in chelator design, specifically for Ac-225. One promising direction involves designing decadentate bispidine-based ligands (e.g., L24), incorporating three picolinic acid groups at N3 and N7,115 aimed at providing an optimal fit for the larger actinium(III) ion (Fig. 8), which requires a more spacious coordination environment for robust binding.116
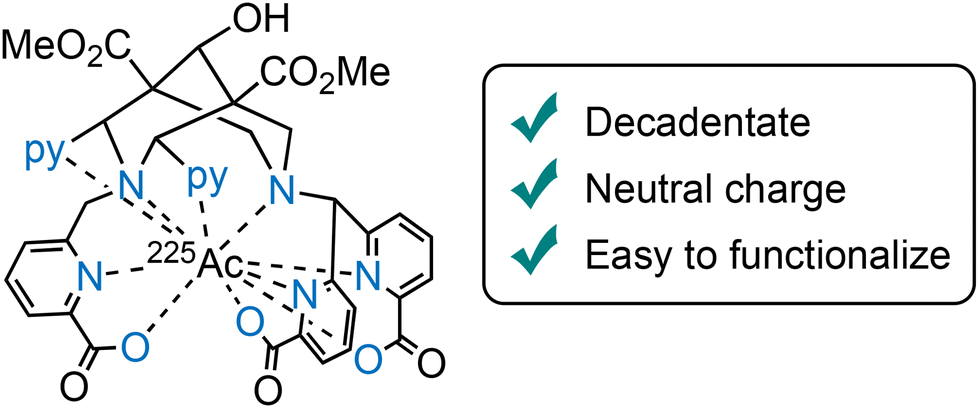 | ||
| Fig. 8 Ac-225 complex of the decadentate bispidine chelator L24 for the selective coordination of AcIII. | ||
Optical imaging
Lanthanide(III) complexes are gaining attention in optical imaging due to their unique photophysical properties, such as distinct, sharp emission bands and large shifts relative to the excitation wavelengths (pseudo-Stokes shifts). However, lanthanide(III) ions inherently exhibit low absorption intensity because of the Laporte exclusion, and this prohibits direct excitation of the lanthanide centers. A common solution involves absorption at the ligand, followed by energy transfer to the metal ions’ excited states. This antenna effect is often explored in molecular probes.117 Initial studies with the octadentate bispidine ligand L17 coordinated to various lanthanide(III) ions demonstrated the potential of the bispidines not only as privileged chelators but also as efficient energy transfer antennas.19 However, the octadentate ligand allowed for water molecules to coordinate to the nonacoordinate complex, confirmed by X-ray crystallography and fluorescence lifetime measurements in water and D2O. A recent study on TbIII complexes of a derivative of L17 (hydrolyzed ester groups at C1 and C5 and inverted stereochemistry at C9 [not correctly drawn in the manuscript]) and the corresponding bis-phosphonate ligand L19 as expected show very similar thermodynamic and kinetic properties of the “new” bis-picolinate ligand and similar thermodynamic but inferior kinetic properties of the bis-phosphonate chelator, with the expected relatively low excited state quantum yields due to the coordinated water molecule.97 As the coordination of water can quench luminescence, the nonadentate bispidine ligands L20 and L21 were developed to fully encapsulate the lanthanides – in addition to preventing fast radiationless decay of the excited states this obviously also increases complex stability and inertness. X-ray structural analysis of the nonadentate ligands coordinated to various LnIII ions (see Fig. 9a) shows identical coordination geometries with all accessible metal ions.20 This similarity suggests that changing the central lanthanide ion does not alter the coordination and that no water is coordinated to any of these probes in solution, ultimately enhancing luminescence efficiency (note also that, for theranostic radiometal probes, this is of importance with respect to matched pairs).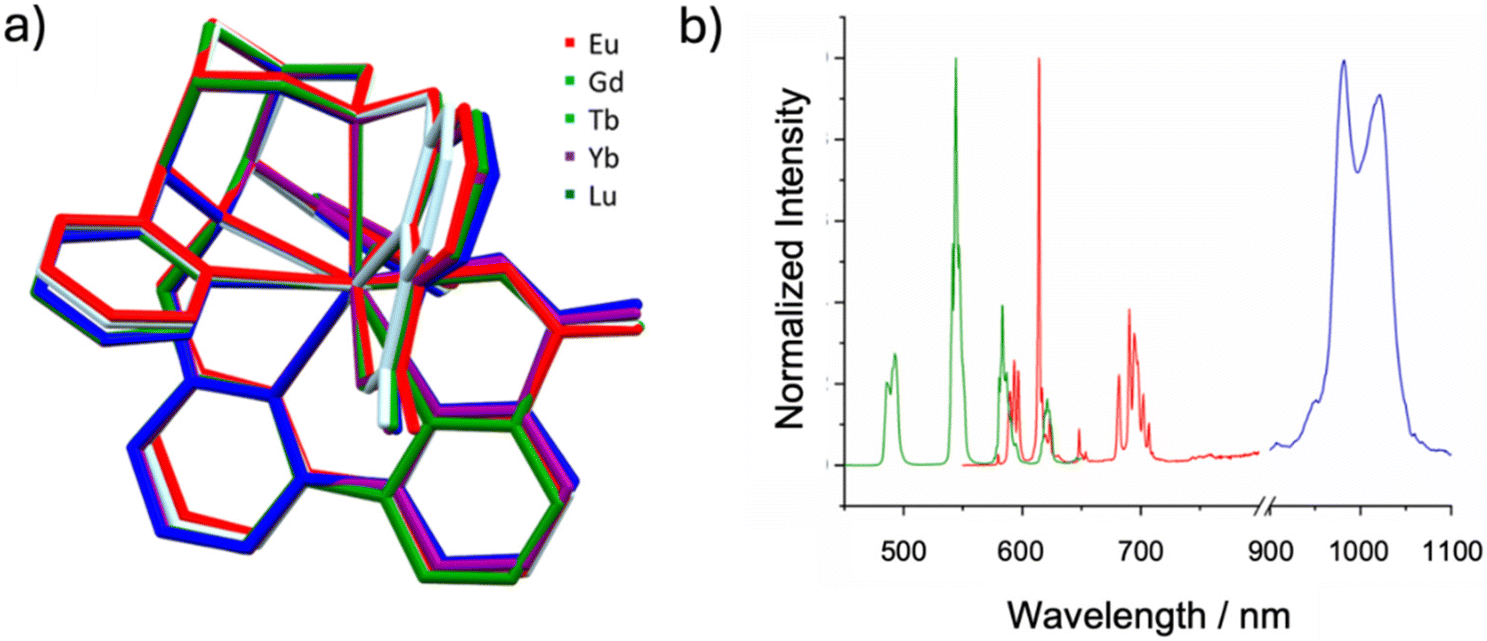 | ||
| Fig. 9 (a) Overlay of [Ln(L20)]+ complex cations from single crystal X-ray structures (non-coordinating residues, hydrogen atoms and counter ions are omitted for clarity); (b) normalized emission spectra of the EuIII (red), TbIII (green) and YbIII (blue) complexes of L20 in water at room temperature, when excited at their respective absorption maximum (see Table 3). | ||
The nonacoordinate metal complexes of EuIII and TbIII exhibit excellent photophysical characteristics, including high luminescent lifetimes and favorable quantum yields (Table 3). With excitation at 313 nm, they are effectively sensitized at near double the energy of the emission in the biological window, typically defined as starting around 650 nm (Fig. 9b). This suggests that with slight red-shifting of the excitation wavelength, these compounds could become highly valuable for optical imaging applications as two-photon luminescent probes in biological contexts. Modifying the ligand at N7 by substituting the picolinic acid group with an analogue featuring a larger π-conjugated system can shift the excitation wavelengths above 350 nm, expanding their applicability towards two-photon lanthanide luminescent probes (2P-LLBs).118–120 This adjustment allows the complexes to be excited in the biological window, a critical feature for applications like two-photon excitation microscopy, where such imaging probes are desirable for their minimized photodamage and deeper tissue penetration. Despite a reduction in quantum yield, the brightness of these complexes remains constant, underscoring their suitability for high-sensitivity imaging without significant trade-offs in optical performance.18–20,121
| Compound | Medium | λ abs max (nm) | ε (L mol−1 cm−1) | λ em max (nm) | τ (ms) | Φ |
|---|---|---|---|---|---|---|
| [Eu(L20)][CF3CO2] | H2O (D2O) | 261 | 13500 |
614 | ||
| 314 | 8000 | 1.51 (2.15) | 0.35 | |||
| [Tb(L20)][NO3] | H2O (D2O) | 261 | 16000 |
|||
| 313 | 9000 | 544 | 1.95 (2.21) | 0.68 | ||
| [Yb(L20)][NO3] | H2O | 262 | 15000 |
3.54 | ||
| 312 | 9500 | 979 |
In addition, the bispidine scaffold has been functionalized with organic dyes, such as Cy5.5 and BODIPY, introducing additional optical properties to the scaffold. These modified structures demonstrate potential as bimodal imaging probes: when complexed to Cu-64, these probes can serve as PET imaging agents, while the fluorophore component provides a luminescent signal for optical imaging. This results in dual functionality, combining complementary imaging capabilities within a single probe.18,20
Reactivity
Tetra- and pentadentate bispidines provide a rigid cavity for metal ions and partially shield them, while leaving an open site for the interaction with reactants and therefore are appropriate for metal-ion-initiated organic transformations. The rigidity and relatively large cavity size stabilize relatively large and therefore rather low- than high-valent metal ions, i.e., for redox processes (e.g., C–H activation and heteroatom group transfer by high-valent metal compounds), bispidine-based transition metal catalysts generally are very efficient oxidants, i.e. often excellent catalysts. This has also been termed a “bispidine effect”. The reactivity of oxidants with tetradentate bispidines generally differs significantly from those with pentadentate derivatives. This is partly due to differences in the shielding of the metal center from oxidants and substrates but, particularly with copper and iron centers, there is a shift of the redox potentials that strongly increases the reactivity of the metal-based oxidants with tetradentate bispidines. With the less reactive systems with pentadentate bispidines, often reactive intermediates are stabilized, allowing for their characterization with various spectroscopic tools. In the following, we will briefly discuss various examples and analyze the influence of the bispidine scaffold on the reactivity and pathways.Copper mediated reactions
The coordination of copper by bispidines, especially the tetradentate “Mannich bispidine” L1 (see Scheme 1) holds a special significance: the extremely rigid bispidine ligands are preorganized to support tetragonal geometries, stabilizing (pseudo)-Jahn–Teller active systems. This is evident in the structurally characterized complexes, where the bond lengths of the metal ion to the aliphatic amines N3 and N7 vary significantly (see Fig. 2).46–50 The usually short bond to N3 results in co-ligands preferring to coordinate in the pyridine/N3 plane, leading to short and strong bonds to the coordinating monodentate co-ligand. Moreover, the relatively inflexible ligand cavity is not well-suited for coordinating copper(I), and as a result, it is likely to produce rather unstable, meaning highly reactive, copper(I) compounds, particularly with redox active co-ligands, e.g., dioxygen. Due to the easily customizable synthesis of bispidine ligands, this offers unique opportunities to modify the ligand scaffold to alter the electronic properties at the copper site (see Table 4 for bispidines used for oxygen activation with copper complexes).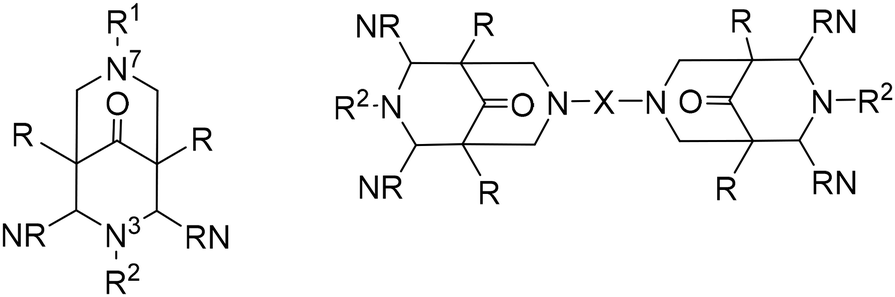
