
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Shining light on the ferrous analogue: excited state dynamics of an Fe(II) hexa-carbene scorpionate complex†
Catherine E. Johnson a,
Mawuli Deegbeyb,
Aleksandra Ilicc,
Nidhi Kaula,
Om Prakashc,
Kenneth Wärnmark*c,
Elena Jakubikova*b and
Reiner Lomoth*a
a,
Mawuli Deegbeyb,
Aleksandra Ilicc,
Nidhi Kaula,
Om Prakashc,
Kenneth Wärnmark*c,
Elena Jakubikova*b and
Reiner Lomoth*a
aDepartment of Chemistry–Ångström Laboratory, Uppsala University, SE-75120 Uppsala, Sweden. E-mail: reiner.lomoth@kemi.uu.se
bDepartment of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, USA. E-mail: ejakubi@ncsu.edu
cCenter for Analysis and Synthesis (CAS), Department of Chemistry, Lund University, SE-22100 Lund, Sweden. E-mail: kenneth.warnmark@chem.lu.se
First published on 12th February 2025
Abstract
A ferrous complex bearing tris(carbene)borate ligands with imidazol-2-ylidene donors has been characterized by experimental and computational methods. Despite the pronounced destabilization of metal centered states by the exceptionally σ-donating ligand, the high-energy 3MLCT state of [Fe(II)(phtmeimb)2] is rapidly deactivated by the barrierless conversion to the 3MC state.
In recent years, complexes of Earth-abundant transition metals have garnered increasingly more attention as light harvesters for both photovoltaic as well as photocatalytic applications.1,2 One very successful approach to overcome the rapid deactivation of charge transfer (CT) states in iron complexes has been the introduction of strongly σ-donating N-heterocyclic carbene (NHC) ligands.3–5 The most notable progress was made with hexa-NHC ligand sets in ferric complexes, namely [Fe(III)(phtmeimb)]+ (phtmeimb = tris(3-methylimidazolin-2-ylidene)(phenyl)borate)6 and its derivatives7 as well as [Fe(III)(btz)3]3+ (btz = 3,3′-dimethyl-1,1′-bis(p-tolyl)-4,4′-bis(1,2,3-triazol-5-ylidene))8 that benefit from particularly strong destabilization of metal centered (MC) states. As a result, deactivation of their doublet ligand-to-metal CT (2LMCT) state via MC states is exceptionally disfavored, and with 2LMCT lifetimes of 100 ps ([Fe(III)(btz)3]3+) and 2 ns ([Fe(III)(phtmeimb)]+) the Fe(III) hexa-NHC complexes exhibit room temperature photoluminescence and engage in bimolecular excited state electron transfer (EET) which has resulted in first photocatalytic applications.9–14 Moreover, for [Fe(III)(btz)3]3+ we have previously shown that the same NHC ligand set results in sizeable ES lifetimes not only for the Fe(III)- but also the Fe(II)-complex.8,15 The 3MLCT state with a lifetime of 528 ps has recently been used as a photo-reductant in a two photon-excitation mechanism for a photoredox reaction, where both the Fe(III)-2LMCT and the Fe(II)-3MLCT partake in bimolecular EET reactions.11 Generally, the ferrous complexes can be expected to offer attractive complementary properties such as more reducing excited states for applications in e.g., photoredox catalysis or higher cage escape yields due to the spin forbidden recombination of triplet radical pairs.16,17 However, ferrous tetra-NHC complexes with complementary pyridyl18–25 or cyclometalating ligands26,27 feature 3MLCT or 3MC ESs with lifetimes on the order of 10 ps which preclude efficient bimolecular reactions. Hence, hexa-NHC complex [Fe(II)(btz)3]2+ with its 528 ps ES life time and demonstrated EET reactivity stands out among all Fe(II)-NHC complexes. We were therefore intrigued whether also the ferrous analogue of hexa-NHC complex [Fe(III)(phtmeimb)]+ could benefit from the superior ligand field splitting by the even more electron donating phtmeimb− ligand. The correspondingly high-energy MC states might ideally prevent rapid 3MLCT → MC deactivation or could possibly be photoactive themselves.28 However, the facile oxidation of the [Fe(II)(phtmeimb)2] ground state prevents its isolation which has hitherto hindered investigations into its excited state dynamics. Here, we report on the in situ characterization of [Fe(II)(phtmeimb)2] by transient absorption spectroscopy that revealed excited state deactivation on the ps time scale, and on computational results that rationalize the origins of its short 3MLCT lifetime.
