
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical synthesis of palladium N-heterocyclic carbene (NHC) complexes†
Roman V.
Larkovich
 a,
Francis
Bru
a,
Maxime R.
Vitale
b,
Laurence
Grimaud
b and
Catherine S. J.
Cazin
*a
a,
Francis
Bru
a,
Maxime R.
Vitale
b,
Laurence
Grimaud
b and
Catherine S. J.
Cazin
*a
aDepartment of Chemistry and Centre for Sustainable Chemistry, Ghent University, Krijgslaan 281-S3, 9000 Ghent, Belgium. E-mail: catherine.cazin@ugent.be
bLaboratoire des Biomolécules (LBM), Département de Chimie, Ecole Normale Supérieure, PSL University, Sorbonne Université, CNRS, 75005 Paris, France
First published on 18th June 2025
Abstract
A novel electrochemical method for the preparation of Pd–NHC (N-heterocyclic carbene) catalysts has been developed. Unlike previously reported procedures, the present method does not employ soluble sacrificial anodes as a metal source but makes use of well-defined Pd-containing precursors instead. Importantly, oxygen was observed to play a key role acting as an electrogenerated base. A superoxide anion is formed via the reduction of oxygen at the cathode. The subsequent deprotonation of the imidazolium salts and reaction of NHCs with the Pd species occurs either in a sequential or in a concerted manner resulting in the formation of the final organopalladium product.
Introduction
N-Heterocyclic carbenes (NHCs) hold a prevalent position amongst privileged ligands in precisely defined organometallic complexes utilized in academia and industry.1 Over the last 30 years, namely since the discovery of the first stable carbene,2 significant advances have been achieved in the use of NHCs as a “privileged” class of ligands for transition metal-based catalyst preparation. Their tuneable stereo-electronic properties have enabled this ligand family to be used to stabilise various metal centres.3–5 Once bound to a metal, they have been shown to significantly influence catalytic performance.1,6 Within families of transition metal catalysts, NHCs have played a pivotal role in palladium cross-coupling catalysis.7–9 It is worth noting that well-defined Pd pre-catalysts bearing NHC ligands display high stability, a defined Pd/ligand ratio, and permit an understanding of the nature of the catalytically active species.10 They currently hold an important position in the state-of-the-art of such catalytic reactions.Four main approaches have been deployed leading to the isolation of Pd–NHC complexes (Scheme 1). The first involves the generation of the free carbene obtained by deprotonation of an imidazolium salt using a strong base, followed by a subsequent reaction with a Pd precursor.11 The second strategy, known as the transmetallation route, is based on the use of Cu or Ag–NHC complexes from which the NHC is transferred to a Pd source.12 In the third route (built-in base), a ligand on a Pd precursor acts as an internal base delivering the product without the need for an external base.13 The most recent route, the weak base route,14 uses a mild base (e.g. K2CO3) and offers a number of advantages over the previously mentioned approaches, including the possibility of performing the reaction under aerobic conditions and is quite cost-effective. Furthermore, the methodology has been shown operative in flow,15 as well as under solventless conditions by implementing ball-milling or grinding of solid reactants.16,17 Nevertheless, state-of-the-art synthetic pathways to NHC–Pd complexes rely on the use of a base to abstract the hydrogen atom located at the C2 position of the imidazolium salt.
 | ||
| Scheme 1 Most common synthetic pathways to Pd–NHC complexes. | ||
Over the last 10 years, organic electrochemistry has experienced a revival leading to the development of now established synthetic methodologies.18,19 Despite these advances, relatively little progress has been made in applying these methodologies to the synthesis of more intricate transition metal complexes.
Admittedly, electroreductive C–H cleavage occurring at the cathode and forming corresponding anions, also called electrogenerated bases (EGB), has been described in numerous reports.20–22 Similarly, the cathodic electrochemical reduction of an imidazolium cation leads to the scission of the C–H bond in 2-position, between the two nitrogen atoms, and the formation of free carbenes with concomitant formation of molecular hydrogen.23,24 Although at first this reaction was studied focusing on the imidazolium electrochemical behaviour, the possibility of using the formed carbene as an organocatalyst or base was rapidly recognized.25–28 Additionally, the described methodologies were found particularly valuable when using imidazolium salts as ionic liquids.29,30
Interestingly, it has been shown that some NHC-ligated metal complexes can be prepared using an electrosynthetic procedure involving an electrochemical reduction of the imidazolium moiety at the cathode, producing a free carbene, while in parallel, anode oxidation liberates a metal cation in solution, and subsequent binding of both species leads to the formation of M–NHC compounds. For instance, the first report showcasing this methodology was published in 2011, by Chen and co-workers, where this electrochemical methodology was applied to the synthesis of cationic copper complexes bearing NHC-ligands with N-pyridine or N-pyrimidine substituents (Scheme 2).31 The reaction was later expanded by Willans and co-workers by using ligands that do not have a pendant donor-arm and delivered both neutral and cationic complexes (Scheme 2).32,33 Furthermore, this approach was elaborated in flow (continuous mode),34 and expanded to other metal complexes, (e.g. Au, Ni and Fe-based compounds) (Scheme 2).35,36
 | ||
| Scheme 2 Electrochemical pathways to M–NHC complexes. | ||
Nevertheless, all described reports are restricted to a number of examples since: (a) only few metals possess an appropriate oxidation potential that allows them to act as a sacrificial anode, with most noble metals being far too inert to act in this manner (e.g. Pd and Pt); (b) in the aforementioned examples metals are used in the form of their corresponding cations allowing NHC ligands to bind them. The said approach is limited to the synthesis of complexes containing only pyridine and NHC co-ligands requiring the use of alternative metal precursors with ligands that are initially pre-installed.
