
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalization of a Ru(II) polypyridine complex with an aldehyde group as a synthetic precursor for photodynamic therapy†
Lisa-Marie
Servos
,
Hung Manh
Tran
,
Nicolás
Montesdeoca
,
Zisis
Papadopoulos
,
Eun
Sakong
and
Johannes
Karges
 *
*
Faculty of Chemistry and Biochemistry, Ruhr-University Bochum, Universitätsstrasse 150, 44780 Bochum, Germany. E-mail: johannes.karges@ruhr-uni-bochum.de; Tel: +49 2343224187 Web: https://www.kargesgroup.ruhr-uni-bochum.de
First published on 3rd April 2025
Abstract
Photodynamic therapy has garnered significant attention over the past decades for its potential in treating various types of cancer, as well as bacterial, fungal, and viral infections. However, current clinically approved photosensitizers based on a tetrapyrrolic scaffold face notable limitations, including low water solubility, slow body clearance, and photobleaching. As a promising alternative, Ru(II) polypyridyl complexes have emerged due to their favorable photophysical and biological properties (i.e., reactive oxygen species generation, high water solubility, and biocompatibility). Despite these attractive properties, the vast majority of compounds are associated with poor tumor accumulation, representing a major hurdle for therapeutic applications. To overcome this limitation, herein, the chemical synthesis and photophysical evaluation of the functionalization of a Ru(II) polypyridyl complex with an aldehyde group, as a synthetic precursor for further conjugation, is reported. To ensure that the intrinsic chemical reactivity of the aldehyde group remains unaffected by the coordination environment to the metal center, a phenyl spacer was strategically introduced between the central ligand framework and the aldehyde functionality. Computational studies indicated that upon excitation of the metal complex, an excited state electron from the ruthenium t2g orbital is transferred to the π* ligand orbital in a metal-to-ligand charge transfer transition. The compound was found to be highly stable under physiological conditions as well as upon irradiation. Upon light exposure, the metal complex was found to efficiently convert molecular oxygen to singlet oxygen. These findings highlight the potential of the aldehyde functionalized Ru(II) polypyridyl complex as a versatile precursor for photodynamic therapy.
Introduction
Cancer, a disease caused by genetic mutations leading to uncontrolled cell division, can develop in any tissue, spread through metastasis, and result in organ failure.1 Despite rising cancer mortality rates since the 1970s, advances in targeted treatments have slowed the rate of increase, with lung, prostate, and breast cancer remaining leading causes of death globally. Modern cancer treatments primarily include chemotherapy, radiotherapy,2 and immunotherapy,3 each with unique mechanisms to target cancer cells. However, these methods often affect normal cells as well, leading to significant side effects, highlighting the need for more selective and less toxic approaches.4–7As a complementary medicinal technique, photodynamic therapy (PDT) is emerging for the treatment of various types of cancer (i.e., lung, bladder, esophageal, and brain cancer), as well as bacterial, fungal, and viral infections.8,9 The therapeutic mechanism of PDT involves a photosensitizer (PS), oxygen, and light.10 Upon light irradiation, the PS is uplifted to an excited singlet state and subsequently undergoes intersystem crossing to an excited triplet state.11 In this state, the PS can facilitate the generation of reactive oxygen species (ROS) by two pathways.12 In the type I pathway, the PS interacts with nearby substrates, transferring electrons to produce radicals such as superoxide or hydroxyl radicals. In the type II pathway, energy is transferred to molecular oxygen (3O2), generating singlet oxygen (1O2).13,14 Most photosensitizers interact with their biological environment predominantly through the type II mechanism.15 Due to their high reactivity and short lifetime, ROS are capable of impacting the biological milieu within an approximate radius of 100 nm.16 To date, the majority of investigated and clinically approved PSs are based on a tetrapyrrolic scaffold (porphyrin, phthalocyanine, chlorin, bacteriochlorin).17,18 Despite their high clinical success, these compounds are associated with several limitations including poor water solubility, poor (photo-)stability, and slow clearance from the body, leading to prolonged photosensitivity in patients.19 Among emerging alternatives, Ru(II) polypyridine complexes have gained increasing attention due to their attractive photophysical and biological properties, such as high water solubility, excellent chemical and photostability, strong luminescence, large Stokes shifts, and efficient 1O2 production.20–25 Notably, the PS TLD-1433, developed by the McFarland group, is currently studied in phase II clinical trial as a PDT agent for the treatment of bladder cancer.26,27 Despite their excellent potential as PSs, both tetrapyrrolic and Ru(II) polypyridyl-based PSs generally suffer from poor selectivity toward cancer cells.28 As a result, high drug doses are often required, increasing the risk of side effects.29 Ideally, the PS should remain non-toxic in the absence of light, but induce cellular damage upon irradiation.30 However, due to the strong scattering of light by skin and tissue during treatment, and the practical difficulty of precisely irradiating only the tumor site, surrounding healthy tissue is often affected.31 To address these challenges, conjugating PSs to tumor-targeting groups is essential to improve selectivity and reduce off-target effects. While several conjugates of PSs to tumor-targeting groups have been previously reported, the conjugation of these inherently different molecules remains challenging.
