
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A revised understanding of the speciation of gold(III) dithiocarbamate complexes in solution†
Ryan K.
Brown‡
a,
Joseph N.
Bunyan‡
a,
Ashi
Agrawal
a,
Guoyu
Li
a,
Dominykas
Dautoras
 a,
Jagodish C.
Sarker
ab,
Terng Tor
Keat
a,
Thomas
Hicks
a,
Graeme
Hogarth
a and
David
Pugh
*a
a,
Jagodish C.
Sarker
ab,
Terng Tor
Keat
a,
Thomas
Hicks
a,
Graeme
Hogarth
a and
David
Pugh
*a
aDepartment of Chemistry, King's College London, Britannia House, 7 Trinity Street, London, SE1 1DB, UK. E-mail: David.Pugh@kcl.ac.uk
bDepartment of Chemistry, Jagannath University, Dhaka 1100, Bangladesh
First published on 18th March 2025
Abstract
The cytotoxic series of cis-platin mimics “[AuX2(dtc)]” (X = Cl, Br; dtc = dithiocarbamate) were recently patented as promising anticancer metallotherapeutics. Using a range of dialkyl-, cyclic alkyl- and diaryl-dithiocarbamate ligands, we have discovered that “[AuX2(dtc)]” actually exist in solution as a mixture containing neutral [AuX2(dtc)] and cationic [Au(dtc)2]+. For the latter, single crystal X-ray crystallography proved that a variety of halide-containing anions such as [AuX4]−, [AuX2]− and even X− balanced the charge. Based on a thorough investigation into the synthesis of these compounds, we discovered that literature syntheses which claim to produce pure material in fact generate mixtures. In some cases the major component of the mixture is actually the cationic [Au(dtc)2]+ rather than the claimed neutral [AuX2(dtc)]. Refinement of the synthetic conditions led to a mixture where the neutral [AuX2(dtc)] was the dominant component, from which pure solid [AuX2(dtc)] could be obtained by fractional crystallisation. However, the isomerisation process immediately restarted upon dissolution of the crystalline material, thus it is not possible to obtain pure [AuX2(dtc)] in solution. This discovery has important ramifications for any future use of these compounds, especially as therapeutics since the solution-phase speciation means that “pure” [AuX2(dtc)] cannot exist under biologically relevant conditions. A critical reinterpretation of existing literature data demonstrates that there is already significant uncertainty surrounding which component(s) of this mixture are biologically active.
Introduction
Since the Thalidomide disaster it has been discovered that many pharmaceutically active molecules have isomers which are highly toxic, and there are plenty of documented cases where a licensed pharmaceutical can convert to a toxic isomer in vivo. Thus, a thorough understanding of the solution-phase speciation of therapeutic molecules is required to ensure that no toxic isomers are formed under biologically relevant conditions. Recently, compounds of the general formulation [AuX2(dtc)] (X = Cl, Br; dtc = dithiocarbamate; Fig. 1) have gained a lot of attention as potential new metallopharmaceuticals.1 Structurally these compounds are very similar to cis-platin and they are considered extremely active antitumour agents against cultured human cancer cell lines.2–6 Although this family of compounds has been known for decades, their applications have only just started to emerge: in addition to metallopharmaceuticals, they have also found potential uses as 11C-radiolabelled PET tracers,7 homogeneous catalysts,8 and molecular precursors for the deposition of gold monolayers by Atomic Layer Deposition.9 | ||
| Fig. 1 A selection of “[AuX2(dtc)]” complexes which have demonstrated anti-cancer activity (X = Cl, Br). | ||
The established syntheses of “[AuX2(dtc)]” are supposedly straightforward and have been known since the 1960s (Scheme 1).10 The generally accepted understanding of the reactivity, across a vast range of dtc ligands, posits that the addition of one molar equivalent of M(dtc) (M = Li+, Na+, etc.) to a solution of “AuX3” (X = Cl, Br)§ leads to the sole formation of neutral “[AuX2(dtc)]”, whereas addition of two molar equivalents of M(dtc) results in formation of the cationic “[Au(dtc)2]+”, supposedly with X− as anion.11 Alternatively, oxidation of the Au(I) species [Au(dtc)]n with elemental X2 is claimed to generate “[AuX2(dtc)]” as a single product.10 Reported yields are almost quantitative but characterisation data are often sparse, especially in the older literature where high resolution NMR spectroscopy was not routinely available.
 | ||
| Scheme 1 The two most common synthetic routes towards “[AuX2(dtc)]” by salt metathesis (top) and oxidative addition (bottom). X = Cl or Br; R2 = dialkyl, cyclic alkyl, diaryl. | ||
Although the existing data are generally consistent with the proposed product formulations, solution-phase anomalies have been noted in some recent publications. For example, “[AuBr2(MSDT)]” (MSDT = methylsarcosinedithiocarbamate) clearly showed two sets of resonances in its 1H and 13C{1H} NMR spectra.12 The second set of resonances were initially misassigned as a “hypothesized isomerism in solution” caused by restricted rotation around the C–N bond of the dtc ligand,13 but were later reassigned as the putative dimeric species [AuBr2(μ2-dtc)]2 on the basis of infra-red spectroscopic evidence.14 Similarly, the reported synthesis of “[AuCl2(Bz2dtc)]” (Bz2dtc = dibenzyldithiocarbamate) clearly showed two sets of dtc-based resonances in the 1H and 13C{1H} NMR spectra (see Fig. S5–S7 from the ESI of ref. 8) although no explanation was given.8 Finally, [Au(dtc)2]+ was observed in the mass spectra of some [AuX2(dtc)] species; this was explained as decomposition in solution, although no NMR data was presented to corroborate this.15
We were seeking a high-yielding synthesis of pure [AuX2(dtc)] using dialkyl-, cyclic alkyl-, and diaryl-dithiocarbamate ligands (Fig. 2). Following the published synthetic procedures we also observed anomalies with the generally understood reactivity of this family of compounds. Hence, we conducted a detailed study into the synthesis of “[AuX2(dtc)]”, the results of which are reported herein. The solution-phase behaviour of this long-established series of supposedly pure compounds is not as simple or as well-understood as previously thought, and our findings have important implications for any future application of these compounds, especially as metallopharmaceuticals.
 | ||
| Fig. 2 “[AuX2(dtc)]” complexes synthesized in this paper (X = Cl, Br). | ||
Results and discussion
Identifying the mixture
There are many reported syntheses of “[AuX2(dtc)]” using a range of reaction conditions. Chlorinated solvents e.g. CH2Cl2, CHCl3,10 and CCl4,8 can be employed (sometimes in combination with other solvents such as acetone), but addition of an antisolvent is required to precipitate the “[AuX2(dtc)]” product. Alternatively, water can be used as solvent: a yellow powder, claimed to be “[AuX2(dtc)]”, precipitated upon addition of an aqueous solution of Na(dtc) to an aqueous solution of “AuX3”.12–14Thus, following the method of Nilakantan et al.,16 addition of an aqueous solution of Na(Et2dtc) to aqueous K[AuCl4] immediately led to the precipitation of a yellow solid (Scheme 2). However, close analysis of the 1H NMR spectrum of this solid clearly showed the presence of two overlapping Et2dtc-containing products at very similar chemical shift values (Fig. 3). The major set of peaks matched the data reported by Nilakantan et al. for “[AuCl2(Et2dtc)]”16 but the minor set did not match any residual starting material (Na(Et2dtc)), or plausible byproducts such as tetraethylthiuram disulfide. Repeated syntheses varying conditions such as concentration and temperature always resulted in a mixture of products being formed (albeit in slightly differing ratios), and the same product mixture was also observed in the NMR spectra obtained in a variety of deuterated solvents, confirming this mixture was the outcome of the synthetic procedure.