Herein, we report a versatile electrochemical method for the synthesis of Pd–NHC complexes making use of an electrogenerated base originating from O2, which, to the best of our knowledge, is the first report of the synthesis of transition metal–NHC complexes without the use of a sacrificial anode as the metal source (Scheme 2).27
Results and discussion
Inspired by recent work on the weak base route,17,37 where HCl elimination is facilitated by reaction with a base, we targeted the removal of an HCl molecule electrochemically.20–22 The electrochemical cell configuration used was that of an undivided cell with Zn electrodes operating under aerobic conditions and using the organometallic [IPrH][Pd(η3-cin)Cl2] (1b) reagent as a palladium source.38 The palladate was selected to enable optimization with a smaller number of variables, not initially considering the imidazolium salt and a palladium precursor but rather an already assembled adduct combining the two components. First the reaction was carried out at constant current (galvanostatic mode) with a Zn electrode chosen as anode so that its oxidation could serve as an anodic counter-reaction for our reductive process. Gratifyingly, the reaction under these initial conditions delivered the product in 51% yield (Table 1, entry 1). Reasoning that organometallic intermediates were involved, and that oxygen sensitivity/reactivity might be at the origin of this initial moderate product yield, our next trials were performed in the absence of oxygen in an inert atmosphere glovebox. When conducting the reaction under an Ar atmosphere, significant decomposition of the Pd precursor to palladium black was observed, (Table 1, entry 2). All subsequent reactions conducted in the undivided cell arrangement using different electrodes delivered the product in yields not surpassing 50% (Table S6†). We resorted to performing future reactions in air. Bubbling air through the reaction mixture throughout the experiment did not lead to improved yields (Table 1, entry 3). It appeared at this stage that performing the electrochemistry in air and in an undivided cell could only afford a maximum of 50% yield. The next step was to proceed with a different cell design, and a split cell equipped with a Nafion™ proton-exchange membrane was used and permitted the reaction to reach a 79% yield as determined by NMR spectroscopy (Table 1, entry 4). Again, as the cell configuration was modified, a series of experiments were conducted under an inert atmosphere, and confirmed the superior reaction outcome when oxygen is present (Table 1, entry 5). The number of electrons of 1.6 F mol−1 was initially selected to ensure full conversion. This amount can be increased to 2.1 F mol−1 and lead to the complete conversion of the palladate (Table 1, entry 6). Remarkably the conversion does not significantly decrease while the isolated yield remains high when a smaller number of electrons (1 F mol−1) is used (Table 1, entry 7). Changing the electrolyte from n-Bu4NBF4 to n-Bu4NCl did not lead to any increase in yield (Table 1, entry 8). However, Me4NBF4 and Et4NOTs proved to be much less effective electrolytes for this reaction (Table 1, entries 9 and 10). A change in the current density also affects the reaction outcome (Table 1, entries 4, 11 and 12) and our initial setting of 6 A m−2 appears optimum. The use of a glassy carbon cathode in lieu of a Zn cathode led to somewhat poorer results (Table 1, entry 13).|
|
||
|---|---|---|
| Entry | Deviation from optimal conditionsa | Conv. (yield)b, % |
| a Optimal reaction conditions: IPr·HCl (0.05 mmol), [Pd(η3-cin)(μ-Cl)]2 (0.025 mmol), n-Bu4NBF4 (0.05 M), split cell, anode = cathode = Zn, MeCN, j = 6.0 A m−2, t = 52 min (1.6 F mol−1), r.t., in air. b Conversion of IPr·HCl/yield of product determined by 1H NMR (1,3,5-trimethoxybenzene as internal standard). c Solvent saturated with air. d Pre-formed palladate [IPr·H][Pd(η3-cin)Cl2]. | ||
| 1d | Undivided cell | 69 (51) |
| 2d | Undivided cell, Ar atm. (glovebox) | 25 (18) |
| 3c,d | Undivided cell | 59 (48) |
| 4d | [IPr·H][Pd(η3-cin)Cl2] | 100 (79) |
| 5d | Ar atm. (glovebox) | 5 (<5) |
| 6d | n (e) = 2.1 F mol−1 | 100 (79) |
| 7d | n (e) = 1 F mol−1 | 94 (85) |
| 8d | n-Bu4NCl as a support. electrolyte | 100 (76) |
| 9d | Me4NBF4 as a support. electrolyte | 92 (61) |
