
AdcA lipoprotein involved in Zn(II) transport in Streptococcus mutans – is it as metal-specific as expected?†
Kinga
Garstka
a,
Aleksandra
Hecel
*a,
Henryk
Kozłowski
ab,
Alicia
Dominguez-Martin
 c,
Krzysztof
Szewczyk
d and
Magdalena
Rowińska-Żyrek
*a
c,
Krzysztof
Szewczyk
d and
Magdalena
Rowińska-Żyrek
*a
aFaculty of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: magdalena.rowinska-zyrek@uwr.edu.pl; aleksandra.hecel2@uwr.edu.pl
bInstitute of Health Sciences, University of Opole, Katowicka 68 St, 45-060 Opole, Poland
cDepartment of Inorganic Chemistry, Faculty of Pharmacy, University of Granada, E-18071 Granada, Spain
dDepartment of Oncology, Wrocław Medical University, pl. L. Hirszfelda 12, 53-413 Wrocław, Poland
First published on 4th March 2025
Abstract
Streptococcus mutans, a Gram-positive pathogen, is a primary causative agent of dental caries. It modifies the oral biofilm architecture on tooth enamel and, like other bacteria, requires transition metal ions such as Zn(II), Cu(II), and Ni(II) for survival and virulence. Physiological salivary Zn(II) levels are insufficient for optimal bacterial growth, prompting S. mutans to develop a specialized ABC transport system comprising AdcA, AdcB, and AdcC. Among these, the lipoprotein AdcA plays a pivotal role in Zn(II) acquisition. In this study, we examined two probable Zn(II)-binding sites in AdcA—EGHGHKGHHHA and HGIKSQKAEHFH—and their Zn(II), Cu(II), and Ni(II) complexes, keeping in mind that Cu(II) and Ni(II) are essential nutrients for bacterial enzymes and can compete with Zn(II) for its binding sites. At physiological pH, in the Zn(II)–Ac–EGHGHKGHHHA–NH2 species, Zn(II) binds to histidine residues, forming complexes with up to four coordinated imidazole nitrogens, while in the Zn(II)–Ac–HGIKSQKAEHFH–NH2 complex, we found three coordinated histidine side chains. The same regions of the AdcA lipoprotein are able to bind Cu(II) with even higher affinity. The stability of Zn(II) and Ni(II) complexes, on the other hand, is more comparable, with a slight advantage for Ni(II). In this case, at pH 7.4, the coordination spheres of both Zn(II) and Ni(II) consist of the same set of donor atoms. The metal binding preferences align with the Irving–Williams series; however, given the significantly higher Zn(II) concentrations in saliva and dental plaques, Zn(II) occupies the AdcA binding sites in vivo, highlighting its critical role in S. mutans virulence and metal ion homeostasis.
Introduction
Streptococcus mutans is a Gram-positive, key pathogen of dental caries,1,2 capable of modifying the architecture of the oral biofilm formed on hard surfaces of the tooth and environment.3,4S. mutans does not act alone to cause dental caries but interacts with other organisms to assemble a cariogenic biofilm in the presence of a sugar-rich diet,3 which changes the microenvironment for other acidogenic and aciduric bacteria, making it easier for them to establish themselves.4S. mutans causes infective endocarditis, responsible for life-threatening infection of heart valves5 and is strongly associated with sclerosis multiplex (MS), especially in early childhood caries.6 During its colonization, S. mutans has to respond and adapt to changes in the environment, like for example, oxidative and acid stress tolerance, carbohydrate utilization or the maintenance of intracellular metal ion homeostasis.7 Its neutral habitat is human saliva – a unique body fluid, critical for oral health, due to its organic and inorganic components,8,9 such as, among others, the divalent zinc ion.10Under physiological conditions, Zn(II) is an essential element for both eukaryotic and prokaryotic cells, functioning as a structural or catalytic component of numerous proteins.11 It is estimated that Zn(II) is present in up to 10% of all proteins in humans,12 while in bacteria, it is found in about 5–6% of them13 (mostly due to the smaller number of zinc finger transcription factors). Like in the case of other transition metal ions, the excess of Zn(II) might cause growth arrest and defects in the virulence of microorganisms.7 For this reason, the intercellular concentration of bacterial Zn(II) should be strictly regulated for their survival and virulence in the human oral cavity.14–17 Zn(II) is critical to the infection process, while the physiological salivary level of Zn(II) fluctuates. Below pH 6.0, most free zinc present in saliva is in the Zn(II) form, but the increase of pH drastically reduces its availability.18 The salivary Zn(II) level also fluctuates throughout the day – between meals, the zinc concentration is limited, whereas during mealtime, its concentration increases.7 Zn(II) is also present in the tooth enamel surface and in the dental plaque, where its concentration is much higher, reaching millimolar levels.19 There are many different types of metal transport systems, and several of them are involved in Zn(II) homeostasis in streptococci20 (discussed in Fig. 1).