To address this limitation, this study reports on the chemical synthesis and photophysical evaluation of a Ru(II) polypyridyl complex functionalized with an aldehyde group, serving as a synthetic precursor for further conjugation. To preserve the intrinsic chemical reactivity of the aldehyde group, a phenyl spacer was strategically introduced between the central ligand framework and the aldehyde functionality to mitigate any influence from the metal coordination environment. Computational studies revealed that, upon excitation, an electron from the ruthenium t2g orbital is transferred to the ligand's π* orbital by a metal-to-ligand charge transfer transition. Experimental analyses demonstrated that the complex exhibits high stability under physiological conditions and upon light irradiation. Importantly, the metal complex efficiently generated 1O2 from 3O2 upon light exposure. These findings underscore the potential of the aldehyde functionalized Ru(II) polypyridine complex as a versatile precursor for photodynamic therapy applications.
Results and discussion
Synthesis and characterization
Ru(III) chloride was reduced to Ru(II) using ethanol as a reducing agent and solvent. The coordination sphere of the metal was further saturated with dimethyl sulfoxide to yield the metal complex [Ru(dimethyl sulfoxide)4Cl2] (1). The resulting compound was then refluxed in N,N-dimethylformamide with two equivalents of 1,10-phenanthroline and an excess of lithium chloride. The product [Ru(1,10-phenanthroline)2(Cl)2] (2) was obtained upon precipitation with acetone. The extended aldehyde functionalized bipyridine ligand (3) was prepared by Suzuki–Miyaura coupling reaction from 4-Bromo-2,2′-bipyridine and 4-formylphenyl boronic acid in the presence of [1,1′-Bis(diphenylphosphino)ferrocene]palladium(II) dichloride using a previously reported protocol.32,33 The desired Ru(II) polypyridine complex [Ru(1,10-phenanthroline)2(4-(2,2′-bipyridyl)benzaldehyde)][PF6]2 (4) was synthesized by heating a mixture of compounds 2 and 3 at reflux temperature in a water/ethanol mixture overnight (Scheme 1). The metal complex was isolated upon precipitation as a hexafluorophosphate salt from water using ammonium hexafluorophosphate. All compounds were characterized using NMR spectroscopy, mass spectrometry, elemental analysis and analytical HPLC (Fig. S1–S12†).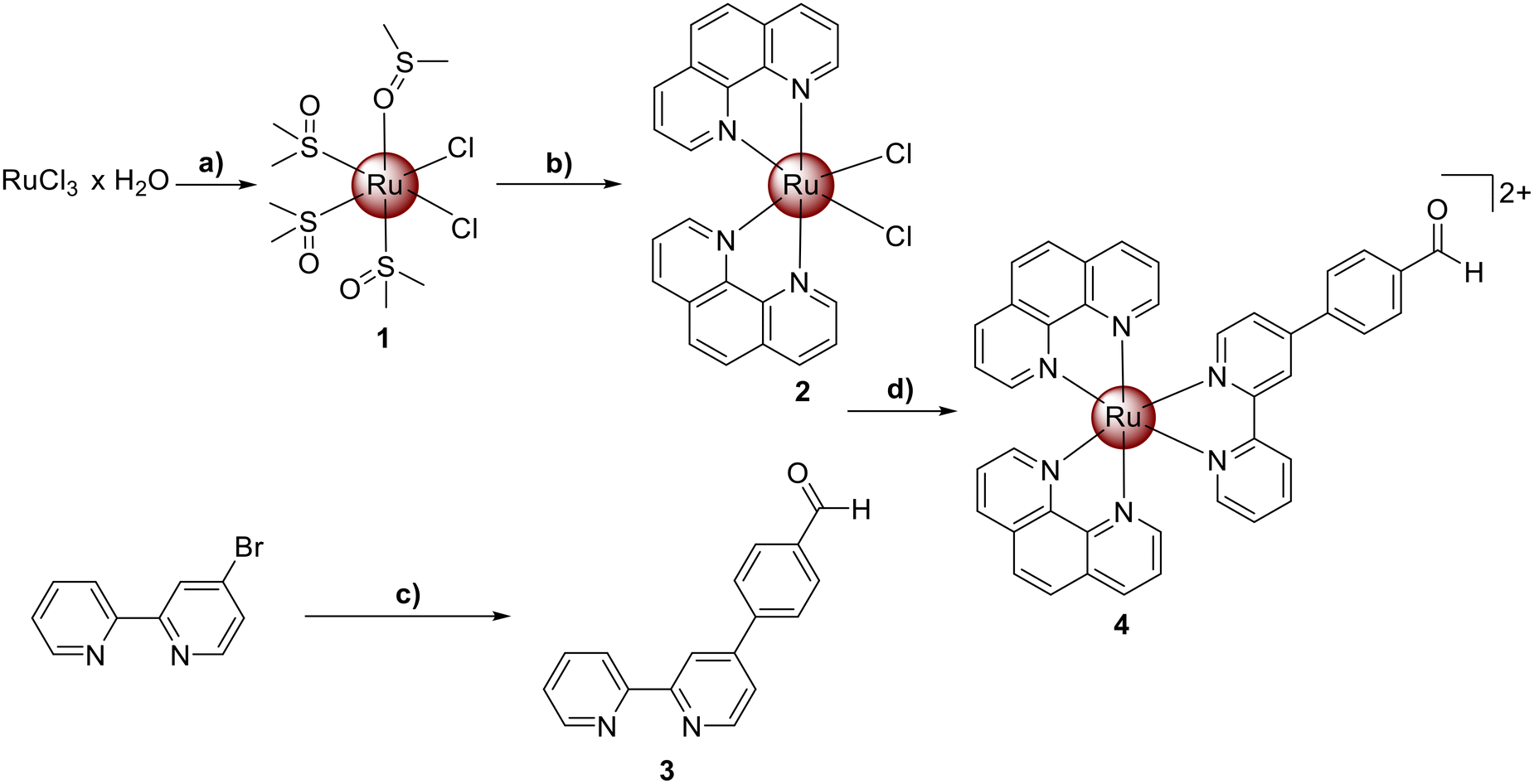 | ||
Scheme 1 Synthesis of the aldehyde functionalized Ru(II) polypyridine complex [Ru(1,10-phenanthroline)2(4-(2,2′-bipyridyl)benzaldehyde)][PF6]2 (4). a) (i) EtOH, 3 h, reflux; (ii) DMSO, 2 h, 150 °C; b) 1,10-phenanthroline, LiCl, DMF, overnight, reflux; c) water/dioxane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), 4-formylboronic acid, Pd(dfpp)2·DCM, K2CO3, 6 h, reflux; d) H2O/EtOH (1:1), overnight, reflux. All metal complexes were isolated as the respective hexafluorophosphate salts. :4), 4-formylboronic acid, Pd(dfpp)2·DCM, K2CO3, 6 h, reflux; d) H2O/EtOH (1:1), overnight, reflux. All metal complexes were isolated as the respective hexafluorophosphate salts. | ||
Pharmacological properties
The solubility of the metal complex was assessed using dynamic light scattering measurements. A stock solution of the metal complex was prepared in dimethyl sulfoxide and subsequently diluted in phosphate-buffered saline (PBS) to achieve a final dimethyl sulfoxide concentration of 0.1%. Dynamic light scattering measurements were conducted to monitor the formation of any precipitates or particles. No evidence of particle formation or aggregation was detected, indicating that 4 exhibited good aqueous solubility under physiological conditions. A solubility test in 9:1 PBS/DMSO revealed a maximum solubility of 25 mg mL−1 in aqueous solution (Fig. S13†). Therefore, 4 possesses good water solubility compared to tetrapyrrolic clinically approved photosensitizers (Table S1†).
