
C–C bond-forming reactions of 2-isocyanobiphenyl·BX3 adducts: spontaneous construction of polycyclic heteroaromatics†
Yu-Tsen
Kuo
 a,
Chia-Yu
Yao
b,
Yi-Hung
Liu
a,
Mu-Jeng
Cheng
b and
Jeffrey M.
Farrell
*ac
a,
Chia-Yu
Yao
b,
Yi-Hung
Liu
a,
Mu-Jeng
Cheng
b and
Jeffrey M.
Farrell
*ac
aDepartment of Chemistry, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd., Taipei 10617, Taiwan. E-mail: farrell@ntu.edu.tw
bDepartment of Chemistry, National Cheng Kung University, Tainan 701, Taiwan
cCenter for Emerging Materials and Advanced Devices, National Taiwan University, No. 1, Sec. 4, Roosevelt Rd., Taipei 10617, Taiwan
First published on 19th March 2025
Abstract
The syntheses and reactivities of 2-isocyanobiphenyl·BX3 adducts (X = I, Br, Cl) are reported. These adducts undergo unexpected C–H bond-functionalizing cyclization upon heating, yielding phenanthridinium-6-trihaloborate zwitterions. Where X = Cl, an unexpected helical polycyclic boronium salt is formed via a competing pathway. Mechanistic details are probed experimentally and computationally.
The importance of C–C bond formation to synthetic chemistry has lent prominence to organoboranes and their reactions. Accordingly, new synthetic possibilities arise from new pathways to boron-substituted products. Reactions that increase molecular complexity through concomitant C–C and C–B bond formations, such as borylative cyclizations, hold particular appeal.1
Isonitrile-BX3 adducts (X = alkyl, silyl, H, or halide) generally undergo facile insertion of isonitrile into B–X bonds to form (boryl)iminomethanes,2 which often dimerize via dual imine-borane coordination. Indeed, this was observed for isonitrile-BX3 adducts (where X = Br or Cl) by Meller and Batka (Fig. 1a).2h Nevertheless, alternative reactions of isonitriles and boranes (via (boryl)iminomethanes) have recently been exploited in synthesis. For