 | ||
| Fig. 1 A summary diagram of all known types of transporters for Zn(II) in streptococci, with arrows indicating metal import or export: histidine triad proteins (Pht), ABC transporter, CDF pump (cation diffusion facilitator): CzcD. The ZccE (P-type ATPase) is only found in S. mutans. | ||
Differently from other pathogenic bacteria that encode multiple Zn(II) import systems, S. mutans encodes a single, highly conserved and high affinity zinc ABC-type importer known as AdcABC.21,22 ABC transporters (ATP-binding cassette transporters) consist of the following: AdcA, a zinc-binding lipoprotein that in Gram-positive bacteria is localized at the cell surface,23 AdcB – a membrane permease and AdcC – a cytoplasmic ATPase22,24–26 (adenosine 5′-triphosphatase), and their genes are regulated in response to environmental zinc availability by an adhesin competence repressor, AdcR.27 Moreover, S. mutans encodes a ZccE protein, which is a P-type ATPase, responsible for Zn(II) export.7,21,28 Also, in contrast to most streptococcal genomes, which encode two copies of adcA (called adcA and adcAII), the genome of S. mutans encodes a single adcA gene,22 whose size is more similar to adcBC than to adcAII.21 The streptococcal adcAII is genetically coupled to the pht genes (metal-binding poly-histidine triad genes) and is present in proteins that scavenge zinc, where it binds zinc outside the cell and then most probably shuttles it to AdcA.29–31 Thus, the polyhistidine triad proteins (Pht) contain multiple copies of the histidine triad motif (HxxHxH, where x may stand for any amino acid).32 Despite the fact that the AdcABC transporter is critical to the cariogenic potential of S. mutans and zinc is a growth-limiting factor for producing the oral biofilm, the genome of S. mutans lacks pht homologs21,32 and S. mutans is the only Streptococcus species that does not express Pht proteins.23 Thus, inactivating adcA or adcCB would inhibit the growth of S. mutans.33–35
For Zn(II) uptake, the Streptococcus species synthesizes the double-domain AdcA protein with two zinc-binding sites: the N-terminal and C-terminal domains (Fig. 2). The N-terminal domain, in the presence of Zn(II), quickly and strongly binds the metal and then changes its conformation, which stabilizes the second, C-terminal domain (H36, H122, H186, and E261).36 A tight zinc-binding pocket facilitates zinc binding, and the relatively less stable binding and conformation of the C-terminal domain (H436, H445, and H447) ensure a rapid transfer of Zn(II) from AdcA to the transmembrane AdcB protein.36 As in other streptococci, AdcA from S. mutans has two zinc-binding domains: the N-terminal domain, which consists of five histidine residues (H132, H134, H137, H138, and H139 – Fig. S1, ESI†), and the C-terminal domain, which contains three histidine residues in close proximity (H457, H466, H468 – Fig. S1, ESI†).21
 | ||
| Fig. 2 The model of Zn(II) uptake in Streptococcus via AdcA in a Zn(II)-deficient environment. The N- and C-terminal domains of AdcA are marked green and yellow, respectively. The two binding sites are represented as an open state without metal ions (left part) and a half-closed or closed state in Zn(II)-bound AdcA (right part). The AdcA structures are based on the files from the Protein Data Bank (accession codes: 7JJ9, 7JJ8, 7JJA and 7JJB). | ||
Another well-conserved and potentially metal-binding site contains three conserved histidine residues (H70, H144, and H208) and one glutamic acid residue (E294).21 This AdcA-based high-affinity Zn(II) transport system is homologous to the one from Escherichia coli ZnuA,37 ZnuA and AztC solute binding proteins from Paracoccus denitrificans38 or to the fungal zincophore-based transport systems, comprising proteins with high Zn(II) affinity that are synthesized in the cytoplasm and then exported to bind Zn(II).39–42
During infection, the host limits the availability of micronutrients for pathogens at the colonization surface in a process referred to as nutritional immunity43–45 and plays a role in host defence against pathogenic microorganisms, constituting an ideal therapeutic target in times of increasing antibiotic resistance. Antimicrobial peptides are a promising direction to find new, effective, and specific drugs, and their use in STAMP technology (specifically targeted antimicrobial peptides) increases the chances of winning the fight against microorganisms. STAMP is based on two domains: an antimicrobial domain (an antimicrobial peptide – AMP, small host defence peptides that form part of the innate immune response46) and a targeting domain (a fragment of the protein from the pathogen), linked via a short flexible linker.47 A precise understanding of the Zn(II) transport mechanism present in pathogens is an important research direction towards the design of a targeting domain for new, highly specific therapeutics against antibiotic-resistant bacterial species.
Because biofilm formation is a survival strategy by which S. mutans, an important factor for the initiation of dental caries, adapts to its environment, we focused on Zn(II), Cu(II) and Ni(II) complexes of two regions of the AdcA lipoprotein involved in Zn(II) transport in Streptococcus mutans: Ac–EGHGHKGHHHA–NH2 and Ac–HGIKSQKAEHFH–NH2 (highlighted in red in Fig. S1, ESI†). We checked their structural and thermodynamic properties using a number of complementary methods: potentiometry, EPR, UV-Vis and CD spectroscopy. The results allowed us to provide information on the protonation and stability constants, identify the number and types of donor atoms involved in Zn(II), Cu(II), and Ni(II) binding and determine the influence of metal ions on the secondary structure of the analyzed ligands.
