
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymerisation isomerism of tungsten(VI) oxomethoxide: new insights into the structure and reactivity of non-cluster metal oxoalkoxide aggregates†
Gulaim A.
Seisenbaeva
 a,
Lars
Kloo
b,
Peter
Agback
a and
Vadim G.
Kessler
*a
a,
Lars
Kloo
b,
Peter
Agback
a and
Vadim G.
Kessler
*a
aDepartment of Molecular Sciences, BioCenter, Swedish University of Agricultural Sciences, Box 7015, SE-750 07 Uppsala, Sweden. E-mail: gulaim.seisenbaeva@slu.se; vadim.kessler@slu.se
bDivision of Applied Physical Chemistry, Royal Institute of Technology (KTH), Teknikringen 30, 100 44 Stockholm, Sweden
First published on 18th April 2025
Abstract
Polymerization isomerism is potentially important for metal alkoxides as precursors of oxide materials. Here, we present this phenomenon for tungsten oxo-methoxide, reporting the molecular and crystal structure of its polymeric form [WO(OMe)4]∞(1), its dimeric form W2O2(OMe)8(2), and a higher extent oxo-substituted by-product of the synthesis, Li2(MeOH)6W12O29(OMe)16(3).
The structure and reactivity of metal (oxo)alkoxide complexes have been highly attractive and are frequently addressed topics since the middle of the 1950s, starting with the pioneering work of D.C. Bradley,1 when the utility of alkoxides as precursors in the soft chemistry synthesis2 of metal oxides was realized. In the early years, the dominating hypothesis regarded these compounds as typical metal–organic compounds with the stress on organic, and assumed they should predominantly follow a kinetically driven reactivity.3 This indicates that the structure of the resulting oxides produced by low-energy chemical treatment, such as hydrolysis, should be related to and dependent on the structure of the precursor species.4
As metal alkoxides often exist in the form of oligonuclear aggregates, it was expected that derivatives of many metals should display polymerization isomerism, sometimes denoted also as coordination polymerism. They were expected to exist simultaneously in several kinds of individual molecular species with different sizes and reactivities, and undergo kinetically resolved transformations among each other.5 The search for examples of this phenomenon revealed it to be extremely rare. Among the few examples proved by unequivocal evidence were a few species of aluminium; in particular, iso-propoxide existing as a liquid trimeric form and a solid crystalline tetrameric one at room temperature.6 Another small family, in which this phenomenon was observed includes the dimeric methoxides of rhenium(V,VI), Re2O3(OMe)6 and ReMoO2(OMe)7, transforming in time into tetranuclear Re4O6(OMe)12 and analogous mixed-metal Re–Mo species.7 The iron and gallium alkoxides, initially reported as displaying polymerization isomerism, turned out to simply be contaminated by halide impurities.8,9
Conversely, the belief in the kinetic stability of the oligonuclear oxo-alkoxide species became rather widespread, leading to the introduction of controversial terms, such as “titanium oxo-alkoxide clusters”.10,11
Therefore, it is of interest to gain deeper structural and quantitative insights into the reactivity of metal oxo-alkoxides. We chose to address the tungsten oxo-methoxide, WO(OMe)4, system; a compound with an apparent discrepancy between descriptions of the forms obtained from different solvents. When precipitated from a toluene solution upon freezing, it forms bulky crystals with a monoclinic structure, space group P21/c, a = 10.449(1); b = 11.688(1); c = 13.908(1); β = 98.831(5), consisting of dimeric [WO(OMe)4]2 molecules.12 Conversely, when methanol is used as the solvent, a very different material precipitates, which forms tiny, thin needles.13 An X-ray powder diffraction study of the latter type of crystals permitted the proposal of a tetragonal model with an oxo-bridged polymeric structure, [WO(OMe)4]∞, a = 10.04; c = 3.99 Å.13 Limitations of the X-ray equipment at that time made X-ray single crystal studies impossible, leaving the question about the existence and stability of different crystal forms open.