| 10d | Et4NOTs as a support. electrolyte | 78 (69) |
| 11d | j = 12.0 A m−2 | 90 (75) |
| 12d | j = 2.4 A m−2 | 91 (73) |
| 13d | Cathode = glassy carbon | 100 (74) |
| 14 | None | 100 (85) |
| 15 | [IPrH]BF4 + ½ [Pd(η3-cin)(μ-Cl)]2 | 88 (67) |

As the formation of the palladate (from the imidazolium salt and the palladium dimer) is instantaneous in solution as gauged by NMR spectroscopic studies, the protocol can be evolved by launching the electrochemical reaction using 1 eq. of the imidazolium salt (IPr·HCl) and 0.5 eq. of the palladium dimer [Pd(η3-cin)(μ-Cl)]2 (Table 1, entry 14). Finally, the effect of the nature of the imidazolium counter-anion was examined, and it was shown that the reaction using a non-coordinating counter-anion BF4− proceeds (Table 1, entry 15) with a lower conversion and yield of [Pd(η3-cin)(Cl)(IPr)] in comparison with those of the reaction using Cl− derivatives.
With these optimal reaction conditions in hand, the scope of the reaction was assessed, and a series of Pd complexes 1–8 was obtained in high isolated yields (Scheme 3). Both imidazolylidene and imidazolinylidene complexes of palladium were successfully synthesised in good to excellent yields (66–85%). Interestingly, new Pd pre-catalysts bearing substitution on the backbone of the NHCs, containing electron-withdrawing Cl substituents (6) and an expanded π-conjugated system (8), were also fully compatible with the electrochemical synthetic protocol. To further showcase the applicability of the developed procedure we tested Pd precursors bearing various allyl-substituted ligands, namely [Pd(η3-allyl)(μ-Cl)]2 and [Pd(η3-1-tBu-indenyl)(μ-Cl)]2. While the reaction with [Pd(η3-allyl)(μ-Cl)]2 led to the final product 2 with a yield of 70%, the analogous transformation using [Pd(1-tBu-indenyl)(μ-Cl)]2 failed to deliver any of the corresponding product.
 | ||
Scheme 3 Electrochemical synthesis of Pd–NHC complexes. Reaction conditions: imidazol(in)ium (0.05 mmol), [Pd(η3-R-allyl)(μ-Cl)]2 (0.025 mmol), n-Bu4NBF4 (0.125 mmol), MeCN (2.5 mL) [Zn cathode]; n-Bu4NBF4 (0.125 mmol), MeCN (2.5 mL) [Zn anode]; split cell with a Nafion™ 117 membrane, constant current density (j) = 6.0 A m−2, r.t., 52 min (1.6 F mol−1). Isolated yields. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Acetone was used instead of acetonitrile. b1.1 F mol−1. Acetone was used instead of acetonitrile. b1.1 F mol−1. | ||
These results, along with previously observed reaction decomposition in the absence of oxygen, prompted us to explore the role of oxygen in this transformation through additional cyclic voltammetry (CV) experiments. To gain insight into the mechanism of the process, CV experiments were conducted in acetonitrile under inert atmosphere. The imidazolium salt exhibited a reduction at Ep = −2.19 V vs. Ag/AgCl/KCl (3N) (Fig. S2†), whereas both the palladate [IPrH][Pd(η3-cin)Cl2] and the product [Pd(η3-cin)(Cl)(IPr)] display higher reduction potentials, with values of Ep = −1.41 V (R2) and −1.46 V vs. Ag/AgCl (Fig. S1 and S3†), respectively. In air, due to the presence of oxygen, the CV displays a quasi-reversible reduction peak R1 at Ep = −0.89 V vs. Ag/AgCl, which could be attributed to the reduction of oxygen to the superoxide anion (Fig. 1).39–41 The gradual addition of the palladate precursor to an aerobic MeCN solution provoked a substantial nearly two-fold increase in R1, accompanied by the consistent disappearance of the oxidation peak (O1) on the reverse scan, indicative of superoxide anion consumption in the presence of the palladate (Fig. 1, Fig. S10†). Under these conditions, in an aprotic dipolar solvent, superoxide anions are recognized as highly reactive and soluble naked bases.42,43 Similarly, increasing the concentration of the [IPrH]BF4/[Pd(η3-cin)(μ-Cl)]2 1/0.5 mixture in MeCN led to an increase of R1 with loss of O1 (Fig. S11†). However, the further addition of IPr·HCl, even at four times the concentration (16 mM), did not result in the complete disappearance of the reverse peak O1 (Fig. S12†). This confirms the need of Pd for an efficient reaction with oxygen superoxide to proceed, as well as the required presence of the imidazolium cation to form the product. CVs were also recorded for [IPrH][Pd(η3-allyl)Cl2] (Fig. S7†) and [IPrH][Pd(η3-1-tBu-indenyl)Cl2] (Fig. S9†).
