
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced electron transfer cascade between Mo- and W-based polyoxometalates†
Manu
Sánchez
 ab,
Ana
González
a,
Emma
Guerrero-Ortega
a,
Vipul
Bansal
*c and
Jose M.
Dominguez-Vera
*a
ab,
Ana
González
a,
Emma
Guerrero-Ortega
a,
Vipul
Bansal
*c and
Jose M.
Dominguez-Vera
*a
aDepartamento de Química Inorgánica and Instituto de Biotecnología, Universidad de Granada, Granada 18071, Spain. E-mail: josema@ugr.es
bDonostia International Physics Center, DIPC, Donostia 20018, Gipuzkoa, Spain
cSir Ian Potter NanoBiosensing Facility, NanoBiotechnology Research Laboratory, RMIT University, Melbourne, VIC 3000, Australia. E-mail: vipul.bansal@rmit.edu.au
First published on 5th May 2025
Abstract
The Keggin-type polyoxometalates phosphotungstic acid (PTA, H3PW12O40) and phosphomolybdic acid (PMA, H3PMo12O40) can undergo reduction through chemical and photochemical methods. However, the higher electron affinity of Mo(VI) relative to W(VI) and the higher LUMO delocalisation in PTA result in different redox behaviours of the two polyoxometalates. We have identified specific experimental conditions that allow for the photoreduction of PTA in the presence of UV radiation and an electron donor, such as isopropanol (IPA), while PMA does not undergo this reaction. This distinct redox behaviour of polyoxometalates has been leveraged to develop a photoinduced electron transfer cascade from PTA to PMA that can be switched on and off by light. To demonstrate the unique capabilities of this transfer, the mimicry of the early stages of photosynthesis has been achieved by introducing the redox pair benzoquinone (BQ)-hydroquinone (HQ) between PTA and PMA. The transient photoexcitation of the four-component PTA–BQ–HQ–PMA system in the presence of IPA, triggered a cascade of reactions. Initially, PTA was reduced to PTAred, which, under dark conditions, could reduce BQ to HQ. HQ then reduced PMA to PMAred. This cascade of reactions, akin to the first stage of photosynthesis, was facilitated by the generation of a proton (H+) gradient through the spatial separation of the positive holes created at the PTA terminal and the excited electron migrated across the cascade and localized at the PMAred terminal. This represents the first example of a unidirectional photo-induced electron transfer cascade between four molecular components.
Introduction
The study of photoinduced electron transfer reactions has been a prominent area of research due to their crucial involvement in the biological process of photosynthesis and the design of solar cells. In the first stage of photosynthesis, as the incident photon from the sunlight is absorbed by the light-harvesting complex (LHC) in chlorophyll, the electrons transferred to LHC from the oxygen-evolving complex (OEC) are promoted to higher energy state.1,2 These high-energy electrons now enter a redox cascade process, resulting in their transfer to plastoquinone (PQ) and two protons to form plastoquinol (PQH2), which is released as a lipid-soluble mobile electron carrier.3 Through a cascade of intermediate reactions, these electrons are transferred from PQH2 to water-soluble electron carriers, finally reducing NADP+ to NADPH, the latter being the universal agent to reduce CO2 into sugars.4 The process creates a proton gradient that continues to drive the reaction in the forward direction, as water molecules continue to serve as sacrificial electron donors to feed electrons into this cascade redox process, producing diatomic oxygen as the by-product (Scheme 1).1,2 | ||
| Scheme 1 Redox reactions in the early stages of photosynthesis. | ||
Inspired by natural photosynthesis, the scientific community has developed the concept of artificial photosynthesis,5 which attempts to mimic natural photosynthesis to create an efficient way to convert sunlight into storable energy forms, primarily hydrogen. One of the keys to successful artificial photosynthesis systems is the ability to resist recombination of the charge carriers generated during photoexcitation for a sufficient period to allow the excited electrons and electron vacancies (holes) to become available for reduction and oxidation reactions, respectively. The most common configuration for an artificial photosynthetic system is one in which a chromophore is covalently bound to the electron acceptor, or to both the electron acceptor and electron donor molecules.6–11 An alternative approach involves the use of semiconductors, which, upon photoexcitation, produce an excited electron at the conduction band and a positive hole in their ground state.12–14
The primary limitation of both chromophore- and semiconductor-based systems is the rapid charge recombination.15–19 The avoidance of undesired charge recombination represents a fundamental challenge in the pursuit of functional artificial photosynthesis. In photosynthesis, this recombination does not occur, as the excited electrons enter an electron cascade through a series of acceptor-donor multi-redox systems, which spatially separate the excited electron and electron vacancy sites.