Experimental
Synthesis
Peptides (Ac–EGHGHKGHHHA–NH2 and Ac–HGIKSQKAEHFH–NH2) were purchased from KareBay Biochem (certified purity: 98%) and were used as received. The carbonate-free stock solution of 0.1 M NaOH was purchased from Sigma-Aldrich and then potentiometrically standardized with potassium hydrogen phthalate. Zinc perchlorate, copper perchlorate and nickel chloride were extra-pure products (Sigma-Aldrich). The concentrations of salt solutions were determined by inductively coupled plasma (ICP) mass spectrometry.Potentiometric measurements
The stability constants for Ac–EGHGHKGHHHA–NH2 and Ac–HGIKSQKAEHFH–NH2 ligands and their Zn(II), Cu(II) and Ni(II) complexes were calculated from titration curves carried out in the pH range of 2–11 at 298 K in 0.1 M NaClO4. The total volume of the solution used was 3.0 cm3. The potentiometric titrations were performed using a Dosimat 800 Metrohm titrator connected to a Metrohm 905 pH meter and a Mettler Toledo pH inLab Science electrode. The glass cell was equipped with a magnetic stirring system, a microbiuret delivery tube and an inlet–outlet tube for argon. Solutions were titrated with 0.1 M carbonate-free NaOH. The electrodes were calibrated daily for hydrogen ion concentration by titrating HClO4 with NaOH using a total volume of 3.0 cm3. The purities and the exact concentrations of the ligand solutions were determined using the Gran method. The ligand concentration was 0.5 mM. The Zn(II), Cu(II) and Ni(II) to ligand ratio was 0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The HYPERQUAD 2006 program was used for the stability constant calculations.48 The standard deviations were computed using HYPERQUAD 2006 and referenced to random errors only. The constants for hydrolytic Zn(II) and Ni(II) species were used in these calculations.49,50 The speciation and competition diagrams were computed using the HYSS program.51
:1. The HYPERQUAD 2006 program was used for the stability constant calculations.48 The standard deviations were computed using HYPERQUAD 2006 and referenced to random errors only. The constants for hydrolytic Zn(II) and Ni(II) species were used in these calculations.49,50 The speciation and competition diagrams were computed using the HYSS program.51
Spectroscopic studies
The absorption spectra were recorded on a Jasco-730 spectrophotometer in the range 200–800 nm, using a quartz cuvette with an optical path of 1 cm. Circular dichroism spectra were recorded on a Jasco J-1500 CD spectrometer in the 250–800 nm and 180–250 nm ranges, using a quartz cuvette with an optical path of 1 cm and 0.01 cm in the visible and near-UV range. The concentration of sample solutions used for spectroscopic studies was similar to that employed in the potentiometric experiment. The metal:ligand ratio was 0.8:1. All spectroscopic measurements were recorded in the pH range 2.5–10.5. The pH of the samples was adjusted with appropriate amounts of concentrated HClO4 and NaOH solutions. OrginPro 2016 was used to process and visualize the obtained spectra.52 Electron paramagnetic resonance (EPR) spectra were recorded in liquid nitrogen on a Bruker ELEXSYS E500 CW-EPR spectrometer at an X-band frequency (9.5 GHz) and equipped with an ER 036TM NMR teslameter and an E41 FC frequency counter. The ligands were prepared in an aqueous solution of HClO4 at I = 0.1 M (NaClO4). The concentration of Cu(II) was 1 mM, and the metal:ligand ratio was 0.8:1. Ethylene glycol (25%) was used as a cryoprotectant for EPR measurements. The EPR parameters were analyzed by computer simulation of the experimental spectra using WIN-EPR SIMFONIA software, version 1.2 (Bruker). The pH was adjusted with appropriate amounts of HClO4 and NaOH solutions.
Results
Ligand protonation