 | ||
| Fig. 1 CVs towards reduction potentials of MeCN and [IPrH][Pd(η3-cin)Cl2] (1–4 mM, in air and under Ar) in acetonitrile (0.1 M n-Bu4NPF6 as supporting electrolyte) at a glassy carbon disk electrode (d = 3 mm) with a scan rate of 0.1 V s−1 at 25 °C. | ||
Based on these observations, a mechanism for this transformation is proposed in Scheme 4: the cathodic half-reaction includes the reduction of molecular oxygen dissolved in the solvent at the cathode forming oxygen superoxide radical anions which can react with imidazolium cations by abstracting the H2-proton (evident from the disappearance of the reverse peak on the CV), with the resultant radical being further reduced to a hydroperoxide anion thereafter. Anodic half-reaction involves Zn sacrificial anode oxidation liberating Zn2+ cations to the anodic compartment. The presence of a hydroperoxide anion was confirmed by the qualitative KI/starch reaction (Fig. S13 and S14†). The oxygen superoxide anion is probably not a strong enough base to deprotonate the imidazolium C2 atom.44,45 Nonetheless, in the presence of the palladate anion, which intercepts accumulated free-carbene species, the equilibrium is shifted, which delivers the final desired product, with the entire sequence corresponding to the stepwise deprotonation/coordination mechanism.46 As the overall reaction occurs in air and does not darken as would be expected if carbene decomposition had taken place, we conclude that the reaction might proceed via a concerted mechanism, where both proton abstraction and reaction with the Pd centre occur simultaneously.47,48 Importantly, the proposed mechanism shows that the reaction is more likely to be efficient if both the metal precursor and the reaction product are stable under experimental conditions, that is, the precursor undergoes reduction at lower reduction potentials and the product does not.
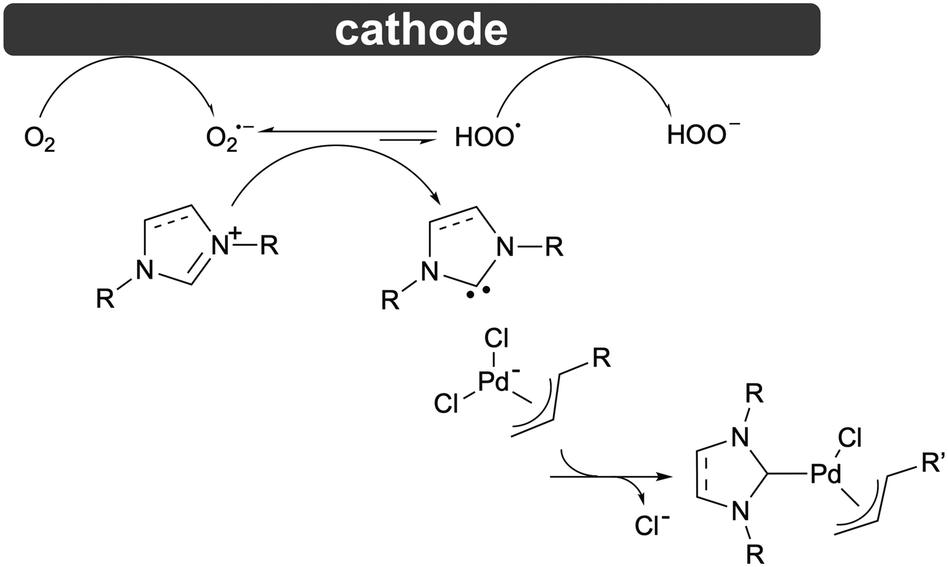 | ||
| Scheme 4 Proposed mechanism of [Pd(η3-cin)(Cl)(NHC)] formation at cathode (anode: Zn → Zn2+) in a split cell Zn|Zn. | ||
In summary, a novel electrochemical synthesis of Pd-cross-coupling pre-catalysts bearing NHC-ligands has been reported. The reaction occurs in air and makes use of superoxide anion-radicals as an electrogenerated base formed at the cathode as the product of oxygen reduction. We have implemented the developed procedure for the synthesis of eight palladium–NHC complexes. Ongoing work in our laboratories aims at exploring the scope of this protocol for the synthesis of other organometallic complexes.