Inspired by photosynthesis, the transport of both holes and electrons to spatially separate them appears as one of the most promising strategies to avoid charge recombination.
It has been demonstrated that certain polyoxometalates (POMs) exhibit certain characteristics like those observed in semiconducting particles.20 POMs possess distinctive advantages as photocatalysts due to their chemical stability in electron transfer processes.21 Indeed, POMs have been demonstrated to function as photocatalysts in response to light irradiation. This is achieved by utilizing the positive holes (h+) and excited electrons (e−) (eqn (1)) for oxidizing and reducing reactions, respectively.22
| POM + hv ⇌ POM*(e− + h+) | (1) |
A particularly noteworthy attribute of POMs is their photochromic behaviour, whereby they undergo a chemical reduction from colourless to a coloured form under light irradiation in the presence of a sacrificial electron donor.23,24 These reduced POMs, commonly known as ‘heteropoly blues’, exhibit characteristic deep blue or green colours with high extinction coefficients.25,26
A large variety of POMs with a combination of different transition metals (especially Mo and W) and heteroatoms (especially P and Si) in their structures have been reported.27–31 Each POM displays reducibility characteristics depending on the transition metal cluster embedded in the structure. For instance, Keggin-type Mo-based POMs are generally more effective chemical redox catalysts,32 whereas W-based POMs are more powerful oxidizing agents under UV and visible light radiations.23,24 In this paper, the distinct redox behaviors of two Keggin W- and Mo-POMs, phosphotungstic acid (PTA) and phosphomolybdic acid (PMA), respectively, have been exploited to develop a photoinduced electron transfer from PTA to PMA. This transfer is switched on and off by light. The redox pair BQ/HQ was then introduced between the PTA and PMA, creating a unidirectional photo-induced electron cascade system, designated PTA–BQ–HQ–PMA. To the best of the authors’ knowledge, no examples of a photo-induced multi-redox system containing four molecular components have been reported.
Experimental
Reagents and instruments
The following chemical compounds used in this work were procured from Sigma-Aldrich: phosphotungstic acid hydrate (H3PW12O40·xH2O), phosphomolybdic acid hydrate (H3PMo12O40·xH2O), cysteine, ascorbic acid, 1,4-benzoquinone (p-BQ), hydroquinone (HQ), and all alcohol reagents. The list of alcohols subjected to testing included ethanol (EtOH), methanol (MeOH), 2-propanol (IPA), cyclohexanol (CyOH) and tert-butyl alcohol (t-BuOH). The reagents were weighed using a BOECO Balance BAS 31 plus analytical balance with a sensitivity of 0.1 mg.The ultraviolet (UV) light source was a Jiadi 36-watt nail gel curing UV lamp, comprising four 9-watt halogen tubes with a maximum wavelength of 365 nm (UVA). The UV-Vis absorbance spectra of the solutions were recorded using a Cary 3500 Multicell UV-Vis Spectrophotometer. A Hewlett Packard 7890A Agilent gas chromatographer (USA) coupled to a mass spectrometer triple quadrupole Quattro microGC from Waters (USA) was used for HS-GC analysis.