 | ||
| Scheme 2 Synthetic route to “[AuCl2(Et2dtc)]” in aqueous conditions. | ||
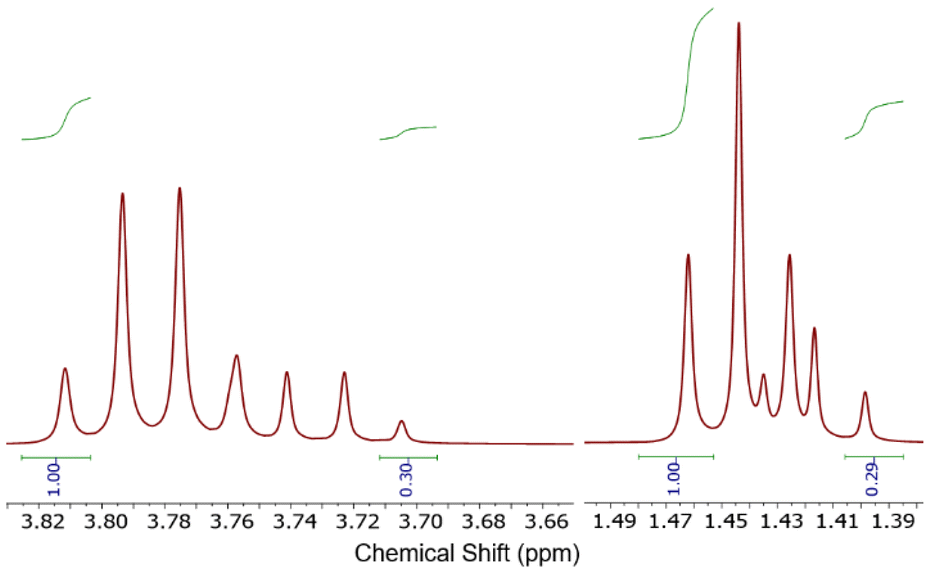 | ||
| Fig. 3 The 1H NMR spectrum (in CDCl3) of the as-synthesized [AuCl2(Et2dtc)] mixture focused on the CH2 and CH3 regions of the spectrum. | ||
Attempts to purify this mixture by column chromatography were partially successful. The components were clearly separable on silica, forming distinct orange and yellow bands which eluted at different retention times. However, 1H NMR spectroscopic analysis of the “pure” fractions obtained from the column revealed that a mixture was still present, albeit the ratio of products had altered significantly to favour one component of the mixture. When these NMR sample solutions were rerun after standing for several days, the ratio of products had changed again. Repeated separations on silica resulted in the same outcome. This indicated that “[AuCl2(Et2dtc)]” isomerises in solution, rather than undergoing a decomposition process.
To investigate this anomaly further, syntheses of some analogous complexes were conducted under the same reaction conditions. A yellow solid was isolated from the synthesis of “[AuCl2(Me2dtc)]” in water, and the 1H NMR spectrum (DMSO-d6) contained two resonances which overlapped with the residual HDO peak. In other deuterated solvents (acetone-d6, CD3CN) it was clear that two distinct products at very similar chemical shift values were present (see ESI, Fig. S5–S7†). Similarly, the aqueous synthesis of “[AuCl2(iPr2dtc)]” and “[AuCl2(iBu2dtc)]” both yielded products with 1H NMR spectra clearly showing two distinct components. For “[AuCl2(iPr2dtc)]”, comparison of our data to that reported by Angeloski et al. suggested that the mixture contained [AuCl2(iPr2dtc)] and [Au(iPr2dtc)2][AuCl4],17 and an X-ray crystal structure of the former (see ESI, Fig. S1†) was obtained.18 Fractional crystallization of the “[AuCl2(iBu2dtc)]” product mixture afforded two distinct coloured crystals with different morphologies (orange rods and yellow blades) as viewed under an optical microscope. The orange rods corresponded to [AuCl2(iBu2dtc)] (Fig. 4a) whereas some of the yellow blades were [Au(iBu2dtc)2][AuCl2] (Fig. 4b).
 | ||
| Fig. 4 (a) Solid-state molecular structure of [AuCl2(iBu2dtc)] showing the Au1-centred molecule. Selected bond lengths (Å): Au–S1 2.2946(14); Au–S2 2.2948(13); Au–Cl1 2.3240(14); Au–Cl2 2.3092(14); (b) solid-state molecular structure of [Au(iBu2dtc)2][AuCl2]. Range of Au–S bond lengths (Å): 2.3236(6)–2.3414(6). | ||
[AuCl2(iBu2dtc)] is a 4-coordinate square planar species with two symmetry-independent molecules in the asymmetric unit. The Au–S and Au–Cl distances are consistent with analogous distances in other [AuCl2(dtc)] structures,15 but there are no aurophilic interactions.19 Instead, there are short CH⋯Au intermolecular contacts between neighbouring pairs of molecules. The approximate CH⋯Au distances of 2.951 and 3.223 Å are 0.3–0.6 Å less than the sum of van der Waals radii for Au and H,20 and are consistent with stated literature examples of CH⋯Au(III) hydrogen bonding.21 The concept of CH⋯Au(III) hydrogen bonding is still controversial,22–24 but the approximate CH⋯Au angles in [AuCl2(iBu2dtc)] of 153.16° and 149.50° indicate significant directionality to this interaction.
[Au(iBu2dtc)2][AuCl2] contains two symmetry-independent ion pairs in the asymmetric unit, with distinct square planar Au(III) and linear Au(I) environments. The Au–S bonds are slightly longer than those found in [AuCl2(iBu2dtc)], but are consistent with other Au–S bonds from various [Au(dtc)2]+ cations.15 No CH⋯Au interactions or aurophilic interactions are present in this structure. Both compounds are unknown in the literature, but the related species [Au(iBu2dtc)2][AuCl4] was isolated by Rodina et al. from the reaction of [Cd(iBu2dtc)2] with “AuCl3” and we were able to match the unit cell parameters from other yellow blade crystals in the same batch to this structure.25 The [AuCl2]− anion likely originated from a background redox process where the dithiocarbamate ligand is oxidized to a thiuram disulfide with concomitant reduction of Au(III) to Au(I).
Similar experiments with other types of dtc ligand were conducted: “[AuCl2(pyrr-dtc)]” and “[AuCl2(pip-dtc)]”, with cyclic dtc ligands based on pyrrolidine and piperidine respectively, were synthesized by reacting K[AuCl4] with the respective Na(dtc) salts in water.26 The complex multiplets observed for the methylene protons in the 1H NMR spectra of the products precluded the identification of a mixture, although they were consistent with literature data for “[AuCl2(dtc)]”.26 However, the 13C{1H}NMR spectra of both mixtures clearly showed the presence of two distinct environments for each of the methylene carbons in the ring (see ESI, Fig. S16 and S18†). It was also possible to obtain crystals from both reaction products and the synthesis of [AuCl2(dtc)] was confirmed by matching our unit cell parameters to literature data.27,28 Poor-quality crystals of the corresponding [Au(dtc)2]+ cations with anions such as [AuCl4]−, [AuCl2]− and Cl− were also grown. These datasets were not of great quality, but the atomic connectivity was unambiguous and we were able to match our unit cell parameters to existing data for some cationic structures.28 Therefore “[AuCl2(dtc)]” with cyclic dtc ligands also exist as mixtures of products rather than a single species.