A suitable compound for biological applications should possess both lipophilic and hydrophilic properties.34 Hydrophilicity is crucial for ensuring the compound's solubility in biological media, while lipophilicity aids its transportation across cell membranes, facilitating efficient cellular uptake.35 To assess the lipophilic and hydrophilic characteristics of compound 4, the octanol/PBS partition coefficient (LogP) was determined. The metal complex 4 was found mainly in the octanol phase, indicating lipophilic properties (LogP = 1.67 ± 0.04).
(Photo-)stability
The stability of compounds under physiological conditions is essential for their biological applications to ensure a safe and effective therapeutic outcome, as undesired degradation can reduce efficacy and cause side effects. To assess the stability of the metal complex under such conditions, compound 4 was incubated in water for up to 48 hours and the absorption profile consistently monitored. The absorption spectrum showed no noticeable changes (Fig. S15†), indicating that the metal complex remained stable under physiological conditions.The potential decomposition upon light irradiation was subsequently investigated, as previous studies have shown that the stability of metal complexes can be affected by light exposure.36 This evaluation is particularly important, as the majority of currently approved PDT agents exhibit significant photobleaching effects. The complexes were subjected to continuous irradiation at 420 nm, and their absorption spectra was monitored over time. [Ru(2,2′-bipyridine)3]2+ was used as a positive control while Photofrin served as a negative control. A comparison of the spectra revealed that metal complex 4 exhibited photobleaching effects within a similar range to those observed for [Ru(2,2′-bipyridine)3]2+ (Fig. S16–S18†).
Photophysical properties
To evaluate the potential of compound 4 as a PS for PDT, its absorption characteristics were investigated using UV/Vis spectroscopy. In PDT, the excitation wavelength of a photosensitizer is a critical factor, as it directly influences the tissue penetration depth of light. Longer excitation wavelengths allow for deeper tissue penetration, which is particularly important for targeting large or deeply located tumors. The absorption spectrum of compound 4 in acetonitrile revealed an intense absorption band centered at 290 nm (ε = 20.6 M−1cm−1 × 103), along with two weaker bands at 385 nm (ε = 5.7 M−1 cm−1 × 103) and 455 nm (ε = 8.1 M−1 cm−1 × 103) (acetonitrile: Fig. 1, PBS: Fig. S14†). A direct comparison with the parent complex [Ru(2,2′-bipyridine)3]2+ demonstrated that compound 4 exhibits an approximately 45 nm stronger red-shifted absorption peak.37 Notably, compound 4 also displayed a significant absorption tail extending into the red region, which could be advantageous for enhanced light penetration into the tissue. | ||
| Fig. 1 (left) Normalized absorption spectrum of 4 in acetonitrile. (right) Emission spectrum of 4 in acetonitrile upon excitation at 290 nm. | ||
The emission of the metal complex in acetonitrile or PBS was measured following excitation at 290 nm. Complex 4 exhibited an emission maximum centered at 620 nm, corresponding to a large Stokes shift, which indicates minimal overlap between excitation and emission (acetonitrile: Fig. 1, PBS: Fig. S14†).
Theoretical studies
To gain a deeper understanding of the photophysical mechanism, density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations were performed using B3LYP functional. Organic atoms were modelled with 6-31G(d) and ruthenium with LANL2DZ basis set. These calculations enabled the assignment of the respective bands in the absorption spectrum. The metal complex displayed a prominent absorption peak centered at 290 nm, which was attributed to a ligand-centered (LC) transition. In contrast, the lowest energy transitions at 385 and 455 nm were identified as having a distinct metal-to-ligand charge transfer (MLCT) character, as confirmed by the analysis of the excited state density (Fig. 2).38 | ||
| Fig. 2 Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) ± 4 of 4. The molecular orbitals were obtained using the triple-zeta functional B3LYP. Organic atoms were modelled with 6-31G(d) and ruthenium with LANL2DZ basis set. | ||
To get an understanding of the charge transfer length of the MLCT the difference in density between ground state and vertical excited states was computed (Fig. 3).39 This allows the visualization of areas undergoing density increase/decrease (green/red) during the excitation process. Hereby, an increase in density is representative for the acceptor regions of the molecule while a density decrease depicts donor moieties. To obtain quantitative information about the charge transfer distance, the barycenters of the density depletion and increase regions were calculated. These barycenters offer a straightforward visualization of the hole and electron positions for the examined transitions. The electron density hole is localized in the ruthenium center as expected, while the density increases on the ligand bearing the aldehyde functionality, which is consistent with the characteristics of an MLCT transition (ES1,† ET1). In the case of ES3 a MLCT to the phenanthroline ligand can be observed, showcasing a shorter DCT.