Photoinduced (UV) reduction of PTA in the presence of alcohol as sacrificial electron donors
Mixtures of degassed water solutions of PTA (5 mM) and alcohols (50 mM) were exposed to UVA light for up to 60 min. The characteristic UV-Vis bands of reduced PTA (PTAred) at 490 and 754 nm developed with time.Chemical reduction of PTA and PMA
A study was conducted using UV-Vis spectroscopy to analyse the effect of ascorbic acid and cysteine (5 and 10 mM) on the colour development of water solutions of PTA or PMA (5 mM) in the absence of light. The results demonstrated that only the PMA solutions exhibited coloration, indicating the formation of reduced PMA (PMAred), as evidenced by a characteristic UV-Vis band centred at 840 nm.Electron transfer from PTAred to PMA
The initial step involved the preparation of a degassed solution of PTA (5 mM) and its photoreduction in the presence of IPA, as previously outlined. Subsequently, a degassed solution of PMA (5 mM) was added with a syringe, and the light was deactivated. The UV-Vis spectrum of the mixture matched that of PMAred, with an absorption band centred at 840 nm. The same result was achieved when a degassed solution containing IPA and both PTA (5 mM) and PMA (5 mM) was first irradiated with UV for 60 min and then switched off. The UV-Vis spectrum of the irradiated mixture matched that of PMAred, while the spectrum collected in the absence of light corresponded to that of the initial colourless PTA and PMA mixture.Electron transfer PTA–BQ–HQ–PMA
The initial step involved the preparation of a degassed solution of PTA (5 mM) in the presence of IPA, as previously outlined. Subsequently, a solution of BQ was added to the airtight cuvette via syringe to a final concentration of 2 mM. The blue colour of PTAred was immediately replaced by a clear solution. Subsequently, a solution of PMA (5 mM) was added to the mixture. The addition of the solution resulted in the immediate development of a blue colour, which matched the UV-Vis spectrum of PMAred, exhibiting a large absorption band at 840 nm.Gas chromatography-mass spectrometry (GC-MS)
To compare the different samples prepared for the HS-GC analysis, we used standards of IPA, acetone, EtOH, acetaldehyde, cyclohexanol, cyclohexanone and t-BuOH. The samples for each alcohol were as follows: (1) PTA:alcohol non-irradiated with UV light (t0), (2) PTA:alcohol irradiated with UV light for 90 min (t90), and (3) PTA:alcohol irradiated with UV light for 720 min (t720). For the PTA:IPA/BQ/PMA samples, they were as follows: (1) PTA:IPA/BQ/PMA non-irradiated with UV light (t0), (2) PTA:IPA/BQ/PMA irradiated with UV light for 90 min (t90), and (3) PTA:IPA/BQ/PMA irradiated with UV light for 720 min (t720). PTA and alcohols concentrations were 5 mM and 50 mM, respectively. To prevent reoxidation by air, samples were gradually injected into the chromatography column immediately after irradiation. The vials were stored at room temperature until they were inserted into the autosampler, at which point they were heated to equilibrium at 70 °C for 10 min with an agitation speed of 500 rpm. A 0.5 mL sample of the headspace was injected into the column (a capillary column VOCOL from Supelco, USA) with the injector and transfer line both maintained at 220 °C. The GC conditions were as follows: the column temperature began at 40 °C, was held for 2 min, then increased to 200 °C at a rate of 8 °C min−1 for a total of 20 min. The GC was operated with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 split ratio and a 7 mL min−1 continuous column flow rate, using helium as the carrier gas. The mass spectrometer was operated in electron impact ionization (EI+) mode at 70 eV. The full scan was 20–250 Da, and the source temperature was 220 °C.
:1 split ratio and a 7 mL min−1 continuous column flow rate, using helium as the carrier gas. The mass spectrometer was operated in electron impact ionization (EI+) mode at 70 eV. The full scan was 20–250 Da, and the source temperature was 220 °C.