Tungsten oxo-methoxide, WO(OMe)4, was produced by the anodic oxidation of tungsten metal in methanol in the presence of LiCl. The hexa-methoxide, W(OMe)6, formed as an admixture, was separated by crystallization from the initial electrolyte at −18 °C. Then, the mother liquor was concentrated by solvent evaporation under vacuum, and the tiny thin white needles were crystallized on storage at +8 °C over several months. The precipitated crystalline material was washed by cold methanol and dried in vacuum. Re-dissolution in MeOH under reflux and subsequent cooling to room temperature offered a crop of slightly thicker, needle-shaped crystals (ca. 40 μm in diameter).
Investigation of this material by single crystal X-ray diffraction revealed a linear chain structure, [WO(OMe)4]∞(1) (Fig. 1a), with a tetragonal symmetry (space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) ), but crystallizing as a racemic twin mimicking P4/mmm symmetry. The unit cell parameters are a = 7.1477(2), b = 4.0139(2) Å. These results match the ones reported in ref. 13, assuming that the powder data were recorded for a textured sample, resulting in an overestimation of the basal unit cell parameter with the factor of the square root of 2. The experimental powder X-ray diffraction pattern for the obtained material perfectly matches the theoretically calculated one (see Fig. 1b and c), indicating that the bulk of the material displays the same crystalline structure. The latter is truly exciting, as the bond lengths in the W(1)–O(2)–W(1′) bridge are unexpectedly totally equal in length and very well defined (2.00695(10) Å). This means that the structure is not disordered with respect to the atoms generating the metal-oxo chain. The alkoxide ligand is disordered between two equivalent positions, where the W(1)–O(3) distance is 1.880(14), with only one (coinciding) position for the carbon atom.
), but crystallizing as a racemic twin mimicking P4/mmm symmetry. The unit cell parameters are a = 7.1477(2), b = 4.0139(2) Å. These results match the ones reported in ref. 13, assuming that the powder data were recorded for a textured sample, resulting in an overestimation of the basal unit cell parameter with the factor of the square root of 2. The experimental powder X-ray diffraction pattern for the obtained material perfectly matches the theoretically calculated one (see Fig. 1b and c), indicating that the bulk of the material displays the same crystalline structure. The latter is truly exciting, as the bond lengths in the W(1)–O(2)–W(1′) bridge are unexpectedly totally equal in length and very well defined (2.00695(10) Å). This means that the structure is not disordered with respect to the atoms generating the metal-oxo chain. The alkoxide ligand is disordered between two equivalent positions, where the W(1)–O(3) distance is 1.880(14), with only one (coinciding) position for the carbon atom.
 | ||
| Fig. 1 Molecular structure of [WO(OMe)4]∞, showing the three formula units (a) and its experimental (b) and theoretical (c) powder diffraction patterns (Mo Kα radiation, λ = 0.71073 Å). | ||
The produced needle-shaped crystals were removed by decantation, washed with a minimal amount of anhydrous methanol and dried in vacuum. A fresh methanol solution was prepared with 60 mg ml−1 concentration and placed in a freezer at −18 °C. Massive crystallization in this case offered a crop of crystals with very different features, i.e., transparent blocks (Fig. S1†).
An X-ray single crystal study of the latter showed them to be identical to the structure reported by Errington, Clegg et al.,12 W2O2(OMe)8 (2), featuring dimeric molecules (two centrosymmetric, but symmetrically independent ones) with terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds, W(1)–O(5) 1.700(3) and W(2)–O(15) 1.703(4) Å, and a combination of bridging oxygen atoms, such as W(1)–O(4) 2.033(3) and W(1)–O(4′) 2.231(3) Å, and essentially equally long terminal (1.869(3)–1.898(4) Å) alkoxide contacts (see Fig. 2). This molecular structure was earlier found to be closely analogous to that in the only phase known for the molybdenum derivative [MoO(OMe)4]2,14 which could be obtained by crystallization from both the parent alcohol and the hydrocarbon solvents. However, the latter displayed a different crystal structure following higher symmetry, P212121. The powder diffraction pattern of 2 matches the one predicted from the single crystal data (Fig. S2†), but features some apparent texture with a few strong reflections at higher diffraction angles. It should be noted that if left under a minor amount of methanol at room temperature overnight, the block-shaped crystals split into needles (Fig. S3†). These needles give a powder diffraction pattern corresponding to 1, but of rather low intensity, because of the small crystallite size.