 | ||
| Fig. 3 Plots representing the difference in density between excited singlet, vertical excited triplet state and ground state. Depicted are the first (ES = 1, λ = 490 nm, f = 0.013 a.u.) and third (ES = 3 λ = 457 nm, f = 0.001 a.u.) vertical singlet transition as well as the first vertical triplet transition (ET = 1; λ = 530 nm, f = 0.000 a.u.) together with the corresponding DCT values (in Å). Green regions show the increase in electron density upon excitation and red regions the density decrease. Corresponding barycenters are depicted in green (increase) and red (decrease). Atoms color scheme: C-grey, H-white, N-blue, O-red, Ru-light blue. | ||
To maximize the quantum yield and thereby the likelihood of an energy transfer between the excited triplet state sensitizer and ground-state molecular oxygen the intersystem crossing energy is of fundamental importance.40 Only if the intersystem crossing energy is greater than 0.2 eV, the triplet state cannot equilibrate back to the excited singlet state.41 To calculate if the energy gap of 4 is big enough, the energy of the excited singlet state was calculated to be 2.53 eV (λ = 490 nm, blue light excitation), whilst the energy of the excited triplet state is 2.09 eV (593 nm), yielding an ISC energy of 0.46 eV (Fig. 4). Therefore, the obtained ISC energy is above the 0.2 eV benchmark. The obtained T1 → S0 transition energy corresponds to a wavelength of 593 nm predicting emissive properties with dark yellow color. These results match the obtained emission maxima of around 600 nm (Fig. 1). This gap is energetically distinctive enough to enable effective interaction between molecular oxygen and the metal complex to generate ROS.
 | ||
| Fig. 4 Jablonski diagram of 4. | ||
To confirm the potential for further functionalization of the metal complex at the aldehyde group, an electrostatic potential map was calculated. The results revealed that the coordinatively saturated ruthenium center carries a positive charge (blue), while the negative charge (red) is concentrated at the aldehyde group. These observations confirm the aldehyde functionality as a reactive site, making it well-suited for further chemical modifications (Fig. 5).
 | ||
| Fig. 5 Electrostatic potential map of 4, calculated using the Hirshfeld method. Regions of negative charge are depicted in red, while positive charge is shown in blue. | ||
Singlet oxygen production
The ability of complex 4 to transfer energy from its excited triplet state to molecular oxygen, leading to the photo-catalytic production of singlet oxygen, was investigated. Singlet oxygen formation was spectroscopically monitored in acetonitrile and PBS by tracking the decrease in absorbance of N,N-dimethyl-4-nitrosoaniline at 420 nm (acetonitrile) and 440 nm (PBS) in the presence of complex 4, both in the dark and under light irradiation (450 nm). No significant changes in the absorption spectrum were observed when N,N-dimethyl-4-nitrosoaniline was incubated with complex 4 in the dark or when only N,N-dimethyl-4-nitrosoaniline alone was irradiated. However, when complex 4 was present, the absorbance intensity of N,N-dimethyl-4-nitrosoaniline rapidly decreased upon irradiation (Fig. 6), indicating efficient singlet oxygen generation. A quantum yield of ΦMeCN > 0.95 in acetonitrile and ΦPBS = 0.25 in PBS was obtained for 4. Therefore, complex 4 demonstrated a higher singlet oxygen production than the reference and parent complex [Ru(2,2′-bipyridine)3][Cl]2. These findings emphasize complex 4's ability to generate singlet oxygen upon light irradiation, suggesting its potential as a photosensitizer for photodynamic therapy applications. | ||
| Fig. 6 Changes in the absorption spectrum of the singlet oxygen scavenger system N,N-dimethyl-4-nitrosoaniline and imidazole (in acetonitrile)/histidine (in PBS). Insets represent the correlation of absorption changes with irradiation time, fitted using linear regression (full size: Fig. S19†). Top left: Complex 4 in acetonitrile. Top right: Reference in acetonitrile. Bottom left: Complex 4 in PBS. Bottom right: Reference in PBS. | ||