Results and discussion
The capacity for reduction exhibited by each POM is contingent upon the nature and energy of its lowest unoccupied molecular orbital (LUMO). It has been demonstrated that as the electronegativity of the metal centre in the POM increases, the energy of the LUMO decreases, thereby facilitating chemical reduction.33,34 The electron affinity of Mo(VI) is significantly higher than that of W(VI), making PMA more readily reducible with a standard reducing agent than PTA. On the other hand, although PMA has a smaller HOMO–LUMO gap compared to PTA,35 it is not necessarily more efficient at photoreduction, as this efficiency also depends on LUMO delocalisation, and PTA has more delocalised LUMO than PMA, which favours its photoactivity.36Therefore, while both PTA23 and PMA24,37 can be reduced chemically and photochemically, it is plausible to identify conditions under which their distinct redox behaviours become discernible. For instance, we found that while PTA (H3PW12O40) is readily reduced by UV irradiation in the presence of IPA, a sacrificial electron donor, PMA (H3PMo12O40) does not undergo efficient photoreduction in the presence of the same electron donor. Conversely, PMA is readily reduced by conventional chemical reducing agents (e.g., ascorbic acid, AA, or cysteine, Cys) in the absence of light, whereas PTA is not. Fig. 1 presents absorption spectroscopy data that supports the selective photo- vs. chemo-reducibility of PTA and PMA, respectively. A deoxygenated water solution of PTA containing an excess of IPA was reduced to heteropoly blue following UV exposure. The UV-Vis spectrum displayed a shift in the UV band (Fig. S1†) together with the appearance of the characteristic absorption bands of the 2e−PTAred (Fig. 1), a small band centred at 490 nm (intra-intervalence charge transfer, IVCT W(V)–W(VI)) and a large one at 754 nm (inter-IVCT, W(V)–W(VI)) with an extinction coefficient of 21.000 M−1.25,27 In contrast, the aqueous solution of PMA containing IPA exhibited minimal reduction under similar photoexcitation conditions (Fig. 1B). Alternatively, the reduction of PMA to heteropoly blues was achieved efficiently when chemical reducing agents, such as AA and Cys, were employed. The UV-Vis spectra of PMAred does not exhibit the band at 490 nm, in contrast to PTAred. However, two IVCT absorption bands associated with the 2e−PMAred and 4e−PMAred products are present. As demonstrated in Fig. 1B, the absorption of these reduction products occurs at 700 and 840 nm, respectively.37 The extinction coefficient of the band due to the 4-electron reduction (26.000 M−1) is significantly higher than that of the 2-electron reduction (11.000 M−1).38 This finding provides evidence to support the experimental observation that the absorbances values of the spectra corresponding to the reduction of PMA with AA are higher than those with Cys, as AA is a stronger reducing agent than Cys, with standard reduction potentials of −81 mV (ref. 39) and −34 mV,40 respectively.
 | ||
| Fig. 1 Selective photo- vs. chemo-reducibility of PTA and PMA. (A) Spectra of UV-irradiated PTA in the absence or presence of IPA (50 mM) for 60 min. (B) UV-Vis absorbance spectra of 5 mM PTA in the presence of cysteine (Cys, 5 and 10 mM) or ascorbic acid (AA, 5 and 10 mM) conducted without photoexcitation. (C) Spectra of UV-irradiated PMA in the presence of IPA (50 mM) for 60 min. (D) UV-Vis spectra of 5 mM PMA mixed with Cys (5 and 10 mM) or AA (5 and 10 mM) conducted without photoexcitation. | ||
We exploited this astonishing photo- vs. chemo-selective redox behaviours of PTA and PMA to design a photoinduced electron transfer system by controlling the switch-on/off of the light stimulus (Fig. 2). First, PTA was photo-reduced (switch-on) in the presence of IPA, then PMA was introduced, and the light was switched off. This resulted in the absorbance profile of the system to swiftly shift from the typical spectra observed for PTAred to that of PMAred. These results reveal the ability of this system to undergo spontaneous electron transfer from PTAred to PMA, to produce PMAred. The same result was obtained when both PTA and PMA were co-dissolved in water containing IPA and then, a UV switch on/switch off sequence was applied (see Experimental). The resulting UV-Vis spectrum of the mixture was that corresponding to PMAred. Notably, this electron transfer system is not reversible, as once PMAred is formed through either of the above two exemplified cases, the exposure of this system to UV radiation could not produce PTAred.
