
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkylamido lutetium complexes as prospective lutetium imido precursors: synthesis, characterization and ligand design†
Jackson P.
Knott
,
Shou-Jen
Hsiang
and
Paul G.
Hayes
 *
*
Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive, Lethbridge, AB, Canada T1K 3M4. E-mail: p.hayes@uleth.ca; Fax: +1403 329 2057; Tel: +1 403 329 2313
First published on 20th March 2025
Abstract
Mixed alkylamido lutetium complexes, LiPrLu(CH2SiMe3)(NHCPh3) (7CPh3) and LiPrLu(CH2SiMe3)(NHDipp) (7Dipp) (LiPr = 2,5-[iPr2P = N(4-iPrC6H4)]2C4H2N−), were synthesized by addition of a bulky primary amine, NH2R (R = CPh3, Dipp) (Dipp = 2,6-iPr2C6H3) to the dialkyl complex LiPrLu(CH2SiMe3)2 (6). Unlike complexes supported by the related pincer ligand LPh (LPh = 2,5-[Ph2P = N(4-iPrC6H4)]2C4H2N−) these species proved resistant to C–H cyclometalative processes. Attempts to access lutetium imdes via addition of 4-dimethylaminopyridine (DMAP) to 7CPh3 and 7Dipp promoted disproportionation, affording 0.5 equivalents of the corresponding bisamide complexes LiPrLu(NHCPh3)2 (8CPh3) and LiPrLu(NHDipp)2 (8Dipp), respectively, as well as 0.5 equivalents of LiPrLu(CH2SiMe3)2, which decomposed in the presence of DMAP. Incorporation of internal Lewis bases was accomplished by replacing the N-aryl substituents in LiPr with 4,6-dimethylpyrimidine groups (LPm, 11). The correspondng dialkyl lutetium complex LPmLu(CH2SiMe3)2 (12) was prepared, from which loss of SiMe4 occured over a period of hours in benzene-d6 solution.
Introduction
Complexes that feature multiple bonding between transition metals and carbon, nitrogen, or other main group elements have led to remarkable new stoichiometric and catalytic transformations that have found great utility across numerous disciplines.1 Although such success prompted tremendous efforts to isolate comparable rare earth analogues, they remained largely elusive until the past two decades. In particular, rare earth complexes bearing a terminal imido functionality (RE = NR, RE = group III and lanthanide elements) were targeted with great enthusiasm, yet prior to 2010 only μ2- and μ3-bridging species,2–4 and complexes wherein a transient RE = NR moiety activates a C–H bond, were reported.5–7 Accordingly, the discovery by Chen and co-workers that addition of one equivalent of DMAP to the scandium alkylamido complex, L1Sc(NHDipp)(CH3) (L1 = DippNC(CH3)CHC(CH3)NCH2CH2N(CH3)2, Dipp = 2,6-iPr2C6H3, DMAP = 4-(CH3)2NC5H4N), affords the terminal scandium imido L1(DMAP)Sc = NDipp (1, Fig. 1) drew considerable attention.8 Notably, DMAP can be removed by reaction of complex 1 with 9-BBN (9-BBN = 9-borabicyclo(3.3.1)nonane), generating the unstable base-free imide L1Sc = NDipp, which readily participates in [2 + 2] cycloaddition reactions and activation of Csp2–H, Csp2–F, and B–H bonds.9 The following year Chen demonstrated that methane elimination can be induced in the absence of DMAP if a tethered Lewis base is incorporated into the β-diketiminato ancillary ligand.10 The corresponding scandium imide, L2Sc = NDipp (2, L2 = DippNC(CH3)CHC(CH3)NCH2CH2N(CH3)CH2CH2N(CH3)2, Fig. 1), has exhibited rich reaction chemistry with unsaturated organic molecules,11 main group elements,10 transition metal coordination complexes,12 and more.13 Ultimately, seminal contributions from Chen,13 Mindiola,5,6 Piers7 and Cui14 demonstrated that given the appropriate system, introduction of either internal or external Lewis bases to a scandium alkylamido complex is an effective strategy for accessing terminal imido functionalities. | ||
| Fig. 1 Selected examples of structurally characterized terminal rare earth imido and alkylamido complexes. | ||
Following Chen's approach, Anwander prepared DMAP-stabilized imide complexes of the larger rare earth metals, yttrium and lutetium (TptBu,MeY = N-2,6-(CH3)2C6H3(DMAP) (3a, Fig. 1) and TptBu,MeLu = N-3,5-(CF3)2C6H3(DMAP)) (3b, Fig. 1) (TptBu,Me = HB[3-CH3-5-tBu-N2C3H]3−).15 More recently, Schelter expanded the list of terminal rare earth imides to include the cerium species K[(TriNOx)Ce = N-3,5(CF3)2C6H3] (TriNOx = N(CH2-2-C6H4N(tBu)O)3); Anwander added the dysprosium and holmium complexes TptBu,MeRE(N-2,6-iPr2C6H3)(DMAP) (RE = Dy, Ho).16–18 Notably, these examples utilize starting materials that include a mixed methyl/tetramethylgallato system, TptBu,MeRE(Me)(GaMe4) (RE = Y, Dy, Ho), that reacts with H2NDipp to yield mixed methylamido species primed for imide-formation.
It is important to note that all reported rare earth complexes bearing a terminal imido functionality contain an aromatic group on the imide nitrogen.4–8,10,14–18 The apparent requirement for aryl substitution piqued our interest, particularly given that neither TptBu,MeLu(CH3)(NHAd) (4a, Fig. 1) nor TptBu,MeLu(CH3)(NHtBu) (4b, Fig. 1), served as appropriate precursors for a lutetium imide.19 Our group previously synthesized LPhLu(CH2SiMe3)(NHCPh3), (LPh = 2,5-[Ph2P = N(Pipp)]2C4H2N−, Pipp = 4-iPrC6H4) in hopes of observing similar reactivity;20 however, exhaustive efforts (e.g. addition of Lewis or Brønsted bases, heating, etc.) failed to yield the targeted imido LPhLu = NCPh3. Instead, Csp2–H activation of the CPh3 substituent occurred.20 Herein we utilize the more electron rich NNN-pincer ligand LiPr (LiPr = 2,5-[iPr2P = N(Pipp)]2C4H2N−)21 to alleviate unwanted activations and probe the impact that the amine bound substituent has on rare earth imido synthesis. Furthermore, with the goal of expanding the library of Lewis bases that can promote alkane loss from rare earth alkylamido species, a second NNN-pincer that features 4,6-dimethyl pyrimidine groups, LPm = [iPr2P = N(4,6-(Me)2C4H4N2)]2C4H2N−, was synthesized. Unfortunately, mixed alkylamido lutetium species were unable to be isolated with this ligand, due to spontaneous loss of SiMe4.
Results and discussion
Synthesis of LiPrSc(CH2SiMe3)2 and LiPrLu(CH2SiMe3)2
The dialkyl complexes LiPrRE(CH2SiMe3)2 (LiPr = 2,5-[iPr2P = N(Pipp)]2C4H2N−), 5 (RE = Sc) and 6 (RE = Lu), were prepared by reaction of HLiPr with RE(CH2SiMe3)3THF2 in toluene for 1 hour (Scheme 1). Production of LiPrSc(CH2SiMe3)2 and LiPrLu(CH2SiMe3)2 was supported by the consumption of 1H and 31P NMR resonances attributed to HLiPr, as well as the emergence of a resonance at δ 0.00 in the 1H NMR spectra that integrated for 12H, attributed to eliminated SiMe4. New single resonances in the 31P{1H} NMR spectra (5δ 48.0, 6δ 49.3) similarly support the synthesis of discrete metal complexes in high purity. As previously observed,21 the P–(CH(CH3)2)2 methyl groups are chemically inequivalent on the NMR timescale (5: δ 0.93, 0.88, 6: δ 0.96, 0.85). Like LPhLu(CH2SiMe3)2, no Csp2–H bond activation was observed at ambient temperature in solution.22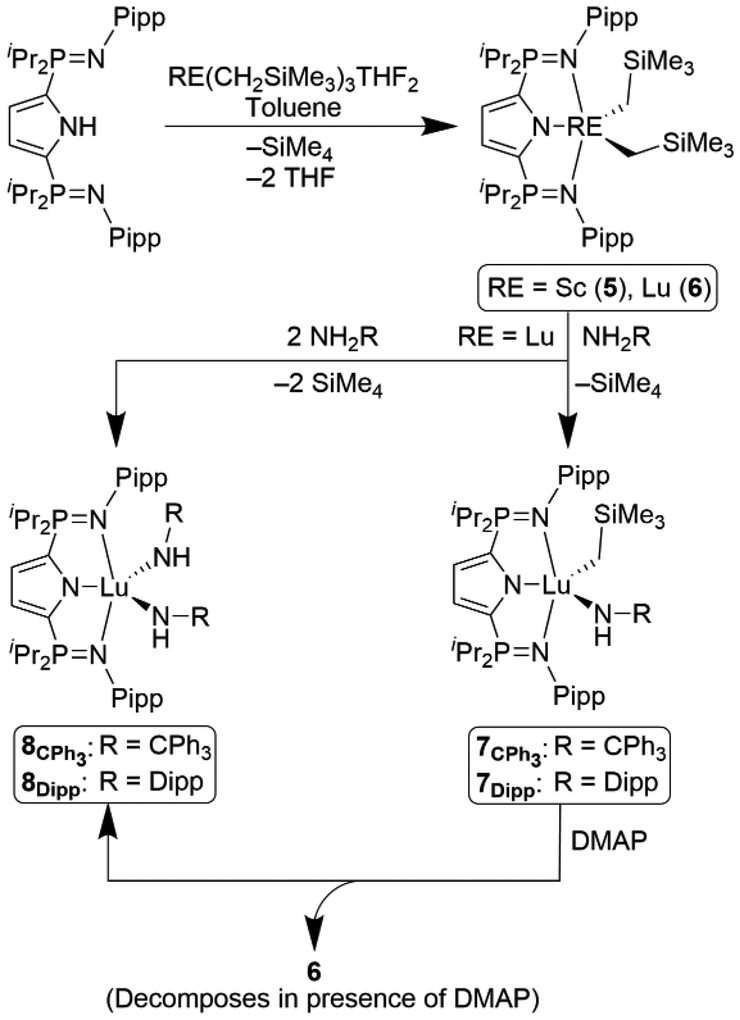 | ||
| Scheme 1 Synthesis and reactivity of complexes 5 and 6. | ||
X-Ray diffraction analysis of complexes 5 and 6
Single crystals suitable for diffraction analysis of both complexes 5 and 6 were obtained by slowly cooling warm (50 °C) heptane solutions of each to ambient temperature. Both 5 and 6 crystallized in the P21/n space group (Fig. 2) and the geometry about each metal centre is best described as distorted square pyramidal (τ5 = 0.37 (5) τ5 = 0.31 (6)). The solid-state structures revealed Lu–C35, Lu–C39, and Lu–N1 distances that were, on average, 0.117 Å longer than those in the scandium congener. As expected, the phosphinimine P–N bond lengths are similar in both complexes (5: 1.619(2) Å, 6: 1.611(3) Å) and elongated relative to HLiPr.21 These contacts are also comparable to those observed in rare earth complexes supported by the related pincer ligand, LPh (1.606(4)–1.610(3) Å).22,23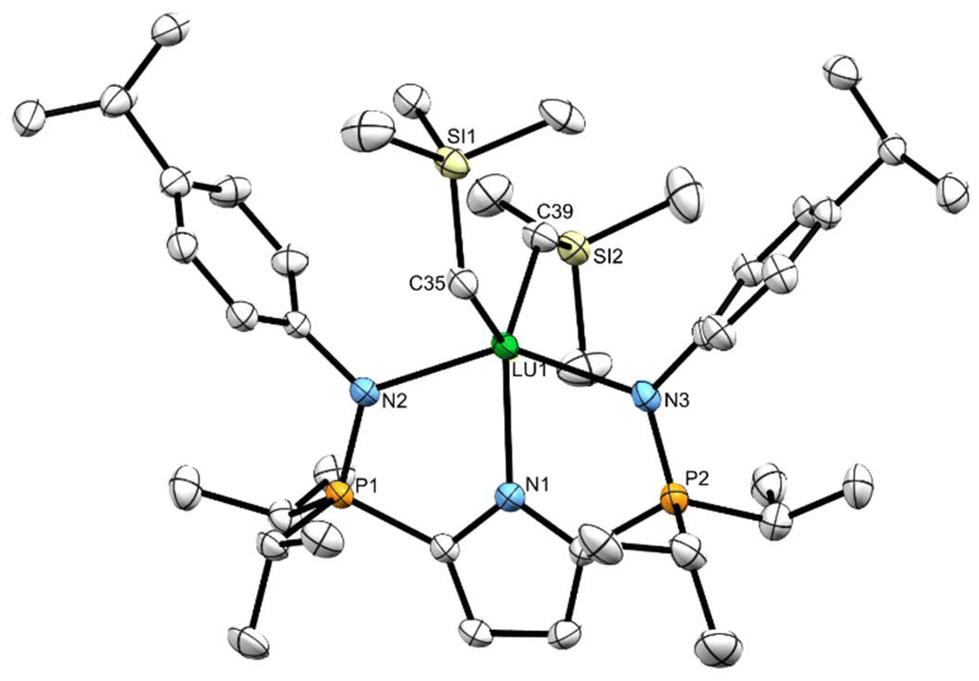 | ||