This becomes particularly evident in the reaction with molecular oxygen. The reaction of molecular oxygen with CuI coordinated to L1 forms a dinuclear end-on-[Cu2O2]2+ species, which does not rearrange to a side-on-[Cu2O2]2+ species:27 the pyridines in the plane perpendicular to the Cu–N7 pseudo-Jahn–Teller axis prevent short distances between the two CuII centers, favoring the end-on configuration over the side-on isomer. In addition to steric hindrance, the very short and strong copper(II)–oxygen bond contributes to the stabilization of this species. These features can be adjusted through changes in the ligand structure. Since the peroxido species is dinuclear, it is plausible to use a preorganized ethyl- or propyl-bridged ligand to accelerate the formation and stabilize the dinuclear species.58 The [Cu2O2]2+ complex obtained with L25 differs spectroscopically and energetically from the non-bridged ligand systems because the orientation of the individual copper sites to each other varies slightly. As a result of the introduced bridge, this complex is one of the most stable [Cu2O2]2+ complexes reported so far.58 Due to the similarity to type III copper enzymes, bridged bispidine ligand dicopper complexes have been used as catecholase mimics. Although there are structural similarities between the enzymes and the dinuclear copper-bispidine complexes with L25, L26, L27, L28, L29, L30 and L31, the underlying mechanism in oxidizing substrates proceeds differently. With the copper bispidine compounds, each catalytic cycle converts one substrate and produces one equivalent of H2O2 (see Fig. 10), whereas in the enzyme, oxygen is reduced to water, and two substrates are oxidized in one catalytic cycle.28,56 The catalytic activity in the bispidine based model is further reduced by the formation of the inactive resting state (i) depicted in Fig. 10, limiting the conversion rate of the substrate.54
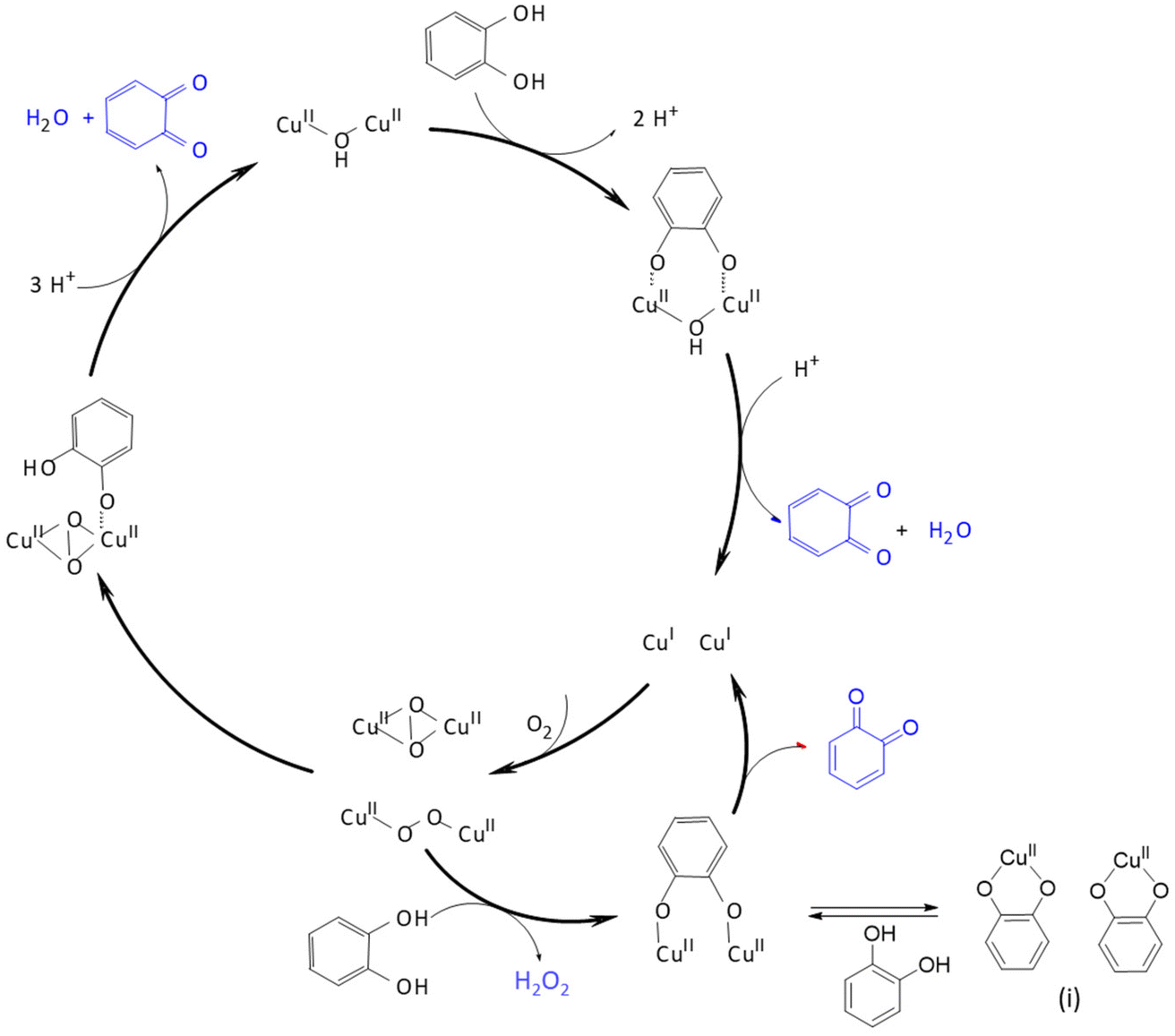 | ||
| Fig. 10 Reaction mechanism of catechol to quinone, catalyzed by a dinuclear copper(II)–peroxido complex. Adapted from ref. 54. | ||
In addition to promoting the formation of dinuclear species, efforts were also made to prevent them. For this purpose, sterically demanding substituents were introduced into the bispidine scaffold. One approach involved the methylation of the pyridine donors in ortho-position. This resulted in substantial steric bulk in the equatorial plane (L2, see also Fig. 2): the corresponding copper(I) complex did not oxygenate to a dicopper(II) peroxido species, but was oxidized directly to the mononuclear CuII compound.49 This is also due to a destabilization of the CuII peroxide species as a result of the fact that the coordination of a monodentate ligand in the equatorial plane is hindered, resulting in a weaker Cu–O bond (see also Fig. 2).
Another approach focused on varying the coordination geometry. For this purpose, ligand L32 was developed (see Table 3): this tetradentate ligand does not have the usual in-plane pyridine groups but two additional tertiary amines substituted at N3, leading to an enforced trigonal bipyramidal coordination geometry. This lead to hindered dimerization and the possibility to trap and characterize a mononuclear superoxido species.122
In addition to reactions involving oxygen, the transfer of nitrogen groups is also facilitated by copper bispidine species. The aziridination of styrene by PhINTs as oxidant has been particularly thoroughly studied with bispidine based systems60,61,123,124 (see Fig. 11). Here too, positioning of the nitrene group in the complexes with tetradentate ligand systems predominantly occurs trans to N3 (X in the complex of Fig. 11), resulting in relatively short copper–nitrene bonds. To optimize the reaction, various aspects of the ligand were modified to influence parameters such as the coordination geometry and the redox potential. For example, sterically demanding groups were introduced to force the nitrene into the axial position (Y in the complex of Fig. 11), and the pyridine donors were also substituted by groups to alter their electronic properties and therefore influence the efficiency of the aziridination. However, while the effects were as predicted, all these changes had only minor effects on yields and turnover frequencies (TOFs) – the steric influence of ortho-substituted pyridine groups, forcing the nitrene group into an axial position generally had the greatest impact (see Table 5).10,31,60,123
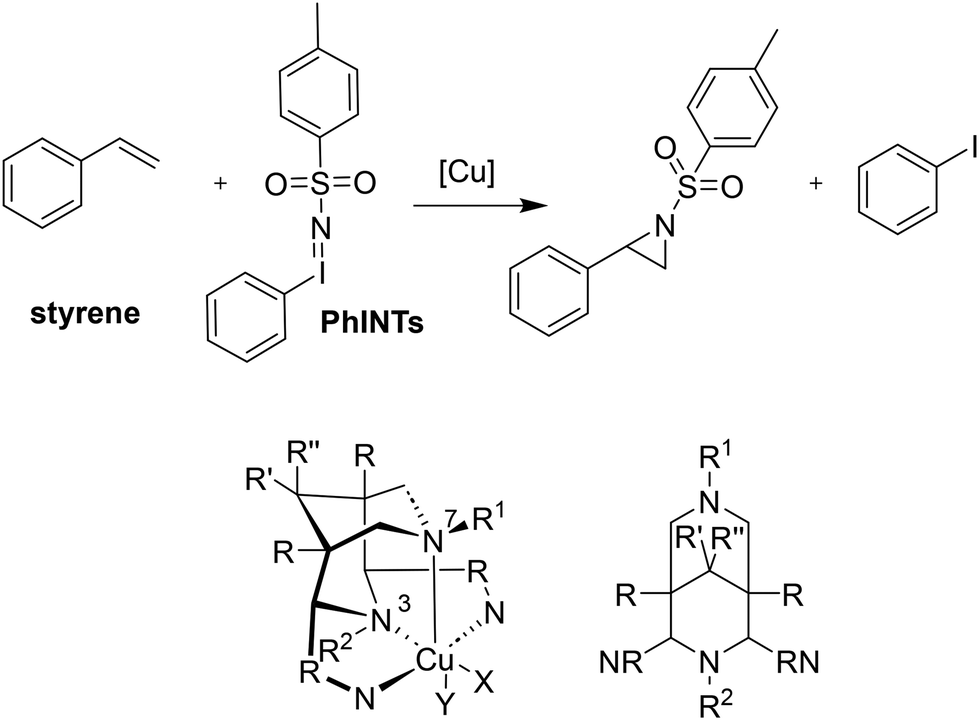 | ||
| Fig. 11 Catalytic conversion of styrene with PhINTs as the oxidizing agent to the corresponding aziridine. The bispidine ligand and its corresponding copper complex are modified with various substituents to alter the bispidine framework. X and Y represent potential co-ligands or solvent molecules. | ||
:PhINTs:styrene = 0.05:1:20; yields relative to PhINTs; TOFs calculated based on time until a clear solution is obtained (poor solubility of PhINTs in MeCN, see ref. 61, standard deviations are given in brackets; yields from other sources adjusted accordingly)
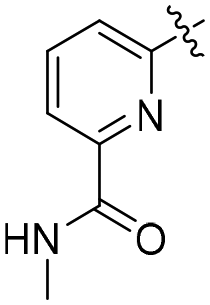
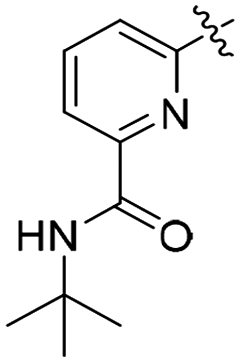
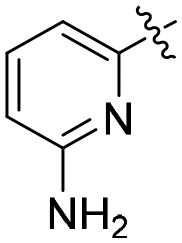
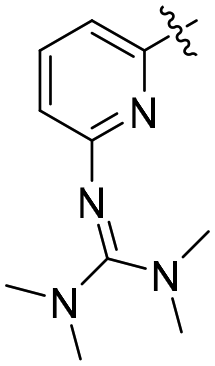
| R1 | R2 | R | R′/R′′ | RN | TOF (h−1) | Yield (%) | Lit | |
|---|---|---|---|---|---|---|---|---|
| a 0.4 mol PhINTs, 5 mol% catalyst, 1 mL styrene. b 0.5 eq. copper complex used. c Not completely water free conditions. | ||||||||
| L2 | Me | Me | COOMe | O |
py | 1.3 (0.0) | 35.0 (0.0) | 61 |
| L9 | Me | Me | COOMe | OH/H | py | 1.4 (0.0) | 40.5 (0.5) | 61 |
| L37 | Me | Me | COOH | OH/H | py | 3.2 (0.1) | 49.0 (1.0) | 61 |
| L38 | Me | Me | CH2OH | OH/H | py | 2.1 (0.0) | 49.0 (1.0) | 61 |
| L39 | py | Me | COOMe | O |
py | 0.9 (0.0) | 22.0 (2.0) | 61 |
| L40 | Me | py | COOMe | O |
py | 1.4 (0.0) | 33.5 (0.5) | 61 |
| L41 | Et | Me | COOMe | O |
py | 6.0 (0.3) | 66.5 (1.5) | 61 |
| L42 | iPr | Me | COOMe | O |
py | 9.1 (0.2) | 66.5 (1.5) | 61 |
| L43 | iBu | Me | COOMe | O |
py | 2.0 (0.0) | 55.5 (2.5) | 61 |
| L44 | t Bu | Me | COOMe | O |
py | 7.2 (0.1) | 67.5 (1.5) | 61 |
| L45 | Bn | Me | COOMe | O |
py | 7.0 (0.3) | 73.0 (0.0) | 61 |
| L46 | H | Me | COOMe | O |
py | 8.7 (0.5) | 71.5 (0.5) | 61 |
| L47 | H | Me | COOMe | OH/H | py | 5.0 (0.0) | 59.0 (2.0) | 61 |
| L48 | Me | H | COOMe | O |
py | 527 (14) | 81.5 (1.5) | 61 |
| L49 | Me | H | COOMe | OH/H | py | 505 (27) | 81.0 (1.0) | 61 |
| L50 | H | H | COOMe | OH/H | py | 500 (14) | 84.0 (3.0) | 61 |
| L51 | Me | Me | COOMe | O |
|
40.0 (—) | 94.0 (—) | 123 |
| L26 | Me | Me | COOMe | O |
|
10.0 (—) | 51.5 (—) | 60 |
| L52 | Me | Me | COOMe | O |
|
3 (—) | 29 (—)a | 60 |
| L53 | Me | Me | COOMe | O |
|
3 (—) | 4.0(—)c | 123 |
| L54 | Me | Me | COOMe | O |
|
3 (—) | 38.5(—)c | 123 |
| L55 | Me | Me | COOMe | O |
|
10 (—) | 59.0 (—)c | 123 |
| L56 | Me | Me | COOMe | O |
|
10 (—) | 55.5 (—)c | 123 |
| L57 |
|
Me | COOMe | O |
py | 3 (—) | 63.0 (—)c | 123 |
| L58 | py | Me | COOMe | O |
|
3 (—) | 60.5 (—)c | 123 |
| L59 |
|
Me | COOMe | O |
|
10 (—) | 74.5 (—) | 123 |
| L34 | Me |
|
Ph | O |
H | 30 (—) | 85 (—)b | 124 |
Interestingly, introducing a secondary amine at N3 (L48–L50) resulted in drastically increased TOFs and yields. It appears that a reversible deprotonation during the catalytic cycle changes the electron density at the copper center and therefore the nitrene character: due to the higher electron density at the copper center caused by the negative charge on the deprotonated ligand, the nitrene character shifts towards a radical Fischer-type nitrene, leading to higher activity.61 The deprotonated amine exhibits π backbonding, reminiscent of type-1 blue copper proteins (see Fig. 12 for spectroscopic changes upon deprotonation).125 Indeed, the observed charge transfer transitions are astonishingly similar and, as expected, also comparable to those of bispidine-copper(II)-peroxido complexes.54 Deprotonation of coordinated amines usually only occurs at very high pH and often is not possible in aqueous solution, with only few examples to the contrary.126,127 For the CuII complex with L48 a pKa value of 18.35 ± 0.06 in acetonitrile (MeCN) was determined (approx. 10.3 in H2O).61,128 It appears that the tetradentate bispidines with secondary amines at N3 and/or N7 offer interesting_ possibilities to change the electronics and reactivities of the corresponding metal complexes (i.e. also of the corresponding high-valent iron oxidants discussed below), with the possibility to switch certain reactions on or off. The aziridination with copper bispidines nicely demonstrates how various modifications can be made to the nitrogen donors to enhance reactivity through steric, geometric, or electronic changes.