Diaryl dithiocarbamates are much less widely studied than their dialkyl analogues, although they are of interest as ligands in coordination chemistry, notably as single-source precursors for metal sulfide materials.29–31 The reaction of Li(p-tolyl2dtc) with K[AuCl4] afforded a brown solid with a 1H NMR spectrum clearly showing two distinct compounds. From this mixture two crystal morphologies, orange blocks and yellow plates, were grown. The orange blocks corresponded to [AuCl2(p-tolyl2dtc)], whereas the yellow plates gave the structure of [Au(p-tolyl2dtc)2][AuCl2] (Fig. 5).
 | ||
| Fig. 5 Solid-state molecular structure of (a) [AuCl2(p-tolyl2dtc)2]. Selected bond lengths: Au–Cl 2.3167(11), Au–S 2.2979(12) Å; (b) [Au(p-tolyl2dtc)2][AuCl2]. Range of Au–S bond lengths (Å): 2.3326(6)–2.3399(6). | ||
[AuCl2(p-tolyl2dtc)] exists as a square planar Au(III) species with Au–S and Au–Cl bond lengths consistent with bond lengths from other [AuCl2(dtc)] structures.15 [Au(p-tolyl2dtc)2][AuCl2] contains two symmetry-independent square planar Au(III) cations and two linear Au(I) anions in the asymmetric unit, with Au–S bond lengths being consistent with other examples of Au–S bonds in gold dialkyldithiocarbamate complexes. Neither complex exhibits aurophilic interactions, but more interestingly the Au(I) anions appear to be acting as hydrogen bond acceptors to an aromatic CH on a neighbouring cation. The approximate CH⋯Au distance of 2.760 Å is substantially within the sum of van der Waals radii for H and Au (3.52 Å),20 albeit the approximate CH⋯Au angle of 148.64° is not consistent with a strongly directional interaction. It is, however, a significantly more linear interaction than the majority of CH⋯Au(I) hydrogen bonds which have been noted in the literature.22
A similar reaction between AuBr3 and Li(p-tolyl2dtc) also afforded a brown precipitate of “[AuBr2(p-tolyl2dtc)]” with two distinct compounds in its 1H NMR spectrum. Two crystalline products were isolated, orange rods and yellow plates, with both morphologies giving viable structure solutions. Further crystallographic discussion of these compounds is in the ESI (Fig. S3†).
In light of the X-ray diffraction results, it is clear that both the neutral (orange) [AuX2(dtc)] and the cationic (yellow) [Au(dtc)2]+ can be co-crystallized from the same reaction mixture with a variety of different dtc and halide ligands. To the best of our knowledge, both series of compounds are indefinitely stable in the solid phase.
Assigning the components of the mixture
With these results in mind, we re-examined the 1H NMR spectroscopic data of the “[AuCl2(Et2dtc)]” mixture (Fig. 3). Although 1H NMR spectroscopic data for [Au(Et2dtc)2]Cl had been reported in the literature,15 to avoid any potential isomerisation issues we synthesized [Au(Et2dtc)2][BF4] as a single pure compound, including its crystal structure (see ESI, Fig. S4†), by addition of two equivalents of Na(Et2dtc) to K[AuCl4] in the presence of Na[BF4] as a halide abstractor (Scheme 3).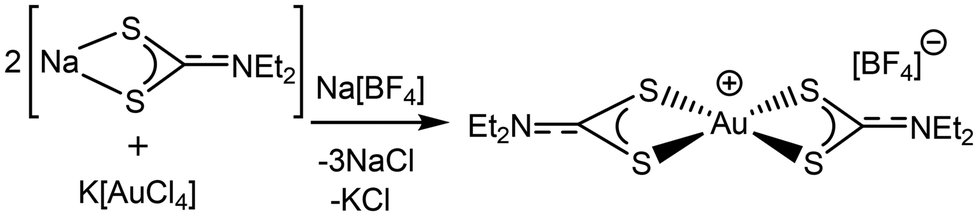 | ||
| Scheme 3 Synthesis of [Au(Et2dtc)2][BF4] (reaction conducted in acetone). | ||
We monitored [Au(Et2dtc)2][BF4] in solution and discovered that it is indefinitely stable as a single species (i.e. it does not isomerise in solution), but to our surprise we matched its 1H NMR spectrum to the major peaks in the “[AuCl2(dtc)]” mixture (Fig. 6). This indicated that – under the reaction conditions reported by Nilakantan et al. – the major product which precipitated from aqueous solution is actually cationic [Au(Et2dtc)2]+ rather than the stated neutral product [AuCl2(Et2dtc)].16 It is worth noting that the solid product which precipitated from water was yellow (both in our hands and also as reported by Nilakantan et al.), and all crystals of cationic [Au(dtc)2]+ that we obtained were also yellow. This contrasts to the neutral species which formed orange crystals. Partial integration of the non-overlapping peaks suggested that the ratio of products was roughly 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 [Au(Et2dtc)2]+:[AuCl2(Et2dtc)].
:3 [Au(Et2dtc)2]+:[AuCl2(Et2dtc)].
 | ||
| Fig. 6 Portions of the 1H NMR spectra (acetone-d6) of [Au(Et2dtc)2][BF4] (blue, top); [AuCl2(Et2dtc)] (green, middle) and the as-synthesized mixture which precipitated from water (red, bottom) with the coloured circles indicating which peaks correspond to which component of the mixture. | ||
Although we had spectroscopic evidence for the formation of a mixture in the “[AuCl2(Me2dtc)]” system, it is noteworthy that the reported 1H NMR data for ionic [Au(Me2dtc)2]Cl is identical to that for neutral [AuCl2(Me2dtc)],15 and it also overlaps with the HDO signal in DMSO-d6. Given the importance of both [AuCl2(Me2dtc)] and [AuBr2(Me2dtc)] as potential metallopharmaceuticals, we revisited the “[AuX2(Me2dtc)]” (X = Cl, Br) system using AuBr3 rather than K[AuCl4] as starting material. Two distinct peaks in the 1H NMR spectrum of the “[AuBr2(Me2dtc)]” mixture synthesized by precipitation from water, as well as two CH3 peaks in the 13C{1H} NMR spectrum (which were correlated by HMBC spectra) were observed, confirming that the Me2dtc system also forms a mixture (see ESI, Fig. S21 and 22; also Fig. S6 for X-ray structures†). Here we note that the “[AuX2(Me2dtc)]” system is not particularly soluble and we were unable to see the 13C signal for the dithiocarbamate carbon by 13C{1H} NMR spectroscopy. However, HSQC spectra on the “[AuCl2(Me2dtc)]” system exhibited two distinct correlations between a broad peak for the CH3 protons (overlapping neutral and cationic) and the separate dithiocarbamate carbons which could not be seen by 13C{1H} NMR spectroscopy (see ESI, Fig. S49†).
To explain these findings, we monitored the synthesis of “[AuCl2(Et2dtc)]” by 1H NMR spectroscopy. A solution of Na(Et2dtc) in DMSO-d6 was added portionwise to a solution of K[AuCl4] in DMSO-d6 at RT, and a series of 1H NMR spectra were obtained (Fig. 7). The spectra clearly show that as soon as Na(Et2dtc) is added a mixture of neutral [AuCl2(Et2dtc)] and cationic [Au(Et2dtc)2]+ is formed, even though K[AuCl4] is in excess, and that the majority product is the cationic species [Au(Et2dtc)2]+ rather than the desired neutral [AuCl2(Et2dtc)]. Further additions of Na(Et2dtc) do not significantly affect the ratio of products formed, and after complete addition of Na(Et2dtc) a product ratio of 7:5 cationic:neutral was obtained.
 | ||
| Fig. 7 Stacked plot of 1H NMR spectra (DMSO-d6) following the portionwise addition of Na(Et2dtc) to Na[AuCl4]. The majority product is [Au(Et2dtc)2]+ and the minority product is [AuCl2(Et2dtc)]. | ||
A critical reinterpretation of existing literature data shows that there are indications, even in the older literature, that the synthesis of “[AuX2(dtc)]” always forms a mixture rather than a pure compound. For example, van der Kerk and co-workers identified that the oxidation product of [Au(R2dtc)]n with elemental I2 (R = Me, Et, nPr, nBu) led to the formation of a putative Au(II) species [AuI(R2dtc)].10 In reality this was likely to be a mixture of [AuI2(R2dtc)] and [Au(R2dtc)2][AuI2]. Oxidation of [Au(R2dtc)]n with an excess of the other halogens led to the isolation of yellow solid material, assigned as “[AuX2(R2dtc)]”.10 Based on our observations (above), yellow compounds indicated the presence of cationic [Au(dtc)2]+ hence we repeated this reaction using [Au(Et2dtc)]n and an excess of Br2 in CH2Cl2. After complete addition of Br2, the 1H NMR spectrum of the product mixture clearly showed two distinct sets of peaks, one of which was matched to [Au(Et2dtc)2][BF4] (vide supra). We were able to grow two distinct crystal morphologies from the mixture: orange rods corresponding to [AuBr2(Et2dtc)] and yellow plates of [Au(Et2dtc)2][AuBr2] (Fig. 8, also see ESI† for further discussion). These structures provide further confirmation that both the neutral and cationic products are always present when “[AuX2(dtc)]” is synthesized.