example, Figueroa and co-workers demonstrated reactivities of sterically encumbered (boryl)iminomethanes that included “frustrated Lewis pair” (FLP) small molecule activations (Fig. 1b).3 Erker and co-workers utilized (boryl)iminomethane intermediates to construct heterocycles such as 1,3-dihydro-1,3-azaborinines via bora-Nazarov reactions,4 and to construct various heteroboroles via reactions with carbon–heteroatom double bonds (Fig. 1c).5 Reactions of isonitriles with B–B single6 and multiple7 bonds, bis-diazidoboranes,8 metal boryls,9 metal borylenes,9b,10 and N-heterocyclic boryl anions11 have also revealed many unique reaction pathways to complex products.12
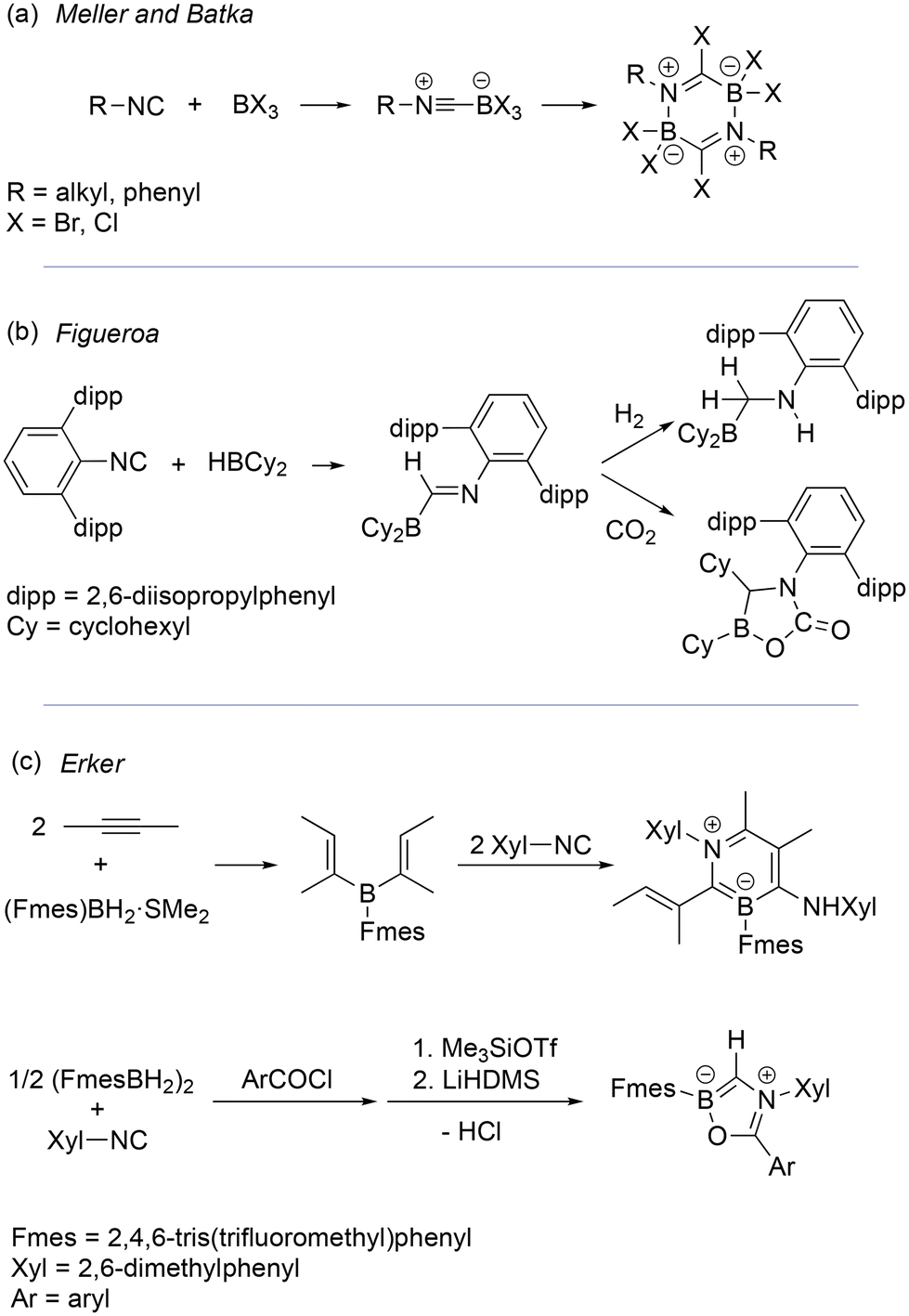 | ||
| Fig. 1 Examples of reactivities of boranes and isonitriles via presumed or isolated (boryl)iminomethane intermediates. | ||
Herein, we describe the synthesis and reactivity of 2-isocyanobiphenyl·BX3 (X = I, Br, Cl) adducts. Instead of anticipated insertion and dimerization, these compounds underwent C–C bond-forming borylative cyclization upon heating, via functionalization of a biphenyl C–H bond, to form phenanthridinium-6-trihaloborate zwitterions. For X = I or Br, the reactions proceeded cleanly without apparent by-products. On the other hand, where X = Cl, the phenanthridinium-6-trichloroborate product was accompanied by an unexpected helical 6,6′-bisphenanthridine-chelated dichloroboronium salt. The latter reaction involves three C–C bond formations, one of which is intermolecular. All isonitrile–borane adducts and cyclization products were isolated, characterized, and studied by single crystal X-ray crystallography. NMR, EPR, and density functional theory (DFT) studies were undertaken to examine reaction mechanisms. These surprising reactivities offer new prospects for C–C bond construction enroute to polycyclic aromatic scaffolds.