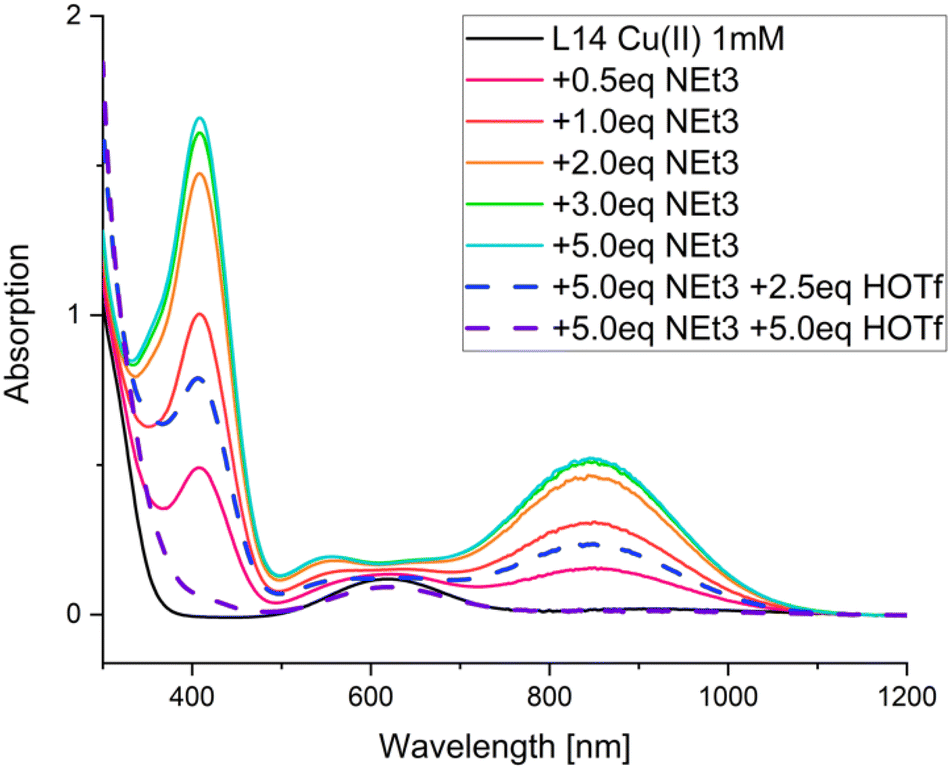 | ||
| Fig. 12 UV-vis-NIR spectra of a [CuII(L48)(MeCN)]2+ solution with different amounts of NEt3 (solid lines) and trifluoromethanesulfonic acid. Conditions: absolute MeCN, argon, rt, [CuII(L48)(MeCN)]2+ (1 mM), NEt3 (0, 0.5, 1.0, 2.0, 3.0, 5.0 mM), HOTf (0, 2.5, 5.0 mM). Reprinted from ref. 61 with permission from Elsevier. | ||
In addition to facilitating the formation of C–O or C–N bonds, copper bispidine complexes have also been used for inducing C–C coupling, specifically in copper-catalyzed electrochemical atom transfer radical additions (eATRRA).129
High valent iron mediated reactions
Nonheme iron(IV)–oxido chemistry has been extensively explored over the past 20 years, where numerous different ligands have been designed to imitate the reactive species found in various enzymes such as TauD and SyrB2.130 The benefit of using nonheme iron(IV)–oxido mimetics is that, due to the variable coordination geometry, a broad range of different reactions can be catalyzed, and a wide variety of substrates can be converted.131–134 The iron(IV)–oxido complexes can catalyze group transfers to heteroatoms and alkenes as well as performing C–H activation (see Scheme 4). C–H activation occurs through a hydrogen atom abstraction step (HAA), where the iron(IV)–oxido group is reduced to an iron(III)–hydroxido site, resulting in the formation of an organic radical. This radical can then either recombine in a rebound step with the existing co-ligands of the iron(III) species, forming, e.g., an alcohol when recombining with the hydroxyl group, or it can undergo a so-called cage escape, where the organic radical leaves the immediate vicinity of the iron complex, in order to react further in the reaction solution. Although the active species as well as pathways to various products have been studied extensively, there are open questions, in particular also regarding variations in efficiency and selectivity of synthetic systems. Even though different donor sets and geometries have been considered in the design of ligands, the reactivity and selectivity of enzymes generally is unattained by enzyme mimetics.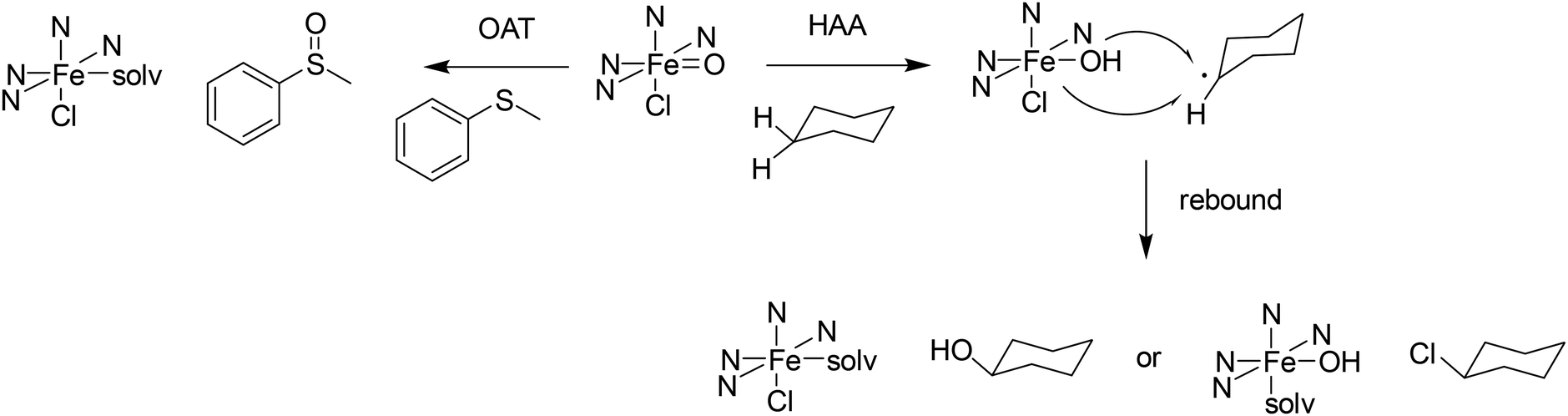 | ||
| Scheme 4 Possible reaction pathways starting from an iron(IV)–oxido species include oxygen atom transfer (OAT) to a heteroatom and C–H activation (HAA) in cyclohexane. | ||
Apart from generally acknowledged effects of proteins in providing a confined space with well-defined shape, charge distribution, electric field, hydrophobicity, H-bonding network etc., the limitation of synthetic models has been attributed to the spin ground state of the iron center and the energy gap to other low lying spin states. In enzymes, the ground state of the ferryl complex is typically high-spin (S = 2), with so far one reported exception,135 whereas in synthetic complexes, the ground state usually is S = 1, although a few high-spin iron(IV)–oxido species are known.136,137 The synthetic iron(IV)–oxido complexes are often produced by oxygen atom transfer to an iron(II) precursor, typically by derivatives of iodosylbenzene (PhIO), as this results in a clean conversion of the iron(II) precursor. Molecular oxygen (as in nature), peroxides and other oxidants have been used to generate high-valent iron-oxido complexes but often mixture of various products are formed, also including superoxides, peroxides and hydroperoxides.138,139
For C–H activation, the HAA step usually is rate limiting, and it generally proceeds over an S = 2 transition state (with a tetrakis-carbene ligand enforcing a very strong in-plane ligand field as a notable exception).140 That is, most of the model systems require a spin crossover to the quintet surface, and the size of the triplet–quintet gap is an important parameter for the rate of the substrate oxidation (see Fig. 13). The driving force, i.e., the redox potential of the FeIVO species, is the other decisive parameter for the rate and efficiency of ferryl oxidants. Looking at the bispidine systems, the very rigid binding pocket of the ligand scaffold is too large for high-valent iron cations, especially iron(IV), and this is an important difference to the copper-bispidine assisted oxygen activation discussed above. Therefore, bispidine-based FeIVO oxidants have exceedingly large driving forces (they are believed to be among the high-valent nonheme iron species with the highest redox potentials).68–71 This is one reason, why they are among the most efficient ferryl oxidants, and this is another “bispidine effect” (see inertness of bispidine-based chelators for medicinal applications above),141 also due to the rigid bispidine scaffold.44 The other is the small and tunable triplet–quintet gap. Population of the quintet state requires lowering the dx2–y2 orbital, which for bispidines is already relatively low due to the large cavity, (see Fig. 14) and, especially, the relatively long Fe–N7 bond (see Introduction). Importantly, it is further tunable by the monodentate coligand X on the molecular y axis. Experimentally, this was shown by d–d spectra along the series of the three ferryl oxidants [(L1)FeIVO(NCMe)]2+, [(L1)FeIVO(Cl)]+ and [(L1)FeIVO(Br)]+ with increasingly lower energy transitions to the dx2–y2 orbital (see Fig. 14). A thorough ligand field analysis allowed to calculate the triplet–quintet gaps as well as the zero field splitting parameters, which are experimentally accessible – but have not been determined so far.62 Importantly, the decrease of the triplet–quintet gap from the MeCN to the Cl− and Br− species leads to a large acceleration of the HAA of cyclohexane (see Table 6),62 and [(L1)FeIVO(Cl)]+ is the fastest FeIV oxido complex known to date, with rates as fast as those of enzymes (the full kinetics of [(L1)FeIVO(Br)]+ and temperature dependent kinetics have not yet been reported), see Table 6.62,142,143
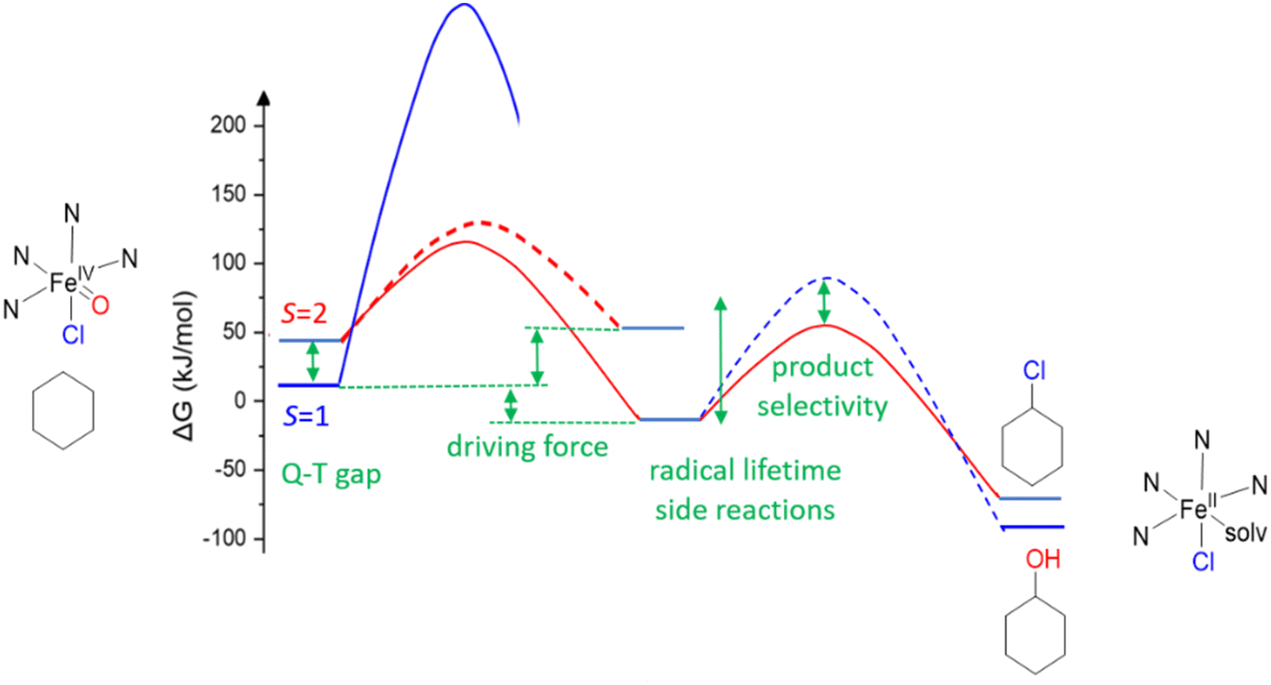 | ||
| Fig. 13 General potential energy surface (PES) of the oxidation of cyclohexane by a nonheme iron(IV) oxido oxidant (HAA). | ||
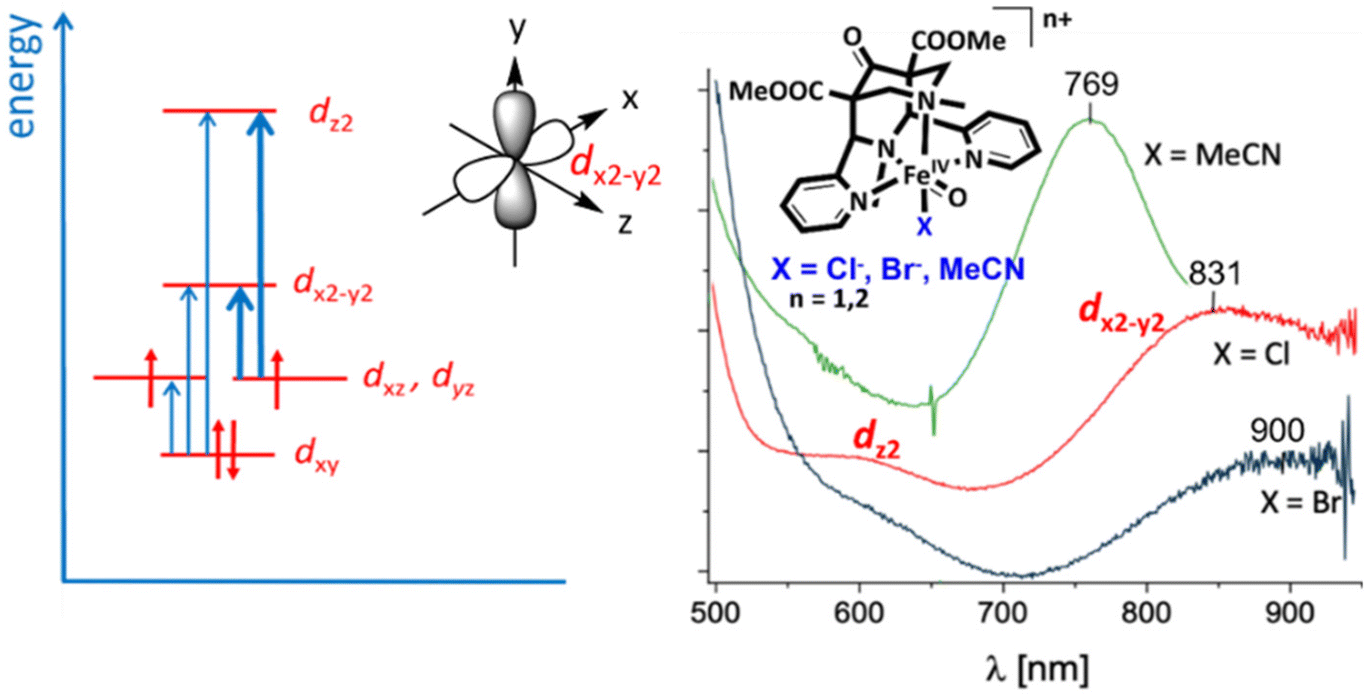 | ||
| Fig. 14 Ligand field scheme for the d–d transitions of FeIVO complexes, coordinate system and orientation of the dx2–y2 orbital for the example of [(L1)FeIVO(X)]n+ with the corresponding experimental spectra.62 | ||
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R′/R′′ | X | Y | k 2 (M−1 s−1) thioanisole | k 2 (M−1 s−1) cyclohexane (×10−3) | k 2 (M−1 s−1) bzOH | Lit | |
| L1 | Me | Me | O |
O |
MeCN | 2.9 × 10−1, −35 °C | 70 | ||
| Cl− | (7.55 ± 0.27) × 102, −90 °C, EtCN | 143 | |||||||
| Br− | approx. 10× faster than with Y = Cl− | 143 | |||||||
| L46 | H | Me | O |
O |
Cl− | 142 | |||
| Br− | 143 | ||||||||
| L39 | py | Me | O |