O double bonds, W(1)–O(5) 1.700(3) and W(2)–O(15) 1.703(4) Å, and a combination of bridging oxygen atoms, such as W(1)–O(4) 2.033(3) and W(1)–O(4′) 2.231(3) Å, and essentially equally long terminal (1.869(3)–1.898(4) Å) alkoxide contacts (see Fig. 2). This molecular structure was earlier found to be closely analogous to that in the only phase known for the molybdenum derivative [MoO(OMe)4]2,14 which could be obtained by crystallization from both the parent alcohol and the hydrocarbon solvents. However, the latter displayed a different crystal structure following higher symmetry, P212121. The powder diffraction pattern of 2 matches the one predicted from the single crystal data (Fig. S2†), but features some apparent texture with a few strong reflections at higher diffraction angles. It should be noted that if left under a minor amount of methanol at room temperature overnight, the block-shaped crystals split into needles (Fig. S3†). These needles give a powder diffraction pattern corresponding to 1, but of rather low intensity, because of the small crystallite size.
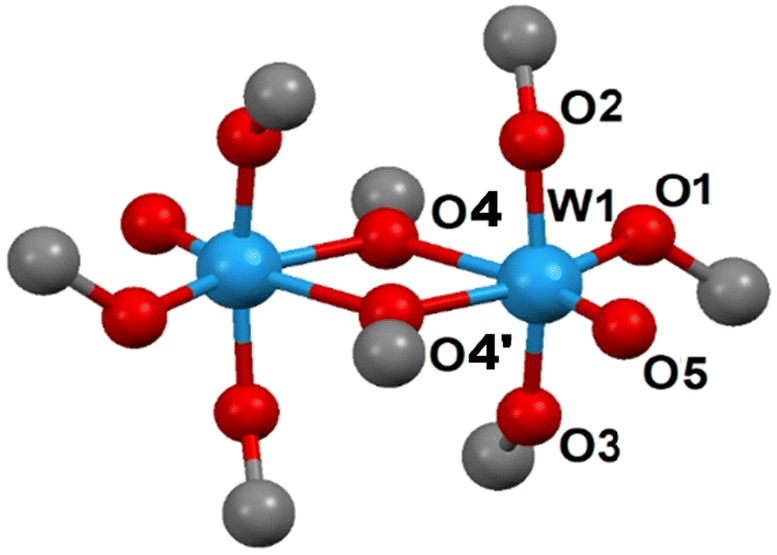 | ||
| Fig. 2 Molecular structure of [WO(OMe)4]2, displaying one of the symmetrically independent dimers in the crystal structure. | ||
To gain insight into the effect of temperature on the behavior of this compound, apparently featuring polymerization isomerism, we have carried out a variable temperature 1H-NMR study of tungsten oxo-methoxide, both forms 1 and 2, in methanol-D4 and benzene-D6. Earlier studies of this compound in toluene-D8, reported by Errington et al.,12 demonstrated that it was present in a well-defined dimeric form in the temperature range of 210–270 K, while a further increase in temperature resulted in enhanced exchange between the ligands, eventually leading to dissociation into a monomer form at 360 K. The use of methanol as a solvent was expected to facilitate both the exchange of ligands and dissociation, with the formation of a methanol-solvated monomer, giving spectral peaks that are indistinguishable from the dominating signal of the residual non-isotope-substituted methanol. The experiments were carried out with solutions of 10 mg ml−1 concentration, prepared by room temperature dissolution of both 1 and 2 forms, producing identical spectra in both peak positions and temperature behavior. However, the observed behavior turned out to be even more exciting (see Fig. 3).