 | ||
| Fig. 2 A PTA–PMA photo switchable redox system. (A) Schematic representation of the PTA–PMA photo switch showing the ability of PMA to reoxidise PTAred while being reduced to PMAred in the absence of any additional photo- or chemical reducing agent, and (B) UV-Vis absorbance spectra of PTAred formed by UV-induced reduction of 5 mM PTA for 60 min (switch-on) compared with that of the PTAred–PMA redox system at 1 and 10 h after PMA addition to PTAred under ambient, non-photo irradiated conditions (switch-off). | ||
Following confirmation of the photo-induced electron transfer between these POMs, we investigated the potential for the construction of a spontaneously driven redox cascade through the introduction of a two-molecule redox pair between the PTA–PMA system. The objective was to emulate the process of natural photosynthesis, whereby the excited electron and electron vacancy sites are separated. This would be achieved by selecting a molecule that can be reduced and then oxidized, allowing electrons to be extracted from PTAred and transferred to PMA. Given the exceptional redox properties of quinones during the light reaction of photosynthesis (Scheme 1), we selected the benzoquinone/hydroquinone (BQ/HQ) pair as the basis for our electron transfer cascade (Fig. 3A). Therefore, when BQ was introduced to an aqueous solution containing PTAred, the dark heteropoly blue colour of PTAred promptly vanished (Fig. 3B) upon the termination of illumination, accompanied by a complete reduction in the characteristic absorbance bands of PTAred (490 and 754 nm) (Fig. 3C). These observations support that BQ could oxidize PTAred to PTA, while being simultaneously reduced to HQ (Fig. 3A). Upon introduction of PMA to the mixture, a change in colour was observed, indicating a reduction of PMA to PMAred. This conclusion is supported by the appearance of the typical absorbance spectra of PMAred. These observations lend support to the hypothesis that the PTA–BQ–HQ–PMA system functions as a unidirectional, spontaneous, photo-induced electron transport system (Fig. 3A).
 | ||
| Fig. 3 (A) The schematic representation of the photo-induced PTA–BQ–HQ–PMA redox cascade system. (B) Photographs of reactions during each step of the multi-redox cascade process; and (C) UV-Vis absorbance spectra supporting the operation of the multi-redox cascade system, as evidenced from the appearance of typical PTAred heteropoly blue spectrum (blue line) after UV irradiation of an aqueous solution of PTA containing IPA (black line). When the UV was turned off and BQ was added (purple line for pure BQ), the bands of PTAred disappeared (red line). Subsequent addition of PMA resulted in PMAred (green line). | ||
After these results, we investigated the fate of the species produced at the positive holes generated at PTA after its photoexcitation. The reaction at the PTA terminal was probed by analysing the oxidation of different alcohols introduced to the PTA–BQ–HQ–PMA cascade before the UV excitation. To begin with, we employed gas chromatography to investigate the chemical species formed from the hole-mediated oxidation of IPA, which revealed conversion of IPA to acetone. Fig. S2† shows the chromatograms of the mixture PTA/BQ/PMA in an aqueous solution containing IPA when exposed to UV at different times. Meanwhile IPA has a peak at 5.92 min, a new peak at 6.19 min develops with light irradiation and becomes predominant after 720 min. The mass spectrum of this peak confirmed the presence of acetone formed via the oxidation of IPA, with peaks corresponding to the entire molecule (molecular weight of 58 g mol−1) and its characteristic fragmentation (43 g mol−1). These results confirm the ability of the hole generated in the PTA–BQ–HQ–PMA system after UV irradiation to oxidize IPA.