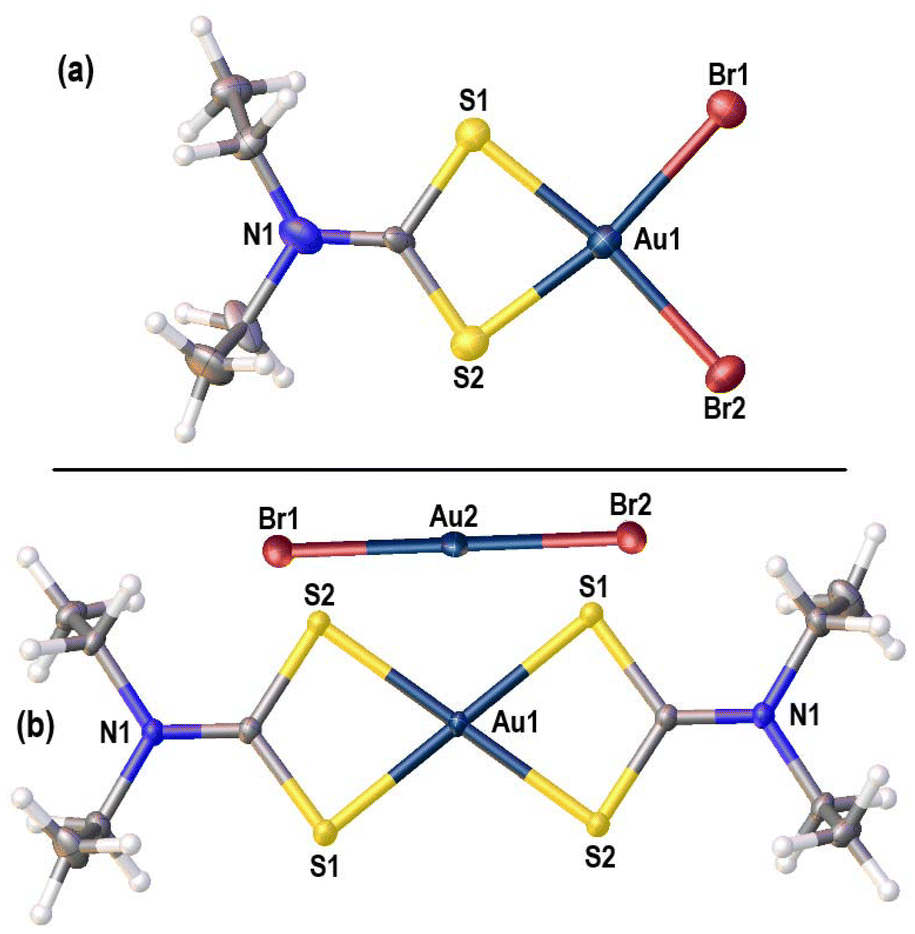 | ||
| Fig. 8 (a) Solid-state molecular structure of [AuBr2(Et2dtc)] showing the Au1-centred molecule. Range of selected bond lengths (Å): Au–S 2.288(6)–2.329(6); Au–Br 2.440(3)–2.451(3); (b) solid-state molecular structure of [Au(Et2dtc)2][AuBr2]. Selected bond lengths (Å): Au1–S1 2.3261(6); Au1–S2 2.3216(5). | ||
Other authors have isolated both neutral [AuX2(dtc)] and cationic [Au(dtc)2]+ components from the same reaction mixture. Radanović et al. obtained [AuX2(Me2dtc)] and [Au(Me2dtc)2]X (X = Cl, Br) from the oxidative addition of tetramethylthiuram disulfide to [AuX3(N-methylimidazole)].32 Loseva et al. structurally characterized a co-crystal containing two molecules of [AuCl2(iPr2dtc)] and three [Au(iPr2dtc)2]+ cations, with one [AuCl2]− anion and two [ClO4]− anions to balance the charge.33 Finally, during the course of this work, Angeloski et al. reported that the addition of auric acid to a solution of Na(iPr2dtc) in a 1:1 ratio formed “[Au(iPr2dtc)2][AuCl4]”. Their 1H NMR spectra clearly show a mixture of the cationic species and [AuCl2(iPr2dtc)] was formed, with the cationic species as the majority product.17
We were able to conclusively demonstrate that the “[AuX2(dtc)]” system always exists as a mixture by reacting pure [Au(Et2dtc)2][BF4] (synthesized as above) with one equivalent of K[AuCl4], forming [Au(Et2dtc)2][AuCl4] in situ (Scheme 4). The reaction was monitored by 1H NMR spectroscopy: after 1 day small shoulders corresponding to the neutral species had appeared and, over time, the peaks corresponding to the cationic species dropped in intensity and peaks corresponding to the neutral species grew (Fig. 9). This indicates that the [AuX2(dtc)]/[Au(dtc)2]+ system – with Au-containing anions that can interconvert – always exists as a mixture rather than as a single pure species, irrespective of how it is synthesized.
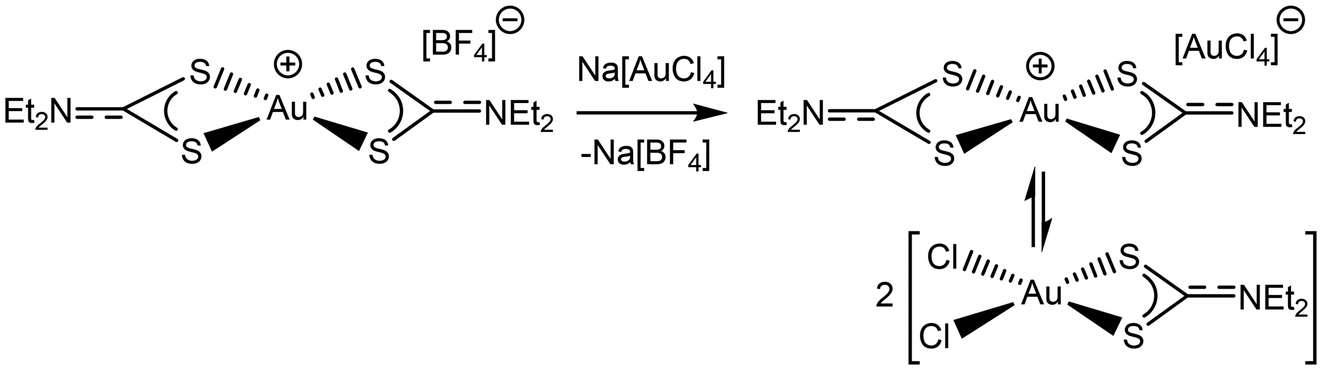 | ||
| Scheme 4 Regeneration of the [AuCl2(Et2dtc)]/[Au(Et2dtc)2]+ mixture by metathesizing stable [Au(Et2dtc)2][BF4] with Na[AuCl4]. | ||
 | ||
| Fig. 9 Stacked plot of 1H NMR spectra in acetone-d6 following the addition of K[AuCl4] to [Au(Et2dtc)2][BF4] (red, bottom) at 4 day intervals. | ||
It is clear that – in solution – the series of supposedly “pure” compounds “[AuX2(dtc)]” actually exist as a mixture containing [AuX2(dtc)] and [Au(dtc)2]+ (with the charge balanced by a variety of anions), presumably a result of the dissociation of labile dithiocarbamate ligands. A critical reappraisal of existing literature data, coupled to repeated syntheses of selected published procedures, is strongly indicative that the series of compounds “[AuX2(dtc)]” have always been synthesized as mixtures rather than as pure individual species, despite many claims to the contrary. Indeed, some reported literature syntheses of “pure” [AuX2(dtc)] actually generate a mixture where the [Au(dtc)2]+ cation is the major component!