Reactions of 2-isocyanobiphenyl and boron trihalides (BI3, BBr3, or BCl3) in dichloromethane at room temperature furnished analytically pure 2-isocyanobiphenyl·BX3 adducts (1: X = I, 40% yield; 2: X = Br, 32% yield; 3: X = Cl, 36% yield) after recrystallization from n-hexane (Scheme 1). Adducts 1–3 are stable at room temperature under N2, and each were characterized by 1H, 13C, and 11B NMR spectroscopies, HRMS, and single crystal X-ray crystallography (e.g.Fig. 2a). Nearly identical packing arrangements are adopted by 1, 2, and 3 in the solid state, and distorted linear B–C–N and C–N–C bond geometries are observed for each. B–C bond lengths ascend from 1 (1.562(3) Å), to 2 (1.591(3) Å), to 3 (1.613(2) Å), in line with the relative Lewis acidities of BI3, BBr3, and BCl3, respectively. Isonitrile N–C bonds are 1.145(3) Å (1), 1.141(3) Å (2), and 1.138(2) Å (3), conforming with reported bond lengths between isonitriles and strong boron Lewis acids.13
 | ||
| Scheme 1 Synthesis of 2-biphenylisocyanide·BX3 adducts 1–3 and cyclized products 4–6. (i) BX3, CH2Cl2, r.t. or −78 °C – r.t., 2–18 h, (ii) C2H2Cl4 or C2D2Cl4, 130 °C, 24–48 h. | ||
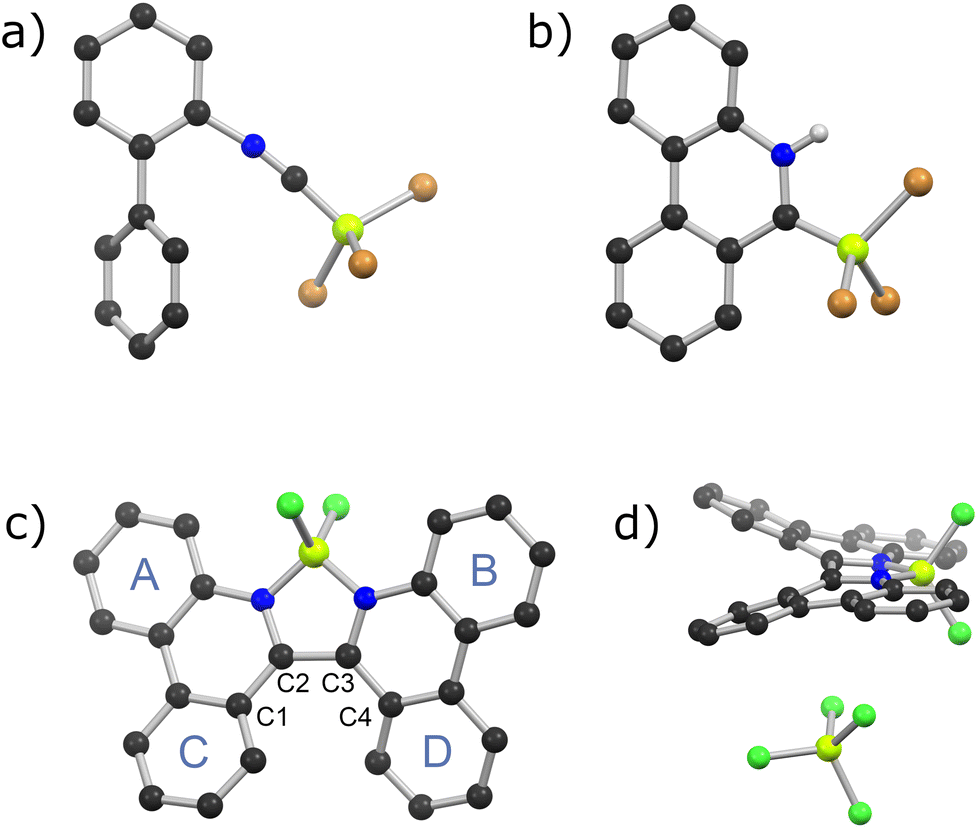 | ||
| Fig. 2 Solid-state structures of (a) 2, (b) 5a, (c) the cation of 6b, and (d) 6b (side-on view). C: black, N: blue, B: yellow-green, Cl: green, Br: orange, H: grey. Selected H atoms omitted for clarity. | ||
Transformations of 1 and 2 at elevated temperatures were studied by 1H and 11B NMR spectroscopy in C2D2Cl4. At 130 °C, each cleanly converted to a single product without observable intermediates (see ESI, Fig. S1–S4†). These products, 4a and 5a, crystallized from solution and were isolated in 75% and 70% yields, respectively. X-ray crystallography revealed their structures as phenanthridinium-6-trihaloborate zwitterions, indicating C–H functionalization of 2-isocyanobiphenyl 2′-positions (Scheme 1). Interestingly, no direct evidence for isonitrile insertion into B–X bonds was observed in either of these reactions. Analytically pure 4a or 5a could also be isolated in 52% (4a) or 67% (5a) yields from reactions of 2-isocyanobiphenyl and BI3 or BBr3, without rigorous isolation of intermediates 1 or 2.