Both peptides (Ac–EGHGHKGHHHA–NH2 and Ac–HGIKSQKAEHFH–NH2) are protected on the N-terminus by acetylation but also on the C-terminus by amidation. The Ac–EGHGHKGHHHA–NH2 peptide behaves as an LH7 acid with the protonating groups corresponding to the deprotonation of the carboxylic side chain of glutamic acid (pKa = 3.87), five histidine imidazole side chain groups (pKa values of 5.48, 5.73, 6.47, 6.51 and 7.46, respectively) and to the deprotonation of the lysine side chain group with a pKa value of 10.07 (Table 1). The Ac–HGIKSQKAEHFH–NH2 peptide has six protonation constants. The first one (pKa = 3.90) can be assigned to the deprotonation of the carboxylic side chain of glutamic acid. The next three constants (pKa = 5.84, 6.35 and 7.08) are related to the deprotonation of three histidine imidazole groups and the last two (pKa = 9.88 and 10.56) correspond to the deprotonation of two lysine side chain groups (Table 1).| Species | Ac–EGHGHKGHHHA–NH2 | Ac–HGIKSQKAEHFH–NH2 | ||||
|---|---|---|---|---|---|---|
| Logβ |
LogK |
Residue | Logβ |
LogK |
Residue | |
| HL | 10.07(1) | 10.07 | (K) | 10.56(1) | 10.56 | (K) |
| H2L | 17.54(3) | 7.46 | (H) | 20.43(1) | 9.88 | (K) |
| H3L | 24.05(3) | 6.51 | (H) | 27.51(1) | 7.08 | (H) |
| H4L | 30.52(3) | 6.47 | (H) | 33.86(1) | 6.35 | (H) |
| H5L | 36.25(3) | 5.73 | (H) | 39.71(2) | 5.84 | (H) |
| H6L | 41.72(3) | 5.48 | (H) | 43.60(2) | 3.90 | (E) |
| H7L | 45.59(3) | 3.87 | (E) | |||
| Zn( II ) complexes | ||||||
| ZnH3L | 29.00(1) | 30.88(1) | ||||
| ZnH2L | 22.85(1) | 6.14 | (H) | 25.13(1) | 5.75 | (H) |
| ZnHL | 15.92(2) | 6.93 | (H) | 17.37(2) | 7.76 | (H2O) |
| ZnL | 7.46(2) | 8.46 | (H2O) | 9.24(2) | 8.13 | (H2O) |
| ZnH−1L | −2.11(3) | 9.58 | (K) | −0.60(3) | 9.84 | (K) |
| ZnH−2L | −11.19(4) | 10.51 | (K) | |||
| Cu( II ) complexes | ||||||
| CuH4L | 36.62(1) | |||||
| CuH3L | 32.28(1) | 4.34 | (H) | 33.73(3) | ||
| CuH2L | 26.91(2) | 5.37 | (H) | 28.18(5) | 5.55 | (H) |
| CuHL | 20.35(2) | 6.56 | (am) | 22.49(4) | 5.69 | (am) |
| CuL | 12.69(3) | 7.66 | (am) | 15.68(5) | 6.81 | (am) |
| CuH−1L | 5.23(3) | 7.46 | (am) | 8.24(4) | 7.44 | (am) |
| CuH−2L | −4.40(5) | 9.63 | (K) | −1.00(7) | 9.24 | (K) |
| CuH−3L | −10.60(5) | 9.60 | (K) | |||
| Ni( II ) complexes | ||||||
| NiH4L | 36.56(8) | |||||
| NiH3L | 29.02(1) | 31.45(1) | 5.11 | (H) | ||
| NiH2L | 22.87(1) | 6.15 | (H) | 25.48(1) | 5.98 | (H) |
| NiHL | 16.00(1) | 6.87 | (H) | |||
| NiL | 7.61(2) | 8.39 | (am) | 8.61(1) | ||
| NiH−1L | −1.90(2) | 9.51 | (am) | −0.78(1) | 9.38 | (am) |
| NiH−2L | −11.04(1) | 10.27 | (K) | |||
| NiH−3L | −23.65(4) | (K) | −21.59(1) | 10.55 | (K) | |
Metal complexes of Ac–EGHGHKGHHHA–NH2
Metal complexes of Ac–HGIKSQKAEHFH–NH2
Secondary structure
The coordination of Zn(II) and Cu(II) influences pronounced secondary structural changes of both Ac–EGHGHKGHHHA–NH2 and Ac–HGIKSQKAEHFH–NH2 AdcA regions (Fig. 3). At pH 7.4, both peptides are random coils, with a positive absorption band around 215 nm and a negative one below 200 nm.54 Metal coordination changes the CD spectra and therefore also the secondary structure of the complexed ligands – for both Zn(II) and Cu(II) complexes, the intensity of the spectral minima becomes smaller and the minima and maxima shift to ca. 195 nm and 210 nm, respectively. Although these parameters are not typical for any particular structure (they are sometimes inadequately assigned to PPII like helices in the literature), it is clear that metal coordination significantly induces structural changes. | ||
| Fig. 3 Far UV CD spectra of the (A) Ac–EGHGHKGHHHA–NH2 and (B) Ac–HGIKSQKAEHFH–NH2 fragments with Zn(II), Cu(II) and Ni(II) in the range of 180–250 nm, pH 7.4, T = 298 K; optical path = 0.01 cm; [L] = 0.0005 M; M(II)/L = 0.8:1. | ||
Ni(II) coordination is an interesting case – its binding appears to have almost no structural impact on the overall structure of Ac–HGIKSQKAEHFH–NH2, while in the case of Ac–EGHGHKGHHHA–NH2, it induces a significant structural rearrangement, enhancing and shifting the CD maximum at 215 nm and making the spectral minimum at 195 nm much more shallow.
Discussion