As shown previously, electrochemical one-electron reduction of [Fe(III)(phtmeimb)2]+ (−1.16 V vs. Fc in acetonitrile) yields an EPR silent product attributed to the Fe(II) complex [Fe(II)(phtmeimb)2].6 Here we show that that the same product can be obtained by chemical reduction of [Fe(III)(phtmeimb)2]+ by LiAlH4 (Fig. 1) and the in situ reduced complex could be characterized by NMR (ESI).
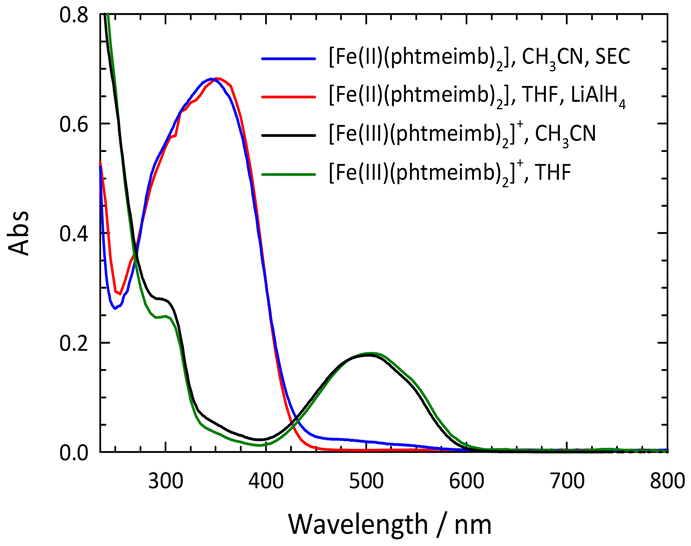 | ||
| Fig. 1 Electronic absorption spectra of [Fe(III)(phtmeimb)2]+ and [Fe(II)(phtmeimb)2] obtained by spectroelectrochemistry (SEC) in acetonitrile (0.6 mM, l = 0.1 cm) or reduction by LiAlH4 in THF solution. (Spectra in THF scaled at the maxima of the LMCT and MLCT bands, respectively.) | ||
The 1H NMR spectrum exhibited well-resolved signals as would be expected for a diamagnetic compound (Fig. S1†) and peak assignments were made possible by 2D NMR spectroscopy (COSY, HMQC, HMBC, see ESI†). In the 13C NMR spectrum (Fig. S2†), a strong downfield shift (216.9 ppm) of the carbene-carbon signal is observed. This value exceeds previously reported values for a tetra-carbene Fe(II) complex (201.2 ppm)3 but also for a hexa-carbene complex [Fe(II)(btz)3]2+ (206.7 ppm)15 and can be attributed to the particularly strong electron donation of the six imidazol-2-ylidene donors and the double negative charge of the two phtmeimb− ligands. Correspondingly large ligand-field splitting in the title complex is in accordance with computational results for the 3MC energies of [Fe(II)(phtmeimb)2] and [Fe(II)(btz)3]2+ (see below).
The lowest energy absorption band of [Fe(II)(phtmeimb)2] falls into the UV range (λmax = 348 nm, εmax = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 850 M−1 cm−1). Consistent with electrochemical data (E1/2(FeIII/II) = −1.16 V, E1/2(L/L˙−) < −3 V), this band can be tentatively attributed to an MLCT transition. Support for this assignment was obtained from computational results for the ground state electronic structure. The QC calculations were performed at the B3LYP+D2/6-311G*(B,C,H,N),29–32 SDD(Fe)33 level of theory using the Gaussian 16 Revision A.03 software34 (see ESI† for details). The resulting molecular orbitals for the singlet ground state (1GS) of [Fe(II)(phtmeimb)2] are shown in Fig. 2 and an orbital energy diagram is found in Fig. S13.† The highest occupied molecular orbitals (HOMO through HOMO−2) are predominantly iron t2g-based. Occupied MOs at lower energies (HOMO−3 through HOMO−12) are phtmeimb−-based orbitals, with HOMO−3 through HOMO−9 predominantly localized on carbene moieties and HOMO−10 through HOMO−12 localized on aryl groups. The lowest unoccupied molecular orbitals (LUMO–LUMO+3) are aryl-based π* orbitals delocalized over both phtmeimb− ligands. Carbene π* orbitals are observed from LUMO+5 to LUMO+7. With the first three HOMOs being metal t2g-based and the LUMOs (LUMO to LUMO+7) being ligand π*-orbitals, the computational results support the notion that the lowest energy transitions would in fact be MLCT.