Identification of this mixture is not straightforward, even with routine access to high-resolution NMR spectroscopy, as the signals for neutral and cationic species often overlap. However, 2D NMR spectroscopy (notably HSQC) allows for the observation of distinct correlations between the alkyl/aryl substituents and the dithiocarbamate carbons for both neutral and cationic compounds, and this can be used as a diagnostic indicator for the presence of both species in a sample even if the dithiocarbamate carbon cannot be observed using 1D 13C{1H} NMR spectroscopy. UV-vis spectroscopy can also be used as a diagnostic tool since the UV-vis spectrum of the cationic species has a characteristic secondary maximum absorption (λmax ∼ 287 nm) which is much less pronounced in the neutral complex (see ESI, Fig. S66–S68†). However, it cannot be used to accurately quantify the components of the mixture since the mixture contains multiple anions ([AuCl4]−, [AuCl2]−, Cl−) in unknown concentrations.
Here, we also note that elemental analysis (EA) is not an appropriate bulk characterisation method for these compounds since the neutral and cationic species with [AuX4]− anion have identical empirical formulae, thus they cannot be distinguished by EA. We demonstrated this by obtaining ‘acceptable’ EA data (i.e. ±0.4%) for “[AuCl2(dtc)]” using iPr2dtc and iBu2dtc ligands, despite submitting a mixture of [AuCl2(dtc)] and [Au(dtc)2][AuCl4] for analysis. A recent patent from Nardon et al. also acknowledged this fact: “However, the syntheses therein proposed [of [AuX2(dtc)]] do not permit to obtain the compounds in pure form, but as a mixture of two different complexes having the same empirical formula. This result was not evident from the elemental analysis”.34
Selective formation of [AuCl2(dtc)]
In 2005, Ronconi et al. observed a minor impurity in their 1H NMR spectrum of “[AuX2(dtc)]” (dtc = MSDT or ESDT),13 which was assigned as the dimeric species [AuBr2(μ2-dtc)]2 based on the observation of a linear “AuBr2” moiety in infra-red spectral data.14 Given our findings above, it is clear that the minor impurity is actually [Au(dtc)2]+, most likely with an [AuBr2]− anion which would account for the linear “AuBr2” moiety observed by IR spectroscopy. Interestingly, this supposed “dimer” slowly converted to the “monomer” upon leaving a solution of the mixture in acetone-d6 to stand for 24 hours. Complete conversion did not occur, but a strong solvent dependence on the interconversion between majority and minority products was noted. Certain solvents (CDCl3, CD2Cl2, acetone-d6) enabled conversion to the majority product but other solvents (DMSO-d6, CD3OD) showed no obvious change after 24 hours.14 This observation prompted us to investigate the “[AuCl2(Et2dtc)]” system in more detail with the aim of converting the mixture towards the desired [AuCl2(Et2dtc)] complex, and ultimately isolating this compound as a single species.Our as-synthesized mixture of “[AuCl2(Et2dtc)]” (with a product ratio of 5:3 cationic:neutral) was dissolved in both acetone-d6 and DMSO-d6, and the behaviour of the system was monitored by 1H NMR spectroscopy at room temperature (Fig. 10). After 24 hours a shift in the ratio of the two components had taken place, although complete conversion to [AuCl2(Et2dtc)] had not occurred. Long-term monitoring by 1H NMR spectroscopy indicated that the system stopped converting after 39 days in acetone-d6 (final product ratio 1:2.6 cationic:neutral) but for DMSO-d6 the process was faster, reaching the end point in 19 days (final ratio 1:7.3 cationic:neutral). We were unable to completely convert the system to [AuCl2(Et2dtc)] in either solvent, although it was noted that heating the mixture did increase the rate of conversion.
 | ||
| Fig. 10 Comparison of 1H NMR spectra (in DMSO-d6) following conversion of the “[AuCl2(Et2dtc)]” mixture, synthesized from water. Blue dots correspond to peaks from the cationic [Au(Et2dtc)2]+ species and green dots correspond to peaks from neutral [AuCl2(Et2dtc)]. | ||
The precipitation of orange crystals was observed during the isomerisation process in acetone-d6 (and also CD2Cl2) but not in DMSO-d6. Unit cell parameters of all the crystals we measured matched the cell parameters of [AuCl2(Et2dtc)] and no evidence for any other compound was observed. This indicated the neutral species [AuCl2(Et2dtc)] was less soluble in acetone-d6 than the cationic [Au(Et2dtc)2]+, potentially enabling the synthesis of pure [AuCl2(Et2dtc)]. Thus, addition of an acetone solution of Na(Et2dtc) to an acetone solution of Na[AuCl4] at 30 mM concentration led to the formation of a dark yellow precipitate. Although the amount of precipitate was less than that obtained from the reaction conducted in water, it was clear from the 1H NMR spectrum of the product that a mixture of [AuCl2(Et2dtc)] and [Au(Et2dtc)2]+ was still present. Gratifyingly, the ratio was now 9:4 in favour of the neutral complex.
The synthesis of “[AuCl2(Et2dtc)]” reported by Cordón et al. was also conducted in acetone, but notably no precipitate was observed when Na(Et2dtc) was added to in situ-generated “AuCl3”.8 Repeating their procedure did not result in the formation of any precipitate, but 1H NMR spectroscopy showed the product we obtained was clearly a mixture of [AuCl2(Et2dtc)] and [Au(Et2dtc)2]+, contrary to the claim of a pure product by Cordón et al. Close examination of the 1H NMR spectra supplied in their ESI (Fig. S3 and S4 from ref. 8) does appear to show small shoulders on the signals associated with the ethyl groups, likely corresponding to minor amounts of [Au(Et2dtc)2]+ which indicates that they did not obtain pure [AuCl2(Et2dtc)] either.
We speculated that the lack of precipitate was likely due to a co-solvent (CCl4) which was able to solubilise all the reaction products, rather than 3 mL of residual acetone. We adapted this synthesis to replace the toxic acetone/CCl4 mix with DMSO, since both [AuCl2(Et2dtc)] and [Au(Et2dtc)2]+ exhibited high solubility in DMSO-d6, as well as exhibiting a high neutral:cationic ratio. Thus, upon addition of Na(Et2dtc) to K[AuCl4] in DMSO, no precipitate was observed. To precipitate the product a significant amount of water was added, but this resulted in a reduced isolated yield (41%). The 1H NMR spectrum of the precipitated product showed that a mixture was still present, albeit the ratio of [AuCl2(Et2dtc)]:[Au(Et2dtc)]+ was now 9:1, higher than our previous syntheses conducted in water and acetone.