Materials and methods
General considerations
1H and 13C–{1H} APT Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker Avance 300 and 400 Ultrashield spectrometers at 298 K. Chemical shifts (expressed in parts per million) are referenced to residual solvent peaks: (CHCl3: δH = 7.26 ppm, δC = 77.16 ppm) at 298 K in CDCl3. [Pd(η3-cin)(μ-Cl)]2 was used as received. n-Bu4NBF4 was dried at 110 °C for 6 h and then stored under dry nitrogen atmosphere. Acetonitrile was dried over MW-activated 3 Å molecular sieves for 14 h. Other solvents and commercially available reagents were used without preliminary purification. BIAN-IMes·HCl49 and SIPr(OMe)·HCl50 were synthesised following corresponding literature procedures. Elemental analyses were performed at Université de Namur. All reactions were carried out using an IKA ElectraSyn 2.0 device, a Metrohm Multi Autolab M204 potentiostat or an Origaflex OGF 500 potentiostat. When using a split cell two compartments were separated by an anion exchange membrane – a DuPont Nafion 117 perfluorinated membrane of 430 μm thickness (Aldrich). Cyclic voltammograms (CV) were recorded using a Metrohm Multi Autolab M204 potentiostat connected to a Nova software interface in a three-electrode cell in a glovebox at 25 °C with a scan rate of 0.1 V s−1 using a glassy carbon disk (d = 3 mm) as a working electrode, a platinum wire as a counter electrode and an Ag electrode as a quasi-reference electrode in 10 mL of a 0.1 M solution of n-Bu4NPF6 in the solvent. Ferrocene was added to the background solution at the end of the experiment to reference recorded peak potentials to the Ag/AgCl/3M KCl reference electrode potential with the ferrocene redox potential Fc+/Fc equal to 0.43 V (vs. Ag/AgCl/3M KCl).General procedure for the electrochemical preparation of Pd complexes
The cathodic compartment of a split electrochemical cell was loaded with NHC·HCl (0.05 mmol), [Pd(η3-cin)(μ-Cl)]2 or [Pd(η3-allyl)(μ-Cl)]2 (0.025 mmol), and n-Bu4NBF4 (0.125 mmol; 41.1 mg), whereas the anodic chamber was loaded with n-Bu4NBF4 (0.125 mmol; 41.1 mg). Acetonitrile (2.5 mL) was added to each compartment. An anode and a cathode (Zn [8 × 52.5 × 2 mm]; Simmersed = 4.2 cm2) were immersed into the solution in both compartments. Constant current electrolysis (I = 2.5 mA) was carried out at room temperature with the reaction mixture being constantly stirred. After completion of the reaction, the content of cathodic compartment was evaporated, transferred into a separation funnel containing EtOAc (15 mL) and washed 3 times with water (3 × 15 mL) portions. The organic phase was dried over MgSO4 and filtered. The solvent was removed on a rotary evaporator. Pentane (5 mL) was added and the suspension was subjected to sonication (10–15 min). The product was collected by filtration and washed with hexane (5 mL).Syntheses of novel compounds
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 4.35 (d, J = 13.0 Hz, 1H, cin PhCHCH), 3.96 (s, 4H, NCH2CH2N), 3.85 (s, 6H, OCH3), 3.40 (br s, 4 H, CH–CH3), 2.90 (br s, 1H, cin CH2 Hsyn), 1.62 (br s, 1H, cin CH2 Hanti), 1.40 (d, J = 6.7 Hz, 12H, CH–CH3), 1.25 (d, J = 6.7 Hz, 12H, CH–CH3). 13C{1H} NMR (100 MHz, CDCl3): δC (ppm) = 213.0 (NCN), 159.6 (C–OCH3), 148.7 (o-CHArOMe), 137.8 (i-CHArOMe), 129.8 (i-CAr), 128.4 (o-CHAr), 127.4 (m-CHAr), 126.8 (p-CHAr), 109.6 (m-CHArOMe), 109.2 (PhCHCH–CH2), 91.9 (PhCHCH–CH2), 55.3 (OCH3), 54.2(NCH2CH2N), 46.0 (cin PhCHCH–CH2), 28.9 (CH–CH3), 26.7 (CH–CH3). Elemental analysis: calculated: C 64.31, H 7.24, N 3.95. Found: C 64.08, H 7.41, N 3.85.
CH), 4.44 (d, J = 12.5 Hz, 1H, cin PhCHCH), 3.28 (d, J = 6.8 Hz, 1H, cin CH2 Hsyn), 2.42 (s, 6H, CH3), 2.34 (s, 6H, CH3), 2.29 (s, 6H, CH3), 2.05 (d, J = 11.9 Hz, 1H, cin CH2 Hanti).