850 M−1 cm−1). Consistent with electrochemical data (E1/2(FeIII/II) = −1.16 V, E1/2(L/L˙−) < −3 V), this band can be tentatively attributed to an MLCT transition. Support for this assignment was obtained from computational results for the ground state electronic structure. The QC calculations were performed at the B3LYP+D2/6-311G*(B,C,H,N),29–32 SDD(Fe)33 level of theory using the Gaussian 16 Revision A.03 software34 (see ESI† for details). The resulting molecular orbitals for the singlet ground state (1GS) of [Fe(II)(phtmeimb)2] are shown in Fig. 2 and an orbital energy diagram is found in Fig. S13.† The highest occupied molecular orbitals (HOMO through HOMO−2) are predominantly iron t2g-based. Occupied MOs at lower energies (HOMO−3 through HOMO−12) are phtmeimb−-based orbitals, with HOMO−3 through HOMO−9 predominantly localized on carbene moieties and HOMO−10 through HOMO−12 localized on aryl groups. The lowest unoccupied molecular orbitals (LUMO–LUMO+3) are aryl-based π* orbitals delocalized over both phtmeimb− ligands. Carbene π* orbitals are observed from LUMO+5 to LUMO+7. With the first three HOMOs being metal t2g-based and the LUMOs (LUMO to LUMO+7) being ligand π*-orbitals, the computational results support the notion that the lowest energy transitions would in fact be MLCT.
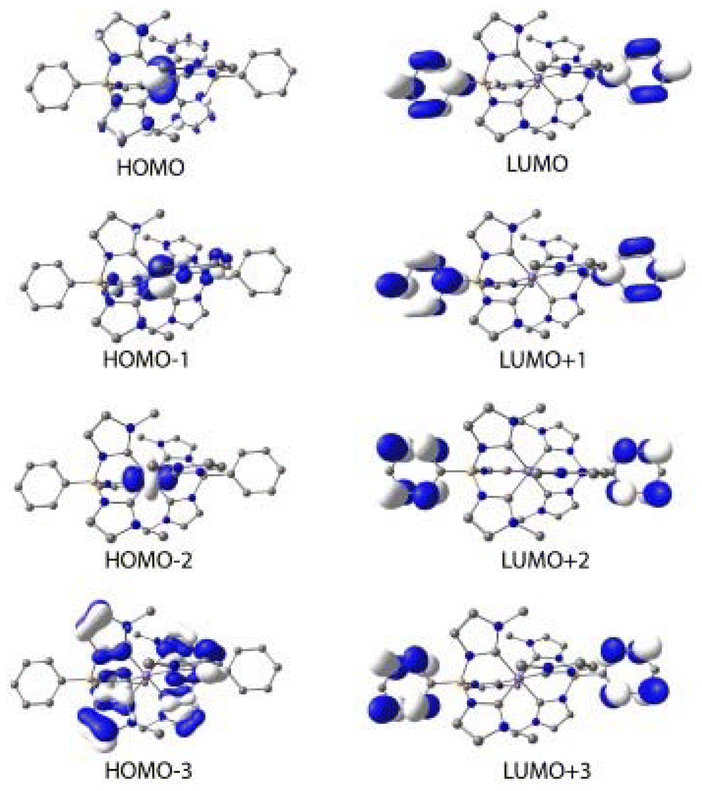 | ||
| Fig. 2 Calculated molecular orbitals (HOMO−3 to LUMO+3) of the singlet ground state of [Fe(II)(phtmeimb)2] using B3LYP+D2/6-311G*, SDD(Fe) in acetonitrile. Contour isovalue of 0.04 e Å−3. | ||
The excited state dynamics of [Fe(II)(phtmeimb)2], obtained by in situ LiAlH4 reduction of the Fe(III) congener, upon MLCT excitation was studied by femtosecond transient absorption (TA) spectroscopy. Spectra at selected delay times and kinetic traces at selected wavelengths are shown in Fig. 3.