 | ||
| Fig. 3 Variable temperature 1H-NMR spectra of the WO(OMe)4 solution in methanol-D4. Asterisk (*) indicates the signal corresponding to W(OMe)6. | ||
The quick exchange in the dimer structure was traceable already at 273 K, where the spectrum features two broadened signals at 4.43 and 4.38 ppm in an approximate intensity ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. At 283 K, the signals broadened further, indicating accelerated exchange. However, further development of the signals on heating showed a much more complex pattern. The signal corresponding to W(OMe)6 started to increase in intensity (Fig. S4†). Meanwhile, the signals corresponding to the oxo-methoxide, instead of becoming sharper and shifting slightly downfield (which would be typical for the formation of a free monomer), broadened drastically. This indicated that there was an exchange with a more complex oxo-alkoxide structure when free methanol was the solvent. This behavior is comparable with that of tungsten oxo-halides, where upon heating, the mono oxo species remain in equilibrium with the hexa-halide and dioxo-halide ones.15 It is thus logical to propose that the co-existence of two parallel equilibria already occurs at a temperature as low as 20 °C in methanol solution:
:1. At 283 K, the signals broadened further, indicating accelerated exchange. However, further development of the signals on heating showed a much more complex pattern. The signal corresponding to W(OMe)6 started to increase in intensity (Fig. S4†). Meanwhile, the signals corresponding to the oxo-methoxide, instead of becoming sharper and shifting slightly downfield (which would be typical for the formation of a free monomer), broadened drastically. This indicated that there was an exchange with a more complex oxo-alkoxide structure when free methanol was the solvent. This behavior is comparable with that of tungsten oxo-halides, where upon heating, the mono oxo species remain in equilibrium with the hexa-halide and dioxo-halide ones.15 It is thus logical to propose that the co-existence of two parallel equilibria already occurs at a temperature as low as 20 °C in methanol solution:
| [WO(OMe)4]2 ⇄ 2WO(OMe)4 | (1) |
| WO(OMe)4 ⇄ W(OMe)6 + “[WO2(OMe)2]x” | (2) |
DOSY 1H spectra indicated the presence of two major forms: (i) W(OMe)6 with a slightly lower diffusion coefficient and thus larger hydrodynamic radius; and (ii) a signal, strongly broadened by exchange, with higher diffusion coefficient and lower radius, corresponding to the non-solvated WO(OMe)4 monomer. The exact determination of the hydrodynamic radii was hindered by the exchange phenomenon. We were not able to locate a separate signal for the larger oligonuclear aggregate, but this was not unexpected in view of its low intensity and strong exchange with the monomer. Insight into the possible structure of this more oxo-substituted and larger complex was provided by the X-ray single crystal results of the third phase, which was found as a minor admixture upon crystallization of the electrolyte after removal of the excess W(OMe)6, identified as [W12O29(OMe)16]2−[Li(MeOH)3]2+ (3) (see Fig. 4). This structure displays a dodeca-tungstate anion, unprecedented so far. It is centrosymmetric, and all tungsten atoms are octahedrally coordinated. Five of the six symmetrically independent ones have a doubly bonded terminal oxygen atom for W(4) further bound to a solvated Li(MeOH)3+ cation. Only W(6) carries a single terminal alkoxide group, and is involved in five bridging W–O–W linkages that are comparable in length. Of the six W atoms, only three form a triangle with the edge-sharing octahedra joining at a central oxygen atom, which is otherwise a typical building block in molybdate and tungstate polyoxometallates (POM).16 Dioxo alkoxides of tungsten have been reported earlier,17 but no direct structural evidence is yet available for homometallic derivatives.