Is it theoretically possible for other biologically relevant metal ions, Cu(II) and Ni(II), necessary for proper growth and replication of microorganisms, to compete with Zn(II) for the binding sites in the AdcA protein?In order to identify the AdcA fragment that binds Zn(II), Cu(II) and Ni(II) with the highest affinity, we used the thermodynamic data for the metal complexes to simulate a theoretical situation in which equimolar amounts of Zn(II) and both studied unstructured AdcA fragments were mixed. This allows a direct comparison of the calculated constants at different pH values (Fig. 4A) and shows that in almost the entire pH range, the Ac–EGHGHKGHHHA–NH2 ligand is preferred over the Ac–HGIKSQKAEHFH–NH2 one, which is not surprising, since the Ac–EGHGHKGHHHA–NH2 region contains more histidine residues (five vs. three in the Ac–HGIKSQKAEHFH–NH2 ligand). At pH 7.4, more than 70% of available Zn(II) would be bound to Ac–EGHGHKGHHHA–NH2.
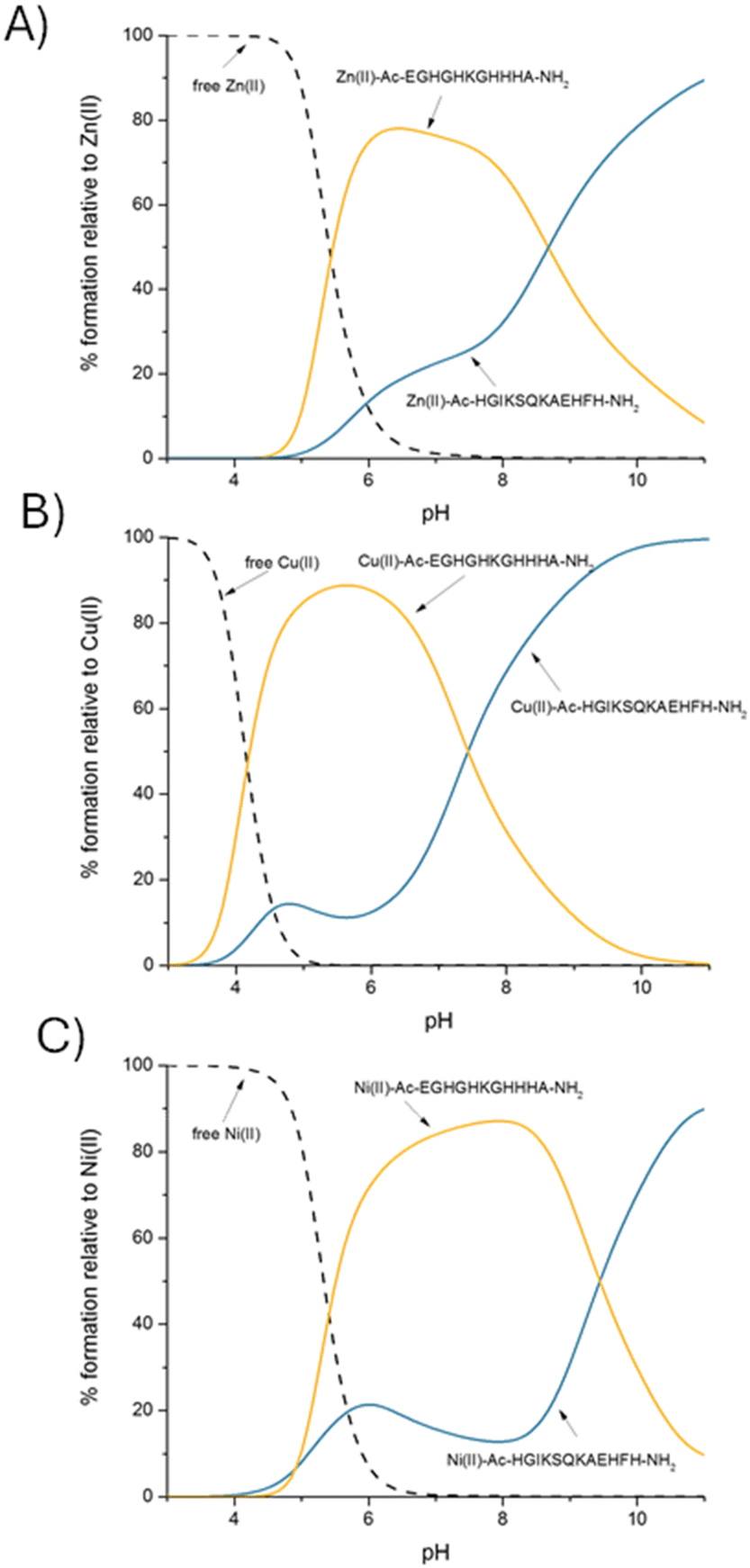 | ||
Fig. 4 A competition plot between AdcA fragments: Ac–EGHGHKGHHHA–NH2 and Ac–HGIKSQKAEHFH–NH2 and (A) Zn(II), (B) Cu(II) and (C) Ni(II), describing complex formation at different pH values in a hypothetical situation in which equimolar amounts of all reagents are mixed. Calculations are based on binding constants from Table 1. Conditions: T = 298 K, I = 0.1 M NaClO4, [Ac–EGHGHKGHHHA–NH2]![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) [Ac–HGIKSQKAEHFH–NH2][Zn(II)][Cu(II)][Ni(II)] = 0.001 M. [Ac–HGIKSQKAEHFH–NH2][Zn(II)][Cu(II)][Ni(II)] = 0.001 M. | ||
A very interesting case is observed for Cu(II) complexes with the studied ligands; at acidic pH, Cu(II) prefers to bind to the more His-rich Ac–EGHGHKGHHHA–NH2 region, while at basic pH, in which amide nitrogens dominate in the Cu(II) coordination sphere, the Ac–HGIKSQKAEHFH–NH2 fragment is preferred. Interestingly, at pH 7.4, the two complexes are almost equally stable (Fig. 4B). Ni(II) also chooses Ac–EGHGHKGHHHA–NH2 over Ac–HGIKSQKAEHFH–NH2 almost in the entire pH range, in which His imidazoles are the metal binding sites; at pH 7.4, in our hypothetical situation, more than 85% of the metal would be bound to the His-rich Ac–EGHGHKGHHHA–NH2 region. As in the case of Cu(II), the situation changes dramatically in an alkaline environment when amide nitrogens are involved in coordination – the Ni(II)–Ac–HGIKSQKAEHFH–NH2 complex becomes more stable (Fig. 4C). To summarize, poly-His complexes are more tempting metal binding sites at lower pH values, while at higher pH, when amide nitrogens are involved in Cu(II) and Ni(II) coordination, the affinity of poly-His sites turns out to be lower than that of the sequence with a lower number of His residues, which is more likely to involve an amide in binding.
The Ac–EGHGHKGHHHA–NH2 region itself strongly prefers Cu(II) over Zn(II) and Ni(II), which is consistent with the Irving–Williams series.55,56 Even in a theoretical situation in which equimolar amounts of Zn(II), Cu(II) and Ni(II) would be available, in the whole pH range, the sequence strongly prefers to bind Cu(II) (Fig. 5A). As far as Zn(II) and Ni(II) binding preferences are concerned, up to pH 7.4, no significant differences in the stability of both complexes are observed – the two metals bind with almost identical affinity, which is quite surprising. The situation changes in an alkaline environment when amide nitrogens are involved in the coordination of Ni(II), and the Ni(II)–Ac–EGHGHKGHHHA–NH2 complex becomes significantly more stable than the Zn(II) one (Fig. 5B).
 | ||
| Fig. 5 A competition plot between (A) the AdcA fragment: Ac–EGHGHKGHHHA–NH2, Zn(II), Cu(II) and Ni(II) and (B) the AdcA fragment: Ac–EGHGHKGHHHA–NH2, Zn(II), and Ni(II), describing complex formation at different pH values in a hypothetical situation in which equimolar amounts of all reagents are mixed. Calculations are based on binding constants from Table 1. Conditions: T = 298 K, I = 0.1 M NaClO4, (A) [Ac–EGHGHKGHHHA–NH2][Zn(II)][Cu(II)][Ni(II)] = 0.001 M; (B) [Ac–EGHGHKGHHHA–NH2][Zn(II)][Ni(II)] = 0.001 M. | ||
A very analogous situation is observed for Ac–HGIKSQKAEHFH–NH2: consistent with the Irving–Williams series,55,56 the Cu(II)–Ac–HGIKSQKAEHFH–NH2 complex is the most stable one (Fig. 6A). As for the Zn(II) and Ni(II) specificity of this region, it seems to have no strong metal preference up to pH 9. At basic pH, the coordination of amide nitrogens in Ni(II) complexes gives them greater stability than Zn(II) complexes (Fig. 6B), as in the case of the previous ligand (Fig. 5B).