Conclusion
In summary, this study reports on the chemical synthesis and photophysical evaluation of a novel Ru(II) polypyridyl complex that incorporates an aldehyde moiety, positioning it as a promising synthetic precursor for photodynamic therapy applications. The compound was found with a high water solubility and (photo-)stability, ensuring its structural integrity under biological conditions. Computational studies offered valuable insights into the complex's electronic structure and excited state properties. The metal complex displayed a strong absorption peak around 290 nm, which was attributed to a ligand-centered transition. The lower energy transitions centered at 385 and 455 nm were identified as metal-to-ligand charge transfer transitions. In the excited triplet state, the electron density was shifted from the ruthenium center on the extended ligand containing the aldehyde functionality. Importantly, the complex demonstrated to efficiently transfer energy from its excited triplet state to molecular oxygen, thereby catalyzing the production of singlet oxygen. Furthermore, an electrostatic potential map was calculated, revealing important details about the charge distribution within the complex. The ruthenium center was a positively polarized while the aldehyde group was negative polarized. These observations underscore the aldehyde group as a reactive site, making it a suitable synthetic handle for further chemical modifications. Future research will focus on conjugating this complex with other therapeutic agents or tumor-targeting groups to enhance its anticancer efficacy.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
J. Karges gratefully acknowledges the financial support provided by the Liebig fellowship from the Chemical Industry Fund of the German Chemical Industry Association, the Life Sciences Bridge Award from the Aventis Foundation, and the Paul-Ehrlich-Gesellschaft Stiftung Early Career Award 2024 – a prize awarded by the Paul Ehrlich Foundation, Germany.References
- R. F. Brissos, P. Clavero, A. Gallen, A. Grabulosa, L. A. Barrios, A. B. Caballero, L. Korrodi-Gregório, R. Pérez-Tomás, G. Muller, V. Soto-Cerrato and P. Gamez, Inorg. Chem., 2018, 57, 14786–14797 CAS.
- S. S. Ahmad, S. Duke, R. Jena, M. V. Williams and N. G. Burnet, Br. Med. J., 2012, 345, e7765 Search PubMed.
- M. Schuster, A. Nechansky and R. Kircheis, Biotechnol. J., 2006, 1, 138–147 CAS.
- D. Yerramilli, A. J. Xu, E. F. Gillespie, A. F. Shepherd, K. Beal, D. Gomez, J. Yamada, C. J. Tsai and T. J. Yang, Adv. Radiat. Oncol., 2020, 5, 589–594 Search PubMed.
- M. S. Aslam, S. Naveed, A. Ahmed, Z. Abbas, I. Gull and M. A. Athar, J. Cancer Ther., 2014, 5, 817–822 Search PubMed.
- F. Niemeier, L.-M. Servos, Z. Papadopoulos, N. Montesdeoca, K. Ni, S. Heinrich and J. Karges, J. Med. Chem., 2025, 68, 1316–1327 Search PubMed.
- J. Schleisiek, E. Michaltsis, S. Mayer, N. Montesdeoca and J. Karges, Dalton Trans., 2025, 54, 942–950 CAS.
- L. K. McKenzie, H. E. Bryant and J. A. Weinstein, Coord. Chem. Rev., 2019, 379, 2–29 CAS , ISSN 0010-8545.
- V. Brabec and O. Nováková, Drug Resistance Updates, 2006, 9, 111–122 CrossRef CAS PubMed.
- J. Karges, Angew. Chem., Int. Ed., 2022, 61, e202112236 Search PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 889–905 CrossRef CAS PubMed.
- L. He, M.-F. Zhang, Z.-Y. Pan, K.-N. Wang, Z.-J. Zhao, Y. Li and Z.-W. Mao, Chem. Commun., 2019, 55, 10472–10475 RSC.
- L. M. Lifshits, J. A. Roque III, P. Konda, S. Monro, H. D. Cole, D. Von Dohlen, S. Kim, G. Deep, R. P. Thummel, C. G. Cameron, S. Gujar and S. A. McFarland, Chem. Sci., 2020, 11, 11740–11762 RSC.
- M. Zheng, X. Lin, K. Xiong, X. Zhang, Y. Chen, L. Ji and H. Chao, Chem. Commun., 2024, 60, 2776–2779 RSC.