py R1 | O |
480 ± 2; 2.4, −10 °C | 4.91 ± 0.08 | 68 and 70 | |
| NTs |
117 ± 16 | 4.9 ± 8 × 10−3 | 144 | ||||||
| L10 | py | Me | OH/H | py R1 | O |
129 ± 2 | 2.56 ± 0.05 | 68 and 70 | |
| L40 | Me | py | O |
O |
py R2 | 5.65 ± 0.10; 0.024, −10 °C | 0.130 ± 0.003 | 70 | |
| NTs |
9.77 ± 0.29 | 0.13 ± 3 × 10−3 | 144 | ||||||
| L60 | Me | py | OH/H | O |
py R2 | 145 | |||
| L61 | Me |
|
OH/H | O |
py R2 | 5.9 | 145 | ||
| L62 | Me |
|
OH/H | O |
py R2 | 145 | |||
| L63 | Me |
|
OH/H | O |
py R2 | 0.4 | 145 | ||
| L64 | Me |
|
OH/H | O |
py R2 | 70 | 145 | ||
| L65 | Me |
|
OH/H | O |
py R2 | ||||
| L66 | Me |
|
OH/H | –OO | py R2 | ||||
| L67 | O |
0.49; 0.014, −10 °C, 0.05 ± 0.004, 0 °C | 8.3 × 10−2, 20 °C | 68 and 146 | |||||
| NTs |
2.6; 0.26 ± 0.003, 0 °C | 5.5 × 10−2 | 146 | ||||||
| L68 | O |
8.2; 0.33–10 °C | 68 and 146 | ||||||
| NTs |
0.054 | 0.39 | 146 and 147 | ||||||
| L69 | O |
0.004–10 °C, 0.091 ± 0.004, 20 °C | 68 | ||||||
| NTs |
0.012; (7.9 ± 0.8)10−3, 20 °C | No reaction | 148 | ||||||
With [(L1)FeIVO(Cl)]+ and cyclohexane, chlorocyclohexane is the only product.142 That is, rebound does not take place at the hydroxyl but selectively at the halide coligand (see Scheme 4 and Fig. 13). According to a DFT analysis, this is attributed to steric effects and primarily is due to the relatively long lifetime of the rebound precursor [(L1)FeIII(OH)(Cl)]+/substrate radical. The long life-time is supported by the observed differences of product distributions under inert and non-oxygen-free conditions, where oxygen reacts with the radical due to its relatively long life-time.142,149
The enormous reactivity of the [(L1)FeIVO(X)]n+ complexes also has disadvantages: there are several side reactions, and these are also very efficient.143 One is the formation of an oxido-bridged dinuclear iron(III) complex, the second an N-centered demethylation that occurs at the nitrogen atom N7, and the reduction of Cl− by the ferryl oxidant is another one. The N-centered demethylation can be suppressed by removing the corresponding N-methyl substituent from the precursor (ligand L46vs. L1). However, this also changes the ligand field at the FeIVO site and experimentally has been shown to lead to a slightly slower oxidation of cyclohexane.143 To avoid formation of dinuclear species, substituents in ortho position of the pyridine rings, as in the previously discussed copper–dioxygen reactions, might prevent reaction of the ferryl complex with excess iron(II) precursor but the changes of the electronic properties might also influence other pathways.
How variations at the bispidine backbone, designed to influence the electronics and stabilities of high-valent iron complexes, may lead to unforeseen side-reactions is demonstrated in a study, where the more stable pentadentate bispidine ligands (derivatives of L60) were equipped with various substituents to shield and stabilize the oxido group, as has been done successfully with several other ligand systems.150–152 Unfortunately, introducing various residues to the bispidine did not result in an efficient stabilization of the system.145 However, with a permethylated guanidine group introduced in the ortho position of the pyridine appended to N3, it was observed that, after the formation of the iron(IV)–oxido species, unexpectedly a rapid transformation to a peroxido species occurred. Based on spectroscopy (EPR, UV-vis-NIR, Mössbauer) and mass spectrometry of various intermediates, kinetics and an analysis of the reactivities of the iron-based oxidants, it emerges that the highly reactive iron(IV)–oxido species A (see Scheme 5) undergoes an intramolecular C–H abstraction, involving a methyl group of the guanidine residue, followed by a rebound to the FeIII–OH intermediate B to lead to an FeII species with a hydroxylated guanidine substituent C. With excess 1-(tert-butylsulfonyl)-2-iodosylbenzene (sPhIO), this is reoxidized to the corresponding FeIVO species and eventually forms the iron(III)–peroxido product D.153
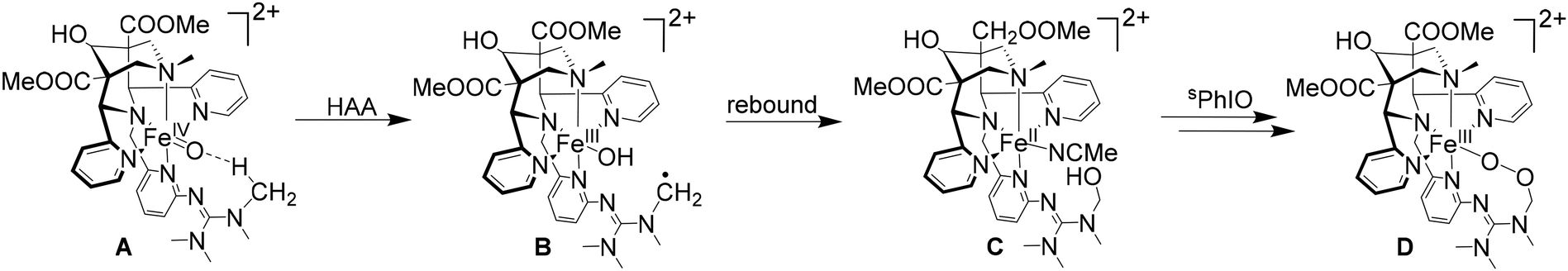 | ||
| Scheme 5 Formation of an FeIII alkylperoxido complex from the FeII precursor under strictly oxygen- and water-free conditions, i.e., O–O bond formation in the coordination sphere of an FeIVO species.153 | ||
Generally, iron(III)–peroxido complexes are sluggish oxidants and only few oxidation reactions of organic substrates have been studied in detail.154,155 Possible decay pathways of iron(III)–peroxido species are assembled in Scheme 6, showing the resulting iron based oxidants FeIVO, FeVO (homolysis and heterolysis of iron(III)–peroxido intermediates have been studied experimentally and computationally),29,156,157 and iron(III)–superoxido species. Note that in specific cases, iron(III)–peroxido species may exist as the more reactive iron(II)–superoxido valence tautomer, and this has recently been analyzed with a combination of experiments and computational work.158 Homolysis of the O–C bond of a coordinated alkylperoxide is an additional but uncommon process that leads to highly reactive iron(III)–superoxido species – note that this is the intermediate of oxygen activation by iron(II), i.e., this is biologically relevant but rarely observed with synthetic nonheme iron species.159
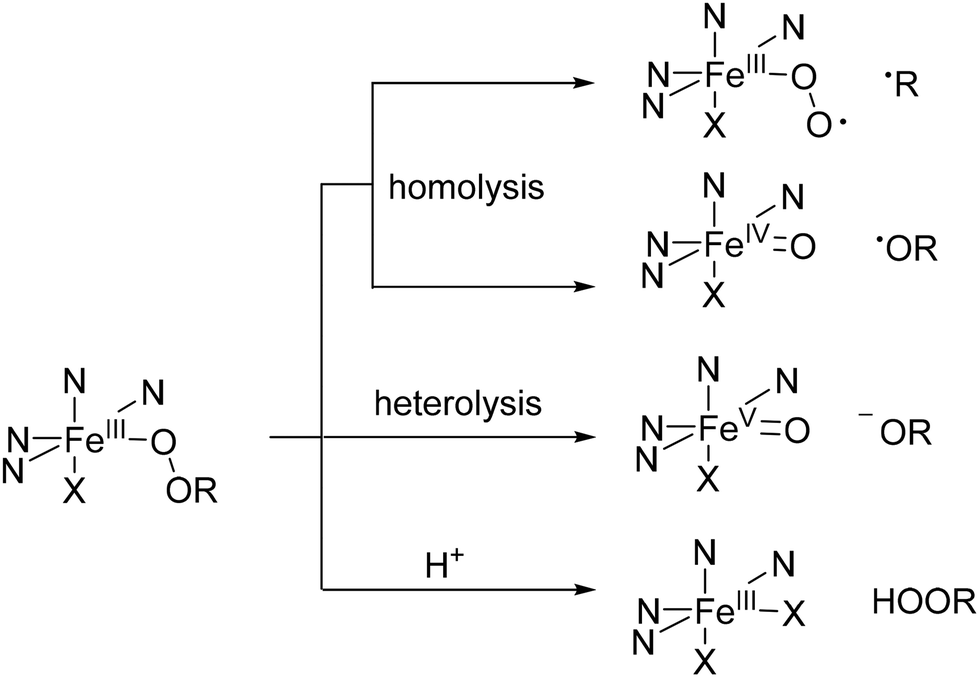 | ||
| Scheme 6 Various possible decomposition pathways of an iron(III)–peroxido species. | ||
Among these possible decay pathways of iron(III)–peroxido species, O–C homolysis has only recently been observed. This reaction was identified in a bispidine–iron(III)–alkylperoxido complex by a combined experimental and DFT-based computational study.159 The ligand used was the pentadentate bispidine L70, where the usual pendent pyridine at N7 (see L39) was substituted by a tertiary amine (see Scheme 7). The corresponding iron(III)–alkylperoxido species was generated by reaction of tBuOOH with the iron(II) precursor. The preference for O–C over O–O cleavage was shown to be due to hydrogen bonding, involving the N-methyl groups and the distal oxygen atom of the peroxide.159
 | ||
| Scheme 7 A series of modified ligands for chlorite oxidation. The ligand with five nitrogen donors forms a superoxido species in the reaction with tBuOOH. | ||
The same ligand scaffold was further modified by changing the donor set from N5 to N4S and N4O (Scheme 7), and complexes of these ligands were used for the oxidation of chlorite by the corresponding iron(IV)–oxido species. A significant increase in reactivity was observed, with the TON rising from 30 to 232 and 242, respectively, for the ligands with mixed donor sets. The modification of the donor set in the equatorial plane had no impact on the reaction mechanism – the preferred oxygen atom transfer still occurred. However, with the mixed donor bispidines, the yields and reactivity of the FeIVO complexes increased significantly.160