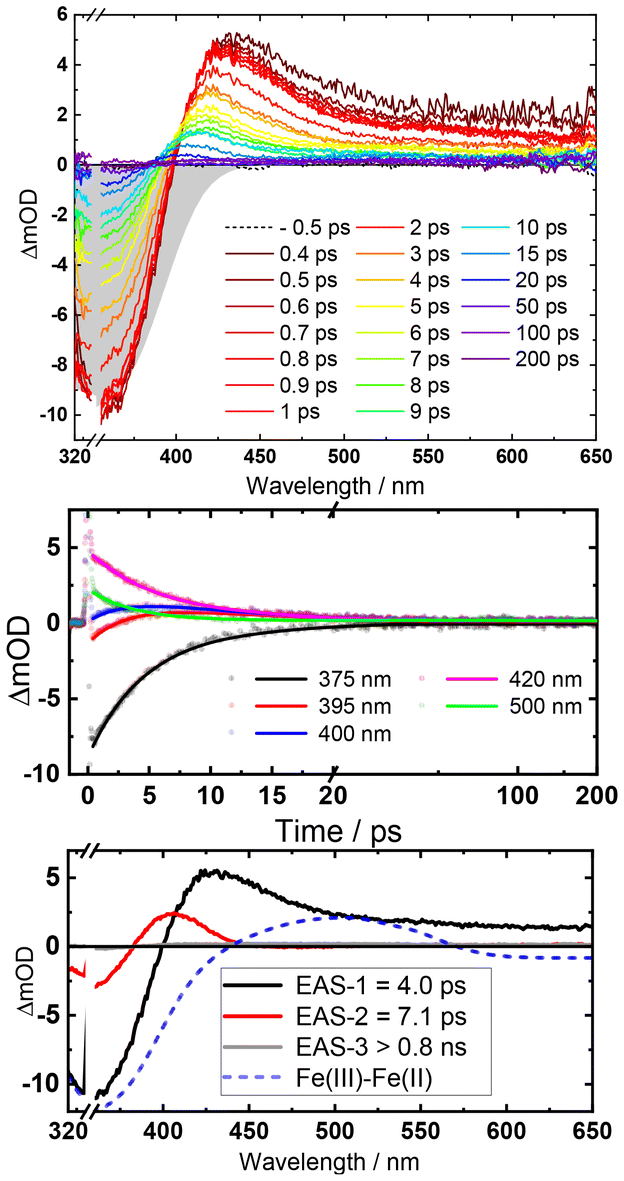 | ||
| Fig. 3 Top: fs-TA spectra of [Fe(II)(phtmeimb)2] at selected delay times after 350 nm excitation (gray-shaded area indicates the inverted ground-state absorption). Middle: kinetic traces at the indicated wavelengths (dots) and fit results (solid lines). Bottom: evolution associated spectra (EAS). | ||
The initial TA spectra are composed of a ground state bleach (GSB) signal spanning from 320 nm to around 400 nm, and broad excited state absorption (ESA) above 400 nm, peaking at around 430 nm. Both features decay to a large extent within the first ten ps, along with a blue shift of the ESA signal to around 405 nm. The TA data is accurately described by three exponential terms for which a global fit returned lifetimes of 4.0 ps and 7.1 ps for the dominating components. The third term (>800 ps) was included to account for some very minor, essentially non-decaying features probably related to the limited photostability of the complex (Fig. S6†). Kinetic traces at selected wavelengths together with the fit results are shown in Fig. 3, middle. Corresponding evolution associated spectra (EAS, Fig. 3, bottom) apply to a simple sequential decay model (see Fig. S9† for decay and species associated spectra for other decay models). Complementary TA measurements performed on [Fe(II)(phtmeimb)2] obtained by electrochemical in situ reduction in MeCN solution yielded very similar results in terms of spectra and lifetime (Fig. S10†). The agreement corroborates the notion that the observed excited state deactivation is intrinsic to the complex and the lifetime not restricted by reactions with solvent, excess reductant or products of the latter. Since intersystem crossing from the initially populated 1MLCT state can be expected to occur within the time resolution of the experiment,35 EAS-1 might be tentatively assigned to the 3MLCT state that returns to the GS via a similarly short-lived MC state described by EAS-2 (3MLCT → 3MC → 1GS). Computational results (see below) confirm the notion of efficient 3MLCT deactivation via the 3MC state while additional formation of 3MC states directly from a short-lived MLCT precursor, as previously reported for complexes with py2NHC4 ligand sets,21–23 cannot be excluded from our TA data (ESI). The broad absorption of EAS-1 resembles spectra recently assigned to 3MLCT states of