Contrary to 1 and 2, heating compound 3 at 130 °C in C2D2Cl4 leads to complex 1H and 11B NMR spectra as 3 is consumed over 48 h. We postulated that a phenanthridinium 6-trichloroborate zwitterion (6a) was one of the reaction products, based on similarly shifted 1H NMR peaks to 4a and 5a, and a singlet 11B NMR peak observed at δ = 4.9 ppm. Another product was identified by X-ray analysis of deep red crystals recovered from the reaction mixture as the polycyclic 6,6′-biphenanthridine-chelated dichloroboronium tetrachloroborate salt 6b (Scheme 1 and Fig. 2c, d). This product could be isolated in 16% yield and exhibits 11B NMR singlets at δ = 9.1 and 6.8 ppm attributable to boronium and tetrachloroborate boron atoms. Pure 6b could be isolated from 2-isocyanobiphenyl and BCl3 in 13% yield without rigorous isolation of 3. In separate experiments, the zwitterionic product 6a could be isolated from this reaction using a purification procedure that involves hydrolysis of 6b, followed by extraction of 6a and recrystallization from chloroform/n-hexane. Some tolerance of 6a towards water is indicated by this purification procedure. Both 6a and 6b were fully characterized by 1H, 11B, and 13C NMR spectroscopies, X-ray crystallography, and HRMS. Ambient stabilities of 4a–6a were assessed by 1H NMR spectra of solid samples kept under ambient conditions for 24 hours. Spectra for 5a and 6a show no signs of decomposition, while those of 4a show minor impurities.
With growing interest in B-containing helicenes as chiral functional dyes,14 we were intrigued by the helical structure of the 6b cation revealed by X-ray crystallography. The dihedral angle of 6b defined by C1–C2–C3–C4 (Fig. 2c and d) is 24.8°, considerably less than dihedral angles defined by analogous carbon atoms in recently reported 6,6′-biphenanthridine ruthenium complexes, which lie between 52.3° and 59.5°.15 The dihedral angle between calculated mean planes of rings A and B is only 12.1°, while that between calculated mean planes of rings C and D is 40.6° (Fig. 2c and d). Accordingly, both phenanthridine ring systems of the helical 6b are warped in comparison to the planar phenanthridine ring systems of 4a–6a. The B–N bond lengths of 6b are 1.563(2) Å and 1.569(2) Å, similar to those observed for 2,2′-bipyridyldichloroboronium chloride.16
The thermally induced reactions of 1–3 to form 4a–6a stand in apparent contrast to previously reported reactivities of isonitrile boranes. However, borylative cyclization of 2-isocyanobiphenyl to form 6-borylated phenanthridine has been shown by Wang and co-workers,17 using an N-heterocyclic carbene-borane and a radical initiator. This reactivity was attributed to a radical, Minisci-type18 mechanism involving imidoyl radicals, recalling oxidative cyclizations of 2-isocyanobiphenyl by Tobisu, Chatani, and co-workers.19 On the other hand, ionic Friedel–Crafts mechanisms have been proposed by Currie and Tennant for Lewis acid-catalyzed cyclizations of biphenyl-2-isocyanide dihalides to form 6-halophenanthridines,20 as well as by Yu and co-workers for syntheses of trifluoromethylated phenanthridines from 2-isocyanobiphenyl and synthetic equivalents of [CF3]+.21
To probe possible radical reaction mechanisms, the reactions of 1–3 in thoroughly degassed C2H2Cl4 were studied by EPR spectroscopy, at room temperature and at 100 °C. No signals were observed for reactions of 2 or 3 at either temperature, although a minor signal was observed for reaction of 1 (Fig. S56–S67†). Nonetheless, thermal reaction rates of 1 assessed by 1H NMR spectroscopy in C2D2Cl4 were insensitive to freeze–pump–thaw degassing cycles or protection from light (Fig. S50†). Notably, there is no evidence for Büchner-type ring expansion of 1 during reaction under ambient lighting, whereas pure 2-isocyanobiphenyl can undergo this ring expansion under irradiation.22 We interpreted these combined results to favor ionic mechanisms, rather than radical mechanisms, for the thermal reactions of 1–3 to form 4a–6a. Moreover, ionic mechanisms align with the absence of clear radical initiators for these reactions, and the ionic reactivities of related isontrile-BX3 adducts (where X = Br or Cl) reported by Meller and Batka (Fig. 1a).2h
To investigate the mechanism underlying the conversion of 2-isocyanobiphenyl and BX3 (X = I, Br, Cl) to 4a, 5a, and 6a, density functional theory (DFT) calculations were performed. Focusing first on the reaction between 2-isocyanobiphenyl and BCl3, the DFT results align with experimental observations, indicating the formation of a stable adduct, 3, with a ΔG of −10.7 kcal mol−1 (Fig. 3).