Oxygen activation as in nature, i.e., formation of FeIVO species by O2 is rare and, with bispidine systems, direct oxidation of FeII precursors with dioxygen is impossible due to much too high redox potentials. The experimentally observed formation of FeIII–OOR and FeIVO species with bispidine ligands L1 and L39 under ambient conditions without any exogenous oxidant was shown to be initiated by alkylperoxide formation due to traces of radical impurities in the substrate.161 Iron(II) complexes of various bispidine derivatives, primarily of L39 and close analogues, have been used as bleaching catalysts and for alkyd paint drying with a range of patents published (partly cited in the references), leading to commercial applications.162,163 It appears that, to a large extent, these applications are based on the formation of catalytically active high-valent iron species, initiated by organic radical based peroxide formation as described above.
In addition to the extensively studied iron(IV)–oxido chemistry, nitrogen atom transfer and C–H activation with high-valent iron–nitrogen compounds have also been investigated since the early 2000s.164,165 Further to extensive work on bispidine iron–oxygen chemistry, recently two iron(IV)–nitrene (or more precisely tosylimido) complexes were isolated, characterized and their reactivities tested.144 Similar to the previously studied iron(IV)–nitrene systems in comparison with the corresponding oxido systems, a generally significantly lower reactivity of the nitrene species was also noticed with the bispidine complexes. The combination of a weaker bond, the lower electronegativity of nitrogen, reduced radical character, and, depending on the complex, steric hindrance generally renders the nitrene complexes less reactive than their oxido analogs.146–148,166
Interestingly however, there are two notable exceptions: the iron–nitrene species with the N4py ligand L67 and the bispidine L40 both react faster in nitrene atom transfer (NAT) than in oxygen atom transfer (OAT) to thioanisole (see Table 6).144 A DFT analysis indicates that this can be attributed to longer Fe–N than FeO bonds, resulting in less steric repulsion in the Fe–N⋯substrate than in the Fe–O⋯substrate transition state. The two important points in this respect are: (i) the DFT analysis indicates that upon approach of the substrate, i.e., in the reactant complex, there is a significant amount of electron transfer from the substrate to the nitrene, resulting in partial reduction of the iron(IV) center and therefore to significantly elongated FeIVN (FeIII–N) bonds. (ii) The steric hindrance primarily results from interactions of the α hydrogen atoms of pyridine rings parallel to the Fe–O (or Fe–N) axis, and the complexes with N4py (L67) and the bispidine L40 have more such pyridine rings (four and three, respectively) than the other complexes studied (see Fig. 15).144 This is supported by earlier experiments and computational work with the N4py (L67) based system, indicating that the differences in reaction rates are primarily due to differences in the electron affinity and orbital interactions between the oxido and tosylimido species: the electron affinity of [(L67)FeIVNTs]2+ is so large that it accepts electrons from hydrogen atom transfer (HAT) substrates over a long distance. As a result, the actual HAT is carried out by the reduced [(L67)FeIII–NTs]+ species, which is less catalytically potent than the corresponding iron(IV)–oxido species. On the other hand, heteroatom transfer reactions do not start with long-range electron transfer but proceed through a rate-determining group transfer, where two electrons are transferred to the iron(IV)–imido species, while only one electron is transferred at the same point in the reaction with the iron(IV)–oxido complex. Therefore iron(IV)–tosylimido favors heteroatom transfer, whereas iron(IV)–oxido prefers hydrogen atom transfer.167
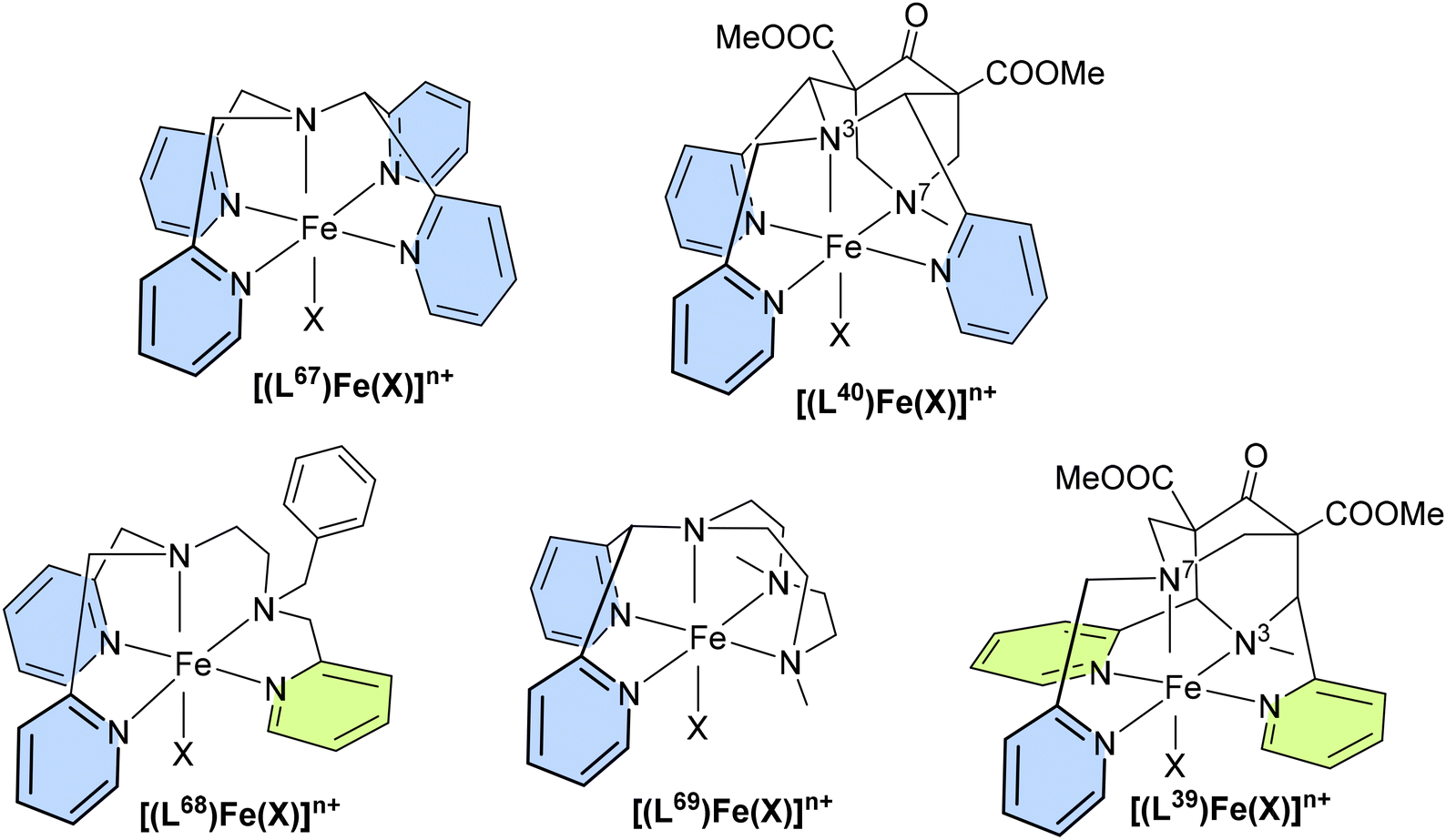 | ||
| Fig. 15 Structural plots of the iron complexes with L39.40,67–69 X is either O or NTs. The complexes are oriented such that the co-ligand is aligned along the z-axis to indicate the orientation of the pyridine donors: blue indicates parallel to the z-axis, green indicates orthogonal to the z-axis.144 | ||
Other metal ions
In addition to the well-studied copper and iron bispidine compounds described above, there are other metal centers that have been described to yield stable and active bispidine based transition metal catalysts.The oxido chemistry of manganese and ruthenium compounds in general has been studied extensively, and related bispidine species have been shown to exhibit relatively high activities. For example, the MnIV–oxido complex with the pentadentate bispidines L39 and L40 achieve second order rate constants for the oxidation of thioanisole of k2 = 1.2 × 10−2 M−1 s−1 and 1.2 × 10−1 M−1 s−1, respectively.168 This is significantly slower than for the corresponding FeIV based systems (see Table 6 for the corresponding FeIV rates). More interesting, however, is the observation that the difference in reactivity between the two isomers is reversed, and this is due to a change in the reaction channel (σ vs. π, i.e., ≈180° vs. ≈120° angle of MO⋯substrate).168 In another study, the focus was on the reactivity of an MnIII–peroxido species with L40, where the methyl substituent at N7 was substituted with a benzyl group.169 The peroxido complex did not lead to deformylation of aldehyde substrates via nucleophilic addition. Instead, the side-on peroxide performed a HAA. This is partially due to the low redox potential, which allows efficient electron transfer from the peroxide to manganese. It emerges that the nucleophilic pathway is disfavored by steric repulsion involving the ligand substituents: the ligand system influences the redox potential as well as the approach of the substrate, leading to a preference for HAA, and both effects are due to the shape of the ligand.169
For the RuIV–oxido chemistry, both OAT to thioanisole and the epoxidation of alkenes, such as styrene or cyclooctene, were investigated using L39. The conversion of thioanisole was quantitative, and the epoxidation reactions were also of high efficiency and generally more efficient but with a slightly lower stereoselectivity than the iron-based systems.170,171 Differences between the iron- and ruthenium-based reactivities are mainly due to ligand field effects (preference for the S = 1 vs. S = 2 spin surface for ruthenium vs. iron) and, in the case of the sulfoxidation, the S/O linkage isomerization of the sulfoxide product.