| Fig. 2 Solid-state structure of complex 6 with thermal ellipsoids represented at 50% probability. hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): Lu1–N1 2.305(2), Lu1–N2 2.333(3), Lu1–C35 2.360(4), P1–N2 1.611(3), C35–Lu1–C39 112.1(1), N2–Lu1–N3 143.79(9), N1–Lu1–C35 125.0(1), N1–Lu1–C39 122.9(1). | ||
Mixed alkylamido lutetium complexes
In order to probe what impact swapping P–Ph2 for P–iPr2 groups has on the ability of our ligands to support rare earth terminal imido precursors, complexes 5 and 6 were independently exposed to one equivalent of various primary amines (Ph3CNH2, DippNH2, AdNH2, or Mes*NH2). Unfortunately, compound 5 did not react with Ph3CNH2, nor Mes*NH2 (Mes* = 2,4,6-tBu3C6H2) even upon exposure to heat. Slow, cold addition of 1-adamantylamine (AdNH2) to complex 5 promoted formation of an inextricable mixture of LiPrSc(CH2SiMe3)2 (5), LiPrSc(CH2SiMe3)(NHAd), and LiPrSc(NHAd)2 as indicated by multinuclear NMR spectroscopy. With complex 5 not undergoing successful protonolysis with primary amines, focus was directed to LiPrLu(CH2SiMe3)2 (6).Addition of Ph3CNH2 and DippNH2 to compound 6 gave rise to complexes 7CPh3, LiPrLu(CH2SiMe3)(NHCPh3), and 7Dipp, LiPrLu(CH2SiMe3)(NHDipp), respectively (Scheme 1). As expected, one equivalent of SiMe4 (δ 0.00) was liberated and observed in both reaction mixtures via1H NMR spectroscopy. Furthermore, a sharp singlet (assigned as the N–H resonance), integrating for one proton, which did not give rise to cross peaks in 1H–1H COSY or 1H–13C HSQC experiments, emerged (7CPh3δ 2.43, 7Dippδ 4.14). Installation of the NHCPh3/NHDipp ligand produced Cs-symmetric complexes, resulting in the appearance of two sets of P–(CH(CH3)2)2 resonances in the 1H NMR spectra of complexes 7CPh3 (δ 2.16, 1.75) and 7Dipp (δ 2.26, 1.93). Further evidence for the formation of these mixed alkylamido compounds was provided by the disappearance of the singlet corresponding to LiPrLu(CH2SiMe3)2 (δ 49.3) in tandem with the emergence of a new, resonance (7CPh3δ 47.9, 7Dippδ 48.7) in the 31P{1H} spectra. Complexes 7CPh3 and 7Dipp were resistant to the intramolecular C–H bond activation reported for our previous generation lutetium alkylamido complex LPhLu(CH2SiMe3)(NHCPh3),20 with no decomposition observed after heating to 70 °C for 72 hours.
X-ray diffraction analysis of 7CPh3 and 7Dipp
Minimal quantities of warm (50 °C) heptane were used to produce saturated solutions of compounds 7CPh3 and 7Dipp. The solutions were allowed to cool to ambient temperature before being placed in a –35 °C freezer for 18 hours, yielding X-ray quality crystalline material. The solid-state geometry about the lutetium centres in 7CPh3 and 7Dipp is best described as distorted square pyramidal (τ5 = 0.15 (7CPh3) τ5 = 0.12 (7Dipp)) with the four nitrogenous atoms forming the square base in each structure (Fig. 3 and 4). Two equivalents of 7CPh3 were found within the asymmetric unit; there were no notable structural differences between them.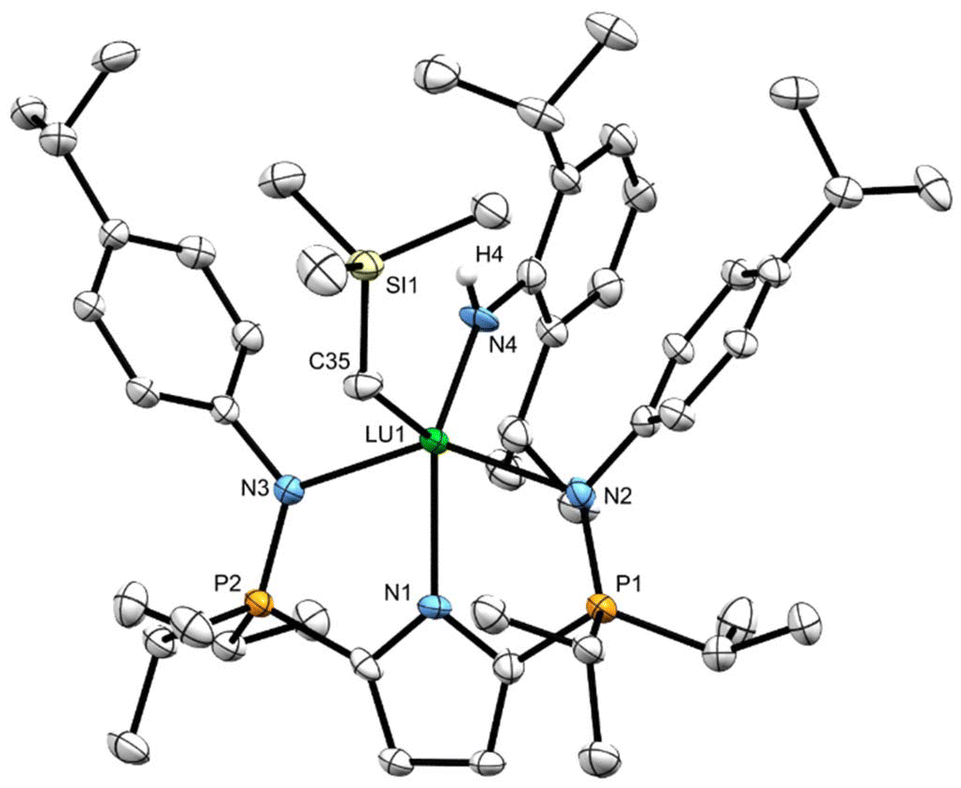 | ||
| Fig. 3 Solid-state structure for 7Dipp with thermal ellipsoids represented at 50% probability. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°) for 7Dipp: Lu1–N1 2.283(5), Lu1–N2 2.352(4), Lu1–N3 2.344(5), Lu1–C35 2.313(5), P1–N2 1.625(5), Lu1–N4 2.195(5), C35–Lu1–N4 110.5(2), N3–Lu1–N2 145.3(2). N1–Lu1–N4 138.4(2). | ||
 | ||
| Fig. 4 Solid-state structure for 7CPh3 with thermal ellipsoids represented at 50% probability. Hydrogen atoms, Pipp isopropyl groups and one equivalent of 7CPh3 have been omitted for clarity. Selected average bond lengths (Å) and angles (°) for 7CPh3: Lu1–N1 2.323(2), Lu–N2 2.342(3), P1–N2 1.617(2), Lu1–C35 2.344(2), Lu1–N4 2.154(2), N3–Lu1–N2 142.9(1), C35–Lu1–N4 105.3(1), N1–Lu1–N4 135.0(1). | ||
The Lu1–C35 bond lengths of complexes 7CPh3 (2.342(3) Å) and 7Dipp (2.313(5) Å) are slightly shorter than the Lu–C distances in LiPrLu(CH2SiMe3)2 (Lu1–C35: 2.360(4) Å, Lu1–C39: 2.366(4) Å). However, these distances lie at the low end of Lu–C bonds for published lutetium alkylamido complexes (2.301(4)–2.40(1) Å).14,15,19,24–29 The phosphinimine P–N bond lengths in both complexes 7CPh3 and 7Dipp are not significantly different than those in 6.
To the best of our knowledge, 4a (TptBu,MeLu(CH3)(NHAd)), 4b (TptBu,MeLu(CH3)(NHtBu)), and LPhLu(NHCPh3)2 represent rare examples of structurally characterized lutetium complexes which possess an amido ligand bearing a non-aromatic substituent.19,20 Complex 7CPh3 exhibits a marginally longer Lu–NHCPh3 bond (2.154(2) Å) than those reported in the literature (2.126(2) Å to 2.144(2) Å for complexes with non-aromatic substituents on nitrogen). Conversely, numerous examples of complexes bearing a monodentate Lu–NHAr (Ar = aromatic) functionality are known, with Lu–NHAr bond lengths ranging from 2.144(3)–2.245(4) Å.14,15,24–28 The Lu1–N4 distance in complex 7Dipp (2.195(5) Å) falls in the middle of this range. Notably, complex 7CPh3 has a shorter Lu1–N4 length of 2.154(2) Å than 7Dipp (2.195(5) Å), despite the large steric presence of the NHCPh3 group (Table 1).
| Parameter | 7CPh3 | 7Dipp | 4b | 8CPh3 | 8Dipp | LPhLu(NHCPh3)2 |
|---|---|---|---|---|---|---|
| Crystal system | Triclinic | Monoclinic | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21 | P21/n |
P |
P21/n |
P |
| R1 (I > 2σ(I)) | 0.0585 | 0.0298 | 0.0211 | 0.0349 | 0.0297 | 0.0311 |
| Selected bond lengths (Å) | ||||||
| RE1–C35 | 2.344(2) | 2.313(5) | 2.362(3) | |||
| RE1–N1 | 2.323(2) | 2.283(5) | 2.327(2) | 2.280(2) | 2.293(4) | |
| P1–N2 | 1.617(2) | 1.625(5) | 1.617(3) | 1.615(2) | 1.620(4) | |
| RE1–N4 | 2.154(2) | 2.195(5) | 2.126(2) | 2.153(2) | 2.176(2) | 2.144(2) |
| Selected bond angles (°) | ||||||
| N2–RE1–N3 | 142.9(1) | 145.3(2) | 143.59(8) | 143.76(7) | 143.7(1) | |
| N1–RE1–C35 | 119.65(2) | 111.1(2) | ||||
| N4–RE1–C35 | 105.3(1) | 110.5(2) | 92.81(9) | |||
| N1–RE1–N4 | 135.0(1) | 138.4(2) | 132.20(8) | 119.68(8) | 107.0(1) | |
Reactivity with 4-dimethylaminopyridine (DMAP)
Reaction of LiPrLu(CH2SiMe3)(NHCPh3), 7CPh3, with one equivalent of DMAP promoted slow conversion (12% of compound 7CPh3 consumed over 68 hours) to a new product which exhibited a single resonance in the 31P{1H} NMR spectrum (δ 45.3). Another 12 hours at ambient temperature did not significantly change the product distribution. Heating this sample to 60 °C for 93 hours produced a dark brown, cloudy solution. This mixture contained a myriad of resonances in the 31P{1H} NMR spectrum, with the major product (δ 45.3) comprising 47% of the phosphorous-containing material.Repeating the experiment at 45 °C resulted in formation of a similar mixture, but this time the resonance at δ 45.3 integrated to only 25% of the material. Increasing the ratio of complex 7CPh3 and DMAP to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 led to slower consumption of 7CPh3 (40% consumed after 4 days), with minimal change to the product mixture.