 | ||
| Fig. 3 DFT-calculated Gibbs free energy profile of the formation of 6a. The structures were optimized in CPCM solvation model in C2H2Cl4. | ||
To convert 3 to 6a, both C–H activation and C–C coupling are required. Our results indicate that the reaction begins with C–C coupling, forming a six-membered ring via the pathway 3 → TS1-Cl → Int1-Cl, with activation and reaction free energies (ΔG‡/ΔG) of 24.1/22.1 kcal mol−1. Subsequently, converting Int1-Cl to 6a requires the migration of a hydrogen atom (marked with blue) from carbon to nitrogen. This migration can occur via three possible mechanisms:
(a) Intramolecular 1,3-H migration: The transition state for this mechanism cannot be located.
(b) Two consecutive intramolecular 1,2-H migrations: The first step (Int1-Cl → TS3-Cl → Int4-Cl) has an activation barrier of ΔG‡ = 15.3 kcal mol−1 and leads to BCl3 dissociation.
(c) Intermolecular hydrogen transfer: In this pathway, two Int1-Cl molecules form a van der Waals complex (Int1-Cl → Int2-Cl, ΔG = −11.8 kcal mol−1), and each molecule transfers its hydrogen atom to the nitrogen of the other. The first hydrogen transfer (Int2-Cl → TS2-Cl → Int3-Cl) has a Gibbs free energy barrier of ΔG‡/ΔG = 3.5/−21.2 kcal mol−1, while the second one (Int3-Cl → ½-(6a)2) is barrierless with ΔG = −35.1 kcal mol−1. The dissociation of ½-(6a)2 to 6a is slightly endergonic, with a ΔG of 0.5 kcal mol−1. Recomputing this energy using the M06 functional, a widely used alternative, we find that ΔG becomes slightly exergonic (ΔG = −2.2 kcal mol−1). These results suggest that the intermolecular interaction between the monomers is weak, facilitating dimer dissociation. Among these mechanisms, the intermolecular process is the most energetically favorable. Overall, the rate-determining step for the conversion of 2-isocyanobiphenyl and BCl3 to 6a is the C–C bond formation, with ΔG‡ = 24.1 kcal mol−1.
After identifying the most favorable pathway for forming 6a from 2-isocyanobiphenyl + BCl3, we computed the Gibbs free energy surfaces for 2-isocyanobiphenyl + BI3 → 4a and 2-isocyanobiphenyl + BBr3 → 5a. Similar to 2-isocyanobiphenyl + BCl3 → 6a, the rate-determining step for both 2-isocyanobiphenyl + BI3 → 4a and 2-isocyanobiphenyl + BBr3 → 5a is also C–C bond formation, with comparable ΔG‡ values of 25.0 and 27.1 kcal mol−1, respectively (Fig. S75 and S76†).