The oxido chemistry with vanadium and cobalt bispidines was also investigated, primarily using L39 and L40 as well as close derivatives. While the oxidation of the vanadyl complexes with H2O2 did not show any outstanding reactivity,172 most cobalt(II) complexes were remarkably reactive and formed superoxido complexes with oxygen. These superoxido complexes tend to undergo N-centered demethylation at various positions, in intra- as well as intermolecular reactions. In particular, tetradentate bispidines L1, L45, L46 and L37 but with a ketone at C9 and derivatives of L40 tend to lead to ligand decay pathways, while complexes of L39 and corresponding derivatives remained stable.173–175 This is attributed in part to the redox potentials that are dependent on the substituents at N3 and N7.
Nickel-catalyzed C–C bond formation was studied using L1, where a broad range of aryl halides were successfully coupled with alkyl zinc compounds. After examining various reaction parameters, a wide range of C(sp2)–C(sp3) bond formations was achievable under mild reaction conditions. Particularly for the transformation of heteroaryl bromides, excellent performance was achieved.176
Enantioselective reactions
Due to their modular synthesis, it is relatively easy to introduce stereo-information into bispidine ligands, which makes their complexes of interest for enantioselective catalysis. There has been growing interest in using bispidine ligands in catalytic asymmetric Henry reactions.13,177 Copper complexes formed with chiral bispidine ligands have shown promising results, with some demonstrating excellent enantiomeric excess and high yields. An asymmetric bispidine ligand was also used for the coupling of diethylzinc compounds with aldehydes. It was hypothesized that the bispidine coordinates to diethylzinc, thereby transferring stereoinformation and simultaneously activating the diethylzinc species for the reaction.178 Very elegant one-pot syntheses of chiral bidentate bispidines, with one of the tertiary amines replaced by sulfur and the other bearing chiral substituents, have been described, and these ligands have been used successfully in palladium-catalyzed arylation reactions.179In addition to metal-based reactions, there is an increasing series of publications describing the use of bispidines as organocatalysts. The corresponding bispidines are equipped with substituents containing stereochemical information, enabling asymmetric reactions such as aldol and Mannich reactions as well as Michael additions.14,180–183
Conclusion
All properties of metal complexes – electronics, thermodynamics, reactivities – depend on the structure of the complex, and this is largely enforced by the ligand.44,53,184,185 With rigid ligands, there is no conformational flexibility – with bispidines coordinated to metal ions there generally is a single isomer. In addition, the missing elasticity leads to a high loss of energy with metal ions that do not fit to the ligand in terms of size and shape. Fit is of importance for stability and metal ion selectivity, and misfit may be of advantage for reactivity, where the driving force and entasis may increase the reactivity. The bispidine backbone ideally fulfills these requirements, and one of the “bispidine effects” that has been discussed relates to preventing microscopic reversibility in complex formation and decay, also including the “mousetrap effect”. Increased reactivity as a result of a large driving force, arising from the rigid bispidine cavity that is too large for high-valent iron, has also been described as a “bispidine effect”. As a note of caution and also to avoid the perception that the bispidine scaffold is matchless, we note that (i) rigidity is not always and not only of advantage and (ii) that other ligand platforms, such as cross-bridged tetra-aza- and diaza-pyridinophane-macrocycles have similar structural properties and, indeed are used in areas covered in this perspective.Importantly, bispidines are easy to prepare and allow a large variation of denticity, donor sets, and charges, and also provide various possibilities for ligand functionalization. The preparative scope has allowed to provide chelators that fully encapsulate a large range of metal ions, to construct metal-based probes of unprecedented stability and inertness, and also to substitute them with biological vectors for diagnostic and therapeutic applications. With open coordination sites bispidine complexes have been designed and prepared to activate small molecules and organic substrates in catalytic transformations. A range of possibilities to tune electronic properties – e.g., the ligand field and spin state energies –, and electron transfer properties, has allowed to optimize the corresponding complexes, particularly in the field of oxidation catalysis. There is an increasing interest for chiral bispidines, and this might lead to a further growth of the field. Also, in terms of denticity, the limit is not yet reached, and in terms of donor sets there are possibilities for further variations.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for financial support of our own work by Heidelberg University, the German Science Foundation (DFG), specifically also by the Research Unit FOR 5215 “Bioinspired Oxidation Catalysis” with Iron Complexes “BioOxCat”, the Max Planck School Matter to Life, supported by the BMBF in collaboration with the Max Planck Society, and computational resources provided by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster).References
- Bispidines with a keto group at C9 are generally named bispidones, those with an alcohol at C9 bispidols – the fully reduced forms with an alcohol at C9 and a methylalcohol at C1 and C5 bispi-triols; for simplicity, for all forms we use the names bispidine derivatives or simply bispidines.
- C. Mannich and P. Mohs, Chem. Ber., 1930, B63, 608–612 CrossRef.
- H. Stetter and R. Merten, Chem. Ber., 1957, 90, 868–875 CrossRef CAS.
- R. Haller, Arch. Pharm., 1969, 302, 113–118 CrossRef CAS PubMed.
- D. S. C. Black, G. B. Deacon and M. Rose, Tetrahedron, 1995, 51, 2055–2076 CrossRef CAS.
- S. Z. Vatsadze, N. V. Zyk, R. D. Rakhimov, K. P. Butin and N. S. Zefirov, Russ. Chem. Bull., 1995, 44, 440–442 CrossRef.
- G. D. Hosken and R. D. Hancock, J. Chem. Soc., Chem. Commun., 1994, 1363–1364 RSC.
- P. Comba, B. Nuber and A. Ramlow, J. Chem. Soc., Dalton Trans., 1997, 347–352 RSC.
- W. Brandt, S. Drosihn, M. Haurand, U. Holzgrabe and C. Nachtsheim, Arch. Pharm. Pharm. Med. Chem., 1996, 329, 311–323 CrossRef CAS PubMed.
- I. Tomassoli and D. Gündisch, Curr. Top. Med. Chem., 2016, 16, 1314–1342 CrossRef CAS PubMed.
- D. Shcherbakov, D. Baev, M. Kalinin, A. Dalinger, V. Chirkova, S. Belenkaya, A. Khvostov, D. Krut'ko, E. Volosnikova, E. Sharlaeva, D. Shanshin, T. Tolstikova, O. Yarovaya, R. Maksyutov, N. Salakhutdinov and S. Vatsadze, ACS Med. Chem. Lett., 2022, 13, 140–147 CrossRef CAS PubMed.
- R. Li, Y. Yan, X. Ren, H. Li and J. Huang, Curr. Org. Chem., 2025, 29, 679–693 Search PubMed.
- M. Breuning and M. Steiner, Synthesis, 2008, 2841–2867 CrossRef CAS.
- H. Sun, L. Huang and J. Huang, J. Org. Chem., 2024, 89, 7225–7232 CrossRef CAS PubMed.
- M. Lippi, H. Wadepohl, P. Comba and M. Cametti, Eur. J. Inorg. Chem., 2022, e202200221 CrossRef CAS.
- P. Comba, M. Kerscher, K. Rück and M. Starke, Dalton Trans., 2018, 47, 9202–9220 RSC.
- H. Cui, R. Goddard, K.-R. Pörschke, A. Hamacher and M. U. Kassack, Inorg. Chem., 2016, 55, 2986–2997 CrossRef CAS PubMed.
- D. Brox, P. Comba, D.-P. Herten, E. Kimmle, M. Morgen, C. L. Rühl, A. Rybina, H. Stephan, G. Storch and H. Wadepohl, J. Inorg. Biochem., 2015, 148, 78–83 CrossRef CAS PubMed.
- F. Braun, P. Comba, L. Grimm, D.-P. Herten, B. Pokrandt and H. Wadepohl, Inorg. Chim. Acta, 2019, 484, 464–468 CrossRef CAS.
- L. Abad-Galan, P. Cieslik, P. Comba, M. Gast, O. Maury, L. Neupert, A. Roux-Gossart and H. Wadepohl, Chem. – Eur. J., 2021, 27, 10303–10312 CrossRef CAS PubMed.
- D. Ndiaye, M. Sy, A. Pallier, S. Meme, I. d. Silva, S. Lacerda, A. M. Nonat, L. J. Charbonniere and E. Toth, Angew. Chem., Int. Ed., 2020, 11958–11963 CrossRef CAS PubMed.
- P. Cieslik, P. Comba, D. Ndiaye, E. Toth, G. Velmurugan and H. Wadepohl, Angew. Chem., Int. Ed., 2022, e202115580 CAS.
- D. Ndiaye, P. Cieslik, H. Wadepohl, A. Pallier, S. Meme, P. Comba and E. Toth, J. Am. Chem. Soc., 2022, 144, 22212–22220 CrossRef CAS PubMed.
- M. Atanasov, C. Busche, P. Comba, F. El Hallak, B. Martin, G. Rajaraman, J. van Slageren and H. Wadepohl, Inorg. Chem., 2008, 47, 8112–8125 CrossRef CAS PubMed.
- M. Atanasov, P. Comba, G. R. Hanson, S. Hausberg, S. Helmle and H. Wadepohl, Inorg. Chem., 2011, 50, 6890–6901 CrossRef CAS PubMed.
- M. Atanasov, P. Comba, S. Helmle, D. Müller and F. Neese, Inorg. Chem., 2012, 51, 12324–12335 CrossRef CAS PubMed.
- H. Börzel, P. Comba, C. Katsichtis, W. Kiefer, A. Lienke, V. Nagel and H. Pritzkow, Chem. – Eur. J., 1999, 5, 1716–1721 CrossRef.
- H. Börzel, P. Comba and H. Pritzkow, J. Chem. Soc., Chem. Commun., 2001, 97–98 RSC.
- J. Bautz, P. Comba, C. Lopez de Laorden, M. Menzel and G. Rajaraman, Angew. Chem., Int. Ed., 2007, 46, 8067–8070 CrossRef CAS PubMed.
- G. Mukherjee and C. V. Sastri, Isr. J. Chem., 2020, 60, 1–18 CrossRef.
- P. Comba, C. Lopez de Laorden and H. Pritzkow, Helv. Chim. Acta, 2005, 88, 647–664 CrossRef CAS.
- P. Comba, M. Morgen and H. Wadepohl, Inorg. Chem., 2013, 52, 6481–6501 CrossRef CAS PubMed.
- P. Comba, L. Daumann, J. Lefebvre, G. Linti, B. Martin, J. Straub and T. Zessin, Aust. J. Chem., 2009, 62, 1238–1245 CrossRef CAS.
- A. V. Medved'ko, B. V. Egorova, A. A. Komarova, R. D. Rakhimov, D. P. Krut'ko, S. N. Kalmykov and S. Z. Vatsadze, ACS Omega, 2016, 1, 854–867 CrossRef PubMed.
- H. Cui, R. Goddard and K.-R. Pörschke, Organometallics, 2011, 30, 6241–6252 CrossRef CAS.
- T. Weber, T. Krönke, M. Köckerling, M. Walther, H.-J. Pietzsch, K. Kopka and C. Mamat, Eur. J. Org. Chem., 2024, 27, e202400258 CrossRef CAS.
- A. V. Medved'ko, D. P. Krut'ko, S. V. Gaisen, A. V. Churakov, M. E. Minyaev, A. A. Moiseeva, D. A. Lemenovsky, H. Yu, L. Wang and S. Z. Vatsadze, J. Organomet. Chem., 2021, 949, 121945 CrossRef.
- P. Comba, M. Kerscher and W. Schiek, Prog. Inorg. Chem., 2007, 55, 613–704 CrossRef CAS.
- A. M. Nonat, A. Roux, M. Sy and L. J. Charbonniere, Dalton Trans., 2019, 48, 16476–16492 RSC.
- S. Norrehed, M. Erdelyi, M. E. Light and A. Gogoll, Org. Biomol. Chem., 2013, 11, 6292–6299 RSC.
- P. Comba, B. Kanellakopulos, C. Katsichtis, A. Lienke, H. Pritzkow and F. Rominger, J. Chem. Soc., Dalton Trans., 1998, 3997–4002 RSC.
- I. Kopp, P. Cieslik, K. Anger, T. Josephy, L. Neupert, M. Gast, H. Wadepohl, K. Kopka, M. Bachmann, H. Stephan, M. Kubeil and P. Comba, Inorg. Chem., 2024, 62, 20754–20768 CrossRef PubMed.
- D. Ndiaye, M. Sy, W. Thor, L. J. Charbonniere, A. M. Nonat and E. Toth, Chem. – Eur. J., 2023, e20301880 Search PubMed.
- P. Comba and W. Schiek, Coord. Chem. Rev., 2003, 238–239, 21–29 CrossRef CAS.
- P. Comba, M. Kerscher, M. Merz, V. Müller, H. Pritzkow, R. Remenyi, W. Schiek and Y. Xiong, Chem. – Eur. J., 2002, 8, 5750–5760 CrossRef CAS PubMed.
- P. Comba, A. Hauser, M. Kerscher and H. Pritzkow, Angew. Chem., Int. Ed., 2003, 42, 4536–4540 CrossRef CAS PubMed.
- A. Bentz, P. Comba, R. J. Deeth, M. Kerscher, H. Pritzkow, B. Seibold and H. Wadepohl, Inorg. Chem., 2008, 47, 9518 CrossRef CAS PubMed.