Next, the ability of different alcohols to serve as potential sacrificial electron donors in PTA/BQ/PMA was tested, as previously reported for single PTA, by studying the photoreduction of PTA in the presence of these alcohols (Fig. 4).41 These alcohols include primary alcohols such as methanol (MeOH) and ethanol (EtOH), secondary alcohols such as 2-propanol (IPA) and cyclohexanol (CyOH) and tertiary alcohols such as tert-butyl alcohol (t-BuOH). As expected, only primary and secondary alcohols played the role of efficient sacrificial electron donor, allowing the photoreduction of PTA with their concomitant oxidation to aldehydes and ketones, respectively (Fig. S3†). While MeOH, EtOH and IPA appear to be equally efficient, CyOH is not able to reduce PTA with a similar efficiency. It is not surprising as cyclic alcohols are more resistant to oxidation compared to their corresponding non-cyclic forms. These results are similar to those previously reported for the simple PTA/alcohol pairs, after UV irradiation,23 which confirms that the PTA/BQ/HQ/PMA system has the same ability as the stand-alone PTA to carry out photo-oxidation reactions.
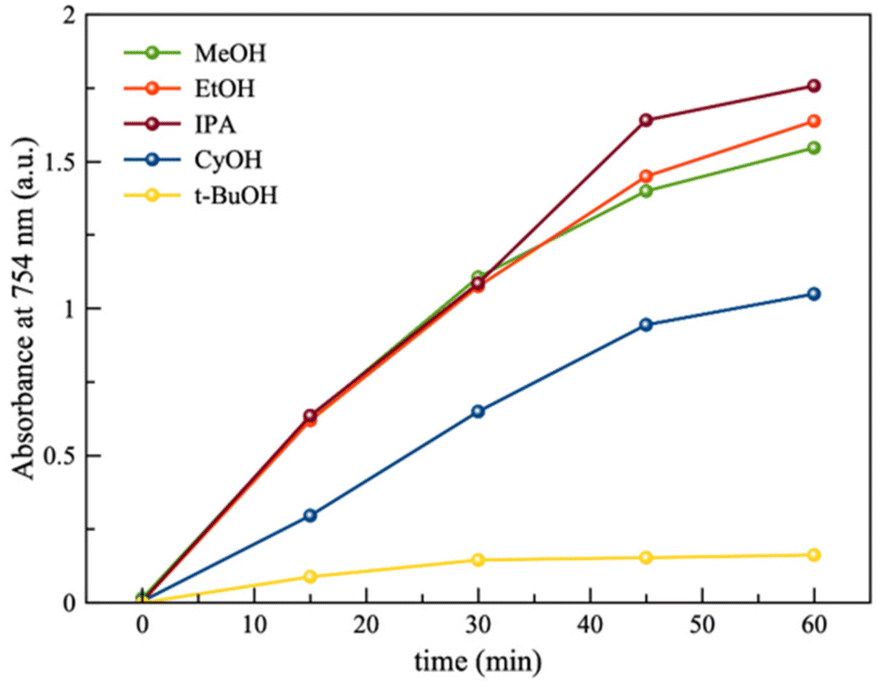 | ||
| Fig. 4 Photooxidation of different alcohols by PTA/BQ–HQ/PMA with time, as probed by the absorbance intensity of the UV-vis spectra at 754 nm (corresponding to PTAred). | ||
Conclusions
We have developed a four-component PTA–BQ–HQ–PMA system that, after UV irradiation (light switch on), generated an electron cascade process starting with the reduction of PTA, followed by electron transfer to benzoquinone to generate hydroquinone (light switch off), which then reduces PMA. On the other hand, we have shown that the positive hole generated on PTA in the PTA–BQ–HQ–PMA system after UV irradiation is able to carry out the oxidation of some alcohols, as previously reported for PTA alone. To the best of the authors’ knowledge, no examples of photo-induced multi-redox polyoxometalate systems containing four molecular components have been reported. Like natural photosynthesis, which involves the oxidation of water to molecular oxygen through a photoinduced electron cascade process, the current work shows the oxidation of isopropanol to acetone via a similar mechanistic process. The next steps will focus on enhancing the efficiency of this process to allow direct water oxidation. Nevertheless, the findings presented here offer a new avenue to expand global efforts in achieving artificial photosynthesis.Author contributions
All authors have given approval to the final version of the manuscript. The manuscript was written by V. B. and J. M. D.-V., M. S.: Synthesis, gas chromatography and mass spectra. A. G: Synthesis and validation. E. G.-O.: synthesis.Data availability
Data for this article are available at Zenodo at https://doi.org/10.5281/zenodo.15013444.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the Spanish Ministerio de Ciencia, Innovación y Universidades (MICIU) (project TED2021-130392A-I00) and Junta de Andalucía (FQM-368). A. G. acknowledge University of Granada for the postdoctoral contract within the Plan Propio UGR.References
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley, 2002 Search PubMed.