:2 led to slower consumption of 7CPh3 (40% consumed after 4 days), with minimal change to the product mixture.
Exposure of LiPrLu(CH2SiMe3)(NHDipp), 7Dipp, to one equivalent of DMAP at 45 °C for 22.5 hours resulted in 20% consumption of 7Dipp and formation of one major product (δ 46.4) according to the 31P{1H} NMR spectrum. Further heating (83.5 hours) resulted in a murky brown solution. In this case, the 31P{1H} NMR spectrum revealed that 60% of compound 7Dipp had been consumed and the new resonance appearing at δ 46.4 accounted for 25% of the phosphorous-containing material. In addition, a second substantial resonance (21% by integration) appeared (δ 44.2), in conjunction with a variety of by-products.
Complex 7Dipp was also reacted with two equivalents of DMAP at 45 °C. After 18 hours the 31P{1H} NMR spectrum revealed 33% of complex 7Dipp had been consumed, along with concomitant formation of one major product (δ 46.4) accounting for 25% of the phosphorous-containing material. Heating this sample for an additional 145.5 hours resulted in almost complete consumption of 7Dipp, and similar to the equimolar reaction, the resonance at δ 46.4 remained the dominant product (43% of all signals).
Many commonalities were noted when comparing the 1H NMR spectra for the reactions of 7Dipp and 7CPh3 across the various conditions. The unaltered chemical shifts attributed to free DMAP (δ 8.45, 6.07, 2.21) suggested that the Lewis base did not coordinate to the metal centres. Approximately one equivalent of SiMe4 was produced (δ 0.00) and there was no evidence for the retention of any –CH2SiMe3 functionalities previously attached to the metal centre (complex 6). The observation of only one P–(CH(CH3)2)2 resonance, coupled with the lone signal in the 31P{1H} spectra, implies the major product formed in these reactions possesses C2v symmetry. Furthermore, a singlet (δ 4.38 and 2.26) with no cross-peaks in 1H–1H COSY experiments grew in each of the reaction mixtures. Diagnostic aromatic resonances pertaining to the NHCPh3 ligand were retained throughout the reaction of complex 7CPh3 and DMAP, eventually doubling in integration with respect to DMAP. Similarly, retention and growth of the isopropyl doublet and septet signals pertaining to the NHDipp ligand were observed in the reaction between complex 7Dipp and DMAP. The combination of this evidence led us to believe that the major products of the reaction between complexes 7 and DMAP were the respective bisamide species, LiPrLu(NHCPh3)2 (8CPh3) and LiPrLu(NHDipp)2 (8Dipp). While attempts at isolating these complexes directly from the crude reaction mixtures were unsuccessful, independent synthesis of both 8CPh3 and 8Dipp was achieved by addition of two equivalents of NH2CPh3 or NH2Dipp, respectively, to stirring toluene solutions of LiPrLu(CH2SiMe3)2. Heat (70 °C), and additional time (69 hours) was required to prepare 8CPh3, compared to 8Dipp, which was generated in 24 hours at ambient temperature. Characterization of complexes 8 revealed 1H and 31P{1H} NMR spectra consistent with the major products formed in the reactions of DMAP with complexes 7CPh3 and 7Dipp. In order to confirm that 8CPh3 and 8Dipp were in fact stable products, rather than long lived intermediates prone to decomposition, a sample of LiPrLu(NHDipp)2 was exposed to 2 equivalents of DMAP at 60 °C for 48 hours, during which no reaction was observed.
In 2004 Piers reported the thermal decomposition of the scandium alkylamido complex LSc(NHtBu)(CH3) (L = DippNC(CH3)CHC(CH3)NDipp).30 Upon being heated to 100 °C LSc(NHtBu)(CH3) underwent a disproportionation reaction yielding half an equivalent of both LSc(NHtBu)2 and LSc(CH3)2, the latter of which decomposed upon formation. A Lewis base induced disproportionation route is proposed as a logical pathway for our systems, with the decomposition of LiPrLu(CH2SiMe3)2 being supported by the elimination of SiMe4. Furthermore, heating (60 °C) a pure sample of LiPrLu(CH2SiMe3)2 (6) and DMAP resulted in decomposition and production of myriad products in the 31P{1H} NMR spectrum which closely matched the product distribution produced when DMAP was added to 7CPh3 or 7Dipp (Scheme 1).
Upon comparison of complexes 7CPh3 and 7Dipp with L1Sc(NHDipp)(CH3), the alkylamido precursor to Chen's imido 1 (vide supra), few substantial structural differences are immediately apparent. The planar arrangement of the three L1 nitrogen donors matches that observed in complexes 7, as does the similar Namido–RE–Calkyl bond angle of 103.7(2)° (cf. 105.3(1)° and 110.5(2)° in 7CPh3 and 7Dipp, respectively).8 Likewise, both 7Dipp and L1Sc(NHDipp)(CH3) bear a sterically demanding Dipp group on the amido nitrogen. While the geometries at the metal centres are similar, it should be noted that Chen's complex features the smaller rare earth element, scandium. In addition, the metal in L1Sc(NHDipp)(CH3) bears a –CH3, rather than the larger –CH2SiMe3.
Although Anwander's TptBu,Me-ligated system supports imidos of different rare earth metals, they too were generated by the elimination of methane. Unfortunately, the substantial differences between the facially capping TptBu,Me ligand and our pincer precludes direct structural comparison.15 With that said, it is worth noting that while Anwander's complexes contain the less sterically demanding 2,6-(CH3)2C6H3 or 3,5-(CF3)2C6H3 groups on the imido nitrogen, aromatic groups were still required.
Perhaps the best comparison to our alkylamido complexes is the phosphazene-based L2Sc(CH2SiMe3)(NHDipp) (L2 = N(Ph2P = NPh)2), reported by Cui in 2013 (Scheme 2).14 In that case, ligand phosphinimine donors, a metal–CH2SiMe3 moiety and an NDipp group were common with 7Dipp. Though the solid-state structure was not obtained for alkylamido L2Sc(CH2SiMe3)(NHDipp), several striking reactivity patterns were observed. Like complexes 7CPh3 and 7Dipp, heat led to mixtures of the corresponding bisamido and dialkyl species, the latter of which decomposed with concomitant generation of the proteo ligand HL2. Nonetheless, addition of DMAP at ambient temperature afforded the terminal scandium imido L2Sc = NDipp, 9, though two equivalents of DMAP were deemed necessary. Of particular interest is the fact that when Cui and co-workers attempted the analogous chemistry with lutetium and yttrium alkylamido complexes, no conversion to the corresponding imido species was observed, even when excess DMAP was added.14 Instead, bisamide species, as well as a mixture of intractable products, formed.