The formation of 6b involves C–H functionalizing cyclization of biphenyl, similar to that observed for the formation of 4a–6a. However, an additional intermolecular C–C bond is formed between precursors, and a series of steps lead to altered bonding environments for boron atoms. Peaks corresponding to 6a and 6b appear to grow simultaneously in our NMR studies (Fig. S5†). We also note that no significant changes are observed by NMR spectroscopy after heating 6a for 3 hours at 130 °C in C2D2Cl4, with or without BCl3. Furthermore, treatment of 6a with 2-isocyanobiphenyl at 130 °C for 24 hours does not yield compound 6b (Fig. S53†). Thus, we suspect that 6a is not an intermediate enroute to 6b, but rather that 6b is formed via an independent pathway. Nevertheless, the mechanism of formation of 6b is still unclear based on preliminary studies (see ESI†). Further investigations of mechanistic details are ongoing in our laboratory.
Conclusions
Adducts between 2-isocyanobiphenyl and BX3 (X = I, Br, Cl) have been prepared. These compounds undergo C–C bond-forming borylative cyclizations at elevated temperatures to give phenanthridinium-6-trihaloborate zwitterions, contrasting with reactivities of other reported isonitrile-borane adducts. Reaction of 2-isocyanobiphenyl·BCl3 forms an additional, unexpected helical 6,6′-bisphenanthridine-chelated dichloroboronium salt via three C–C bond formations. Based on NMR, EPR, and DFT studies, we propose ionic mechanisms for reactions leading to phenanthridinium-6-trihaloborates. The unique reactivities described herein offer new possibilities for isonitrile-boranes in synthesis.Author contributions
Y.-T. K. performed the experimental work. C.-Y. Y. performed DFT calculations. Y.-H L. performed X-ray crystallography. M.-J. C. directed DFT studies. J. M. F. supervised the research and wrote the manuscript. All authors edited the manuscript.Data availability
All experimental details and data supporting this article are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support of the National Science and Technology Council of Taiwan (grant no. 113-2113-M-002-016-MY3) and the Yushan Young Scholar Program of the Ministry of Education of Taiwan (grant no. MOE-110-YSFMS-0003-003-P1). We acknowledge the mass spectrometry technical research services from Consortia of Key Technologies, National Taiwan University and the Instrumentation Center of National Taiwan Normal University. We would like to thank the Instrumentation Center at National Tsing Hua University for EPR measurements.References
- (a) E. Buñuel and D. J. Cárdenas, Eur. J. Org. Chem., 2016, 5446–5464 Search PubMed; (b) K. Kubota, H. Iwamoto and H. Ito, Org. Biomol. Chem., 2017, 15, 285–300 Search PubMed; (c) J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CAS; (d) G. Kehr and G. Erker, Chem. Sci., 2016, 7, 56–65 CAS; (e) S. Kirschner, K. Yuan and M. J. Ingleson, New J. Chem., 2021, 45, 14855–14868 CAS.
- (a) G. Hesse and H. Witte, Angew. Chem., Int. Ed. Engl., 1963, 2, 617–617 Search PubMed; (b) G. Hesse, H. Witte and W. Gulden, Tetrahedron Lett., 1966, 7, 2707–2710 Search PubMed; (c) J. Casanova and R. E. Schuster, Tetrahedron Lett., 1964, 5, 405–409 Search PubMed; (d) J. Casanova Jr. and H. R. Kiefer, J. Org. Chem., 1969, 34, 2579–2583 Search PubMed; (e) S. Bresadola, G. Carraro, C. Pecile and A. Turco, Tetrahedron Lett., 1964, 5, 3185–3188 Search PubMed; (f) S. Bresadola, F. Rossetto and G. Puosi, Tetrahedron Lett., 1965, 6, 4775–4778 CrossRef; (g) J. Tanaka and J. C. Carter, Tetrahedron Lett., 1965, 6, 329–332 CrossRef; (h) A. Meller and H. Batka, Monatsh. Chem., 1969, 100, 1823–1828 CrossRef CAS; (i) K. Škoch, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2020, 56, 12178–12181 RSC; (j) M. Suginome, T. Fukuda and Y. Ito, J. Organomet. Chem., 2002, 643–644, 508–511 CrossRef CAS; (k) M. Suginome, T. Fukuda, H. Nakamura and Y. Ito, Organometallics, 2000, 19, 719–721 CrossRef CAS.
- (a) B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Chem. Commun., 2015, 51, 541–544 RSC; (b) B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2014, 136, 10262–10265 CrossRef CAS PubMed.