 | ||
| Fig. 6 A competition plot between (A) the AdcA fragment: Ac–HGIKSQKAEHFH–NH2, Zn(II), Cu(II) and Ni(II) and (B) the AdcA fragment: Ac–HGIKSQKAEHFH–NH2, Zn(II), and Ni(II), describing complex formation at different pH values in a hypothetical situation in which equimolar amounts of all reagents are mixed. Calculations are based on binding constants from Table 1. Conditions: T = 298 K, I = 0.1 M NaClO4, (A) [Ac–HGIKSQKAEHFH–NH2][Zn(II)][Cu(II)][Ni(II)] = 0.001 M; (B) [Ac–HGIKSQKAEHFH–NH2][Zn(II)][Ni(II)] = 0.001 M. | ||
Metal ions like Zn(II), Cu(II) and Ni(II) are found naturally in saliva, teeth and dental plaques and to compare the stability of Zn(II), Cu(II), and Ni(II) complexes with those naturally found in the human body, we simulated a hypothetical situation in which the studied sequences Ac–EGHGHKGHHHA–NH2, Ac–HGIKSQKAEHFH–NH2 and metals were mixed in appropriate concentrations in which the metal ions could be found in the oral cavity (Fig. 7). Their concentrations are quite variable and depend on the diet, occupational exposure, salivary flow rate, exposure to toxic metals, disease states or quantity of metal-binding proteins. However, the Zn(II) concentration in saliva is within the range of 0.01–0.25 μg ml−1.19 In dental plaques, the Zn(II) concentration is higher and fluctuates from 6 to 20 μg ml−1, while the Cu(II) concentration on the enamel surface is much lower, 0.037 μg ml−1.57 The concentration of Ni(II) in saliva has also been tested and varies between numbers as low as 0.001 and 0.0019 μg ml−1, but may even reach 0.19 μg ml−1 in the case of orthodontic treatment.58
 | ||
| Fig. 7 A competition plot between (A) Ac–EGHGHKGHHHA–NH2, Zn(II), Cu(II) and Ni(II) and (B) Ac–HGIKSQKAEHFH–NH2, Zn(II), Cu(II) and Ni(II), describing complex formation at different pH values in a hypothetical situation in which appropriate amounts of all reagents are mixed. Calculations are based on binding constants from Table 1; hypothetical metal concentrations are taken from the literature.19,57,58 Conditions: T = 298 K, I = 0.1 M NaClO4, (A) [Ac–EGHGHKGHHHA–NH2][Zn(II)] = 0.001 M; (B) [Ac–HGIKSQKAEHFH–NH2][Zn(II)] = 0.001 M. | ||
The results clearly indicate that in a physiological environment, both ligands (Ac–EGHGHKGHHHA–NH2 (Fig. 7A) and Ac–HGIKSQKAEHFH–NH2 (Fig. 7B)) will be found bound to Zn(II) rather than to Cu(II) or Ni(II). The saliva and dental plaque Zn(II) concentrations are much higher than those of Cu(II) and Ni(II); thus, the trend is constant over the entire pH range. This guarantees an efficient Zn(II) transport in S. mutans using the AdcA protein, despite the affinities towards Ni(II) being quite high for both studied regions.
Conclusions
In response to nutritional resistance, bacteria have evolved mechanisms to acquire essential metal ions. One key strategy to mitigate Zn(II) deficiency is the use of ABC transporters. In this study, we identified two likely binding sites in the AdcA lipoprotein of Streptococcus mutans, a bacterial pathogen associated with dental caries. A detailed understanding of this Zn(II) transport mechanism in S. mutans represents a significant step toward elucidating metal transport systems in other pathogens and designing targeted therapeutics against antibiotic-resistant bacterial species.At physiological pH, the Ac–EGHGHKGHHHA–NH2 region binds Zn(II) with higher affinity than the Ac–HGIKSQKAEHFH–NH2 one. The binding preferences of Cu(II) and Ni(II), metal ions that could potentially compete with Zn(II) binding to AdcA, involve not only histidine imidazoles but also amide nitrogen atoms. Despite differences in coordination modes, it seems interesting that almost all metal ions have an influence on the secondary structures of both studied ligands, leading to pronounced changes in their random coil structures. In accordance with the Irving–Williams series, the Cu(II)–AdcA complexes are most stable (compared to Zn(II) and Ni(II) ones; at pH 7.4, at equimolar concentrations, more than 98% of analyzed sequences would bind Cu(II)), while the stability of Zn(II) and Ni(II) complexes is comparable. Given the high Zn(II) concentrations in human saliva and dental plaques, Zn(II)–AdcA complexes are expected to dominate in the oral cavity, rendering Cu(II) and Ni(II) binding affinities less relevant under physiological conditions.
These findings provide valuable insights into the chemistry of Zn(II), Cu(II), and Ni(II), as well as metal transport mechanisms in pathogens. Importantly, they underscore the necessity of considering physiological metal ion concentrations when evaluating competition for biologically relevant ligands. In essence, “high affinity is not enough; high concentration is far more significant”.
Understanding the metal binding specificity of AdcA paves the way for designing specific AdcA inhibitors that selectively reduce S. mutans growth. Several key factors must be considered in this process – the inhibitor must specifically target the AdcA protein in S. mutans, while avoiding disruption of proteins beneficial for the host. Such an inhibitor should block the interaction between AdcA and metals, which would prevent zinc uptake, disrupting the bacterium's zinc homeostasis and leading to its death. Inhibitors could also interfere with the interaction between AdcA and other components of the AdcABC system, such as AdcB and AdcC, potentially inhibiting the entire zinc transport process. Another approach may be effective in the fight against antibiotic resistance: using AdcA as a targeting molecule in the STAMP (specifically targeted antimicrobial peptide) method59 that could involve attaching an antibiotic or antimicrobial peptide to an AdcA peptide fragment that is selectively recognized by its partner on the bacterial cell (specifically by AdcB or AdcC), thereby transporting the antimicrobial agent into the bacteria using the Trojan Horse strategy.