 | ||
| Scheme 2 Terminal imido scandium complex 9 reported by Cui and coworkers. | ||
X-ray diffraction analysis of 8CPh3 and 8Dipp
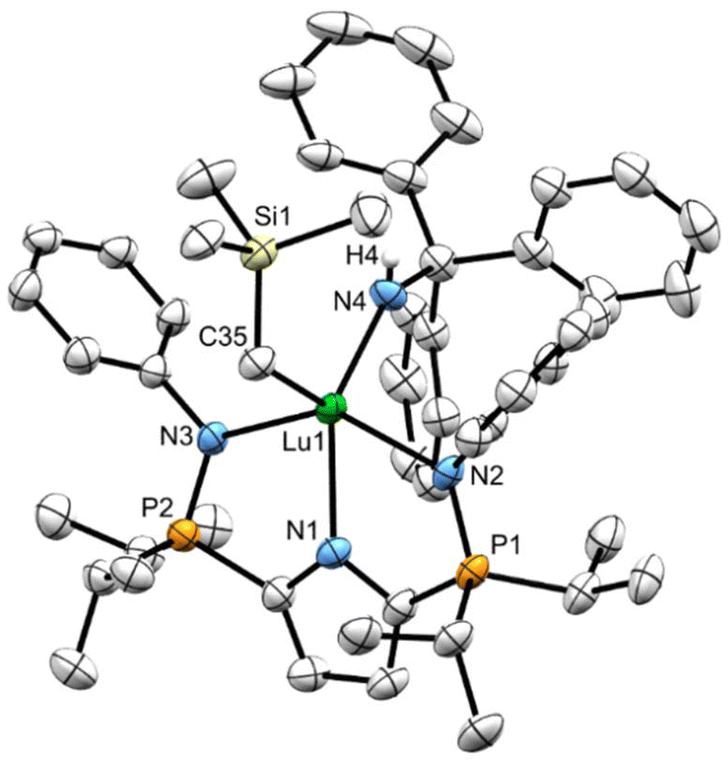
X-ray quality crystals of independently synthesized 8CPh3 and 8Dipp were grown at ambient temperature from saturated warm (50 °C) heptane solutions. Like complexes 7CPh3 and 7Dipp, the geometry at lutetium in complex 8CPh3 (Fig. 5) is best described as distorted square pyramidal (δ5 = 0.19). The square pyramid in complex 8Dipp (Fig. 6), however, is considerably more distorted (δ5 = 0.38). This discrepancy in geometry, and the need for heat when preparing 8CPh3, is attributed to the large steric influence of the NHCPh3 ligand. Complex 8CPh3 possesses a Lu1–N5 bond length of 2.164(3) Å, which is amongst the longest reported for a Lu–NHR species (when R ≠ aromatic group).31 In line with what was observed for complexes 7CPh3 and 7Dipp, the average Lu–NHDipp bond length in complex 8Dipp (2.191(2) Å) is longer than that in 8CPh3 (2.159(2) Å), despite the steric pressure of the CPh3 substituent (Table 1).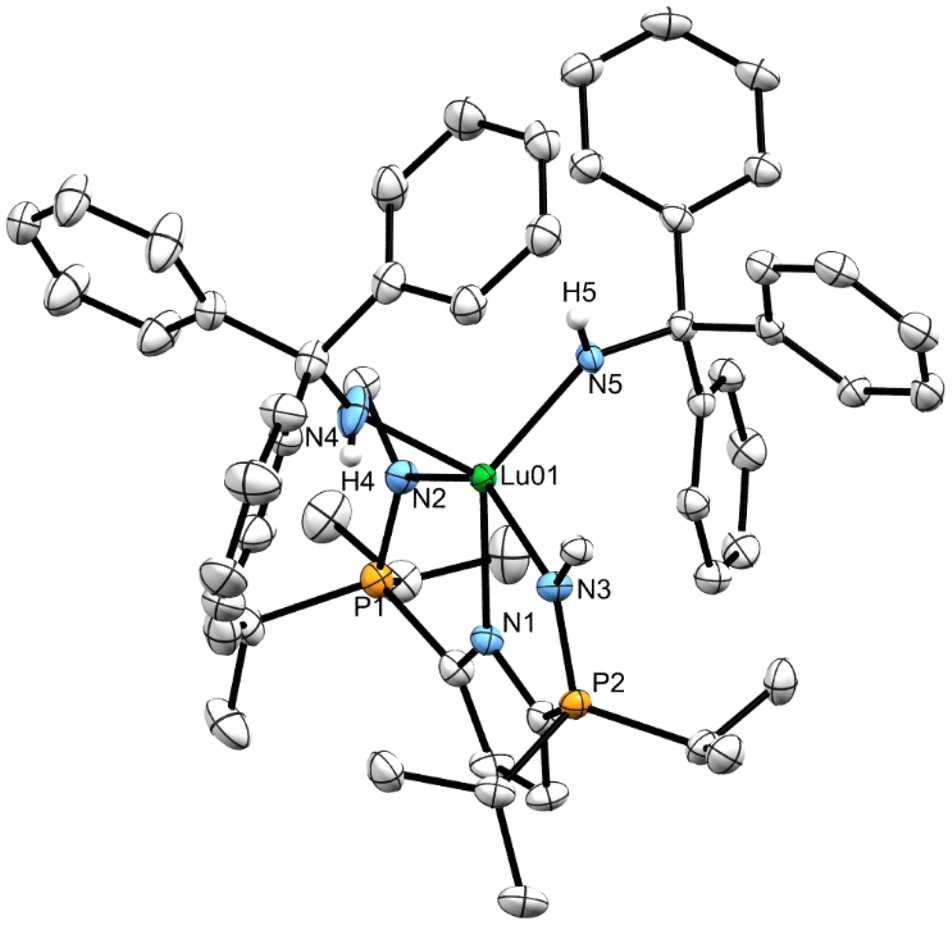 | ||
| Fig. 5 Solid-state structure for complex 8CPh3 with thermal ellipsoids represented at 50% probability.Hydrogen atoms and Pipp groups (except N–Cipso carbon atoms) omitted for clarity. Selected bond lengths (Å) and angles (°) for 8CPh3: Lu1–N1 2.327(2), Lu1–N2 2.375(2), Lu1–N4 2.153(2), Lu1–N5 2.164(3), P1–N2 1.617(3), N5–Lu1–N4 103.58(9), N3–Lu1–N2 143.59(8), N1–Lu1–N4 132.20(8), N1–Lu1–N5 124.01(9). | ||
 | ||
| Fig. 6 Solid-state structure for complex 8Dipp with thermal ellipsoids represented at 50% probability. Hydrogen atoms and Pipp groups (except N–Cipso carbon atoms) omitted for clarity. Selected bond lengths (Å) and angles (°) for 8Dipp: Lu1–N1 2.280(2), Lu1–N2 2.327(2), Lu1–N4 2.176(2), Lu1–N5 2.206(2), P1–N2 1.615(2), N2–Lu1–N3 143.76(7), N1–Lu1–N4 119.68(8), N1–Lu1–N5 119.43(7), N4–Lu1–N5 120.78(8). | ||
Incorporation of internal Lewis bases
In 2011 Chen demonstrated that an internal Lewis base could stabilize a terminal scandium imido (vide supra, L2Sc(NDipp), 2).10 Taking inspiration from those findings, we sought to incorporate internal Lewis bases into our pincer scaffold. Addition of three equivalents of NaN3 to 4,6-dimethyl-2-methylsulfonylpyrimidine in DMF at 100 °C for 3 hours proved an efficient method for producing 2-azido-4,6-dimethylpyrimidine (10a, Scheme 3). The valence tautomerization observed in the 1H NMR spectrum between compound 10a and the dominant tetrazole isomer, 5,7-dimethylpyrazolo[1,5a]pyrimidine (10b) is consistent with previous reports of this molecule.32,33 | ||
| Scheme 3 Synthesis of complex 12. | ||
Formation of HLPm (11) was accomplished by reaction of two equivalents of 10b with 2,5-bis(diisopropylphosphino)-N–H-pyrrole in toluene (Scheme 3). Likely as a consequence of the equilibrium between 10a and 10b, this reaction required additional time compared to the formation of HLiPr (18 hours vs. 1 hour). Removal of volatiles in vacuo afforded compound 11 as a dark beige powder. The 31P{1H} NMR spectrum of the solid revealed one new resonance (δ 29.6). It is worth noting that the methyl substituents of the pyrimidine functionality appear as a singlet in the 1H NMR spectrum (δ 2.35), indicating that free rotation around the NP![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CPm bond occurs on the NMR timescale. A downfield N–H signal (δ 13.2) in the 1H NMR spectrum integrated for one proton and gave rise to no crosspeaks in 1H–1H COSY and 1H–13C{1H} HSQC experiments. This resonance appears substantially further downfield than previously reported N–H chemical shifts for our related protio ligands (δ 10.40–12.53),21,22,34–39 possibly due to hydrogen bonding between N–H and the NPm atoms.
N–CPm bond occurs on the NMR timescale. A downfield N–H signal (δ 13.2) in the 1H NMR spectrum integrated for one proton and gave rise to no crosspeaks in 1H–1H COSY and 1H–13C{1H} HSQC experiments. This resonance appears substantially further downfield than previously reported N–H chemical shifts for our related protio ligands (δ 10.40–12.53),21,22,34–39 possibly due to hydrogen bonding between N–H and the NPm atoms.
X-ray diffraction analysis of compound 11
A sample of compound 11 was dissolved in a warm (60 °C) 5:3 mixture of heptane and toluene, respectively, and allowed to cool to ambient temperature before being placed in a −35 °C freezer for 18 hours, yielding X-ray quality crystalline material. The solid-state structure of compound 11 (Fig. 7), which crystallized in the monoclinic P21/n space group, exhibited pyrimidines that lie out of the plane of the pyrrole ring (C1–P1–N2–C23: −57.20°, C4–P2–N3–C17: 173.89°). The phosphinimine (P1–N2 and P2–N3) distances agree well with previously reported compounds.
 | ||
| Fig. 7 Solid-state structure for compound 11 with thermal ellipsoids represented at 50% probability. Hydrogen atoms (except H1) omitted for clarity. selected bond lengths (Å) and angles (°) for compound 11: N1–N4: 2.859(2), P1–N1: 1.588(1), P2–N3: 1.609(1), P1–N2–C23: 129.3(1), P2–N3–C17: 119.7(1), C23–N4–N1: 109.6(1). | ||
Rare earth complexes of ligand 11
To probe the ability of ligand 11 to support well-defined rare earth complexes, a toluene solution containing one equivalent of Lu(CH2SiMe3)3THF2 was added dropwise to a stirring solution of the compound. Analogous to the preparation of LiPrSc(CH2SiMe3)2 and LiPrLu(CH2SiMe3)2 (5 and 6, respectively), the resulting solution was allowed to stir for one hour at ambient temperature whereupon all volatiles were removed in vacuo, leaving LPmLu(CH2SiMe3)2 (12) as a slightly impure yellow powder (Scheme 3).Evidence for the formation of complex 12 includes the emergence of a new peak in the 31P{1H} NMR spectrum (δ 49.8) along with concomitant disappearance of the phosphorous resonance attributed to 11 (δ 29.6). The pyrimidine-CH3 groups are no longer chemically equivalent (δ 2.67–1.95) in the 1H NMR spectrum suggesting slow rotation about the NPN–Cipso bond on the 1H NMR timescale. A single set of resonances attributed to the lutetium bound alkyl substituents was observed (CH2Si(CH3)3: δ 0.11, CH2Si(CH3)3: δ −1.04), indicative of a Cs or C2v symmetric product.
Unfortunately, complex 12 spontaneously decomposed in solution at ambient temperature. Production of a small quantity of SiMe4 (δ 0.00) was observed in the 1H NMR spectrum after 5 minutes. After 24 hours complex 12 had been 35% consumed. Correspondingly, one third of an equivalent of SiMe4 was observed in the 1H NMR spectrum at that point. Previously we prepared a dialkyl lutetium complex supported by a pincer ligand bearing unsubstituted pyrimidine groups at nitrogen. That species decomposed by alkyl migration from lutetium to pyrimidine, dearomatizing the ring.39 A similar pathway was reported by Kiplinger in 2006.40 However, such alkyl migration does not liberate SiMe4, thereby ruling out that mode of decomposition for complex 12. Unfortunately, repeated attempts to grow X-ray quality crystals of LPmLu(CH2SiMe3)2, even at low temperature, were unsuccessful. Similarly, the instability of complex 12 precluded both subsequent reaction chemistry and satisfactory combustion analysis.
In an effort to ascertain whether the 4,6-dimethylpyrimidine groups in ligand 11 are likely to stabilize a lutetium imido species by coordinating to the metal, we targeted a dichloride complex expecting a more thermally robust product. To this end, ligand 11 was treated with a stoichiometric quantity of NaH, following protocols established for generating NaLiPr.21 Subsequent reaction between the sodiated ligand NaLPm and LuCl3(THF)3 in THF solution afforded the dichloride complex LPmLuCl2 (13) as a pale yellow solid. Complex 13 displays C2v symmetry in solution at ambient temperature, as indicated by one PCH(CH3)2 methine resonance in the 1H NMR spectrum and a single 31P peak (δ 52.2). The pyrimidine CH3 substituents appear as two distinct, signals each integrating to 6H (δ 2.44, 2.05), implying pyrimidine coordination to the metal centre.
X-ray quality crystals of complex 13 were grown from a saturated toluene solution cooled to −35 °C. The solid-state structure determined from X-ray diffraction experiments confirmed that both pyrimidines coordinate to lutetium. Notably, all five nitrogen donors lie approximately in the same plane (P1–N2–C17–N4 torsion angle = 174.3(2)°, P2–N3–C23–N6 torsion angle = 171.46(2)°; Fig. 8), leading to distorted pentagonal bipyramidal geometry at Lu. The two chlorides, which occupy the apical sites, give rise to a Cl–Lu–Cl angle of 162.48(3)°, which, as expected, is substantially greater than the Namido–Lu–Calkyl angles in the mixed alkylamido complexes 7 (105.3(1), 110.5(2)°) and the Namido–Lu–Namido angles in bisamides 8 (124.01(9), 120.78(8)°).