- M. Ju, L. Yang, G. Wang, F. Zong, Y. Shen, S. Wu, X. Tang and D. Yu, Biomater. Sci., 2024, 12, 2831–2840 RSC.
- P. Sharma, A. B. Jha, R. S. Dubey and M. Pessarakli, J. Bot., 2012, 2012, 217037–217063 Search PubMed.
- S. A. McFarland, A. Mandel, R. Dumoulin-White and G. Gasser, Curr. Opin. Chem. Biol., 2020, 56, 23–27 CrossRef CAS PubMed.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- J. Karges, S. Kuang, F. Maschietto, O. Blacque, I. Ciofini, H. Chao and G. Gasser, Nat. Commun., 2020, 11, 3262 CrossRef CAS PubMed.
- S. Bonnet, J. Am. Chem. Soc., 2023, 145, 23397–23415 CrossRef CAS PubMed.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CAS.
- A. Marco, J. Kasparkova, D. Bautista, H. Kostrhunova, N. Cutillas, L. Markova, V. Novohradsky, J. Ruiz and V. Brabec, J. Med. Chem., 2024, 67, 21470–21485 CrossRef CAS PubMed.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 CAS.
- N. Montesdeoca, J. M. Mohr, S. Kruss and J. Karges, J. Biol. Inorg. Chem., 2025, 30, 53–60 CAS.
- K. M. Coombs, K. K. M. Glover, R. Russell, P. Kaspler, M. Roufaiel, D. Graves, P. Pelka, D. Kobasa, R. DuMoulin-White and A. Mandel, Heliyon, 2024, 10, e32140 CAS.
- S. Monro, K. L. Colón, H. Yin, J. I. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CAS.
- Z. Zhu, L. Wei, A. K. Yadav, Z. Fan, A. Kumar, M. Miao, S. Banerjee and H. Huang, J. Org. Chem., 2023, 88, 626–631 CAS.
- J. Ruiz, V. Rodríguez, N. Cutillas, A. Espinosa and M. J. Hannon, Inorg. Chem., 2011, 50, 9164–9171 CAS.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CAS.
- A. Gandioso, E. Izquierdo-García, P. Mesdom, P. Arnoux, N. Demeubayeva, P. Burckel, B. Saubaméa, M. Bosch, C. Frochot, V. Marchán and G. Gasser, Chem. – Eur. J., 2023, 29, e202301742 CAS.
- C. Dolan, A. Byrne, C. Long, K. Czamara, A. Kaczor, M. Baranska and T. E. Keyes, RSC Adv., 2017, 7, 43743–43754 CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 CAS.
- S. Roy, E. Colombo, R. Vinck, C. Mari, R. Rubbiani, M. Patra and G. Gasser, ChemBioChem, 2020, 21, 2966–2973 CAS.
- C. Vraka, S. Mijailovic, V. Fröhlich, M. Zeilinger, E.-M. Klebermass, W. Wadsak, K.-H. Wagner, M. Hacker and M. Mitterhauser, Nucl. Med. Biol., 2018, 58, 20–32 CAS.
- A. V. Chernyshev, N. A. Voloshin, A. V. Metelitsa, V. V. Tkachev, S. M. Aldoshin, E. Solov'eva, I. A. Rostovtseva and V. I. Minkin, J. Photochem. Photobiol., A, 2013, 265, 1–9 CAS.
- Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, vol. 280, 2007 Search PubMed.
- A. Fennes, N. Montesdeoca, Z. Papadopoulos and J. Karges, Chem. Commun., 2024, 60, 10724–10727 RSC.
- J. Karges, F. Heinemann, F. Maschietto, M. Patra, O. Blacque, I. Ciofini, B. Spingler and G. Gasser, Bioorg. Med. Chem., 2019, 27, 2666–2675 CrossRef CAS PubMed.
- X. Miao, W. Hu, T. He, H. Tao, Q. Wang, R. Chen, L. Jin, H. Zhao, X. Lu, Q. Fan and W. Huang, Chem. Sci., 2019, 10, 3096–3103 RSC.
- Y. Yu, S. Mallick, M. Wang and K. Börjesson, Nat. Commun., 2021, 12, 3255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5dt00256g |
| This journal is © The Royal Society of Chemistry 2025 |