Since the strategy of keeping everything dissolved did result in a higher amount of neutral product, albeit a reduced isolated yield with DMSO as solvent, we returned to acetone but at a tenfold-lower concentration of reactants. During the course of this work, Angeloski et al. reported that refluxing a mixture of [AuCl2(iPr2dtc)] and [Au(iPr2dtc)2]+ in acetone rapidly converted the mixture to the point where [AuCl2(iPr2dtc)] was the dominant component.17 Thus, slow addition of Na(Et2dtc) to K[AuCl4] at 3 mM concentration did not generate any precipitate. Refluxing this reaction for 16 hours resulted in a mixture of [AuCl2(Et2dtc)] and [Au(Et2dtc)2]+ with the vast majority (∼17:1) being the neutral species (Fig. 11). Slow evaporation of the solvent led to the precipitation of orange crystals of pure [AuCl2(Et2dtc)]: these are indefinitely stable in the solid state, but upon dissolution the isomerisation resumes and the mixture reforms (see ESI, Fig. S65†).
 | ||
| Fig. 11 1H NMR spectrum (in acetone-d6) of the product obtained from the synthesis of “[AuCl2(Et2dtc)]” in dilute refluxing acetone. The majority product is [AuCl2(Et2dtc)]. | ||
The synthesis detailed above, conducted in dilute refluxing acetone, represents the best approach to generating pure solid [AuX2(dtc)]. We have used this method to demonstrate that pure, crystalline material of all [AuX2(dtc)] mentioned in this paper can be isolated and ‘clean’ NMR spectra can be obtained if the pure crystalline solids are dissolved in a deuterated solvent and data obtained immediately (see ESI† for spectra). This may explain the observation of Pettenuzzo et al. who claim to have isolated a single species of “[AuBr2(dtc)]” (where dtc is derived from the ethyl ester of 4-carboxypyrrolidine) by transmetallation from [Zn(dtc)2] using AuBr3.35
Although the nature of the dtc ligand does have an effect on the ratio of neutral:cationic species observed after heating the reaction mixture (i.e. poorly soluble dtc ligands such as Me2dtc and pyrr-dtc require longer heating to convert the mixture; see ESI† for details), in all cases we saw a majority of the desired neutral compound from which pure material can be fractionally crystallized. However, it is important to note that if the crystallisation process is too quick (e.g. the vessel has too many nucleation points such as scratches on the glass, or the compound is not especially soluble in acetone) the yellow cationic complex co-crystallises with the orange neutral species. In particular, we observed this for the “[AuBr2(Me2dtc)]” system, where fractional crystallisation of a post-refluxed mixture yielded crystals of both neutral [AuBr2(Me2dtc)] and ionic [Au(Me2dtc)2][AuBr2] (see ESI, Fig. S6†).
Finally, we note that gold dithiocarbamate complexes in both the +1 and +3 oxidation state are compounds of interest as potent new metallopharmaceuticals.1 Whilst the speciation of Au(I) dithiocarbamates in solution is not in question,36 we have demonstrated that the same cannot be said about “[AuX2(dtc)]”. The use of “[AuX2(dtc)]” as a structural mimic of cis-platin, using various dtc ligands, has been thoroughly investigated and neutral “[AuX2(dtc)]” compounds with simple ligands such as Me2dtc and Et2dtc have been shown to be active against cultured human cancer cell lines.3,12,26,37,38 Conversely, a recent publication from Fregona and co-workers observed conversion from the neutral “[AuBr2(dtc)]” to cationic [Au(dtc)2]+ (where dtc is derived from proline alkyl esters) in a 1:9 DMSO/H2O mix,39 and it was asserted that the cationic species [Au(dtc)2]+ were more cytotoxic than the neutral species owing to “decomposition” of the neutral species supposedly generating the cationic species in situ.15 Clearly the presence of both components in a product mixture has led to confusing and contradictory cytotoxic data. More work is needed to deconvolute the effects of inadvertently studying the cytotoxicity of a mixture containing both compounds where one, or even both, components of the mixture may be biologically active.
Derivatives of [AuX2(dtc)] where X = Me,9 C6F5,8 mesityl,40 thiolate,5etc. appear to be significantly more stable and do not seem to exist as solution-phase mixtures. Presumably this is due to the reduced stability of the corresponding [AuX4]− and [AuX2]− anions when X is not a halide. Although we have demonstrated that it is possible to isolate pure [AuX2(dtc)] (X = halide) in the solid phase by fractional crystallisation, despite many literature reports to the contrary it is not possible to generate pure [AuX2(dtc)] in solution since mixtures of [AuX2(dtc)] and [Au(dtc)2]+ are always obtained.
Conclusions
A detailed investigation into the synthesis of the supposedly pure series of compounds “[AuX2(dtc)]” (X = Cl, Br; dtc = dithiocarbamate) has been conducted. Contrary to the prevailing understanding of this series of compounds, we discovered that they actually exist as a mixture of multiple species in solution: the desired [AuX2(dtc)] alongside the [Au(dtc)2]+ cation with a variety of different anions such as [AuX4]−, [AuX2]− and X− to balance the charge. In some cases, reported literature syntheses of “pure” neutral [AuX2(dtc)] actually result in the formation of a mixture where cationic [Au(dtc)2]+ is the majority product.It is possible to vary the synthetic conditions to convert the mixture such that neutral [AuX2(dtc)] is the majority product but it is not possible to synthesise it as a single species in solution. Fractional crystallisation does yield pure [AuX2(dtc)] as a crystalline solid. However, it is important to note that [Au(dtc)2]+ is stable as a single species in solution so long as the counteranion is not a haloaurate species. Anion metathesis to [AuCl4]− subsequently results in the mixture reforming. Many of these compounds exhibit CH⋯Au interactions in the solid state which are strongly indicative of hydrogen bonding to both Au(I) and Au(III), and will be of interest to researchers studying these still somewhat controversial interactions.
The supposedly pure “[AuX2(dtc)]” (X = Cl, Br, I) have been employed in a variety of different applications, but most notably as highly promising metallodrugs against cultured human cancer cell lines. In view of our new insights into the synthesis and speciation of this series of compounds in solution, great care needs to be taken with any future studies into this mixture. A critical reappraisal of the literature has already highlighted uncertainties surrounding the precise nature of the biologically active species and future studies may need to focus on analogues which do not isomerise in solution in order to generate reliable cytotoxic data.
Experimental
The aqueous synthesis of [AuX2(dtc)]/[Au(dtc)2]+ mixtures
A slight modification of the method published by Nilakantan et al. was used.16In most cases, two distinct products were observed in NMR spectra. These have simply been assigned as ‘major’ and ‘minor’ components. The analytically pure samples synthesized by a different method have been definitively assigned.
An alternative synthesis for “[AuCl2(Et2dtc)]” was also attempted whereby all reagents were added together in EtOH/water (1:1 volume), then heated to reflux for 1 h at 110 °C.16 The solution was then cooled and the solid isolated by filtration. This was washed with distilled water and Et2O, then dried in vacuo. 1H NMR spectroscopy indicated the presence of the same product mixture.
Note: “[AuBr2(Et2dtc)]” was also synthesized using the general procedure to provide an authentic sample for comparison of NMR spectra: Na(Et2dtc)·3(H2O) (119 mg, 0.67 mmol, 1.0 eq.) was dissolved in water (3 mL) and added dropwise to a vigorously stirred solution of AuBr3 (291 mg, 0.67 mmol, 1.0 eq.) in water (3 mL) over 15 minutes. The suspension was then stirred for 1 h. An orange precipitate was isolated by vacuum filtration, washed with water (3 × 5 mL) and Et2O (2 × 5 mL) then dried in vacuo. Yield: 230 mg of an orange solid.
The acetone synthesis of [AuX2(dtc)]/[Au(dtc)2]+ mixtures (X = Cl, Br) and the isolation of pure [AuX2(dtc)]
Orange crystals of analytically pure [AuX2(dtc)] were obtained by redissolving the crude orange solid in a minimum volume of acetone, then allowing the solvent to slowly evaporate. Before complete solvent loss occurred, the remaining liquid was decanted away from the orange crystals. These were then washed with water and Et2O, and dried in vacuo. Note: the vessel used for slow evaporation needs to be clean and free from excessive nucleation points (e.g. scratches) otherwise yellow crystals of [Au(dtc)2]+ co-crystallize with the desired orange solid.
The analytical data below are based on the orange crystalline material obtained by slow evaporation of an acetone solution. Unless stated otherwise, the percentage yields given are those of pure orange crystalline material based off a crystallization run starting from 50 mg of crude orange solid.
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 23.47 (s, CH2H2CH2).
H2), 23.47 (s, CH2H2CH2).
Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data: CCDC reference numbers 2374277 [AuCl2(iPr2dtc)], 2374278 [AuCl2(iBu2dtc)], 2374279 [Au(iBu2dtc)2][AuCl2], 2374280 [AuCl2(p-tolyl2dtc)], 2374281 [Au(p-tolyl2dtc)][AuCl2], 2374282 [AuBr2(p-tolyl2dtc)], 2374283 [Au(p-tolyl2dtc)][AuBr2], 2374284 [AuBr2(Et2dtc)], 2374285 [Au(Et2dtc)2][AuBr2], 2374286 [Au(Et2dtc)2][BF4], 2374287 [AuBr2(Me2dtc)], 2374288 [Au(Me2dtc)2][AuBr2] contain crystallographic data in CIF format.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an EPSRC New Horizons grant (EP/V04897X/1), and J. C. S. would like to acknowledge the CSC for a Commonwealth Scholarship. King's College London are thanked for funding a PhD student (J. N. B.) and KURF students (A. A.; G. L.; D. D.). We thank the KCL Chemistry Department facilities for technical support, and the EPSRC UK National Crystallography Service at the University of Southampton (in particular, Drs Laura McCormick, Peter Horton, James Orton, and Graham Tizzard) for the collection of the crystallographic data.41 We would also like to thank Prof. Chris Blackman at UCL for contributing some of the Au(III) starting materials and for his input into the early stages of this project.References
- C. K. Adokoh, Therapeutic potential of dithiocarbamate supported gold compounds, RSC Adv., 2020, 10, 2975–2988, 10.1039/C9RA09682E.
- G. Moreno-Alcántar, P. Picchetti and A. Casini, Gold Complexes in Anticancer Therapy: From New Design Principles to Particle-Based Delivery Systems, Angew. Chem., Int. Ed., 2023, 62, e202218000, DOI:10.1002/anie.202218000.
- C. Nardon, S. M. Schmitt, H. Yang, J. Zuo, D. Fregona and Q. P. Dou, Gold(III)-Dithiocarbamato Peptidomimetics in the Forefront of the Targeted Anticancer Therapy: Preclinical Studies against Human Breast Neoplasia, PLoS One, 2014, 9, e84248, DOI:10.1371/journal.pone.0084248.
- C. Nardon, G. Boscutti, C. Gabbiani, L. Massai, N. Pettenuzzo, A. Fassina, L. Messori and D. Fregona, Cell and Cell-Free Mechanistic Studies on Two Gold(III) Complexes with Proven Antitumor Properties, Eur. J. Inorg. Chem., 2017, 1737–1744, DOI:10.1002/ejic.201601215.
- J. Quero, S. Cabello, T. Fuertes, I. Mármol, R. Laplaza, V. Polo, M. C. Gimeno, M. J. Rodriguez-Yoldi and E. Cerrada, Proteasome versus Thioredoxin Reductase Competition as Possible Biological Targets in Antitumor Mixed Thiolate-Dithiocarbamate Gold(III) Complexes, Inorg. Chem., 2018, 57, 10832–10845, DOI:10.1021/acs.inorgchem.8b01464.
- D. Fregona, L. Ronconi, F. Formaggio, Q. P. Dou and D. Aldinucci, Gold(III) complexes with oligopeptides functionalized with sulfur donors and use thereof as antitumor agents, WO2010105691A1, 2010.
- S. Cesarec, F. Edgar, T. Lai, C. Plisson, A. J. P. White and P. W. Miller, Synthesis of carbon-11 radiolabelled transition metal complexes using 11C-dithiocarbamates, Dalton Trans., 2022, 51, 5004–5008, 10.1039/D2DT00266C.
- J. Cordón, G. Jiménez-Osés, J. M. López-de-Luzuriaga, M. Monge, M. Elena Olmos and D. Pascual, Experimental and Theoretical Study of Gold(III)-Catalyzed Hydration of Alkynes, Organometallics, 2014, 33, 3823–3830, DOI:10.1021/om500523r.
- M. Mäkelä, T. Hatanpää, K. Mizohata, J. Räisänen, M. Ritala and M. Leskelä, Thermal Atomic Layer Deposition of Continuous and Highly Conducting Gold Thin Films, Chem. Mater., 2017, 29, 6130–6136, DOI:10.1021/acs.chemmater.7b02167.
- H. J. A. Blaauw, R. J. F. Nivard and G. J. M. van der Kerk, Chemistry of organogold compounds: I. Syntheses and properties of dihalogold(III) N,N-dialkyldithiocarbamates and dialkylgold(III) N,N-dialkyldithiocarbamates, J. Organomet. Chem., 1964, 2, 236–244, DOI:10.1016/S0022-328X(00)80517-3.
- G. Hogarth, Transition Metal Dithiocarbamates: 1978–2003, Prog. Inorg. Chem., 2005, 53, 71–561, DOI:10.1002/0471725587.ch2.
- L. Giovagnini, L. Ronconi, D. Aldinucci, D. Lorenzon, S. Sitran and D. Fregona, Synthesis, Characterization, and Comparative in Vitro Cytotoxicity Studies of Platinum(II), Palladium(II), and Gold(III) Methylsarcosinedithiocarbamate Complexes, J. Med. Chem., 2005, 48, 1588–1595, DOI:10.1021/jm049191x.
- L. Ronconi, L. Giovagnini, C. Marzano, F. Bettio, R. Graziani, G. Pilloni and D. Fregona, Gold Dithiocarbamate Derivatives as Potential Antineoplastic Agents: Design, Spectroscopic Properties, and in Vitro Antitumor Activity, Inorg. Chem., 2005, 44, 1867–1881, DOI:10.1021/ic048260v.
- G. Boscutti, L. Marchiò, L. Ronconi and D. Fregona, Insights into the Reactivity of Gold–Dithiocarbamato Anticancer Agents toward Model Biomolecules by Using Multinuclear NMR Spectroscopy, Chem. – Eur. J., 2013, 19, 13428–13436, DOI:10.1002/chem.201302550.
- M. Altaf, A. A. Isab, J. Vančo, Z. Dvorák, Z. Trávníček and H. Stoeckli-Evans, Synthesis, characterization and in vitro cytotoxicity of gold(III) dialkyl/diaryldithiocarbamato complexes, RSC Adv., 2015, 5, 81599–81607, 10.1039/C5RA15123F.
- L. Nilakantan, D. R. McMillin and P. R. Sharp, Emissive Biphenyl Cyclometalated Gold(III) Diethyl Dithiocarbamate Complexes, Organometallics, 2016, 35, 2339–2347, DOI:10.1021/acs.organomet.6b00275.
- A. Angeloski, K. Flower-Donaldson, F. Matar, D. Hayes, M. Duman, D. Oldfield, M. Westerhausen and A. McDonagh, Gold Microstructures by Thermolysis of Gold(III) Di-isopropyldithiocarbamate Complexes, ChemNanoMat, 2024, 10, e202300514, DOI:10.1002/cnma.202300514.
- O. V. Loseva, T. A. Rodina, A. V. Ivanov, I. A. Lutsenko, E. V. Korneeva, A. V. Gerasimenko and A. I. Smolentsev, Heteroleptic Dithiocarbamato–Chlorido Gold(III) Complexes [Au(S2CNR2)Cl2] (R = CH3, iso-C3H7; R2 = (CH2)6): Synthesis, Supramolecular Structures, and Thermal Behavior, Russ. J. Coord. Chem., 2018, 44, 604–612, DOI:10.1134/S107032841810007X.
- For a useful primer to aurophilic interactions, see: H. Schmidbauer, The aurophilicity phenomenon: A decade of experimental findings, theoretical concepts and emerging applications, Gold Bull., 2000, 33, 3–10, DOI:10.1007/BF03215477.
- H = 1.20 Å; Au = 2.32 Å. See: S. A. Alvarez, A cartography of the van der Waals territories., Dalton Trans., 2013, 42, 8617–8636, 10.1039/C3DT50599E.
- S. Lo Schiavo, F. Nicolo, R. Scopelliti, G. Tresoldi and P. Piraino, Self-assembly of [Bu4N][M(qdt)2] [qdt = quinoxaline-2,3-dithiolate; M = Au and Cu] in a 2D network via combination of CH⋯M and CH⋯S interactions, Inorg. Chim. Acta, 2000, 304, 108–113, DOI:10.1016/S0020-1693(00)00053-0.