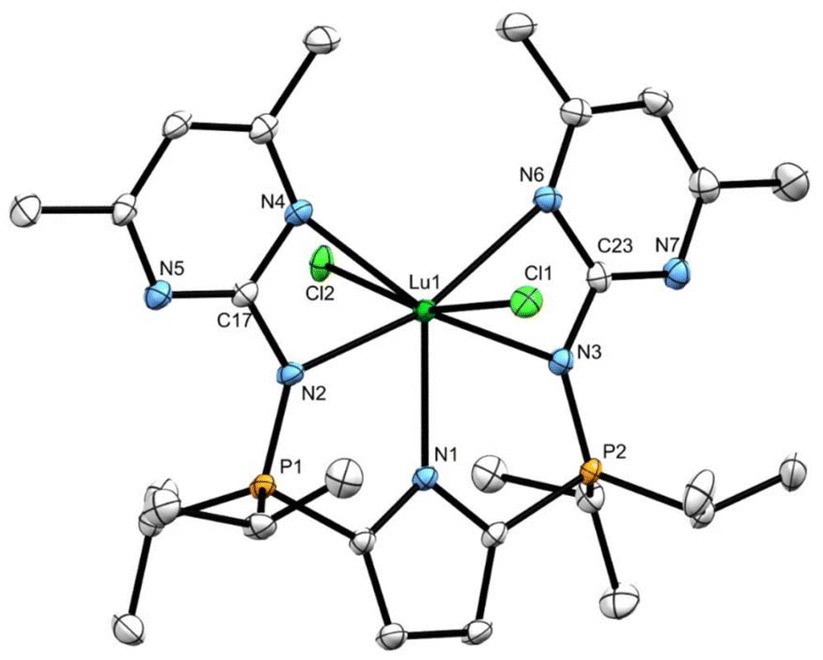 | ||
| Fig. 8 Solid-state structure for compound 13 with thermal ellipsoids represented at 50% probability. Hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°) for compound 13: Lu1–N1: 2.324(2), Lu1–N2: 2.318(2), Lu1–N4: 2.546(2), Lu1–Cl2: 2.5808(8). N2–Lu1–N4: 54.59(6), Cl1–Lu1–Cl2: 162.48(3). | ||
Conclusions
A series of potential precursors for terminal lutetium imido complexes, LiPrLu(CH2SiMe3)(NHCPh3) (7CPh3) and LiPrLu(CH2SiMe3)(NHDipp) (7Dipp) were prepared. Unlike our previously reported lutetium alkylamido complex LPhLu(CH2SiMe3)(NHCPh3), which bears phenyl substituents on the ligand's phosphorus atoms,20 no spontaneous C–H bond activation was observed in solution. Upon addition of DMAP to either 7CPh3 or 7Dipp a disproportionation reaction was induced, yielding half an equivalent of LiPrLu(CH2SiMe3)2 and the corresponding bis-amide species LiPrLu(NHCPh3)2 or LiPrLu(NHDipp)2, respectively. The generated LiPrLu(CH2SiMe3)2 rapidly decomposed in the presence of DMAP. Given the paucity of systems similar to ours it is difficult to draw meaningful conclusions regarding what factors dictate imido formation vs. ligand redistribution, though greater success has been realized with sterically bulky aromatic substituents on the imido nitrogen and with the smaller metal scandium.Installation of 4,6-dimethylpyrimidine groups on the iminophosphorane nitrogen atoms of the pincer framework afforded HLPm, (11), a ligand with the potential to stabilize rare earth imido species by coordination of the pyrimidine groups to the metal centre. While addition of Lu(CH2SiMe3)3THF2 to compound 11 generated the expected dialkyl complex LPmLu(CH2SiMe3)2 (12), rapid decomposition at ambient temperature rendered it impossible to prepare desired alkylamido complexes of the form LPmLu(CH2SiMe3)(NHR). However, preliminary experiments indicate that the rare earth complex LPmLuCl2 is accessible via a salt metathesis route, and notably, both pyrimidine groups coordinate to lutetium in the solution and solid states. The possibility of using the amide salts ([(THF)LiNHCPh3]2 or [(THF)LiNHDipp]2), to ultimately access alkylamido species that may serve as imido precursors, will be the focus of future studies.
Experimental
General procedures
Manipulation of air- and moisture-sensitive materials and reagents was carried out under an argon atmosphere using vacuum line techniques or in an MBraun glove box. Solvents used for air-sensitive materials were purified using an MBraun solvent purification system (SPS), stored in PTFE-sealed glass vessels over “titanocene” (pentane, benzene, and toluene), and freshly distilled at the time of use. Benzene-d6 was dried over sodium benzophenone ketyl, de-gassed via three freeze–pump–thaw cycles, distilled in vacuo and stored over 4 Å molecular sieves in glass bombs under argon. Unless noted, all NMR spectra were recorded at ambient temperature with a Bruker Avance III NMR spectrometer (700.44 MHz for 1H, 176.13 MHz for 13C, and 283.54 MHz for 31P). Chemical shifts are reported in parts per million relative to the external standards SiMe4 (1H, 13C) and 85% H3PO4 (31P); residual H-containing species in C6D6 (δ 7.16 (1H), δ 128.39 (13C)) were used as internal references. Assignments were aided by the use of 13C{1H} DEPT-135, 1H–13C{1H} HSQC, 1H–13C{1H} HMBC and 1H–1H COSY experiments (s = singlet, d = doublet, t = triplet, q = quartet, sp = septet, m = multiplet, br = broad, ov = overlapping signals). Elemental analyses were performed using an Elementar Vario Micro-cube instrument. The reagents Lu(CH2SiMe3)3(THF)2,41 Sc(CH2SiMe3)3(THF)2,42 and 2,5-[iPr2P = N(4-iPrC6H4)]2NH(C4H2)(HLiPr),21 were prepared according to literature methods. The compound 2,6-diisopropylanaline was purchased from Sigma Aldrich and distilled under reduced pressure into a bomb equipped with a Kontes valve before using. Compound 10 was synthesized according to literature procedures with additional details provided in the text above.32,33 All other reagents were purchased from Sigma Aldrich and used without further purification. Unless otherwise specified, reported yields correspond to those obtained for analytically pure samples. When additional purification was required to generate analytically pure compounds for combustion analysis, both the crude and analytical yields are included. In such cases, the reported crude yield corresponds to material that was utilized for successive synthetic steps and was >98% pure as indicated by multinuclear NMR spectroscopy. (N.B. NMR spectra displayed in the ESI† were obtained using these “crude” samples.) It should also be noted that when reported, analytical yields are generally artificially low, as excess crystals grown for X-ray diffraction experiments are used for this purpose. Those recrystallizations were not carried out under conditions that maximize yield, but rather, were optimized for the growth of X-ray quality crystals.X-Ray crystallography
Crystals grown for X-ray diffraction analysis were coated in Paratone oil and placed on a glass slide. Close visual inspection and selection of the crystals was aided by either a standard microscope or polarizing light microscope. The desired crystal chosen for diffraction analysis was placed on a MiTeGen Dual Thickness MicroMount attached to a goniometer head. The crystal was centred on a Rigaku SuperNova diffractometer equipped with a Dectris Pilatus 3R 200K-A detector, Oxford CryoStream 800 cooling system, molybdenum (NOVA) radiation source (Kα = 0.71073 Å), and copper (MOVA) radiation source (Kα = 1.5406 Å). Experiments were performed at 100 K to reduce thermal motion of the atoms. CrysAlisPro software was utilized to determine unit cell parameters and SHELXTL software was utilized using least squares methodology for refinement.Synthesis of LiPrSc(CH2SiMe3)2 (5)
A 100 mL round-bottomed flask was charged with a sample of HLiPr (0.0913 g, 0.161 mmol). Toluene (10 mL) was transferred into this flask via vacuum distillation, yielding a clear, light brown solution. Sc(CH2SiMe3)3(THF)2 (0.0724 g, 0.161 mmol), was added to a 50 mL round-bottomed flask, and dissolved in toluene (10 mL). The Sc(CH2SiMe3)3(THF)2 solution was added dropwise to the stirring HLiPr solution at ambient temperature over 3 minutes. The resulting clear, light brown solution was left to stir at ambient temperature for 1 hour upon which all volatiles were removed in vacuo. The reaction contraption was brought into an inert atmosphere glove box whereupon heptane (10 mL) was added to the light brown solid. The mixture was stirred and heated (50 °C) leaving behind a small amount of a dark, oily solid. This mixture was filtered through a bed of Celite which produced a clear, yellow solution. Removal of the heptane in vacuo liberated the desired product as a light, pale yellow solid (0.113 g, 90.0% (crude)). Facile preparation of analytically pure material was achieved through recrystallization from a minimal amount of warm (50 °C) heptane (0.0521 g, 41.4%). 1H NMR (benzene-d6): δ 7.50 (dd, 3JH–H = 8.4 Hz, 4JH–P = 2.1, 4H, o-Pipp–H), 7.23 (d, 3JH–H = 8.4 Hz, 4H, m-Pipp–H), 6.51 (d, 3JH–P = 2.1 Hz, 2H, pyrrole–H), 2.80 (sp, 3JH–H = 7.0 Hz, 2H, Pipp–(CH(CH3)2)), 2.09 (m, 3JH–H = 6.9 Hz, 2JH–P = 2.7 Hz, 4H, P–(CH(CH3)2))), 1.24 (d, 3JH–H = 7.0 Hz, 12H, Pipp–(CH(CH3)2)), 0.93 (dd, 3JH–P = 32.1 Hz, 3JH–H = 6.9 Hz, 12H, P–(CH(CH3)(CH3))), 0.88 (dd, 3JH–P = 31.4 Hz, 3JH–H = 6.9 Hz, 12H, P–(CH(CH3)(CH3)), 0.31 (s, 4H, Sc–CH2SiMe3), 0.16 (s, 18H, CH2Si(CH3)3). 13C{1H} NMR (benzene-d6): δ 145.54 (s, ipso-Pipp), 144.57 (s, ipso-Pipp), 129.67 (s, o-Pipp C–H), 128.41 (d, 1JC–P = 24.1, ipso-pyrrole) 127.73 (s, m-Pipp C–H), 115.90 (dd, 2JC–P = 24.1 Hz, 3JC–P = 10.1 Hz, pyrrole C–H), 40.00 (br s, Sc–CH2), 34.38 (s, Pipp–CH(CH3)2), 26.30 (d, 1JC–P = 54.0 Hz, P–CH(CH3)(CH3)), 24.79 (s, Pipp–CH(CH3)2), 16.60 (s, P–(CH(CH3)(CH3))), 16.13 (s, P–(CH(CH3)(CH3))), 4.65 (s, SiMe3). 31P{1H} NMR (benzene-d6): δ 48.0. Anal calcd (%) for C42H74ScN3P2Si2 C, 64.33; H, 9.51; N, 5.36. Found: C, 64.00, H, 8.79; N, 5.68.Synthesis of LiPrLu(CH2SiMe3)2 (6)