- P. Comba, L. Grimm, C. Orvig, K. Rück and H. Wadepohl, Inorg. Chem., 2016, 55(24), 12531–12543 CrossRef CAS PubMed.
- H. Börzel, P. Comba, K. S. Hagen, C. Katsichtis and H. Pritzkow, Chem. – Eur. J., 2000, 6, 914–919 CrossRef.
- P. Comba, B. Martin, A. Prikhod'ko, H. Pritzkow and H. Rohwer, C. R. Chim., 2005, 8, 1506–1518 CrossRef CAS.
- P. Comba, C. Haaf and H. Wadepohl, Inorg. Chem., 2009, 48, 6604–6614 CrossRef CAS PubMed.
- K. Born, P. Comba, M. Kerscher, G. Linti, H. Pritzkow and H. Rohwer, Dalton, 2009, 362–367 RSC.
- P. Comba, Coord. Chem. Rev., 2000, 200–202, 217–245 CrossRef CAS.
- P. Comba, B. Martin, A. Muruganantham and J. Straub, Inorg. Chem., 2012, 51, 9214–9225 CrossRef CAS PubMed.
- C. Bleiholder, H. Börzel, P. Comba, R. Ferrari, A. Heydt, M. Kerscher, S. Kuwata, G. Laurenczy, G. A. Lawrance, A. Lienke, B. Martin, M. Merz, B. Nuber and H. Pritzkow, Inorg. Chem., 2005, 44, 8145–8155 CrossRef CAS PubMed.
- K. Born, P. Comba, A. Daubinet, A. Fuchs and H. Wadepohl, J. Biol. Inorg. Chem., 2007, 12, 36 CrossRef CAS PubMed.
- K. Born, P. Comba, R. Ferrari, G. A. Lawrance and H. Wadepohl, Inorg. Chem., 2007, 46, 458–464 CrossRef CAS PubMed.
- H. Börzel, P. Comba, K. S. Hagen, M. Kerscher, H. Pritzkow, M. Schatz, S. Schindler and O. Walter, Inorg. Chem., 2002, 41, 5440 CrossRef PubMed.
- P. Comba, M. Kerscher and A. Roodt, Eur. J. Inorg. Chem., 2004, 23, 4640–4645 CrossRef.
- P. Comba, M. Merz and H. Pritzkow, Eur. J. Inorg. Chem., 2003, 1711–1718 CrossRef CAS.
- K. Bleher, P. Comba, M. Gast, S. Kronenberger and T. Josephy, Inorg. Chim. Acta, 2022, 532, 120752 CrossRef CAS.
- P. Comba, G. Nunn, F. Scherz and P. H. Walton, Faraday Discuss., 2022, 234, 232–244 RSC.
- P. V. Bernhardt and P. Comba, Inorg. Chem., 1992, 31, 2638 CrossRef CAS.
- P. Comba, Coord. Chem. Rev., 1993, 123, 1 CrossRef CAS.
- P. Comba, T. W. Hambley and M. Ströhle, Helv. Chim. Acta, 1995, 78, 2042–2047 CrossRef CAS.
- P. Comba, T. W. Hambley, G. Lauer and N. Okon, MOMEC97, a molecular modeling package for inorganic compounds, Heidelberg, 1997 Search PubMed.
- P. Comba, N. Okon and R. Remenyi, J. Comput. Chem., 1999, 20, 781–785 CrossRef CAS.
- D. Wang, K. Ray, M. J. Collins, E. R. Farquhar, J. R. Frisch, L. Gomez, T. A. Jackson, M. Kerscher, A. Waleska, P. Comba, M. Costas, E. Münck and L. Que, Jr., Chem. Sci., 2013, 4, 282–291 RSC.
- P. Comba, S. Fukuzumi, H. Kotani and S. Wunderlich, Angew. Chem., Int. Ed., 2010, 49, 2622–2625 CrossRef CAS PubMed.
- P. Comba, S. Fukuzumi, C. Koke, A. M. Löhr and J. Straub, Angew. Chem., Int. Ed., 2016, 55, 11129–11133 CrossRef CAS PubMed.
- P. Comba, D. Faltermeier and B. Martin, Z. Anorg. Allg. Chem., 2020, 646, 1839–1845 CrossRef CAS.
- C. T. Lin, D. B. Rorabacher, G. R. Cayley and D. W. Margerum, Inorg. Chem., 1975, 14, 919–925 CrossRef CAS.
- J. A. Drumhiller, F. Montavon, J. M. Lehn and R. W. Taylor, Inorg. Chem., 1986, 25, 3751–3761 CrossRef CAS.
- P. Comba, U. Jermilova, C. Orvig, B. O. Patrick, C. F. Ramogida, K. Rück, C. Schneider and M. Starke, Chem. – Eur. J., 2017, 23, 15945–15956 CrossRef CAS PubMed.
- P. Cieslik, M. Kubeil, K. Zarschler, K. Anger, F. Brandt, M. Ullrich, H. Wadepohl, J. Pietzsch, H. Stephan and P. Comba, J. Am. Chem. Soc., 2022, 144, 21555–21567 CrossRef CAS PubMed.
- B. Drahos, I. Lukes and E. Toth, Eur. J. Inorg. Chem., 2012, 12, 1975–1986 CrossRef.
- A. Gupta, P. Caravan, W. S. Price, C. Platas-Iglesias and E. M. Gale, Inorg. Chem., 2020, 59, 6648–6678 CrossRef CAS PubMed.
- J. M. Omweri, V. Tekin, S. Saini, H. A. Houson, S. B. Jayawardana, D. A. Decato, G. B. Wijeratne and S. E. Lapi, Nucl. Med. Biol., 2024, 128–129, 108874 CrossRef CAS PubMed.
- G. J. Topping, P. Schaffer, C. Hoehr, T. J. Ruth and V. Sossi, Med. Phys., 2013, 40, 042502 CrossRef PubMed.
- A. L. Wooten, T. A. Aweda, B. C. Lewis, R. B. Gross and S. E. Lapi, PLoS One, 2017, 12, e0174351 CrossRef PubMed.
- H. Irving and R. J. P. Williams, Nature, 1948, 162, 746–747 CrossRef CAS.
- M. Sy, D. Ndiaye, I. d. Silva, S. Lacerda, L. J. Charbonniere, E. Toth and A. M. Nonat, Inorg. Chem., 2022, 61, 13421–13432 CrossRef CAS PubMed.
- A. Takacs, R. Napolitano, M. Purgel, A. C. Benyei, L. Zekany, E. Brucher, I. Toth, Z. Baranyai and S. Aime, Inorg. Chem., 2014, 53, 2858–2872 CrossRef CAS PubMed.
- Z. Ye, E.-K. Jeong, X. Wu, M. Tan, S. Yin and Z.-R. Lu, J. Magn. Reson. Imaging, 2012, 35, 737–744 CrossRef PubMed.
- R. Agarwal, S. M. Brunelli, K. Williams, M. D. Mitchell, H. I. Feldman and C. A. Umscheid, Nephrol., Dial., Transplant., 2009, 24, 856–863 CrossRef CAS PubMed.
- T. Grobner, Nephrol., Dial., Transplant., 2006, 21, 1104–1108 CrossRef CAS PubMed.
- D. Ndiaye and E. Toth, C. R. Chim., 2024, 27, 161–177 CrossRef.
- M. Devreux and S. Laurent, Molecules, 2023, 28, 7275 CrossRef PubMed.
- D. H. Powell, O. M. N. Dhubhghaill, D. Pubanz, L. Helm, Y. S. Lebedev, W. Schlaepfer and A. E. Merbach, J. Am. Chem. Soc., 1996, 118, 9333–9346 CrossRef CAS.
- E. Boros and A. B. Packard, Chem. Rev., 2018, 119, 870–901 CrossRef PubMed.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- P. Comba, S. Hunoldt, M. Morgen, J. Pietzsch, H. Stephan and H. Wadepohl, Inorg. Chem., 2013, 52, 8131–8143 CrossRef CAS PubMed.
- P. Comba, M. Kubeil, J. Pietzsch, H. Rudolf, H. Stephan and K. Zarschler, Inorg. Chem., 2014, 53, 6698–6707 CrossRef CAS PubMed.
- N. Choudhary, A. Dimmling, X. Wang, L. Southcott, V. Radchenko, B. O. Patrick, P. Comba and C. Orvig, Inorg. Chem., 2019, 58, 8685–8693 CrossRef CAS PubMed.
- F. Bruchertseifer, P. Comba, B. Martin, A. Morgenstern, J. Notni, M. Starke and H. Wadepohl, ChemMedChem, 2020, 15, 1591–1600 CrossRef CAS PubMed.
- M. Kubeil, C. Neuber, M. Starke, C. Arndt, L. Rodriguez Loureiro, L. Hoffmann, A. Feldmann, M. Bachmann, J. Pietzsch, P. Comba and H. Stephan, Chem. – Eur. J., 2024, e202400366 CrossRef CAS PubMed.
- L. Petitpoisson, A. Mahamoud, V. Mazan, M. Sy, O. Channin, E. Toth, L. J. Charbonniere, M. Elhabiri and A. M. Nonat, Inorg. Chem., 2024, 63, 22829–22844 CrossRef CAS PubMed.
- R. Gillet, A. Roux, S. Huclier-Markai, F. Camerel, O. Jeannin, A. M. Nonat and L. J. Charbonniere, Inorg. Chem., 2017, 56, 11738–11752 CrossRef CAS PubMed.
- A. Roux, R. Gillet, S. Huclier-Markai, L. Sabatier, L. J. Charbonniere and A. Nonat, Org. Biomol. Chem., 2017, 15, 1475–1483 RSC.
- M. Hussain, S. M. Qaim, I. Spahn, M. N. Aslam and B. Neumaier, Front. Chem., 2023, 11, 1270351 CrossRef CAS PubMed.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Reingold, J. Inorg. Chem., 2005, 4829–4833 CAS.
- M. S. Cooper, M. T. Ma, K. Sunassee, K. P. Shaw, J. Williams, R. L. Paul, P. S. Donnelly and P. J. Blower, Bioconjugate Chem., 2012, 23, 1029–1039 CrossRef CAS PubMed.
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651–698 CrossRef CAS PubMed.
- E. A. Ambundo, M.-V. Deydier, A. J. Grall, N. Agnera-Vega, L. T. Dressel, T. H. Cooper, N. J. Heeg, L. A. Ochrymowycz and D. B. Rorabacher, Inorg. Chem., 1999, 38, 4233–4242 CrossRef CAS.
- G. Singh, K. Zarschler, S. Hunoldt, I. I. Santana-Martinez, C. Rühl, M. Matterna, R. Bergmann, D. Mathe, N. Hegedüs, M. Bachmann, P. Comba and H. Stephan, Chem. – Eur. J., 2020, 26, 1989–2001 CrossRef CAS PubMed.
- S. Juran, M. Walther, H. Stephan, R. Bergmann, J. Steinbach, W. Kraus, F. Emmerling and P. Comba, Bioconjugate Chem., 2009, 20, 347–359 CrossRef CAS PubMed.
- H. Stephan, M. Walther, S. Fähnemann, P. Ceroni, J. Molley, G. Bergamini, C. E. Müller, F. Heisig, W. Kraus and P. Comba, Chem. – Eur. J., 2014, 20, 17011–17018 CrossRef CAS PubMed.
- M. F. B. Othman, N. R. Mitry, V. J. Lewington, P. J. Bower and S. Y. A. Terry, Nucl. Med. Biol., 2017, 46, 12–18 CrossRef CAS PubMed.
- T. W. Price, S. Y. Yap, R. Gillet, H. Savoe, L. J. Charbonniere, R. W. Boyle, A. M. Nonat and G. J. Stasiuk, Chem. – Eur. J., 2020, 26, 7602–7608 CrossRef CAS PubMed.
- P. A. Cieslik, S. Klingler, M. Nolff and J. P. Holland, Chem. – Eur. J., 2023, 14, e202303805 Search PubMed.
- S. Franchi, V. D. Marco and M. Tosato, Nucl. Med. Biol., 2022, 114–115, 168–188 CrossRef CAS PubMed.
- J. A. Weil and J. K. Kinnaird, J. Phys. Chem., 1967, 71, 3341 CrossRef CAS.
- A. Ku, V. J. Facca, Z. Cai and R. M. Reilly, EJNMMI Radiopharm. Chem., 2019, 4, 27 CrossRef PubMed.
- S. C. George and E. J. J. Samuel, Front. Chem., 2023, 11, 1218670 CrossRef CAS PubMed.
- P. Cieslik, P. Comba, M. Kubeil and H. Stephan, Bispidine derivatives and the use thereof, European Patent Nr, EP202167391, International Patent Nr.: WO2021EP86407, 2020 Search PubMed.
- J. N. Wacker, J. J. Woods, P. B. Rupert, A. Peterson, M. Allaire, W. W. Lukens, A. N. Gaiser, S. G. Minasian, R. K. Strong and R. J. Abergel, Nat. Commun., 2024, 15, 5741 CrossRef CAS PubMed.