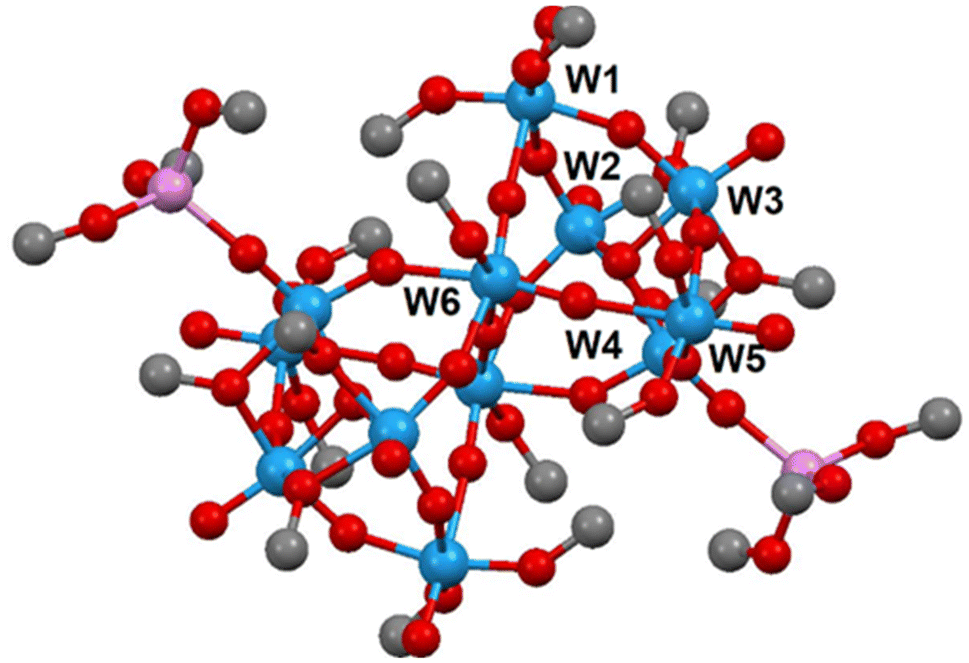 | ||
| Fig. 4 Molecular structure of [W12O29(OMe)16]2−[Li(MeOH)3]2+. | ||
The composition of the isolated bimetallic complex is close to a “W2O5(OR)2” formulation, which has not been reported as a class of tungsten alkoxides so far. For both molybdenum and tungsten derivatives, bimetallic alkoxide complexes have been reported with sodium and lithium ion as counterions.18–20 However, these examples featured either a mononuclear oxo-alkoxide, like [MO(OR)5]−,20 or a dinuclear dioxoalkoxide, as [M2O4(OR)5]−,19 in the structures. The large oligonuclear species described in the present work are rather unusual. The compound 3 turned out to be insoluble in both methanol and benzene, hindering the probing of its solution structure by NMR.
It is interesting to note that when the polymer form 1 is dissolved in benzene-D6, it features a perfectly resolved spectrum corresponding to a dimer at temperatures up to 293 K, and a picture with peaks broadened by exchange at temperatures up to 323 K (Fig. S5†). The spectra are identical to those of solutions produced from dimeric form 2.
To understand the driving forces behind the polymerization isomerism phenomenon, we turned to a comparison of the binding energies in the solid state structures for the polymer and dimer structures using the Crystal 23 program,21 based on a density-functional-level study (for details, please see the ESI, Fig. S6†). As expected, the total energy difference between the two polymorphs turned out to be rather small at 1.49 eV per monomer WO(OMe)4, which favours the dimeric form over the polymeric one. The results are based on approximations of the structures, as described in the computational details.
The binding energies in the two structural forms were estimated by subtracting the total energy of the crystal structure from the monomer(s) in each polymorph. The dimeric structure contains two crystallographically different dimers that formally generate four structurally slightly different monomers. Therefore, the average energy of these four slightly different monomers was used in the comparison (albeit the energy difference between the monomeric structures is quite insignificant). The binding energy of the polymeric form was found to be −1.81 eV per monomer, whereas the binding energy in the dimeric form was found to be −1.76 eV per monomer. The difference (about 0.05 eV, corresponding to less than 5 kJ mol−1) is thus so small that it cannot be used to attribute a thermodynamic driving force to be the predominant factor promoting one crystallographic form over the other. Taking the observed solution equilibria into account, we can conclude that when the solution is dominated by a dimeric form, it undergoes the kinetically facile congruent precipitation/dissolution. Alternatively, when the predominant form in the solution is the (alcohol-solvated) monomeric species, the major product seems to be the polymeric crystal form, supposedly again due to kinetics reasons.
Conclusions
A general conclusion that can be draw from the observations reported in this study is that oligonuclear oxo-alkoxide species apparently do not possess any enhanced stability – both aggregation and composition are significantly influenced by small changes in temperature and by the use of different solvents. It is therefore inappropriate to refer to these species as “clusters”, implying that their core should be sufficiently stable to survive chemical transformations. Their structure is not the inherent property – there is no internal bonding between the metal atoms involved (like between grains in a cluster of wheat or between berries in a cluster of grapes). Instead, the interactions indirectly emerges from the action of external forces. A more adequate description would therefore be the recently proposed term paperbag compounds.22Author contributions
GAS – synthesis and separation of the reaction products, conceptualization; LK – theoretical calculations; PA – NMR experiments, VGK – X-ray studies, conceptualization. All authors contributed to the writing and editing of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Full information on the data collection and refinement of the structures of compounds 1–3, citing the deposition numbers 2402591–2402593.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the Swedish Research Council, Vetenskapsrådet, for support (project 2022-03971_VR).References
- D. C. Bradley and D. G. Carter, Can. J. Chem., 1961, 39, 1434–1443 CrossRef CAS.