A 100 mL round-bottomed flask was charged with a sample of HLiPr (0.1729 g, 0.3040 mmol). Toluene (10 mL) was transferred to this flask via vacuum distillation, yielding a clear, light brown solution. Lu(CH2SiMe3)3(THF)2 (0.1766 g, 0.3040 mmol), was added to a 50 mL round-bottomed flask, and was dissolved in toluene (10 mL). The Lu(CH2SiMe3)3(THF)2 solution was added dropwise to the stirring solution at ambient temperature over 3 minutes. The resulting clear, light brown solution was left to stir at ambient temperature for 1 hour upon which all volatiles were removed in vacuo. The reaction contraption was brought into an inert atmosphere glove box whereupon heptane (10 mL) was added to the light brown solid. The mixture was stirred and heated (50 °C) leaving behind a small amount of a dark, oily solid. This mixture was filtered through a bed of Celite which produced a clear, yellow solution. Removal of the heptane in vacuo liberated the desired product as a pale yellow solid (0.2557 g, 92.01%). Analytically pure material was obtained by recrystallization of the crude sample from a minimal amount of warm (50 °C) heptane (0.1218 g, 43.83%). 1H NMR (benzene-d6): δ 7.43 (dd, 3JH–H = 8.1 Hz, 4JH–P = 1.8 Hz, 4H, o-Pipp–H), 7.22 (d, 3JH–H = 8.1 Hz, 4H, m-Pipp–H), 6.50 (d, 3JH–P = 2.1 Hz, 2H, pyrrole–H), 2.79 (sp, 3JH–H = 6.6 Hz, 2H, Pipp–(CH(CH3)2)), 2.04 (m, 3JH–H = 7.0 Hz, 1JH–P = 2.4 Hz, 4H, P–(CH(CH3)2)), 1.22 (d, 3JH–H = 6.6 Hz, 12H, Pipp–(CH(CH3)2)), 0.96 (dd, 3JH–H = 7.0 Hz, 2JH–P = 16.2 Hz, 12H, P–(CH(CH3)(CH3))), 0.85 (dd, 3JH–H = 7.0 Hz, 2JH–P = 15.5 Hz, 12H, P–(CH(CH3)(CH3))), 0.21 (s, 18H, SiMe3), −0.42 (s, 4H, Lu–CH2SiMe3). 13C{1H} NMR (benzene-d6): δ 144.32 (dd, 1JC–H = 98.6 Hz, 4JC–H = 24.3 Hz, ipso-pyrrole), 129.15 (d, 3JC–P = 13.2 Hz, o-Pipp C–H), 129.11 (d, 2JC–P = 26.4 Hz, ipso-Pipp), 127.89 (s, m-Pipp C–H), 127.40 (s, ipso-Pipp), 116.51 (dd, 2JC–P = 98.6 Hz, 3JC–P = 40.2 Hz, pyrrole C–H), 41.30 (s, Lu–CH2), 34.32 (s, Pipp–(CH(CH3)2)), 26.42 (d, 1JC–P = 54.0 Hz, P–(CH(CH3)2)), 24.76 (s, Pipp–(CH(CH3)2)), 16.51 (s, P–(CH(CH3)(CH3))), 15.99 (s, P–(CH(CH3)(CH3))), 5.19 (s, SiMe3) 31P{1H} NMR (benzene-d6): δ 49.3. Anal. calcd (%) for C42H74LuN3P2Si2 C, 55.18; H, 8.16; N, 4.60. Found: C, 55.01, H, 8.26; N, 4.97.Synthesis of LiPrLu(CH2SiMe3)(NHCPh3) (7CPh3)
A sample of 6 (0.1033 g, 0.1130 mmol) was weighed into a 20 mL scintillation vial containing a stir bar. The powder was dissolved in 6 mL of toluene, yielding a clear yellow solution. One equivalent of NH2CPh3 (0.0286 g, 0.110 mmol) was added into a 5 mL disposable vial and was dissolved using 2 mL of toluene. The amine solution was added dropwise over 3 minutes to the stirring yellow solution. After stirring at ambient temperature for 1 hour the reaction mixture was filtered through a bed of Celite, eliminating insoluble contaminants. All volatiles were removed in vacuo yielding the desired complex as a pale, off-white solid powder (0.103 g, 86.4% yield). X-ray quality crystals were grown at ambient temperature from dissolving the crude powder in a minimal amount of warm heptane (50 °C) (0.0611 g, 51.04% yield). 1H NMR (benzene-d6): δ 7.53 (m, 3JH–H = 3.9 Hz, 6H, o-Ph3), 7.08 (ov m, 3JH–H = 3.9 Hz, 13H, m/p-Ph3, Pipp–H), 6.96 (d, 3JH–H = 6.3 Hz, 4H, Pipp–H), 6.54 (d, 3JH–P = 1.5 Hz, 2H, pyrrole–H), 2.85, (sp, 3JH–H = 6.6 Hz, 2H, Pipp–(CH(CH3)2)), 2.43 (s, 1H, N–H), 2.16 (dsp, 1JH–P = 9.6 Hz, 3JH–H = 6.9 Hz, 2H, P–(CH(CH3)2)), 1.75 (dsp, 1JH–P = 9.6 Hz, 3JH–H = 6.9 Hz, 2H, P–(CH(CH3)2)), 1.32 (d, 3JH–H = 6.6 Hz, 6H, Pipp–(CH(CH3)(CH3))), 1.30 (d, 3JH–H = 6.6 Hz, 6H, Pipp–(CH(CH3)(CH3))), 0.98 (dd, 2JP–H = 7.2 Hz, 3JH–H = 6.9 Hz, 6H, P–(CH(CH3)(CH3))), 0.85 (dd, 3JP–H = 7.2 Hz, 3JH–H = 6.9 Hz, 6H, P–(CH(CH3)(CH3))), 0.77 (d, 2JP–H = 6.9 Hz, 3JH–H = 6.9 Hz, 6H, P–(CH(CH3)(CH3))), 0.61 (dd, 3JP–H = 7.2 Hz, 3JH–H = 6.9 Hz, 6H, P–(CH(CH3)(CH3))), 0.05 (s, 9H, SiMe3), −0.37 (s, 2H, Lu–CH2SiMe3). 13C{1H} NMR (benzene-d6): δ 154.67 (s, ipso-CPh3 C), 144.38 (d, 1JC–P = 88.5 Hz, ipso-pyrrole), 129.60 (s, ipso-Pipp) 129.51 (s, Pipp C–H), 128.68 (s, Pipp C–H) 128.24 (ipso-Pipp) 127.75 (s, CPh3 C–H), 127.69 (s, CPh3 C–H), 125.91 (s, CPh3 C–H), 116.63 (dd, 2JP–C = 24.5 Hz, 3JP–C = 9.6 Hz, pyrrole C–H), 75.74 (s, CPh3), 34.39 (s, Lu–CH2), 32.60 (s, Pipp–(CH(CH3)2)), 26.83 (d, 1JP–C = 54.5 Hz, P–(CH(CH3)2)), 26.33 (d, 1JP–C = 54.5 Hz, P–(CH(CH3)2)), 25.02 (s, Pipp–(CH(CH3)(CH3))), 24.89 (s, Pipp–(CH(CH3)(CH3))), 16.50 (ov s, P–(CH(CH3)(CH3))) and P–(CH(CH3)(CH3)), 16.49 (s, P–(CH(CH3)(CH3))), 16.25 (s, P–(CH(CH3)(CH3))), 4.61 (s, SiMe3). 31P{1H} NMR (benzene-d6): δ 47.8. Anal. calcd (%) for C59H79LuN4P2Si C, 63.08; H, 7.34; N, 5.16. Found: C, 62.86, H, 7.12; N, 5.25.LiPrLu(CH2SiMe3)(NHDipp) (7Dipp)
A sample of 6 (0.1193 g, 0.1305 mmol) was weighed into a 20 mL scintillation vial containing a stir bar. The powder was dissolved in 6 mL of toluene, yielding a clear yellow solution. NH2Dipp (0.0241 g, 0.136 mmol) was weighed into a syringe and added to a vial containing 5 mL of toluene. This amine solution was then added dropwise to the stirring solution of LiPrLu(CH2SiMe3)2 over 5 minutes. The resulting golden yellow solution was left to stir at ambient temperature for 18 hours whereupon all volatiles were removed in vacuo. The resulting pale yellow solid was reconstituted in a minimal amount of a warm (50 °C) 1:1 heptane:benzene mixture and recrystallized at −35 °C (0.0722 g, 55.1% yield). 1H NMR (benzene-d6): δ 7.33 (d, 3JH–H = 7.5 Hz, 2H, m-Dipp C–H), 7.22 (dd, 3JH–H = 8.1 Hz, 4JH–P = 1.8 Hz, 4H, o-Pipp C–H), 7.01 (d, 3JH–H = 8.1 Hz, 4H, m-Pipp C–H), 6.93 (t, 3JH–H = 7.5 Hz, 1H, p-Dipp C–H), 6.52 (d, 3JH–P = 2.1 Hz, 2H, pyrrole C–H), 4.14 (s, 1H, N–H), 3.39 (sp, 3JH–H = 6.6 Hz, 2H, Dipp–(CH(CH3)2)2), 2.73 (sp, 3JH–H = 6.9 Hz, 2H, Pipp–(CH(CH3)2)), 2.26 (m, 3JH–H = 6.9 Hz, 2JH–P = 6.6 Hz, 2H, P–(CH(CH3)2)), 1.93 (m, 3JH–H = 6.9 Hz, 2JH–P = 6.6 Hz, 2H, P–(CH(CH3)2)), 1.35 (d, 3JH–H = 6.6 Hz, 12H, Dipp–(CH(CH3)2)2), 1.20 (ov d, 3JH–H = 6.9 Hz, 6H, Pipp–(CH(CH3)(CH3))), 1.19 (ov d, 3JH–H = 6.9 Hz, 6H, Pipp–(CH(CH3)(CH3))), 1.07 (dd, 3JH–H = 6.9 Hz, 3JH–P = 6.9 Hz, 6H, P–(CH(CH3)(CH3))), 0.94 (dd, 3JH–P = 7.2 Hz, 3JH–H = 6.9 Hz, 6H, P–(CH(CH3)(CH3))), 0.80 ((dd, 3JH–H = 6.9 Hz, 3JH–P = 6.9 Hz, 6H, P–(CH(CH3)(CH3)))), 0.74 (dd, 3JH–P = 7.5 Hz, 3JH–H = 6.9 Hz, 6H, P–(CH(CH3)(CH3))), 0.03 (s, 9H, SiMe3), −0.44 (s, 2H, Lu–CH2SiMe3). 13C{1H} NMR (benzene-d6): δ 154.70 (s, o-ipso-Dipp), 144.56 (s, ipso-Pipp), 144.08 (s, ipso-Pipp), 133.68 (s, ipso-Dipp), 128.33 (dd, 1JC–P = 135.5 Hz, 3JC–P = 14.3 Hz, ipso-pyrrole), 128.28 (s, Pipp C–H), 127.82 (s, Pipp C–H), 123.08 (s, m-Dipp C–H), 117.20, (dd, 2JC–P = 15.6 Hz, 3JC–P = 10.1 Hz, pyrrole C–H), 114.56 (s, p-Dipp C–H), 36.50 (s, Lu–CH2), 34.27 (s, Pipp–(CH(CH3)2)), 29.29 (s, Dipp–(CH(CH3)2)), 27.58 (s, P–(CH(CH3)2)), 26.85 (s, P–(CH(CH3)2)), 24.77 (s, Dipp–(CH(CH3)2)), 24.70 (s, Pipp–(CH(CH3)(CH3))), 24.58 (s, Pipp–(CH(CH3)(CH3))), 16.84 (s, P–(CH(CH3)(CH3))), 16.59 (ov s, P–(CH(CH3)(CH3))) and P–(CH(CH3)(CH3)), 16.38 (s, P–(CH(CH3)(CH3))), 4.44 (s, SiMe3). 31P{1H} NMR (benzene-d6): δ 48.7. Anal. calcd (%) for C50H81LuN4P2Si C, 59.86; H, 8.14; N, 5.58. Found: C, 59.57, H, 8.11; N, 5.68.