Fe(II) (py)2(NHC)4 complexes18,19 based on support from vibrational coherence spectroscopy23 and fluorescence up-conversion studies.24 On the other hand, it has been recognized that pronounced visible absorption can as well emerge from LMCT excitation of 3MC states18–20 and our MLCT assignment of EAS-1 cannot be supported by spectroelectrochemistry36 as the ligand based reduction of [Fe(II)(phtmeimb)2] is electrochemically inaccessible.‡ Some support for the above assignments might be provided by reports on (py)2(NHC)4 complexes where 3MC states have been associated with narrower, blue-shifted absorption more similar to EAS-2.18,19 We note however that EAS-2 could as well be in line with a hot GS invoked in previous studies of (py)2(NHC)4 complexes.21,22 It can hence not be excluded that the 3MLCT state decays on a sub-ps time scale and that it is instead the 3MC state of [Fe(II)(phtmeimb)2] that accounts for EAS-1. While definite assignments will require additional experimental and computational work, it can be safely concluded that the 3MLCT state of [Fe(II)(phtmeimb)2] is considerably shorter-lived than for [Fe(II)(btz)3]2+. Despite the superior σ-donating ability of the phtmeimb− ligand, destabilization of MC states is apparently insufficient relative to the rather high-energy 3MLCT state of [Fe(II)(phtmeimb)2]. This notion was corroborated by computational results. Potential energy curves of GS, 3MLCT, 3MC and 5MC excited states (Tables S1, S2 and Fig. S15†) show that the 3MC state of [Fe(II)(phtmeimb)2] is lower in energy than its 5MC state across all conformations. This result is consistent with a strong ligand field and indicates that deactivation of the 3MLCT state is likely to occur via internal conversion to the 3MC state. Fig. 4 compares potential energy surface diagrams for [Fe(II)(phtmeimb)2] and [Fe(II)(btz)3]2+. The computational results confirm the expectation of significantly higher 3MC energy for the phtmeimb− complex. However, due to its much more energetic 3MLCT state, the driving force (ΔE) for 3MLCT → 3MC internal conversion (IC) is rather exceeding the value for [Fe(II)(btz)3]2+ (Table 1). Together with the lower reorganization energy (λ), IC is predicted to be barrierless (ΔEact = 0) in case of [Fe(II)(phtmeimb)2] for which the calculations (ESI) also result in a larger electronic coupling constant relative to [Fe(II)(btz)3]2+.