- U. Cho and J. K. Chen, Cell Chem. Biol., 2020, 27, 921–936 CrossRef CAS PubMed.
- N. Hamon, A. Roux, M. Beyler, J.-C. Mulatier, C. Andraud, C. Nguyen, M. Maynadier, N. Bettache, A. Duperray, A. Grichine, S. Brasselet, M. Gary-Bobo, O. Maury and R. Tripier, J. Am. Chem. Soc., 2020, 142, 10184–10197 CrossRef CAS PubMed.
- A. D'aleo, A. Picot, A. Beeby, J. A. G. Williams, B. LeGuennic, C. Andraud and O. Maury, Inorg. Chem., 2008, 47, 10258–10268 CrossRef PubMed.
- A. T. Bui, A. Grichine, S. Brasselet, A. Duperray, C. Andraud and O. Maury, Chem. – Eur. J., 2015, 49, 17757–17761 CrossRef PubMed.
- P. A. Cieslik, M. Roux, L. M. A. Ali, S. R. Kiraev, B. Chartier, O. Seneque, S. Brasselet, A. Grichine, M. Gary-Bob, O. Maury and P. Comba, in preparation.
- P. Comba, C. Haaf, S. Helmle, K. D. Karlin, S. Pandian and A. Waleska, Inorg. Chem., 2012, 51, 2841–2851 CrossRef CAS PubMed.
- P. Comba, C. Lang, C. Lopez de Laorden, A. Muruganantham, G. Rajaraman, H. Wadepohl and M. Zajaczkowski, Chem. – Eur. J., 2008, 14, 5313–5328 CrossRef CAS PubMed.
- P. Comba, C. Haaf, A. Lienke, A. Muruganantham and H. Wadepohl, Chem. – Eur. J., 2009, 15, 10880–10887 CrossRef CAS PubMed.
- E. I. Solomon and R. G. Hadt, Coord. Chem. Rev., 2011, 255, 774–789 CrossRef CAS.
- P. Comba and A. M. Sargeson, J. Chem. Soc., Chem. Commun., 1985, 51 RSC.
- L. S. Mello, P. D. Engel, P. Orecchia, K. Bleher, F. Rominger, K. Borate, R. Goetz, P. Deglmann, A. Schäfer, C. Winter, M. Rack, P. Comba, A. S. K. Hashmi and T. Schaub, Chem. – Eur. J., 2024, 30, e202403023 CrossRef CAS PubMed.
- Note that there is a change of 0.9 pH units with respect to the original publication61 due to an arithmetic error therein.
- M. Naher, C. Su, M. A. Gonzalvez, C. M. Williams and P. V. Bernhardt, Organometallics, 2024, 43, 2821–2830 CrossRef CAS.
- C. Krebs, D. Galonici Fujimori, C. T. Walsh and J. M. Bollinger, Jr., Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed.
- E. I. Solomon, T. C. Brunold, M. I. Davis, J. N. Kensley, S.-K. Lee, N. Lehnert, F. Neese, A. J. Skulan, Y.-S. Yang and J. Zhou, Chem. Rev., 2000, 100, 235 CrossRef CAS PubMed.
- M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS PubMed.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed.
- J. Hädeler, G. Velmurugan, R. Lauer, R. Radhamani, F. Keppler and P. Comba, J. Am. Chem. Soc., 2023, 145, 24590–24602 Search PubMed.
- J. C. Paris, S. Hu, A. Wen, A. C. Weitz, R. Cheng, L. B. Gee, Y. Tang, H. Kim, A. Vegas, W.-C. Chang, S. J. Elliott, P. Liu and Y. Guo, Angew. Chem., Int. Ed., 2023, 62, e202309362 CrossRef CAS PubMed.
- A. R. McDonald and L. Que Jr, Coord. Chem. Rev., 2013, 257(2), 414–428 CrossRef CAS.
- X. Engelmann, I. Monte-Perez and K. Ray, Angew. Chem., Int. Ed., 2016, 55, 7632–7649 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2015, 48, 2415–2423 CrossRef CAS PubMed.
- P. Comba, M. Kerscher, M. Krause and H. F. Schöler, Environ. Chem., 2015, 12, 381–395 CrossRef CAS.
- C. Kupper, B. Mondal, J. Serrano-Plana, I. Klawitter, F. Neese, M. Costas, S. Ye and F. Meyer, J. Am. Chem. Soc., 2017, 139, 8939–8949 CrossRef CAS PubMed.
- M. R. Bukowski, P. Comba, C. Limberg, M. Merz, L. Que, Jr. and T. Wistuba, Angew. Chem., Int. Ed., 2004, 43, 1283–1287 CrossRef CAS PubMed.
- K. Bleher, P. Comba, D. Faltermeier, V. Gunasekaran, A. Gupta, M. Kerscher, S. Krieg, B. Martin, H. Wadepohl and S. Yang, Chem. – Eur. J., 2022, 27, e202103452 CrossRef PubMed.
- M. Abu-Odeh, K. Bleher, N. J. Britto, P. Comba, M. Gast, M. Jaccob, M. Kerscher, S. Krieg and M. Kurth, Chem. – Eur. J., 2021, 27, 11377–11390 CrossRef CAS PubMed.
- T. Josephy, R. Kumar, K. Bleher, F. Röhs, T. Glaser, G. Rajaraman and P. Comba, Inorg. Chem., 2024, 63, 12109–12119 CrossRef CAS PubMed.
- K. Bleher, P. Comba, D. Kass, K. Ray and H. Wadepohl, J. Inorg. Biochem., 2023, 241, 112123 CrossRef CAS PubMed.
- A. K. Vardhaman, Y.-M. Lee, J. Jung, K. Ohkubo, W. Nam and S. Fukuzumi, Angew. Chem., Int. Ed., 2016, 55, 3709–3713 CrossRef CAS PubMed.
- E. J. Klinker, T. A. Jackson, M. P. Jensen, A. Stubna, G. Juhasz, E. L. Bominaar, E. Münck and L. Que, Jr., Angew. Chem., Int. Ed., 2006, 45, 7394–7397 CrossRef CAS PubMed.
- G. Coin, R. Patra, S. Rana, J. P. Biswas, P. Dubourdeaux, M. Clemancey, S. P. d. Visser, D. Maiti, P. Maldivi and J.-M. Latour, ACS Catal., 2020, 10, 10010–10020 CrossRef CAS.
- R. Kumar, A. Ansari, P. Comba and G. Rajaraman, Chem. – Eur. J., 2023, e202303300 Search PubMed.
- R. Gupta and A. S. Borovik, J. Am. Chem. Soc., 2003, 125, 13234–13242 CrossRef CAS PubMed.
- E. M. Matson, Y. J. Park and A. R. Fout, J. Am. Chem. Soc., 2014, 17398–17401 CrossRef CAS PubMed.
- F. F. Pfaff, S. Kundu, M. Risch, S. Pandian, F. Heims, I. Pryjomska-Ray, P. Haack, R. Metzinger, E. Bill, H. Dau, P. Comba and K. Ray, Angew. Chem., Int. Ed., 2011, 50, 1711–1715 CrossRef CAS PubMed.
- K. Bleher, R. Rana, T. Josephy, T. Glaser, H. Wadepohl, G. Rajaraman and P. Comba, submitted.
- B. Wang, Y.-M. Lee, M. Clémancey, M. S. Seo, R. Sarangi, J.-M. Latour and W. Nam, J. Am. Chem. Soc., 2016, 138, 2426–2436 CrossRef CAS PubMed.
- G. Velmurugan and P. Comba, Eur. J. Inorg. Chem., 2025, e202400837 CrossRef.
- J. Bautz, P. Comba and L. Que, Jr., Inorg. Chem., 2006, 45, 7077 CrossRef CAS PubMed.
- P. Comba, G. Rajaraman and H. Rohwer, Inorg. Chem., 2007, 46, 3826–3838 CrossRef CAS PubMed.
- W. Zhu, P. Wu, V. A. Larson, A. Kumar, X.-X. Li, M. S. Seo, Y.-M. Lee, B. Wang, N. Lehnert and W. Nam, J. Am. Chem. Soc., 2024, 146, 250–262 CrossRef CAS PubMed.
- G. Mukherjee, G. Velmurugan, M. Kerscher, J. K. Satpathy, C. V. Sastri and P. Comba, Chem. Eur. J., 2023, e202303127 Search PubMed.
- L. Sahoo, P. Panwar, C. V. Sastri and S. P. de Visser, ACS Org. Inorg. Au, 2024, 4, 673–680 CrossRef CAS PubMed.
- P. Comba, Y.-M. Lee, W. Nam and A. Waleska, Chem. Commun., 2014, 50, 412–414 RSC.
- R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2006, 45, 206–222 CrossRef CAS PubMed.
- J. W. de Boer, V. Wesenhagen, E. C. M. Wenker, K. Maaijen, F. Gol, H. Gibbs and R. Hage, Eur. J. Inorg. Chem., 2013, 3581–3591 CrossRef CAS.
- M. P. Jensen, M. P. Mehn and L. Que, Jr., Angew. Chem., Int. Ed., 2003, 43, 4357–4360 CrossRef PubMed.
- F. Avenier and J.-M. Latour, Chem. Commun., 2004, 1544–1545 RSC.
- G. Mukherjee, F. G. Cantu Reinhard, U. K. Bagha, C. V. Sastri and S. P. de Visser, Dalton Trans., 2020, 49, 5921–5931 RSC.
- S. Kumar, A. S. Faponle, P. Barman, A. K. Vardhaman, C. V. Sastri, D. Kumar and S. P. de Visser, J. Am. Chem. Soc., 2014, 136, 17102–17115 CrossRef CAS PubMed.
- P. Barman, A. K. Vardhaman, B. Martin, S. J. Wörner, C. V. Sastri and P. Comba, Angew. Chem., Int. Ed., 2015, 54, 2095–2099 CrossRef CAS PubMed.
- P. Barman, P. Upadhayay, A. S. Faponle, J. Kumar, S. S. Nag, D. Kumar, C. V. Sastri and S. P. d. Visser, Angew. Chem., Int. Ed., 2016, 55, 11091–11095 CrossRef CAS PubMed.
- J. Benet-Buchholz, P. Comba, A. Llobet, S. Roeser, P. Vadivelu, H. Wadepohl and S. Wiesner, Dalton Trans., 2009, 5910–5923 RSC.
- J. Benet-Buchholz, P. Comba, A. Llobet, S. Roeser, P. Vadivelu and S. Wiesner, Dalton Trans., 2010, 39, 3315 RSC.
- P. Comba, S. Kuwata, G. Linti, M. Tarnai and H. Wadepohl, Eur. J. Inorg. Chem., 2007, 657 CrossRef CAS.
- P. Comba, S. Kuwata, G. Linti, M. Tarnai and H. Wadepohl, Chem. Commun., 2006, 2074–2076 RSC.
- P. Comba, M. Kerscher, G. A. Lawrance, B. Martin, H. Wadepohl and S. Wunderlich, Angew. Chem., Int. Ed., 2008, 47, 4740–4743 CrossRef CAS PubMed.
- P. Comba, B. Pokrandt and H. Wadepohl, Aust. J. Chem., 2017, 70, 576–580 CrossRef CAS.
- M. Haberberger, C. I. Someya, A. Company, E. Irran and S. Enthaler, Catal. Lett., 2012, 142, 557–565 CrossRef CAS.
- D. Scharnagel, A. Müller, F. Prause, M. Ecke, J. Goller, W. Milius and M. Breuing, Chem. – Eur. J., 2015, 21, 12488–12500 CrossRef CAS PubMed.
- K. Y. Ponomarev, E. S. Mozhaitsev, N. S. Li-Zhulanov, A. A. Okhina, A. A. Nefedov, A. D. Rogachev, E. V. Suslov, A. I. Dalinger, S. Z. Vatsadze, K. P. Volcho and N. F. Salakhutdinov, Russ. Chem. Bull., 2024, 73(8), 2248–2260 CrossRef CAS.
- G. Li, R. Wang, D. Ye, M. Pu, X. Feng and L. Lin, Eur. J. Org. Chem., 2024, 27, e202400008 CrossRef CAS.
- G. Li, M. Liu, S. Zou, X. Feng and L. Lin, Org. Lett., 2020, 22, 8708–8713 CrossRef CAS PubMed.
- Z. Yang, J. Liu, X. Liu, Z. Wang, X. Feng, Z. Su and C. Hu, Adv. Synth. Catal., 2008, 350, 2001–2006 CrossRef CAS.
- J. Liu, Z. Yang, Z. Wang, F. Wang, X. Chen and X. Liu, J. Am. Chem. Soc., 2016, 130, 5654–5655 CrossRef PubMed.
- G. Li, Y. Zhang, H. Zeng, X. Feng, Z. Su and L. Lin, Chem. Sci., 2022, 13, 4313–4320 RSC.
- P. Comba, Coord. Chem. Rev., 1999, 182, 343–371 CrossRef.
- P. Comba and M. Kerscher, Coord. Chem. Rev., 2009, 253, 564–574 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |