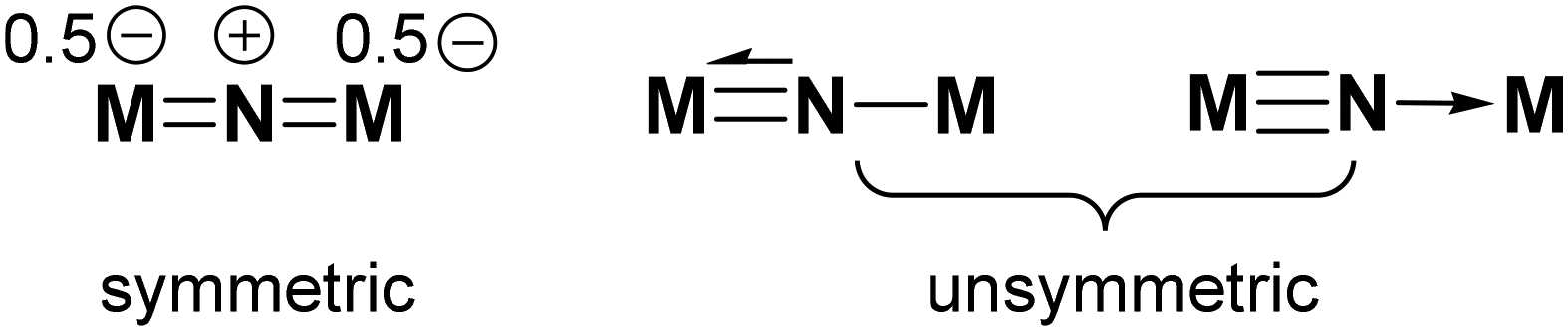DOI:
10.1039/D5DT00712G
(Paper)
Dalton Trans., 2025, Advance Article
A water-soluble heterometallic iron(IV)–ruthenium(IV) μ-nitrido porphyrin complex: synthesis, structure and catalytic activity†
Received
24th March 2025
, Accepted 7th May 2025
First published on 8th May 2025
Abstract
A heterometallic Fe(IV)–Ru(IV) nitrido complex bearing a water-soluble porphyrin ligand has been synthesized, and its redox and catalytic properties have been investigated. Treatment of Na4[Fe(TPPS)(H2O)2] ([TPPS]4− = meso-tetrakis(4-sulfonatophenyl)porphyrinate) with [Ru(N)(LOEt)Cl2] (LOEt− = [Co(η5-C5H5){P(O)(OEt)2}3]−) afforded the water-soluble μ-nitrido complex Na4[(H2O)(TPPS)Fe(μ-N)RuCl2(LOEt)] (Na4[1]). Cation metathesis of Na4[1] with Ph4PCl gave (Ph4P)4[(H2O)(TPPS)Fe(μ-N)RuCl2(LOEt)] ((Ph4P)4[1]), which has been characterized by single-crystal X-ray diffraction. The observed Ru–N(nitride) [1.701(5) Å] and Fe–N(nitride) [1.676(5) Å] distances and the Fe–N–Ru angle [171.4(3)°] of diamagnetic [1]4− are indicative of the Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NRu(IV) bonding behavior. The cyclic voltammogram of Na4[1] in 0.1 M HCl displayed reversible redox couples at +0.66 and +0.94 V versus a saturated calomel electrode, which are assigned metal-centred and porphyrin-based oxidations, respectively. Treatment of Na4[1] with Ag(CF3SO3) and PPh3 resulted in the formation of Na3([(H2O)(TPPS)Fe(μ-N)Ru(LOEt)Cl2{Ag(PPh3)}]) (2). Na4[1] is capable of catalyzing sulfide oxidation with H2O2 in aqueous medium. For example, in MeCN/H2O (1
NRu(IV) bonding behavior. The cyclic voltammogram of Na4[1] in 0.1 M HCl displayed reversible redox couples at +0.66 and +0.94 V versus a saturated calomel electrode, which are assigned metal-centred and porphyrin-based oxidations, respectively. Treatment of Na4[1] with Ag(CF3SO3) and PPh3 resulted in the formation of Na3([(H2O)(TPPS)Fe(μ-N)Ru(LOEt)Cl2{Ag(PPh3)}]) (2). Na4[1] is capable of catalyzing sulfide oxidation with H2O2 in aqueous medium. For example, in MeCN/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), in the presence of 2 mol% of Na4[1], methyl p-tolyl sulfide was oxidized by H2O2 to afford methyl p-tolyl sulfoxide selectively in 95% yield. Electrospray ionization mass spectrometry indicated that the reaction of Na4[1] with H2O2 generated new species with m/z = 430.2228 and 434.2198 corresponding to [1 + O]4− and [1 + 2O]4−, respectively, which are possible active intermediates in the catalytic sulfide oxidation.
:1, v/v), in the presence of 2 mol% of Na4[1], methyl p-tolyl sulfide was oxidized by H2O2 to afford methyl p-tolyl sulfoxide selectively in 95% yield. Electrospray ionization mass spectrometry indicated that the reaction of Na4[1] with H2O2 generated new species with m/z = 430.2228 and 434.2198 corresponding to [1 + O]4− and [1 + 2O]4−, respectively, which are possible active intermediates in the catalytic sulfide oxidation.
Introduction
Nitride (N3−) is a good supporting ligand for high-valent transition metal ions owing to its strong π-donating capability. As a bridging ligand, it can bind to two metal ions in either a symmetric or unsymmetric fashion depending upon the extent of π delocalization in the M–N–M′ unit (Scheme 1).1 Of interest are dinuclear iron complexes with symmetric nitrido bridges, such as [Fe(L)]2(μ-N) (L2− = phthalocyanine or a porphyrin dianion), which are capable of catalyzing C–H oxidation of hydrocarbons, including methane, with H2O2 or alkyl hydroperoxides,2–12 in which high-valent diiron-oxo species (“FeN–FeO”) have been proposed to be the active oxidants. It has been suggested that the enhanced oxidation capability of the diiron-oxo active species originates from the π-donation of the μ-nitrido ligand that increases the basicity of the FeO group and facilitates the H-atom abstraction.13,14 Nevertheless, the catalytic activity of analogous heterometallic Fe–N–M complexes has not been explored.14,15
 |
| | Scheme 1 Dinuclear μ-nitrido complexes with symmetric and unsymmetric nitrido bridges. | |
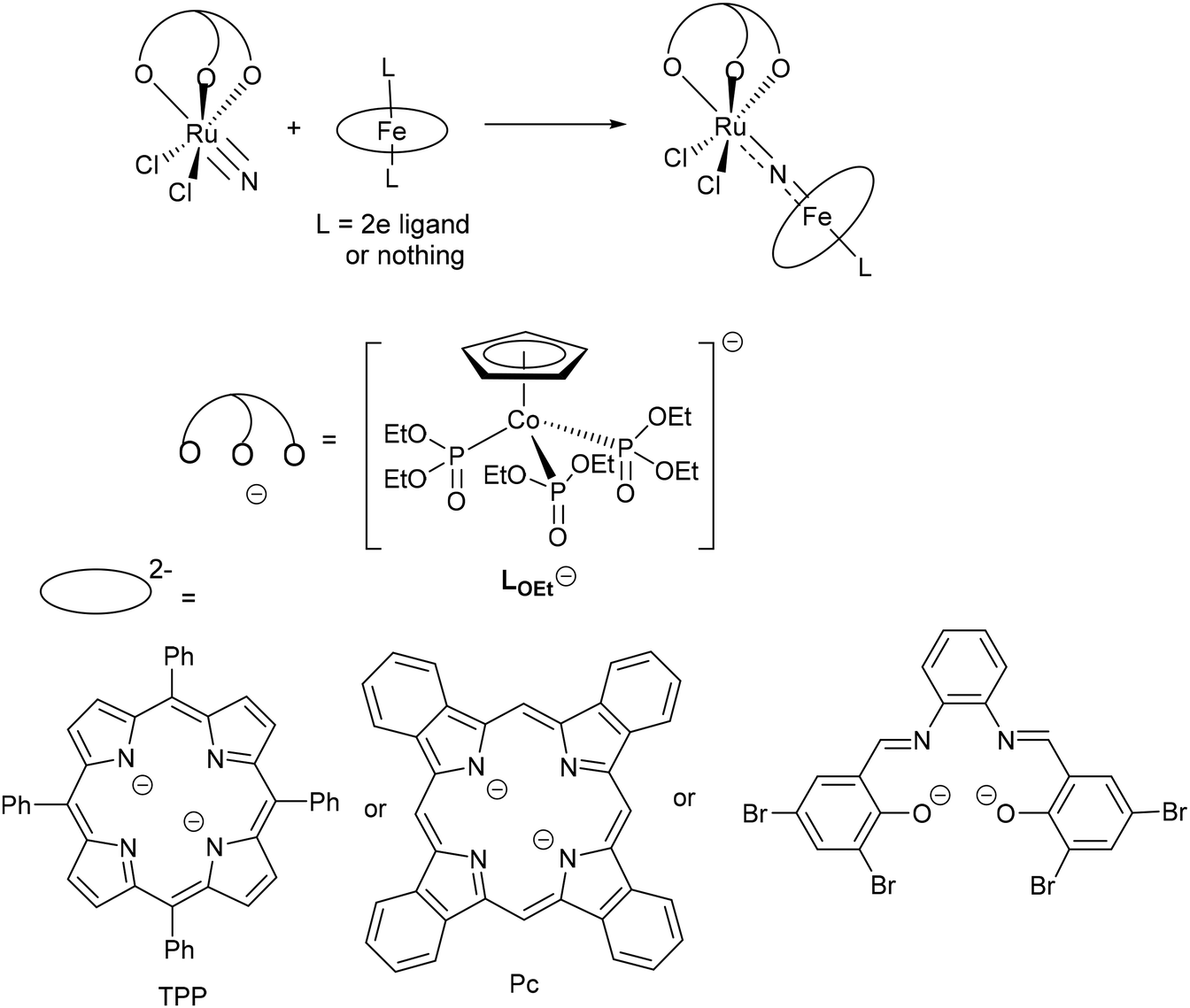
In previous work, we have synthesized a series of heterometallic Fe–N–Ru complexes via the reactions of a Ru(VI) terminal nitride, [Ru(LOEt)(N)Cl2] (LOEt− = [(η5-C5H5)Co{P(O)(OEt)2}3]−), with Fe(II) complexes bearing macrocyclic ligands, including porphyrin, phthalocyanine, and tetradentate Schiff base ligands (Scheme 2).16–18 X-ray diffraction studies revealed that these heterometallic complexes possess MN double bonds that are indicative of the Fe(IV)NRu(IV) resonance structure. Although they exhibit well-defined oxidation couples in the cyclic voltammograms (CVs), attempts to synthesize high-valent heterometallic nitrido complexes were unsuccessful. For instance, the oxidation of [(Pc)Fe–N–Ru(LOEt)Cl2] (Pc2− = phthalocyanine dianion) with NR3SbCl6 (R = 4-bromophenyl) and PhI(CF3CO2)2 led to the isolation of Fe(IV)/Ru(IV) complexes with a Pc cation radical and a hydroxy-Pc ligand, respectively.17