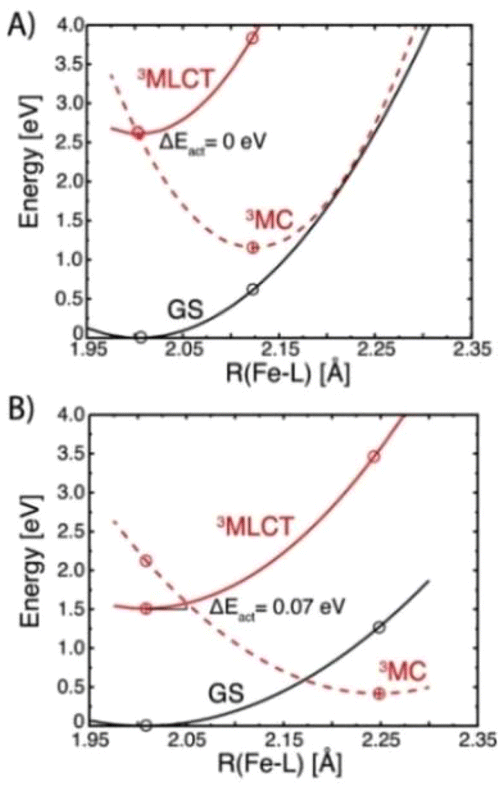 | ||
| Fig. 4 Extrapolated diabatic harmonic singlet and triplet potential energy surfaces for [Fe(II)(pthmeimb)2] (A) and [Fe(II)(btz)3]2+ (B) along an effective one-dimensional average Fe–C reaction coordinate calculated at B3LYP+D2/6-311G*, SDD (Stuttgart/Dresden pseudopotentials and basis set) level of theory in acetonitrile. The energies of the 3MC and 3MLCT states were obtained from TD-DFT (time-dependent density functional theory) calculations using 1GS (singlet ground state) as the reference. | ||
| ΔE (eV) | λ (eV) | ΔEact (eV) | |
|---|---|---|---|
| [Fe(II)(phtmeimb)2] | −1.45 | 1.35 | 0.00 |
| [Fe(II)(btz)]2+ | −1.09 | 1.89 | 0.07 |
In summary, our results indicate that the superior σ-donating ability of the phtmeimb− ligand results in extraordinary destabilization of MC states not only in the previously studied ferric complexes but also in a ferrous analogue investigated in this study. At the same time, the ligand is particularly difficult to reduce and thus the 3MLCT state of [Fe(II)(phtmeimb)2] is also unusually high in energy compared to other Fe(II)NHC complexes. Its short 3MLCT life time can hence be attributed to rapid conversion to the 3MC state. With a close match between driving force and reorganization energy, the 3MLCT → 3MC transition is essentially barrierless and further benefits from strong electronic coupling between these states. Regarding the on-going quest for Fe(II)-NHCs with practically useful ES lifetimes, tuning of these parameters25 will be essential. The directed design of ligands remains however a formidable challenge and computational studies might be an essential asset to further progress.
Data availability
Experimental details and additional experimental and computational data can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support from the Swedish Strategic Research Foundation (EM16-0067), the Knut and Alice Wallenberg Foundation (2018.0074) the Swedish Research Council (2020-03207, 2020-05058), the Swedish Energy Agency (P48747-1), the LMK Foundation, the Sten K Johnson Foundation, the Department of Chemistry at North Carolina State University and the North Carolina State University High Performance Computing Services Core Facility (RRID: SCR 022168).References
- F. Glaser and O. S. Wenger, Coord. Chem. Rev., 2020, 405, 213129 CrossRef CAS.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- Y. Liu, T. Harlang, S. E. Canton, P. Chabera, K. Suarez-Alcantara, A. Fleckhaus, D. A. Vithanage, E. Göransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Chem. Commun., 2013, 49, 6412–6414 RSC.
- Y. Liu, K. S. Kjaer, L. A. Fredin, P. Chábera, T. Harlang, S. E. Canton, S. Lidin, J. Zhang, R. Lomoth, K.-E. E. Bergquist, P. Persson, K. Wärnmark, V. Sundström, K. S. Kjær, L. A. Fredin, P. Chábera, T. Harlang, S. E. Canton, S. Lidin, J. Zhang, R. Lomoth, K.-E. E. Bergquist, P. Persson, K. Wärnmark and V. Sundström, Chem. – Eur. J., 2015, 21, 3628–3639 CrossRef CAS PubMed.
- T. Duchanois, T. Etienne, C. Cebrian, L. Liu, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, Eur. J. Inorg. Chem., 2015, 2469–2477 CrossRef CAS.