- N. Nelson and A. Ben-Shem, Nat. Rev. Mol. Cell Biol., 2004, 5, 971–982 CrossRef CAS PubMed.
- S. Pintscher, R. Pietras, B. Mielecki, M. Szwalec, A. Wójcik-Augustyn, P. Indyka, M. Rawski, Ł. Koziej, M. Jaciuk, G. Ważny, S. Glatt and A. Osyczka, Nat. Plants, 2024, 10, 1814–1825 CrossRef CAS PubMed.
- C. A. Raines, Photosynth. Res., 2003, 75, 1–10 CrossRef CAS PubMed.
- C. Bohne, Q. Pan, P. Ceroni, K. Börjesson, J. Rohacova, F. Lewis, A. Vlcek, D. M. Bassani, F. Würthner, A. Sartorel, A. P. de Silva, D. Nocera, F. Scandola, C. Lemon, C. Allain, G. W. Brudvig, S. Marchesan, V. Sundstrom, S. Campagna, S. W. Sheehan, P.-A. Plötz, F. Monti, J. M. Kelly, E. Gibson, M. Maneiro, A. Harriman, A. Ruggi, E. Galoppini, R. Thummel, J. Weinstein, J. Vos, O. Ishitani, D. Gust and A. Díaz-Moscoso, Faraday Discuss., 2015, 185, 187–217 RSC.
- G. Bottari, G. De La Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768–6816 CrossRef CAS PubMed.
- F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410–1422 CrossRef.
- U. Shee, D. Sinha, S. Mondal and K. K. Rajak, Dalton Trans., 2024, 53, 8254–8263 RSC.
- L. Martín-Gomis, G. Rotas, K. Ohkubo, F. Fernández-Lázaro, S. Fukuzumi, N. Tagmatarchis and Á. Sastre-Santos, Nanoscale, 2015, 7, 7437–7444 RSC.
- M. Marchini, A. Luisa, G. Bergamini, N. Armaroli, B. Ventura, M. Baroncini, N. Demitri, E. Lengo and P. Ceroni, Chem. – Eur. J., 2021, 27, 16250–16259 CrossRef CAS PubMed.
- L. Martín-Gomis, F. Peralta-Ruiz, M. B. Thomas, F. Fernández-Lázaro, F. D'Souza and Á. Sastre-Santos, Chem. – Eur. J., 2017, 23, 3863–3874 CrossRef PubMed.
- K. Achilleos, A. Katsari, E. Nikoloudakis, F. Chatzipetri, D. Tsikritzis, K. Ladomenou, G. Charalambidis, E. Stratakis and A. G. Coutsolelos, Dalton Trans., 2025, 54, 328–336 RSC.
- H. Tada, Dalton Trans., 2022, 51, 3383–3393 RSC.
- N. Zhang and Y. Xiong, Adv. Sens. Energy Mater., 2023, 2, 100047 CrossRef.
- H. Wang, W. Liu, S. Jin, X. Zhang and Y. Xie, ACS Cent. Sci., 2020, 6, 1058–1069 CrossRef CAS PubMed.
- S. Fukuzumi, K. Ohkubo and T. Suenobu, Acc. Chem. Res., 2014, 47, 1455–1464 CrossRef CAS PubMed.
- T. Hasobe, Phys. Chem. Chem. Phys., 2010, 12, 44–57 RSC.