Data availability
The data supporting this article have been included as part of the ESI.† If any additional questions regarding experimental details may arise, the corresponding authors remain at the readers’ disposal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre (UMO2017/26/A/ST5/00363 – H. K.). M. R.-Ż. is supported by 2022/47/O/ST4/01865 and A. H. is supported by NCN (UMO-2023/51/D/ST5/01798), the Polish National Agency for Academic Exchange (Grant BPN/BKK/2022/1/00005), and Excellence Initiative – Research University Grant BPIDUB.13.2024. A. D.-M. acknowledges the Research Thematic Network RED2022-134091-T financed by MCIN/AEI.References
- S. Lory, The Prokaryotes – Firmicutes and Tenericutes, 2014, vol. 4, pp. 367–370 Search PubMed.
- D. Ajdić, W. M. McShan, R. E. McLaughlin, G. Savić, J. Chang, M. C. Carson, C. Primeaux, R. Tian, S. Kenton, H. Jia, S. Lin, Y. Qian, S. Li, H. Zhu, F. Najar, H. Lai, J. White, B. A. Roe and J. J. Ferretti, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14434–14439 CrossRef.
- W. H. Bowen, R. A. Burne, H. Wu and H. Koo, Trends Microbiol., 2018, 26, 229–242 CrossRef PubMed.
- J. A. Lemos, S. R. Palmer, L. Zeng, Z. T. Wen, J. K. Kajfasz, I. A. Freires, J. Abranches and L. J. Brady, Microbiol. Spectrum, 2019, 7, 1–18 Search PubMed.
- S. Pant, N. J. Patel, A. Deshmukh, H. Golwala, N. Patel, A. Badheka, G. A. Hirsch and J. L. Mehta, J. Am. Coll. Cardiol., 2015, 65, 2070–2076 CrossRef PubMed.
- E. Hajishengallis, Y. Parsaei, M. I. Klein and H. Koo, Mol. Oral Microbiol., 2017, 32, 24–34 CrossRef CAS.
- Y. Pan, Y. Chen, J. Chen, Q. Ma, T. Gong, S. Yu, Q. Zhang, J. Zou and Y. Li, Mol. Oral Microbiol., 2021, 36, 278–290 CrossRef CAS.
- G. Ferrazzano, T. Cantile, M. Coda, B. Alcidi, G. Sangianantoni, A. Ingenito, M. Di Stasio and M. Volpe, Molecules, 2016, 21, 1008 CrossRef.
- L. J. Rich and M. Seshadri, Biomed. Opt. Express, 2005, 6, 3157–3162 CrossRef.
- A. M. Uwitonze, N. Ojeh, J. Murererehe, A. Atfi and M. S. Razzaque, Nutrients, 2020, 12, 949, DOI:10.3390/nu12040949.
- J. M. Berg and Y. Shi, Science, 1996, 271, 1081–1085 CrossRef.
- P. Whittaker, Am. J. Clin. Nutr., 1998, 68, 442S–446S CrossRef PubMed.
- D. A. Capdevila, J. Wang and D. P. Giedroc, J. Biol. Chem., 2016, 291, 20858–20868 CrossRef PubMed.
- C. Andreini and I. Bertini, J. Inorg. Biochem., 2012, 111, 150–156 CrossRef PubMed.
- D. Corbett, J. Wang, S. Schuler, G. Lopez-Castejon, S. Glenn, D. Brough, P. W. Andrew, J. S. Cavet and I. S. Roberts, Infect. Immun., 2012, 80, 14–21 CrossRef PubMed.
- M. I. Hood, B. L. Mortensen, J. L. Moore, Y. Zhang, T. E. Kehl-Fie, N. Sugitani, W. J. Chazin, R. M. Caprioli and E. P. Skaar, PLoS Pathog., 2012, 8, e1003068 CrossRef PubMed.
- L. D. Palmer and E. P. Skaar, Annu. Rev. Genet., 2016, 50, 67–91 CrossRef PubMed.
- M. T. Rahman, A. Hossain, C. H. Pin and N. A. Yahya, Biol. Trace Elem. Res., 2019, 187, 51–58 CrossRef PubMed.
- R. J. Lynch, Int. Dent. J., 2011, 61, 46–54 CrossRef PubMed.
- S. Shafeeq, O. P. Kuipers and T. G. Kloosterman, Mol. Microbiol., 2013, 88, 1047–1057 CrossRef.
- T. Ganguly, A. M. Peterson, J. K. Kajfasz, J. Abranches and J. A. Lemos, Mol. Oral Microbiol., 2021, 36, 214–224 CrossRef CAS PubMed.
- L. Bayle, S. Chimalapati, G. Schoehn, J. Brown, T. Vernet and C. Durmort, Mol. Microbiol., 2011, 82, 904–916 CrossRef CAS PubMed.
- M. S. Akbari, K. S. Doran and L. R. Burcham, Microorganisms, 2022, 10, 1501 CrossRef CAS PubMed.
- L. R. Burcham, B. Y. Le, J. N. Radin, B. L. Spencer, L. Deng, A. Hiron, M. R. Ransom, D. C. Mendonca, A. T. Belew, N. M. El-Sayed, K. S. McIver, T. E. Kehl-Fie and K. S. Doran, mBio, 2020, 11, e02302–e02320 CrossRef CAS PubMed.