- K. S. Kjaer, N. Kaul, O. Prakash, P. Chabera, N. W. Rosemann, A. Honarfar, O. Gordivska, L. A. Fredin, K. E. Bergquist, L. Häggström, T. Ericsson, L. Lindh, A. Yartsev, S. Styring, P. Huang, J. Uhlig, J. Bendix, D. Strand, V. Sundström, P. Persson, R. Lomoth and K. Wärnmark, Science, 2019, 363, 249–253 CrossRef CAS PubMed.
- O. Prakash, L. Lindh, N. Kaul, N. Rosemann, I. Losada, C. Johnson, P. Chabera, A. Ilic, J. Schwarz, A. Gupta, J. Uhlig, T. Ericsson, L. Häggström, P. Huang, J. Bendix, D. Strand, A. Yartsev, R. Lomoth, P. Persson and K. Wärnmark, Inorg. Chem., 2022, 61, 17515–17526 CrossRef CAS PubMed.
- P. Chabera, Y. Liu, O. Prakash, E. Thyrhaug, A. E. Nahhas, A. Honarfar, S. Essen, L. A. Fredin, T. C. Harlang, K. S. Kjaer, K. Handrup, F. Ericson, H. Tatsuno, K. Morgan, J. Schnadt, L. Häggström, T. Ericsson, A. Sobkowiak, S. Lidin, P. Huang, S. Styring, J. Uhlig, J. Bendix, R. Lomoth, V. Sundström, P. Persson and K. Wärnmark, Nature, 2017, 543, 695–699 CrossRef CAS PubMed.
- A. Aydogan, R. E. Bangle, A. Cadranel, M. D. Turlington, D. T. Conroy, E. Cauet, M. L. Singleton, G. J. Meyer, R. N. Sampaio, B. Elias and L. Troian-Gautier, J. Am. Chem. Soc., 2021, 143, 15661–15673 CrossRef CAS PubMed.
- A. Aydogan, R. E. Bangle, S. De Kreijger, J. C. Dickenson, M. L. Singleton, E. Cauet, A. Cadranel, G. J. Meyer, B. Elias, R. N. Sampaio and L. Troian-Gautier, Catal. Sci. Technol., 2021, 11, 8037–8051 RSC.
- A. Ilic, J. Schwarz, C. Johnson, L. H. M. de Groot, S. Kaufhold, R. Lomoth and K. Wärnmark, Chem. Sci., 2022, 13, 9165–9175 RSC.
- Y. J. Jang, H. An, S. Choi, J. Hong, S. H. Lee, K. H. Ahn, Y. You and E. J. Kang, Org. Lett., 2022, 24, 4479–4484 CrossRef CAS PubMed.
- A. Ilic, B. R. Strücker, C. Johnson, S. Hainz, R. Lomoth and K. Wärnmark, Chem. Sci., 2024, 15, 12077–12085 RSC.
- J. Schwarz, A. Ilic, C. Johnson, R. Lomoth and K. Wärnmark, Chem. Commun., 2022, 58, 5351–5354 RSC.
- P. Chabera, K. S. Kjaer, O. Prakash, A. Honarfar, Y. Z. Liu, L. A. Fredin, T. C. B. Harlang, S. Lidin, J. Uhlig, V. Sundström, R. Lomoth, P. Persson and K. Wärnmark, J. Phys. Chem. Lett., 2018, 9, 459–463 CrossRef CAS PubMed.
- M. J. Goodwin, J. C. Dickenson, A. Ripak, A. M. Deetz, J. S. McCarthy, G. J. Meyer and L. Troian-Gautier, Chem. Rev., 2024, 124, 7379–7464 CrossRef CAS PubMed.
- J. Olmsted and T. J. Meyer, J. Phys. Chem., 1987, 91, 1649–1655 CrossRef CAS.
- L. Lindh, T. Pascher, S. Persson, Y. Goriya, K. Wärnmark, J. Uhlig, P. Chábera, P. Persson and A. Yartsev, J. Phys. Chem. A, 2023, 127, 10210–10222 CrossRef CAS PubMed.
- L. Lindh, N. W. Rosemann, I. B. Losada, S. Persson, Y. Goriya, H. Fan, O. Gordivska, K. Wärnmark, J. Uhlig, P. Chábera, A. Yartsev and P. Persson, Coord. Chem. Rev., 2024, 506, 215709 CrossRef CAS.