- V. M. Blas-Ferrando, J. Ortiz, K. Ohkubo, S. Fukuzumi, F. Fernández-Lázaro and Á. Sastre-Santos, Chem. Sci., 2014, 5, 4785–4793 RSC.
- M. Sekita, B. Ballesteros, F. Diederich, D. M. Guldi, G. Bottari and T. Torres, Angew. Chem., Int. Ed., 2016, 55, 5560–5564 CrossRef CAS PubMed.
- A. Hiskia, A. Mylonas and E. Papaconstantinou, Chem. Soc. Rev., 2001, 30, 62–69 RSC.
- Y. Wu and L. Bi, Catalysts, 2021, 11, 524 CrossRef CAS.
- S. Y. Lai, K. H. Ng, C. K. Cheng, H. Nur, M. Nurhadi and M. Arumugam, Chemosphere, 2021, 263, 128244 CrossRef CAS PubMed.
- M. Sánchez, A. González, L. Sabio, W. Zou, R. Ramanathan, V. Bansal and J. M. Dominguez-Vera, Mater. Today Chem., 2021, 21, 1000491 Search PubMed.
- W. Zou, A. González, D. Jampaiah, R. Ramanathan, M. Taha, S. Walia, S. Sriram, M. Bhaskaran, J. M. Dominguez-Vera and V. Bansal, Nat. Commun., 2018, 9, 3743 CrossRef PubMed.
- E. Papaconstantinou, Chem. Soc. Rev., 1989, 18, 1–31 RSC.
- M. T. Pope and G. M. Varga, Inorg. Chem., 1966, 5, 1249–1254 CrossRef CAS.
- M. T. Pope, in Heteropoly and Isopoly Oxomatalates, Springer Berlin Heidelberg, Berlin, 1983 Search PubMed.
- K. Dashtian, S. Shahsavarifar, M. Usman, Y. Joseph, M. R. Ganjali, Z. Yin and M. Rahimi-Nasrabadi, Coord. Chem. Rev., 2024, 504, 215644 CrossRef CAS.
- C. Wang, B. Wang, H. Yang, Y. Wan, H. Fang, W. Bao, W. Wang, N. Wang and Y. Lu, Chem. Eng. J., 2024, 483, 149143 CrossRef CAS.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 1–20 CrossRef.
- T. Yamase and M. T. Pope, Polyoxometalate Chemistry for Nano-Composite Design, Springer Nature, 2002 Search PubMed.
- H. Oh, Y. Choi, C. Shin, T. V. T. Nguyen, Y. Han, H. Kim, Y. H. Kim, J. W. Lee, J. W. Jang and J. Ryu, ACS Catal., 2020, 10, 2060–2068 CrossRef CAS.
- X. López, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2002, 124, 12574–12582 CrossRef PubMed.
- M. Vasilopoulou, A. M. Douvas, L. C. Palilis, S. Kennou and P. Argitis, J. Am. Chem. Soc., 2015, 137, 6844–6856 CrossRef CAS PubMed.
- X. López, J. A. Fernández and J. M. Poblet, Dalton Trans., 2006, 9, 1162–1167 RSC.
- T. Ueda, ChemElectroChem, 2018, 5, 823–838 CrossRef CAS.
- E. Ishikawa and T. Yamase, Bull. Chem. Soc. Jpn., 2000, 73, 641–649 CrossRef CAS.
- E. A. Nagul, I. D. McKelvie, P. Worsfold and S. D. Kolev, Anal. Chim. Acta, 2015, 890, 60–82 CrossRef CAS PubMed.
- G. R. Buettner, Arch. Biochem. Biophys., 1993, 300, 535–543 CrossRef CAS PubMed.
- R. Rossi, A. Milzani, I. Dalle-Donne and D. Giustarini, Clin. Chem., 1987, 33, 40–45 Search PubMed.
- E. Papaconstantinou and J. Chem, Soc., Chem. Commun., 1982, 12–14 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Gas chromatograms and mass spectra. See DOI: https://doi.org/10.1039/d5dt00624d |
| This journal is © The Royal Society of Chemistry 2025 |