CH), 4.35 (d, J = 13.0 Hz, 1H, cin PhCHCH), 3.96 (s, 4H, NCH2CH2N), 3.85 (s, 6H, OCH3), 3.40 (br s, 4 H, CH–CH3), 2.90 (br s, 1H, cin CH2 Hsyn), 1.62 (br s, 1H, cin CH2 Hanti), 1.40 (d, J = 6.7 Hz, 12H, CH–CH3), 1.25 (d, J = 6.7 Hz, 12H, CH–CH3). 13C{1H} NMR (100 MHz, CDCl3): δC (ppm) = 213.0 (NCN), 159.6 (C–OCH3), 148.7 (o-CHArOMe), 137.8 (i-CHArOMe), 129.8 (i-CAr), 128.4 (o-CHAr), 127.4 (m-CHAr), 126.8 (p-CHAr), 109.6 (m-CHArOMe), 109.2 (PhCHCH–CH2), 91.9 (PhCHCH–CH2), 55.3 (OCH3), 54.2(NCH2CH2N), 46.0 (cin PhCHCH–CH2), 28.9 (CH–CH3), 26.7 (CH–CH3). Elemental analysis: calculated: C 64.31, H 7.24, N 3.95. Found: C 64.08, H 7.41, N 3.85.
CH), 4.44 (d, J = 12.5 Hz, 1H, cin PhCHCH), 3.28 (d, J = 6.8 Hz, 1H, cin CH2 Hsyn), 2.42 (s, 6H, CH3), 2.34 (s, 6H, CH3), 2.29 (s, 6H, CH3), 2.05 (d, J = 11.9 Hz, 1H, cin CH2 Hanti).
13C{1H} NMR (100 MHz, CDCl3): δC (ppm) = 189.4 (NCN), 139.1 (CAr), 138.9 (CAr), 138.3 (CAr), 135.5 (CAr), 135.4 (CAr), 134.6 (CAr), 129.7 (CPh), 129.5 (CAr), 128.4 (CPh), 127.9 (CAr), 127.7 (CAr), 127.5 (CPh), 126.8 (CPh), 126.0 (CAr), 120.6 (CAr), 109.4 (PhCHCH–CH2), 90.3 (PhCHCH–CH2), 46.7 (cin PhCHCH–CH2), 21.4 (CH3), 18.6 (CH3), 18.6 (CH3). Elemental analysis: calculated: C 69.87, H 5.42, N 4.07. Found: C 69.60, H 5.53, N 4.46.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the FWO, BOF, CNRS and ENS-PSL for funding.References
- C. S. J. Cazin, N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Springer, London, U.K., 2011 Search PubMed.
- A. J. Arduengo, R. L. Harlow and M. A. Kline, A stable crystalline carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- D. J. Nelson and S. P. Nolan, Quantifying and understanding the electronic properties of N-heterocyclic carbenes, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- H. V. Huynh, Electronic Properties of N-Heterocyclic Carbenes and Their Experimental Determination, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- H. Clavier and S. P. Nolan, Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry, Chem. Commun., 2010, 46, 841–861 RSC.
- E. Peris, Smart N-heterocyclic carbene ligands in catalysis, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- X. Chen, K. M. Engle, D. H. Wang and Y. Jin-Quan, Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- N. Kambe, T. Iwasaki and J. Terao, Pd-catalyzed cross-coupling reactions of alkyl halides, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC.
- J. Rayadurgam, S. Sana, M. Sasikumar and Q. Gu, Palladium catalyzed C–C and C–N bond forming reactions: an update on the synthesis of pharmaceuticals from 2015–2020, Org. Chem. Front., 2021, 8, 384–414 RSC.
- N. Marion and S. P. Nolan, Well-defined N-heterocyclic carbenes–palladium(II) precatalysts for cross-coupling reactions, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- M. S. Viciu, O. Navarro, R. F. Germaneau, R. A. Kelly III, W. Sommer, N. Marion, E. D. Stevens, L. Cavallo and S. P. Nolan, Synthetic and Structural Studies of (NHC)Pd(allyl)Cl Complexes (NHC = N-heterocyclic carbene), Organometallics, 2004, 23, 1629–1635 CrossRef CAS.
- M. R. L. Furst and C. S. J. Cazin, Copper N-heterocyclic carbene (NHC) complexes as carbene transfer reagents, Chem. Commun., 2010, 46, 6924–6925 RSC.
- N. Marion, E. C. Ecarnot, O. Navarro, D. Amoroso, A. Bell and S. P. Nolan, (IPr)Pd(acac)Cl: an easily synthesized, efficient, and versatile precatalyst for C–N and C–C bond formation, J. Org. Chem., 2006, 71, 3816–3821 CrossRef CAS PubMed.
- E. A. Martynova, N. V. Tzouras, G. Pisanò, C. S. J. Cazin and S. P. Nolan, The “weak base route” leading to transition metal–N-heterocyclic carbene complexes, Chem. Commun., 2021, 57, 3836–3856 RSC.