- D. C. Bradley, Chem. Rev., 1989, 89, 1317–1322 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- F. Ribot, P. Toledano and C. Sanchez, Chem. Mater., 1991, 3, 759–764 CrossRef CAS.
- N. Y. Turova, E. P. Turevskaya, M. I. Yanovskaya, A. I. Yanovsky, V. G. Kessler and D. E. Tcheboukov, Polyhedron, 1998, 17, 899–915 CrossRef CAS.
- N. Y. Turova, V. A. Kozunov, A. I. Yanovskii, N. G. Bokii, Y. T. Struchkov and B. L. Tarnopol'skii, J. Inorg. Nucl. Chem., 1979, 41, 5–11 CrossRef CAS.
- G. A. Seisenbaeva, A. V. Shevelkov, J. Tegenfeldt, L. Kloo, D. V. Drobot and V. G. Kessler, J. Chem. Soc., Dalton Trans., 2001, 2762–27688 RSC.
- G. A. Seisenbaeva, S. Gohil, E. V. Suslova, T. V. Rogova, N. Y. Turova and V. G. Kessler, Inorg. Chim. Acta, 2005, 358, 3506–3512 CrossRef CAS.
- E. V. Suslova, V. G. Kessler, S. Gohil and N. Y. Turova, Eur. J. Inorg. Chem., 2007, 5182–5188 CrossRef CAS.
- Q. Y. Zhu and J. Dai, Coord. Chem. Rev., 2021, 430, 213664 CrossRef CAS.
- U. Schubert, J. Sol-Gel Sci. Technol., 2022, 105, 587–595 CrossRef.
- W. Clegg, R. J. Errington, P. Kraxner and C. Redshaw, J. Chem. Soc., Dalton Trans., 1992, 1431–1438 RSC.
- S. I. Kucheiko, N. Y. Turova, N. I. Kozlova and B. V. Zhadanov, Russ. J. Coord. Chem., 1985, 11, 1521–1528 Search PubMed.
- V. G. Kessler, A. V. Mironov, N. Y. Turova, A. I. Yanovsky and Y. T. Struchkov, Polyhedron, 1993, 12, 1573–1576 CrossRef CAS.
- P. C. Crouch, G. W. A. Fowles and R. A. Walton, J. Inorg. Nucl. Chem., 1970, 32, 329–333 CrossRef CAS.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- S. I. Kucheiko, N. Y. Turova and O. M. Soloveichik, Russ. J. Gen. Chem., 1985, 55, 2353–2358 CAS.
- S. I. Kucheiko and N. Y. Turova, Russ. J. Coord. Chem., 1987, 13, 1057–1062 Search PubMed.
- V. G. Kessler, A. N. Panov, N. Y. Turova, Z. A. Starikova, A. I. Yanovsky, F. M. Dolgushin, A. P. Pisarevsky and Y. T. Struchkov, J. Chem. Soc., Dalton Trans., 1998, 21–30 Search PubMed.
- N. Y. Turova, V. G. Kessler and S. I. Kucheiko, Polyhedron, 1991, 10, 2617–2628 CrossRef CAS.
- A. Erba, J. K. Desmarais, S. Casassa, B. Civalleri, L. Donà, I. J. Bush, B. Searle, L. Maschio, L. Edith-Daga, A. Cossard, C. Ribaldone, E. Ascrizzi, N. L. Marana, J. P. Flament and B. Kirtman, J. Chem. Theory Comput., 2023, 19, 6891–6932 CrossRef CAS PubMed.
- V. G. Kessler, J. Sol-Gel Sci. Technol., 2025, 114, 37–44 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of theoretical calculations and single crystal diffraction experiments. CCDC 2402591–2402593. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00348b |
| This journal is © The Royal Society of Chemistry 2025 |