Synthesis of LiPrLu(NHCPh3)2 (8CPh3)
A 20 mL scintillation vial was charged with LiPrLu(CH2SiMe3)2 (0.0513 g, 0.0561 mmol) and a small stir bar. A second vial was charged with NH2CPh3 (0.0291 g, 0.112 mmol). Toluene (7 mL) was added to each flask to dissolve all materials. The NH2CPh3 solution was transferred into the LiPrLu(CH2SiMe3)2 solution dropwise via Pasteur pipette. The resulting light brown solution was allowed to stir at 70 °C for 69 hours whereupon all volatiles were removed in vacuo. The desired product remained as a light brown solid (0.0544 g, 88.7%). X-ray quality crystals were grown by dissolving the mixture in a minimal amount of a warm (50 °C) 1:1 mixture of toluene and heptane. The solution was allowed to cool to ambient temperature over 2 hours and was placed in a −35 °C freezer for 16 hours. The resulting crystals were isolated by filtration, washed with cold pentane and dried for 8 hours under vacuum (0.0197 g, 32.1%). 1H NMR (benzene-d6): δ 7.37 (d, 3JH–H = 7.7 Hz, 12H, o-Ph3), 7.08 (ov m, 18H, m- and p-Ph3), 6.85 (d, 3JH–H = 8.4 Hz, 4H, o-Pipp C–H), 6.50 (dd, 3JH–H = 8.4 Hz, 4JH–P = 1.4 Hz, 4H, m-Pipp C–H), 6.55 (d, 3JH–P = 2.1 Hz, 2H, pyrrole C–H), 2.83 (sp, 3JH–H = 6.6 Hz, 2H, Pipp–CH(CH3)2), 2.26 (s, 2H, N–H), 1.96 (dsp 2JH–P = 6.6 Hz, 3JH–H = 6.6 Hz, 4H, P–CH(CH3)2) 1.29 (d, 3JH–H = 6.8 Hz, 12H, Pipp–CH(CH3)2), 0.81 (dd, 3JH–P = 16.1 Hz, 3JH–H = 6.6 Hz, 12H, P–CH(CH3)(CH3)), 0.79 (dd, 3JH–P = 16.1 Hz, 3JH–H = 6.6 Hz, 12H, P–CH(CH3)(CH3)). 13C{1H} NMR (benzene-d6): δ 154.38 (s, CPh3ipso-C), 146.04 (s, ipso-Pipp), 142.74 (s, ipso-Pipp), 129.98 (s, o-CPh3 C–H), 129.16 (d, 3JC–P = 7.1 Hz, o-Pipp C–H), 128.39 (d, 1JC–P = 125.0 Hz, ipso-pyrrole), 127.82 (s, m-CPh3 C–H), 127.36 (s, m-Pipp C–H), 125.60 (s, p-CPh3 C–H), 116.88 (dd, 2JC–P = 22.9 Hz, 3JC–P = 10.6 Hz, pyrrole C–H), 75.41 (s, CPh3), 34.22 (s, Pipp–CH(CH3)2), 27.03 (d, 1JC–P = 54.6 Hz, P–CH(CH3)2), 24.92 (s, Pipp–CH(CH3)2), 16.65 (s, P–CH(CH3)(CH3)), 16.63 (s, P–CH(CH3)(CH3)). 31P{1H} NMR (benzene-d6): 45.3. Anal. calcd (%) for C72H84LuN5P2 C, 68.83; H, 6.74; N, 5.57. Found: C, 68.60, H, 7.66; N, 6.31.
Synthesis of LiPrLu(NHDipp)2 (8Dipp)
A round-bottomed flask was charged with LiPrLu(CH2SiMe3)2 (0.1835 g, 0.2007 mmol) and a small stir bar. A second round-bottomed flask was charged with NH2Dipp (0.0712 g, 0.402 mmol). Toluene (50 mL) was transferred into each flask at −78 °C. The flasks were warmed to ambient temperature whereupon the NH2Dipp solution was transferred into the LiPrLu(CH2SiMe3)2 solution dropwise via syringe. The resulting light brown solution was allowed to stir at ambient temperature for 24 hours whereupon all volatiles were removed in vacuo. The desired product remained as a light brown solid (0.184 g, 84.1%). X-ray quality crystals were grown by dissolving the solid in a minimal amount of a warm (50 °C) 1:1 mixture of toluene and heptane. The solution was allowed to cool to ambient temperature over 2 hours and was placed in a −35 °C freezer for 16 hours. The resulting crystals were isolated by filtration, washed with cold pentane and dried for 8 hours under vacuum (0.0927 g, 42.3%). 1H NMR (benzene-d6): δ 7.24 (d, 3JH–H = 7.5 Hz, 4H, m-Dipp C–H), 7.06 (dd, 3JH–H = 8.4 Hz, 4JH–P = 2.1 Hz 4H, o-Pipp C–H), 6.97–6.88 (ov m, 6H, m-Pipp C–H and p-Dipp C–H), 6.56 (d, 2H, 3JH–P = 2.4 Hz, pyrrole C–H), 4.38 (s, 2H, N–H), 3.20 (sp, 3JH–H = 6.9 Hz, 4H, Dipp–CH(CH3)2), 2.68 (sp, 2H, 3JH–H = 7.2 Hz, Pipp–CH(CH3)2), 2.09 (dd, 3JH–H = 7.2 Hz, 2JH–P = 1.8 Hz, 4H, P–CH(CH3)2), 1.28 (d, 3JH–H = 6.9 Hz, 24H, Dipp–CH(CH3)2), 1.15 (d, 3JH–H = 7.2 Hz, 12H, Pipp–CH(CH3)2), 0.91 (dd, 3JH–P = 15.7 Hz, 3JH–H = 7.2 Hz, 12H, P–CH(CH3)(CH3)), 0.79 (dd, 3JH–P = 15.7 Hz, 3JH–H = 7.2 Hz, 12H, P–CH(CH3)(CH3)). 13C{1H} NMR (benzene-d6): δ 154.42 (s, ipso-Dipp), 144.04 (ov s, ipso-Pipp and p-ipso-Pipp), 134.04 (s, o-ispo-Dipp), 128.53 (dd, 1JC–P = 134.7 Hz, 3JC–P = 13.7 Hz, ipso-pyrrole), 128.45 (d, 3JC–P = 6.4 Hz, o-Pipp C–H), 127.65 (s, m-Pipp C–H), 122.78 (s, m-Dipp C–H), 117.95 (dd, 2JC–P = 17.5 3JC–P = 9.9 Hz, pyrrole C–H), 114.81 (s, p-Dipp C–H), 34.17 (s, Pipp–CH(CH3)2), 29.86 (s, Dipp–CH(CH3)2), 27.40 (d, 1JC–P = 53.6 Hz, P–CH(CH3)2), 24.63 (s, Pipp–CH(CH3)2), 24.17 (s, Dipp–CH(CH3)2), 16.98 (s, P–CH(CH3)(CH3)), 16.54 (s, P–CH(CH3)(CH3)). 31P{1H} NMR (benzene-d6): δ 46.4. Anal. calcd (%) for C58H88LuN5P2 C, 63.78; H, 8.12; N, 6.41. Found: C, 62.82, H, 7.69; N, 7.23.
Synthesis of HLPm (11)
A sample of 2,5-bis(diisopropylphosphino)-N–H-pyrrole (0.1090 g, 0.3641 mmol) was weighed into a round-bottomed flask and attached to a vacuum line. Two equivalents of 2-azido-4,6-dimethylpyrimidine (10) (0.1050 g, 0.7040 mmol) was weighed into a second round-bottomed flask and attached to a vacuum line. Toluene (25 mL) was transferred into each flask at −78 °C. Both solutions were allowed to warm to ambient temperature whereupon a cannula was used to add the solution of compound 10 to the 2,5-bis(diisopropylphosphino)-N–H-pyrrole solution. Bubbles formed shortly after completion of the addition. The solution was left to stir under a constant flow of argon for 18 hours whereupon all volatiles were removed in vacuo leaving behind the desired product as a brown solid (0.173 g, 88.3%). X-ray quality crystals were obtained by dissolving the crude product in a minimal amount of toluene in a 20 mL scintillation vial at ambient temperature. Pentane (4 mL) was added to a small, 5 mL vial which was placed inside the vial containing the saturated toluene solution of HLPm. The entire system was sealed and placed in a −35 °C freezer for 16 hours. The mother-liquor was removed via Pasteur pipette and cold (−35 °C) pentane (10 mL) was added to the vial to wash the crystals. The pentane was removed via Pasteur pipette and the crystals were dried under vacuum for 2 hours. 1H NMR (benzene-d6): δ 13.21 (s, 1H, N–H), 6.35 (s, 2H, pyrrole C–H), 6.08 (s, 2H, pyrimidine C–H), 2.39 (spd, 4H, 3JH–H = 7.0 Hz, 2JH–P = 2.6 Hz, P–CH(CH3)2), 2.39 (s, 12H, pyrimidine CH3), 1.10 (dd, 3JH–P = 16.1 Hz, 3JH–H = 7.0 Hz, P–(CH(CH3)(CH3))), 1.00 (dd, 3JH–P = 16.1 Hz, 3JH–H = 7.0 Hz, P–(CH(CH3)(CH3))). 13C{1H} NMR (benzene-d6): δ 168.56 (s, m-ipso-pyrimidine), 167.51 (d, 2JC–P = 1.5 Hz, ipso-pyrimidine), 124.8 (d, 1JC–P = 41.5 Hz, ipso-pyrrole), 116.76 (br s, pyrrole C–H), 109.15 (s, pyrimidine C–H), 26.52 (d, 1JC–P = 65.9 Hz, P–CH(CH3)2), 24.58 (s, pyrimidine CH3), 17.22 (s, P–(CH(CH3)(CH3))), 16.85 (s P–(CH(CH3)(CH3))). 31P{1H} NMR (benzene-d6): δ 27.7. Anal. calcd (%) for C28H44N7P2 C, 62.09; H, 8.37; N, 18.10. Found: C, 61.36, H, 8.27; N, 17.88.Synthesis of LPmLu(CH2SiMe3)2 (12)
A sample of HLPm (11) (0.0580 g, 0.0999 mmol) was weighed into a 20 mL scintillation vial. A disposable shell vial was charged with Lu(CH2SiMe3)3THF2 (0.0552 g, 0.102 mmol). Toluene (3 mL) was added to each vial to dissolve all material. The Lu(CH2SiMe3)3THF2 solution was added to the stirring solution of compound 11 dropwise over 1 minute via Pasteur pipette. The clear, pale yellow solution was left to stir at ambient temperature for 1 hour whereupon all volatiles were remove in vacuo. Pentane (5 mL) was added to the remaining waxy solid and the mixture was stirred for 15 minutes. The solvent was removed in vacuo affording a pale yellow solid (0.0737 g). As complex 12 decomposes slowly in solution, the solid material contained a mixture of predominantly 12 and several intractable impurities. 1H NMR (toluene-d8): δ 6.62 (d, 3JH–P = 3.5 Hz, 2H, pyrrole C–H), 6.00 (s, 2H, pyrimidine C–H), 2.62–2.56 (ov m, 10H, pyrimidine CH3 and P–CH(CH3)2), 2.03 (s, 6H, pyrimidine CH3), 1.22 (dd, 3JH–P = 16.9 Hz, 3JH–H = 7.1 Hz, 12H, P–(CH(CH3)(CH3))), 1.02 (dd, 3JH–P = 17.2 Hz, 3JH–H = 7.1 Hz, 12H, P–(CH(CH3)(CH3))), 0.02 (s, 18 h, SiMe3), −1.15 (s, 4H, Lu–CH2SiMe3). 13C{1H} NMR (toluene-d8): δ 167.51 (s, ipso-pyrimidine), 165.56 (s, ipso-pyrimidine), 164.13 (s, ipso-pyrimidine), 126.92 (d, 1JC–P = 15.2 Hz, ipso-pyrrole), 117.12 (dd, 2JC–P = 26.7 Hz, 3JC–P = 11.2 Hz, pyrrole C–H), 110.98 (s, pyrimidine C–H), 26.37 (s, Lu–CH2), 25.85 (d, 1JC–P = 53.8 Hz, P–CH(CH3)2), 23.87 (br s, endo/exo pyrimidine CH3), 17.00 (s, P–(CH(CH3)(CH3))), 16.08 (s, P–(CH(CH3)(CH3))), 5.08 (s, SiMe3). 31P{1H} NMR (benzene-d6): δ 42.4.Synthesis of LPmLuCl2 (13)