- A. Simoens, T. Scattolin, T. Cauwenbergh, G. Pisanò, C. S. J. Cazin, C. V. Stevens and S. P. Nolan, Continuous Flow Synthesis of Metal–NHC Complexes, Chem. – Eur. J., 2021, 27, 5653–5657 CrossRef CAS PubMed.
- G. Pisanò and C. S. J. Cazin, Mechanochemical synthesis of Cu(I)-N-heterocyclic carbene complexes, Green Chem., 2020, 22, 5253–5256 RSC.
- G. Pisanò and C. S. J. Cazin, General Mechanochemical Synthetic Protocol to Late Transition Metal–NHC (N-Heterocyclic Carbene) Complexes, ACS Sustainable Chem. Eng., 2021, 9, 9625–9631 CrossRef.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Making electrochemistry easily accessible to the synthetic chemist, Green Chem., 2020, 22, 3358–3375 RSC.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, A Survival Guide for the “Electro-curious”, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- O. A. Levitskiy, O. I. Aglamazova, Y. K. Grishin, S. E. Nefedov and T. V. Magdesieva, Corey–Chaykovsky cyclopropanation of dehydroalanine in the Ni(II) coordination environment: Electrochemical vs. chemical activation, Electrochim. Acta, 2022, 409, 139980 CrossRef CAS.
- I. Chiarotto, L. Mattiello and M. Feroci, The electrogenerated cyanomethyl anion: an old base still smart, Acc. Chem. Res., 2019, 52, 3297–3308 CrossRef CAS PubMed.
- H. Veisi, A. Maleki and S. Jahangard, Electrogenerated base promoted synthesis of 3-methyl-4-aryl-2,4,5,7-tetrahydropyrazolo[3,4-b]pyridin-6-ones via multicomponent reactions of 5-methylpyrazol-3-amine, aldehydes, and Meldrum's acid, Tetrahedron Lett., 2015, 56, 1882–1886 CrossRef CAS.
- B. Gorodetsky, T. Ramnial, N. R. Branda and J. A. C. Clyburne, Electrochemical reduction of an imidazolium cation: a convenient preparation of imidazol-2-ylidenes and their observation in an ionic liquid, Chem. Commun., 2004, 17, 1972–1973 RSC.
- I. Abdellah, C. Gueugnot Sylvie and C. Pichon, Direct Electrochemical Reduction of Azolium Salts Into N-Heterocyclic Carbenes and Their Subsequent Trapping, Curr. Top. Electrochem., 2011, 16, 81–91 CAS.
- K. A. Ogawa and A. J. Boydston, Electrochemical characterization of azolium salts, Chem. Lett., 2014, 43, 907–909 CrossRef CAS.
- D. Rocco, A. A. Folgueiras-Amador, R. C. D. Brown and M. Feroci, First example of organocatalysis by cathodic N-heterocyclic carbene generation and accumulation using a divided electrochemical flow cell, Beilstein J. Org. Chem., 2022, 18, 979–990 CrossRef CAS PubMed.
- For an overview of the state of the art, see: C. Schotten, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Electrochemical Generation of N–Heterocyclic Carbenes for Use in Synthesis and Catalysis, Adv. Synth. Catal., 2021, 363, 3189–3200 CrossRef CAS.
- D. Rocco, I. Chiarotto, F. D'Anna, L. Mattiello, F. Pandolfi, C. Rizzo and M. Feroci, Cathodic behaviour of dicationic imidazolium bromides: The role of the spacer, ChemElectroChem, 2019, 6, 4275–4283 CrossRef CAS.
- G. Forte, I. Chiarotto, A. Inesi, M. A. Loreto and M. Feroci, Electrogenerated N-Heterocyclic Carbene in Ionic Liquid: An Insight into the Mechanism of the Oxidative Esterification of Aromatic Aldehydes, Adv. Synth. Catal., 2014, 356, 1773–1781 CrossRef CAS.
- F. Vetica, M. Bortolami, R. Petrucci, D. Rocco and M. Feroci, Electrogenerated NHCs in organic synthesis: ionic liquids vs organic solvents effects, Chem. Rec., 2021, 21, 2130–2147 CrossRef CAS PubMed.
- B. Liu, Y. Zhang, D. Xu and W. Chen, Facile synthesis of metal N-heterocyclic carbene complexes, Chem. Commun., 2011, 47, 2883–2885 RSC.
- B. R. M. Lake, E. K. Bullough, T. J. Williams, A. C. Whitwood, M. A. Little and C. E. Willans, Simple and versatile selective synthesis of neutral and cationic copper(I) N-heterocyclic carbene complexes using an electrochemical procedure, Chem. Commun., 2012, 48, 4887–4889 RSC.