 |
| | Scheme 2 Heterometallic Fe(IV)–Ru(IV) nitrido complexes derived from Ru(VI) nitride [Ru(LOEt)(N)Cl2].16–18 | |
It is well-known that transition metal aqua complexes can be oxidized to high-valent hydroxo/oxo species in aqueous media via proton-coupled electron transfer.19,20 This prompted us to synthesize water-soluble heterometallic iron aqua complexes and explore their oxidation chemistry. It may be noted that a water-soluble dinuclear iron nitrido complex with tetrakis(4-sulfophthalocyanine) (SPc), [{Fe(SPc)}2(μ-N)]8−, has been used as a catalyst for oxidative degradation of polychlorophenols with H2O2.21 To our knowledge, iron nitrido complexes with water-soluble porphyrin ligands have not been reported.
Metal complexes with water-soluble porphyrins such as 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin [H2TPPS]4− (Scheme 3) have attracted much attention due to their applications in catalytic oxidations.22–25 In particular, the oxidation chemistry of Fe(TPPS) complexes that can serve as functional models for heme enzymes has been studied extensively.26–35 In non-acidic solutions, Fe(TPPS) complexes tend to dimerize to μ-oxo species, [{FeIII(TPPS)}2(μ-O)]8−,36 rendering the isolation of high-valent Fe-oxo species difficult. Although nitrido-bridged iron porphyrins such as [Fe(TPP)]2(μ-N)37 and [{Fe(TPP)}2(μ-N)](SbCl6)38 (TPP2− = tetraphenylporphyrin dianion) are well documented, water-soluble Fe(TPPS) μ-nitrido complexes have not been isolated.
 |
| | Scheme 3 Synthesis of Fe(IV)–Ru(IV) nitrido TPPS complexes. | |
Herein, we report the synthesis of the first heterometallic Fe(TPPS) μ-nitrido complex, [(H2O)(TPPS)Fe(μ-N)RuCl2(LOEt)]4− ([1]4−), which has been isolated as Na+ as well as Ph4P+ salts. The structure of (Ph4P)4[1] featuring an FeNRu bridge has been established by X-ray crystallography. In 1 M HCl, Na4[1] exhibited a reversible redox couple at +0.66 V versus the standard calomel electrode (SCE), which is ascribed to a metal-centred oxidation. Na4[1] is capable of catalyzing sulfide oxidation with H2O2 in aqueous media. Electrospray ionization (ESI) mass spectrometry indicated that the reaction of Na4[1] with H2O2 generated new species assigned as [1 + 2O]4− and [1 + O]4−, which are proposed to be reactive intermediates in the catalytic sulfide oxidation.