- C. Cebrián, M. Pastore, A. Monari, X. Assfeld, P. C. Gros and S. Haacke, ChemPhysChem, 2022, 23, e202100659 CrossRef PubMed.
- K. Kunnus, M. Vacher, T. C. B. Harlang, K. S. Kjær, K. Haldrup, E. Biasin, T. B. van Driel, M. Pápai, P. Chabera, Y. Liu, H. Tatsuno, C. Timm, E. Källman, M. Delcey, R. W. Hartsock, M. E. Reinhard, S. Koroidov, M. G. Laursen, F. B. Hansen, P. Vester, M. Christensen, L. Sandberg, Z. Németh, D. S. Szemes, É. Bajnóczi, R. Alonso-Mori, J. M. Glownia, S. Nelson, M. Sikorski, D. Sokaras, H. T. Lemke, S. E. Canton, K. B. Møller, M. M. Nielsen, G. Vankó, K. Wärnmark, V. Sundström, P. Persson, M. Lundberg, J. Uhlig and K. J. Gaffney, Nat. Commun., 2020, 11, 634 CrossRef CAS PubMed.
- H. Tatsuno, K. S. Kjær, K. Kunnus, T. C. B. Harlang, C. Timm, M. Guo, P. Chàbera, L. A. Fredin, R. W. Hartsock, M. E. Reinhard, S. Koroidov, L. Li, A. A. Cordones, O. Gordivska, O. Prakash, Y. Liu, M. G. Laursen, E. Biasin, F. B. Hansen, P. Vester, M. Christensen, K. Haldrup, Z. Németh, D. Sárosiné Szemes, É. Bajnóczi, G. Vankó, T. B. Van Driel, R. Alonso-Mori, J. M. Glownia, S. Nelson, M. Sikorski, H. T. Lemke, D. Sokaras, S. E. Canton, A. O. Dohn, K. B. Møller, M. M. Nielsen, K. J. Gaffney, K. Wärnmark, V. Sundström, P. Persson and J. Uhlig, Angew. Chem., Int. Ed., 2020, 59, 364–372 CrossRef CAS PubMed.
- F. Hainer, N. Alagna, A. R. Marri, T. J. Penfold, P. C. Gros, S. Haacke and T. Buckup, J. Phys. Chem. Lett., 2021, 12, 8560–8565 CrossRef CAS PubMed.
- L. Liu, D. Agathangelou, T. Roland, O. Cregut, T. Duchanois, M. Beley, J. Leonard, P. Gros and S. Haacke, EPJ Web Conf., 2019, 205, 09009 CrossRef CAS.
- T. Reuter, D. Zorn, R. Naumann, J. Klett, C. Förster and K. Heinze, Angew. Chem., Int. Ed., 2024, 63, e202406438 CAS.
- C. E. Johnson, J. Schwarz, M. Deegbey, O. Prakash, K. Sharma, P. Huang, T. Ericsson, L. Häggström, J. Bendix, A. K. Gupta, E. Jakubikova, K. Wärnmark and R. Lomoth, Chem. Sci., 2023, 14, 10129–10139 RSC.
- J. Steube, L. Fritsch, A. Kruse, O. S. Bokareva, S. Demeshko, H. Elgabarty, R. Schoch, M. Alaraby, H. Egold, B. Bracht, L. Schmitz, S. Hohloch, T. D. Kühne, F. Meyer, O. Kühn, S. Lochbrunner and M. Bauer, Inorg. Chem., 2024, 63, 16964–16980 CrossRef CAS PubMed.
- C. Förster and K. Heinze, Chem. Phys. Rev., 2022, 3, 041302 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- W. Gawelda, A. Cannizzo, V.-T. Pham, F. van Mourik, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2007, 129, 8199–8206 CrossRef CAS PubMed.
- A. M. Brown, C. E. McCusker and J. K. McCusker, Dalton Trans., 2014, 43, 17635–17646 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dt00139k |
| ‡ Comparison of EAS-1 with the differential spectrum for the metal centered oxidation suggests that absorption of the reduced ligand should be expected below 450 nm and above 550 nm. |
| This journal is © The Royal Society of Chemistry 2025 |