A sample of NaLPm (0.6903 g, 1.225 mmol), synthesized according to the procedure disclosed for preparing NaLiPr,21 was added to a 100 mL two-necked round bottomed flask and attached to a swivel frit apparatus. LuCl3(THF)3 (0.3486 g, 1.239 mmol) was added to a separate 100 mL round bottom flask. THF (50 mL) was added into each flask via vacuum distillation at −78 °C. Each flask was allowed to warm to ambient temperature while stirring. The LuCl3(THF)3 solution was then added dropwise via syringe to the stirring NaLPm solution over 5 minutes at ambient temperature. The reaction mixture was left to stir at ambient temperature for 3 hours whereupon all volatiles were removed in vacuo leaving behind a pale yellow solid. Toluene (50 mL) was transferred into the round bottomed flask via vacuum distillation at −78 °C. The solution was allowed to warm to ambient temperature, at which point the NaCl by-product was removed by filtration, affording a clear yellow solution. All volatiles were removed in vacuo giving the desired product as a pale yellow solid (0.8207 g, 85.2%). 1H NMR (benzene-d6): δ 6.68 (d, 3JH–P = 2.4 Hz, 2H, pyrrole C–H), 5.90 (s, 2H, aromatic pyrimidine C–H), 2.60 (dsp, 3JH–H = 7.2 Hz, 2JH–P = 2.4 Hz, 4H, P–CH(CH3)2), 2.44 (s, 6H, pyrimidine CH3), 2.04 (s, 6H, pyrimidine CH3), 1.29 (dd, 3JH–P = 7.2 Hz, 3JH–P = 17.0 Hz, 12H, P– CH(CH3)(CH3)), 1.07 (dd, 3JH–P = 7.2 Hz, 3JH–P = 17.4 Hz, 12H, P–CH(CH3)(CH3)). 13C{1H} NMR (benzene-d6): δ 168.70 (s, pyrimidine ipso-C), 166.05 (d, 2JC–P = 5.28 Hz, pyrimidine ipso-C), 164.94 (s, pyrimidine ipso-C), 128.42 (m, pyrrole ipso-C), 117.75 (dd, 2JC–P = 26.0 Hz, 3JC–P = 10.5 Hz, pyrrole C–H), 111.64 (s, aromatic pyrimidine C–H), 26.16 (d, 1JC–P = 54.4 Hz, P–CH(CH3)2), 24.29 (ov s, pyrimidine CH3), 17.09 (s, P–CH(CH3)(CH3)), 16.23 (s, P–CH(CH3)(CH3)). 31P{1H} NMR (benzene-d6): δ 52.2. Despite exhaustive efforts, satisfactory combustion data was not obtained.Author contributions
J. P. K. contributed to investigation, methodology, data curation, formal analysis, writing – original draft and writing – reviewing and editing. S.-J. H. contributed to data curation, formal analysis, writing – reviewing and editing. P. G. H. contributed to conceptualization, funding acquisition, project administration, formal analysis, supervision, methodology, writing – original draft and writing – reviewing and editing.Data availability
Original data can be obtained from the authors on request. Deposition Numbers 2417587 (for 5), 2417588 (for 6), 2417589 (for 7CPh3), 2417590 (for 7Dipp), 2417591 (for 8CPh3), 2417592 (for 8Dipp), 2423088 (for 11) and 2429452 (for 13)† contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre (CCDC) and Fachinformationszentrum Karlsruche Access Structures service.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This research was financially supported by a Discovery Grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada. The authors thank Mr Tony Montina and Mr Michael Opyr for expert technical assistance with NMR experiments. Prof. René T. Boeré is gratefully acknowledged for his continued support regarding X-ray diffraction analysis. P. G. H. thanks the University of Lethbridge for a Tier I Board of Governors Research Chair in Organometallic Chemistry.References
- O. T. Summerscales and J. C. Gordon, RSC Adv., 2013, 3, 6682 CAS.
- D. J. Beetstra, A. Meetsma, B. Hessen and J. H. Teuben, Organometallics, 2003, 22, 4372 CAS.
- G. R. Geisbrecht, D. L. Clark, P. J. Hay, D. W. Keogh, R. Poli, B. L. Scott, J. G. Watkin and J. C. Gordon, Organometallics, 2002, 21, 4726 Search PubMed.
- A. A. Trifonov, M. N. Bochkarev, H. Schumann and J. Loebel, Angew. Chem., Int. Ed. Engl., 1991, 30, 1149 CrossRef.
- J. Scott, F. Basuli, A. R. Fout, J. C. Huffman and D. J. Mindiola, Angew. Chem., Int. Ed., 2008, 47, 8502 CrossRef CAS PubMed.
- B. F. Wicker, H. J. Fan, A. K. Hickey, M. G. Crestani, J. Scott, M. Pink and D. J. Mindiola, J. Am. Chem. Soc., 2012, 134, 20081 CrossRef CAS PubMed.
- T. Chu, W. E. Piers, J. L. Dutton and M. Parvez, Organometallics, 2013, 32, 1159 CrossRef CAS.
- E. L. Lu, Y. X. Li and Y. F. Chen, Chem. Commun., 2010, 46, 4469 RSC.
- J. X. Chu, X. H. Han, C. E. Kefalidis, J. L. Zhou, L. Maron, X. B. Leng and Y. F. Chen, J. Am. Chem. Soc., 2014, 136, 10894 CrossRef CAS PubMed.
- E. L. Lu, J. X. Chu, Y. F. Chen, M. V. Borzov and G. Y. Li, Chem. Commun., 2011, 47, 743 RSC.
- J. X. Chu, E. L. Lu, Z. X. Liu, Y. F. Chen, X. B. Leng and H. B. Song, Angew. Chem., Int. Ed., 2011, 50, 7677 CrossRef CAS PubMed.
- J. X. Chu, E. L. Lu, Y. F. Chen and X. B. Leng, Organometallics, 2013, 32, 1137 CrossRef CAS.
- E. Lu, J. Chu and Y. F. Chen, Acc. Chem. Res., 2018, 51, 557 CrossRef CAS PubMed.
- W. Rong, J. Cheng, Z. Mou, H. Xie and D. Cui, Organometallics, 2013, 32, 5523 CAS.
- D. Schädle, M. Meermann-Zimmermann, C. Schädle, C. Maichle-Mössmer and R. Anwander, Eur. J. Inorg. Chem., 2015, 2015, 1334 Search PubMed.
- L. A. Solola, A. V. Zabula, W. L. Dorfner, B. C. Manor, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 6928 CAS.
- T. E. Rieser, D. Schädle, C. Maichle-Mössmer and R. Anwander, Chem. Sci., 2024, 15, 3562–3570 Search PubMed.
- T. E. Rieser, R. Thim-Spöring, D. Schädle, P. Sirsch, R. Litlabø, K. W. Törnroos, C. Maichle-Mössmer and R. Anwander, J. Am. Chem. Soc., 2022, 144, 4102–4113 CrossRef CAS PubMed.
- D. Schädle, C. Maichle-Mössmer, C. Schädle and R. Anwander, Chem. – Eur. J., 2015, 21, 662 Search PubMed.
- J. P. Knott, M. M. Hänninen, J. M. Rautiainen, H. M. Tuononen and P. G. Hayes, J. Organomet. Chem., 2017, 845, 135 CAS.
- C. S. MacNeil, K. E. Glynn and P. G. Hayes, Organometallics, 2018, 37, 3248 CAS.
- K. R. D. Johnson, M. A. Hannon, J. S. Ritch and P. G. Hayes, Dalton Trans., 2012, 41, 7873 CAS.
- M. T. Zamora, K. R. D. Johnson, M. M. Hanninen and P. G. Hayes, Dalton Trans., 2014, 43, 10739 CAS.
- K. R. D. Johnson and P. G. Hayes, Organometallics, 2011, 30, 58 CAS.
- T. M. Cameron, J. C. Gordon and B. L. Scott, Organometallics, 2004, 23, 2995 CrossRef CAS.
- T. M. Cameron, J. C. Gordon, B. L. Scott and W. Tumas, Chem. Commun., 2004, 1398 RSC.
- J. D. Masuda, K. C. Jantunen, B. L. Scott and J. L. Kiplinger, Organometallics, 2008, 27, 1299 CrossRef CAS.
- R. K. Thomson, M. J. Monreal, J. D. Masuda, B. L. Scott and J. L. Kiplinger, J. Organomet. Chem., 2011, 696, 3966 CrossRef CAS.
- S. Zhu, W. Wu, D. Hong, F. Chai, Z. Huang, X. Zhu, S. Zhou and S. Wang, Inorg. Chem., 2024, 63, 14860 CrossRef CAS PubMed.
- L. K. Knight, W. E. Piers, P. Fleurat-Lessard, M. Parvez and R. McDonald, Organometallics, 2004, 23, 2087 CrossRef CAS.
- Z.-J. Lv, M. Zhu, W. Liu, Z. Chai, J. Wei and W.-X. Zhang, Organometallics, 2021, 40, 3992 CrossRef CAS.
- C. Temple and J. A. Montgomery, J. Org. Chem., 1965, 30, 826 CrossRef CAS PubMed.
- C. Temple, M. C. Thorpe, W. C. Coburn and J. A. Montgomery, J. Org. Chem., 1966, 31, 935 CrossRef CAS.
- K. R. D. Johnson and P. G. Hayes, Organometallics, 2009, 28, 6352 Search PubMed.
- K. R. D. Johnson, B. L. Kamenz and P. G. Hayes, Can. J. Chem., 2015, 94, 330 Search PubMed.
- K. R. D. Johnson and P. G. Hayes, Inorg. Chim. Acta, 2014, 422, 209 CrossRef CAS.
- K. R. D. Johnson, B. L. Kamenz and P. G. Hayes, Organometallics, 2014, 33, 3005–3011 CrossRef CAS.
- K. R. D. Johnson and P. G. Hayes, Dalton Trans., 2014, 43, 2448 RSC.
- K. R. D. Johnson and P. G. Hayes, Organometallics, 2013, 32, 4046 CrossRef CAS.
- K. C. Jantunen, B. L. Scott, P. J. Hay, J. C. Gordon and J. L. Kiplinger, J. Am. Chem. Soc., 2006, 128, 6322 CrossRef CAS PubMed.
- A. Arndt, P. Voth, T. P. Spaniol and J. Okuda, Organometallics, 2000, 19, 4690 CrossRef.
- F. Estler, G. Eickerling, E. Herdtweck and R. Anwander, Organometallics, 2003, 22, 1212 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Including NMR spectra of key compounds and X-Ray Crystallographic Data. CCDC 2417587–2417592, 2423088 and 2429452 for compounds 5–8, 11 and 13. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00338e |
| This journal is © The Royal Society of Chemistry 2025 |