- C. Y. Loo, K. Mitrakul, I. B. Voss, C. V. Hughes and N. Ganeshkumar, J. Bacteriol., 2003, 185, 2887–2900 Search PubMed.
- C. Y. Ong, O. Berking, M. J. Walker and A. G. McEwan, Infect. Immun., 2018, 86, e00048–e00018 CrossRef CAS.
- H. Reyes-Caballero, A. J. Guerra, E. F. Jacobsen, K. M. Kazmierczak, D. Cowart, U. M. Koppolu, R. A. Scott, M. E. Winkler and D. P. Giedroc, J. Mol. Biol., 2010, 403, 197–216 Search PubMed.
- T. Ganguly, A. Peterson, M. Burkholder, J. K. Kajfasz, J. Abranches and J. A. Lemos, PLoS Pathog., 2022, 18, e1010477 CrossRef CAS.
- B. Bersch, C. Bougault, L. Roux, A. Favier, T. Vernet and C. Durmort, PLoS One, 2013, 8, e81168 CrossRef PubMed.
- E. Loisel, L. Jacquamet, L. Serre, C. Bauvois, J. L. Ferrer, T. Vernet, A. M. Di Guilmi and C. Durmort, J. Mol. Biol., 2008, 381, 594–606 CrossRef CAS PubMed.
- B. Zheng, Q. Zhang, J. GaO, H. Han, M. Li, J. Zhang, J. Qi, J. Yan and G. F. Gao, PLoS One, 2011, 6, e19510 CrossRef CAS PubMed.
- C. D. Plumptre, A. D. Ogunniyi and J. C. Paton, Trends Microbiol., 2012, 20, 485–493 CrossRef CAS PubMed.
- M. B. Brophy, J. A. Hayden and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18089–18100 CrossRef CAS.
- T. G. Nakashige, B. Zhang, C. Krebs and E. M. Nolan, Nat. Chem. Biol., 2015, 11, 765–771 CrossRef CAS.
- J. P. Zackular, W. J. Chazin and E. P. Skaar, J. Biol. Chem., 2015, 290, 18991–18998 CrossRef CAS.
- K. Cao, N. Li, H. Wang, X. Cao, J. He, B. Zhang, Q. Y. He, G. Zhang and X. Sun, J. Biol. Chem., 2018, 293, 6075–6089 Search PubMed.
- A. Hecel, A. Kola, D. Valensin, H. Kozłowski and M. Rowińska-Żyrek, Inorg. Chem., 2020, 59, 1947–1958 CrossRef PubMed.
- K. Garstka, D. Bellotti, J. Wątły, H. Kozłowski, M. Remelli and M. Rowińska-Żyrek, Dalton Trans., 2023, 52, 16140–16150 RSC.
- D. Łoboda and M. Rowińska-Żyrek, Dalton Trans., 2017, 46, 13695–13703 RSC.
- D. Łoboda and M. Rowińska-Żyrek, Dalton Trans., 2018, 8, 2646–2654 RSC.
- K. Garstka, A. Hecel, H. Kozłowski and M. Rowińska-Żyrek, Metallomics, 2022, 14, mfac042 CrossRef PubMed.
- K. Garstka, G. Potoczniak, H. Kozłowski and M. Rowińska-Żyrek, Dalton Trans., 2024, 53, 2848–2858 RSC.
- B. D. Corbin, E. H. Seeley, A. Raab, J. Feldmann, M. R. Miller, V. J. Torres, K. L. Anderson, B. M. Dattilo, P. M. Dunman, R. Gerads, R. M. Caprioli, W. Nacken, W. J. Chazin and E. P. Skaar, Science, 2008, 319, 962–965 CrossRef PubMed.
- S. M. Damo, T. E. Kehl-Fie, N. Sugitani, M. E. Holt, S. Rathi, W. J. Murphy, Y. Zhang, C. Betz, L. Hench, G. Fritz, E. P. Skaar and W. J. Chazin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3841–3846 CrossRef PubMed.
- T. E. Kehl-Fie and E. P. Skaar, Curr. Opin. Chem. Biol., 2010, 14, 218–224 CrossRef PubMed.
- D. Łoboda, H. Kozłowski and M. Rowińska-Żyrek, New J. Chem., 2018, 42, 7560–7568 Search PubMed.
- W. Aoki and M. Ueda, Pharmaceuticals, 2013, 21, 1055–1081 Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 Search PubMed.
- C. F. Baes and R. E. Mesmer, The Hydrolysis of Cations, Wiley, New York, 1976, p. 246 Search PubMed.
- A. Malakahmad, S. Tan and S. Yavari, J. Chem., 2016, 1, 1–8 Search PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef.
- Origin(Pro), Version 2016 Search PubMed.
- W. Bal, J. Lukszo, M. Jeżowska-Bojczuk and K. S. Kasprzak, Chem. Res. Toxicol., 1995, 8, 683–692 Search PubMed.
- J. M. Mouillon, S. K. Eriksson and P. Harryson, Plant Physiol., 2008, 148, 1925–1937 CrossRef PubMed.
- H. Irving and R. J. P. Williams, J. Chem. Soc., 1953, 3192–3210 RSC.
- H. Irving and R. J. P. Williams, Nature, 1948, 162, 746–747 CrossRef.
- T. Sierpińska, K. Orywal, J. Kuc, M. Golebiewska and M. Szmitkowski, Int. J. Prosthodontics, 2013, 26, 423–428 Search PubMed.
- R. Fors and M. Persson, Eur. J. Orthod., 2006, 28, 292–297 Search PubMed.
- B. Xu, L. Wang, C. Yang, R. Yan, P. Zhang, M. Jin, H. Du and Y. Wang, J. Adv. Res., 2025, 67, 301–315 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dt00131e |
| This journal is © The Royal Society of Chemistry 2025 |