- C. Schotten, C. J. Taylor, R. A. Bourne, T. W. Chamberlain, B. N. Nguyen, N. Kapur and C. E. Willans, Alternating polarity for enhanced electrochemical synthesis, React. Chem. Eng., 2021, 6, 147–151 RSC.
- M. R. Chapman, Y. M. Shafi, N. Kapur, B. N. Nguyen and C. E. Willans, Electrochemical flow-reactor for expedient synthesis of copper-N-heterocyclic carbene complexes, Chem. Commun., 2015, 51, 1282–1284 RSC.
- H. R. Stephen, C. Schotten, T. P. Nicholls, M. Woodward, R. A. Bourne, R. A. Bourne, N. Kapur and C. E. Willans, A versatile electrochemical batch reactor for synthetic organic and inorganic transformations and analytical electrochemistry, Org. Process Res. Dev., 2020, 24, 1084–1089 CrossRef CAS.
- T. P. Nicholls, Z. Jia and J. M. Chalker, Electrochemical Synthesis of Gold-N-Heterocyclic Carbene Complexes, Chem. – Eur. J., 2024, 30, e202303161 CrossRef CAS PubMed.
- C. M. Zinser, F. Nahra, M. Brill, R. E. Meadows, D. B. Cordes, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, A simple synthetic entryway into palladium cross-coupling catalysis, Chem. Commun., 2017, 53, 7990–7993 RSC.
- C. M. Zinser, K. G. Warren, F. Nahra, A. Al-Majid, A. Barakat, M. S. Islam, S. P. Nolan and C. S. J. Cazin, Palladate precatalysts for the formation of C–N and C–C bonds, Organometallics, 2019, 38, 2812–2817 CrossRef CAS.
- D. T. Sawyer and J. L. Roberts Jr., Electrochemistry of oxygen and superoxide ion in dimethylsulfoxide at platinum, gold and mercury electrodes, J. Electroanal. Chem., 1966, 12, 90–101 CAS.
- A. D. Goolsby and D. T. Sawyer, Electrochemical reduction of superoxide ion and oxidation of hydroxide ion in dimethyl sulfoxide, Analyt. Chem., 1968, 40, 83–86 CrossRef CAS.
- D. Bauer and J.-P. Beck, Électrochimie de l'oxygène et de ses produits de réduction dans les solvants et les sels fondus, J. Electroanal. Chem., 1972, 40, 233–254 CrossRef CAS.
- F. Pandolfi, I. Chiarotto, D. Rocco and M. Feroci, Electrogenerated superoxide anion induced oxidative amidation of benzoin, Electrochim. Acta, 2017, 254, 358–367 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. Alnashef, Superoxide ion: generation and chemical implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- C. P. Andrieux, P. Hapiot and J.-M. Saveant, Mechanism of superoxide ion disproportionation in aprotic solvents, J. Am. Chem. Soc., 1987, 109, 121–156 Search PubMed.
- M. H. Dunn, N. Konstandaras, M. L. Cole and J. B. Harper, Targeted and Systematic Approach to the Study of pKa Values of Imidazolium Salts in Dimethyl Sulfoxide, J. Org. Chem., 2017, 82, 7324–7331 CrossRef CAS PubMed.
- R. Saito, A. Prato, A. Rubbi, L. Orian, T. Scattolin and S. P. Nolan, Simple synthesis of [Au(NHC)X] complexes utilizing aqueous ammonia: revisiting the weak base route mechanism, Dalton Trans., 2025, 54, 59–64 RSC.
- T. Scattolin, N. V. Tzouras, L. Falivene, L. Cavallo and S. P. Nolan, Using sodium acetate for the synthesis of [Au(NHC)X] complexes, Dalton Trans., 2020, 49, 9694–9700 RSC.
- N. V. Tzouras, F. Nahra, L. Falivene, L. Cavallo, M. Saab, K. Van Hecke, A. Collado, C. J. Collet, A. D. Smith, C. S. J. Cazin and S. P. Nolan, A Mechanistically and Operationally Simple Route to Metal-N-Heterocyclic Carbene (NHC) Complexes, Chem. – Eur. J., 2020, 26, 4515–4519 CrossRef CAS PubMed.
- T. Tu, Z. Sun, W. Fang, M. Xu and Y. Zhou, Robust acenaphthoimidazolylidene palladium complexes: highly efficient catalysts for Suzuki–Miyaura couplings with sterically hindered substrates, Org. Lett., 2012, 14, 4250–4253 CrossRef CAS PubMed.
- S. Meiries and S. P. Nolan, A new synthetic route to p-methoxy-2,6-disubstituted anilines and their conversion into N-heterocyclic carbene precursors, Synlett, 2014, 393–398 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and mechanistic studies, NMR spectra and cyclic voltammograms. See DOI: https://doi.org/10.1039/d5dt00575b |
| This journal is © The Royal Society of Chemistry 2025 |