- J. Li, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2019, 58, 15377–15380 CrossRef CAS PubMed.
- J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2021, 60, 27053–27061 CrossRef CAS PubMed.
- (a) H. T. W. Shere, H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon and C. L. McMullin, Chem. Sci., 2024, 15, 7999–8007 RSC; (b) H. Asakawa, K.-H. Lee, Z. Lin and M. Yamashita, Nat. Commun., 2014, 5, 4245 CrossRef CAS PubMed; (c) N. Arnold, H. Braunschweig, W. C. Ewing, T. Kupfer, K. Radacki, T. Thiess and A. Trumpp, Chem. – Eur. J., 2016, 22, 11441–11449 CrossRef CAS PubMed; (d) Y. Katsuma, H. Asakawa and M. Yamashita, Chem. Sci., 2018, 9, 1301–1310 RSC; (e) Y. Katsuma, H. Asakawa, K.-H. Lee, Z. Lin and M. Yamashita, Organometallics, 2016, 35, 2563–2566 CrossRef CAS.
- J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 4469–4473 CrossRef PubMed.
- T.-T. Liu and Y.-S. Cui, Chem. – Eur. J., 2023, 29, e202302683 CrossRef CAS PubMed.
- (a) M. Tobisu, H. Fujihara, K. Koh and N. Chatani, J. Org. Chem., 2010, 75, 4841–4847 CrossRef CAS PubMed; (b) H. Braunschweig, W. C. Ewing, K. Ferkinghoff, A. Hermann, T. Kramer, R. Shang, E. Siedler and C. Werner, Chem. Commun., 2015, 51, 13032–13035 RSC.
- (a) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed; (b) H. Braunschweig, T. Herbst, K. Radacki, C. W. Tate and A. Vargas, Chem. Commun., 2013, 49, 1702–1704 RSC; (c) H. Braunschweig, K. Radacki, R. Shang and C. W. Tate, Angew. Chem., Int. Ed., 2013, 52, 729–733 CrossRef CAS PubMed; (d) T. J. Hadlington, T. Szilvási and M. Driess, Chem. Sci., 2018, 9, 2595–2600 Search PubMed; (e) M. Nutz, B. Borthakur, C. Pranckevicius, R. D. Dewhurst, M. Schäfer, T. Dellermann, F. Glaab, M. Thaler, A. K. Phukan and H. Braunschweig, Chem. – Eur. J., 2018, 24, 6843–6847 Search PubMed.
- W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 Search PubMed.
- See also, reactivity of an isonitrile-silaborane made via BH3 insertion: T. Kajiwara, N. Takeda, T. Sasamori and N. Tokitoh, Organometallics, 2004, 23, 4723–4734 CrossRef CAS.
- H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS.
- A. Nowak-Król, P. T. Geppert and K. R. Naveen, Chem. Sci., 2024, 15, 7408–7440 RSC.
- D. B. Nemez, I. B. Lozada, J. D. Braun, J. A. G. Williams and D. E. Herbert, Inorg. Chem., 2022, 61, 13386–13398 Search PubMed.
- S. M. Mansell, C. J. Adams, G. Bramham, M. F. Haddow, W. Kaim, N. C. Norman, J. E. McGrady, C. A. Russell and S. J. Udeen, Chem. Commun., 2010, 46, 5070–5072 Search PubMed.
- Y. Liu, J.-L. Li, X.-G. Liu, J.-Q. Wu, Z.-S. Huang, Q. Li and H. Wang, Org. Lett., 2021, 23, 1891–1897 CrossRef CAS PubMed.
- D. Nanni, P. Pareschi, C. Rizzoli, P. Sgarabotto and A. Tundo, Tetrahedron, 1995, 51, 9045–9062 CrossRef CAS.
- M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363–11366 CAS.
- K. S. Currie and G. Tennant, J. Chem. Soc., Chem. Commun., 1995, 2295–2296 CAS.
- Y. Cheng, H. Jiang, Y. Zhang and S. Yu, Org. Lett., 2013, 15, 5520–5523 Search PubMed.
- J. H. Boyer and J. De Jong, J. Am. Chem. Soc., 1969, 91, 5929–5930 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2418957–2418963. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00210a |
| This journal is © The Royal Society of Chemistry 2025 |