- H. Schmidbaur, H. G. Raubenheimer and L. Dobrzanska, The gold–hydrogen bond, Au–H, and the hydrogen bond to gold, Au⋯H–X, Chem. Soc. Rev., 2014, 43, 345–370, 10.1039/C3CS60251F.
- I. Chambrier, D. L. Hughes, R. J. Jeans, A. J. Welch, P. H. M. Budzelaar and M. Bochmann, Do Gold(III) Complexes Form Hydrogen Bonds? An Exploration of AuIII Dicarboranyl Chemistry, Chem. – Eur. J., 2019, 26, 939–947, DOI:10.1002/chem.201904790.
- L. Rocchigiani and M. Bochmann, Recent Advances in Gold(III) Chemistry: Structure, Bonding, Reactivity, and Role in Homogeneous Catalysis, Chem. Rev., 2021, 121, 8364–8451, DOI:10.1021/acs.chemrev.0c00552.
- T. A. Rodina, A. V. Ivanov, A. V. Gerasimenko, O. V. Loseva, O. N. Antzutkin and V. I. Sergienko, Fixation modes of gold(III) from solutions using cadmium(II) dithiocarbamates. Preparation, supramolecular structure and thermal behaviour of polynuclear and heteropolynuclear gold(III) complexes: Bis(N,N-dialkyldithiocarbamato-S,S’)gold(III) polychlorometallates, [Au(S2CNR2)2]nX (n = 1: X = [AuCl4]−; n = 2: X = [CdCl4]2−, [Cd2Cl6]2−), Polyhedron, 2012, 40, 53–64, DOI:10.1016/j.poly.2012.03.043.
- Y. Shi, W. Chu, Y. Wang, S. Wang, J. Du, J. Zhang, S. Li, G. Zhou, X. Qin and C. Zhang, Synthesis, characterization and cytotoxicity of the Au(III) complexes with cyclic amine-based dithiocarbamate ligands, Inorg. Chem. Commun., 2013, 30, 178–181, DOI:10.1016/j.inoche.2013.02.010.
- R. W.-Y. Sun, M. Zhang, D. Li, M. Li and A. S.-T. Wong, Enhanced anti-cancer activities of a gold(III) pyrrolidinedithiocarbamato complex incorporated in a biodegradable metal-organic framework, J. Inorg. Biochem., 2016, 163, 1–7, DOI:10.1016/j.jinorgbio.2016.06.020.
- A. V. Ivanov, S. A. Zinkin, A. V. Gerasimenko and V. I. Sergienko, Polynuclear complexes of gold(III) of the composition ([Au(S2CNR2)2][AuCl4])n (R = C4H9, R2 = (CH2)5): Synthesis, supramolecular structures, and thermal behavior, Koord. Khim., 2011, 37, 452–459, DOI:10.1134/S1070328411050071.
- J. C. Sarker, R. Nash, S. Boonrungsiman, D. Pugh and G. Hogarth, Diaryl dithiocarbamates: synthesis, oxidation to thiuram disulfides, Co(III) complexes [Co(S2CNAr2)3] and their use as single source precursors to CoS2, Dalton Trans., 2022, 51, 13061–13070, 10.1039/D2DT01767A.
- J. C. Sarker, X. Xu, F. Alam, R. Nash, S. Boonrungsiman, D. Pugh, J. K. Cockroft, D. J. Lewis and G. Hogarth, Copper diaryl-dithiocarbamate complexes and their application as single source precursors (SSPs) for copper sulfide nanomaterials, New J. Chem., 2023, 47, 12718–12727, 10.1039/d3nj01918g.
- J. C. Sarker, F. Alam, P. McNaughter, D. Pugh, J. K. Cockroft, D. J. Lewis and G. Hogarth, Synthesis of diaryl dithiocarbamate complexes of zinc and their uses as single source precursors for nanoscale ZnS, Inorg. Chim. Acta, 2023, 556, 121663, DOI:10.1016/j.ica.2023.121663.
- D. J. Radanović, Z. D. Matović, V. D. Miletić, L. P. Battaglia, S. Ianelli, I. A. Efimenko and G. Ponticelli, Reaction products distribution between square-planar gold(III) complexes and N,N,N′,N′-tetramethylthiuram disulfide. Crystal structure of bis(N,N-dimethyldithiocarbamato)gold(III) bromide dihydrate, [Au(Me2NCS2)2]Br·2H2O, Transition Met. Chem., 1996, 21, 169–175, DOI:10.1007/BF00136550.
- O. Loseva , Private communication to the CCDC, deposition number 1977816.
- C. Nardon, D. Fregona, L. Brustolin and N. Pettenuzzo, Coordination compounds, syntheses, nanoformulation and use thereof in oncology, WO2018100561, 2017.
- A. Pettenuzzo, D. Montagner, P. McArdle and L. Ronconi, An innovative and efficient route to the synthesis of metal-based glycoconjugates: proof-of-concept and potential applications, Dalton Trans., 2018, 47, 10721–10736, 10.1039/C8DT01583J.
- M. Morgen, P. Fabrowski, E. Amtmann, N. Gunkel and A. K. Miller, Inclusion Complexes of Gold(I)-Dithiocarbamates with β-Cyclodextrin: A Journey from Drug Repurposing towards Drug Discovery, Chem. – Eur. J., 2021, 27, 12156–12165, DOI:10.1002/chem.202101366.
- L. Ronconi and D. Fregona, The Midas touch in cancer chemotherapy: from platinum- to gold-dithiocarbamato complexes, Dalton Trans., 2009, 10670–10680, 10.1039/B913597A , and references cited therein.
- D. Aldinucci, D. Lorenzon, L. Stefani, L. Giovagnini, A. Colombatti and D. Fregona, Antiproliferative and apoptotic effects of two new gold(III) methylsarcosinedithiocarbamate derivatives on human acute myeloid leukemia cells in vitro, Anticancer Drugs, 2007, 18, 323–332, DOI:10.1097/CAD.0b013e328011ae98.
- L. Brustolin, N. Pettenuzzo, C. Nardon, S. Quarta, L. Marchiò, B. Biondi, P. Pontisso and D. Fregona, Au(III)-Proline derivatives exhibiting selective antiproliferative activity against HepG2/SB3 apoptosis-resistant cancer cells, Dalton Trans., 2019, 48, 16017–16025, 10.1039/C9DT03036K.
- M. Contel, A. J. Edwards, J. Garrido, M. B. Hursthouse, M. Laguna and R. Terroba, Mesityl gold(III) complexes. X-ray structure of mononuclear [Au(mes)2Cl(PPh3)] and the dimer [Au(mes)2Cl]2, J. Organomet. Chem., 2000, 607, 129–136, DOI:10.1016/S0022-328X(00)00306-5.
- S. J. Coles, D. R. Allan, C. M. Beavers, S. J. Teat and S. J. W. Holgate, Leading edge chemical crystallography service provision and its impact on crystallographic data science in the twenty-first century, in Structure and Bonding, Springer, Berlin, Heidelberg, 2020, pp. 1–72. DOI:10.1007/430_2020_63.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental details (including copies of spectra), extra crystallographic discussion and information. CCDC 2374277–2374288. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00240k |
| ‡ These authors contributed equally. |
| § Where the formula of a compound is given inside quotation marks, it indicates that the exact speciation of this compound is not known. Gold dithiocarbamate complexes are discussed extensively in the main text, but “AuX3” indicates that the exact nature of the gold(III) halide reagent is not clear. Many publications interchangeably refer to anhydrous AuX3, hydrated AuX3, AuX3 dissolved in aqueous HX, H[AuX4], and even [AuX4]− salts, and it is not always clear which of these Au(III) compounds have been used. To avoid lengthy discourse, these have been simplified to “AuX3” as a generic term for a source of gold(III) halide. |
| This journal is © The Royal Society of Chemistry 2025 |