Results and discussion
Synthesis
Treatment of a suspension of Na4[Fe(TPPS)(H2O)2]39 in methanol with [Ru(LOEt)(N)Cl2] resulted in the formation of a heterometallic nitrido complex, Na4[(H2O)(TPPS)Fe(μ-N)RuCl2(LOEt)] (Na4[1]) (Scheme 3). Unlike Na4[Fe(TPPS)(H2O)2], which is sparingly soluble in methanol, Na4[1] is soluble in both water and methanol, but it is insoluble in acetonitrile or acetone. The 1H NMR spectrum of Na4[1] in D2O showed sharp peaks that can be assigned to the TPPS4− and LOEt− ligands in normal regions, which is indicative of its diamagnetic behavior. The UV/visible spectrum in water displayed the porphyrin Soret and Q bands at 425 and 548 nm, respectively, which are red-shifted compared with Na4[Fe(TPPS)(H2O)2] (413 and 517 nm, respectively). The ESI mass spectrum of Na4[1] in water showed a signal at m/z = 426.2221 corresponding to [1]4− (Fig. S13, ESI†). Despite many attempts, we have not been able to obtain single crystals of Na4[1] for structure determination. We have also synthesized the LOMe− analogue of Na4[1], Na4[1-Me], from Na4[Fe(TPPS)(H2O)2] and [Ru(N)(LOMe)Cl2] (LOMe− = [(η5-C5H5)Co{P(O)(OMe)2}3]−). Recrystallization of Na4[1-Me] from MeOH/MeCN/acetone afforded red single crystals, which were identified as the heterometallic Fe–N–Ru complex [H][Na3(Me2CO)(MeOH)(H2O)][(MeOH)(TPPS)Fe(μ-N)RuCl2(LOMe)] according to an X-ray diffraction study (Fig. S1, ESI†). However, the bond lengths and angles of the LOMe− complex have not been analyzed owing to serious disorder problems.
Cation metathesis of Na4[1] with PPh4Cl in methanol led to the isolation of [Ph4P]4[(H2O)(TPPS)Fe(μ-N)RuCl2(LOEt)], (Ph4P)4[1], which is soluble in organic solvents such as CH2Cl2. Recrystallization from CH2Cl2/Et2O/hexane afforded dark red crystals, which were suitable for X-ray diffraction. To our knowledge, (Ph4P)4[1] is the first structurally characterized Fe(TPPS) nitrido complex. It should be noted that very few crystal structures of Fe(TPPS) complexes have been reported in the literature.40 The structure of the complex anion in (Ph4P)4[1] is shown in Fig. 1. Displacements of atoms from the porphyrin N4 plane are given in Fig. 2. The porphyrin macrocycle displays a saddled distortion. The Fe–N(porphyrin) distances [av. 2.003 Å] are similar to those of [(py)(TPP)Fe(μ-N)Ru(LOEt)Cl2] (py = pyridine) (1.997 Å)16 but shorter than those of [{Fe(TPPS)}2(μ-O)]8− (av. 2.079 Å).40 The Fe–OH2 distance of 2.069(4) Å is similar to that of the Pc complex [(H2O)(Pc)Fe(μ-N)RuCl2(LOEt)] [2.046(5) Å].17 The Fe–N(nitrido) [1.676(5) Å] and Ru–N(nitrido) [1.701(5) Å] distances in [1]4− compare well with those of [(py)(TPP)Fe(μ-N)Ru(LOEt)Cl2] [1.683(3) and 1.704(3) Å, respectively].16 The Fe–N–Ru unit is roughly linear with a bond angle of 171.4(3)°. The observed short M–N(nitride) bond distances and diamagnetism of [1]4− are indicative of the Fe(IV)NRu(IV) bonding behavior.
 |
| | Fig. 1 Molecular structure of the tetra-anion [1]4−. Hydrogen atoms are omitted for clarity. The ellipsoids are drawn at the 30% possibility level. Selected bond lengths (Å) and angles (°): Fe1–N1: 1.676(5); Fe1–N2: 2.006(4); Fe1–N3: 1.997(4); Fe1–N4: 2.016(4); Fe1–N5: 1.994(4); Fe1–O10: 2.069(4); Ru1–N1: 1.701(5); Ru1–Cl1: 2.3284(13); Ru1–Cl2: 2.3412(13); Ru1–O7: 2.124(3); Ru1–O8: 2.075(4); Ru1–O9: 2.068(3); Fe1–N1–Ru1 171.4(3). | |
 |
| | Fig. 2 Atomic displacements (10−3 Å) from the porphyrin N4 plane in [1]4−. | |
Electrochemistry
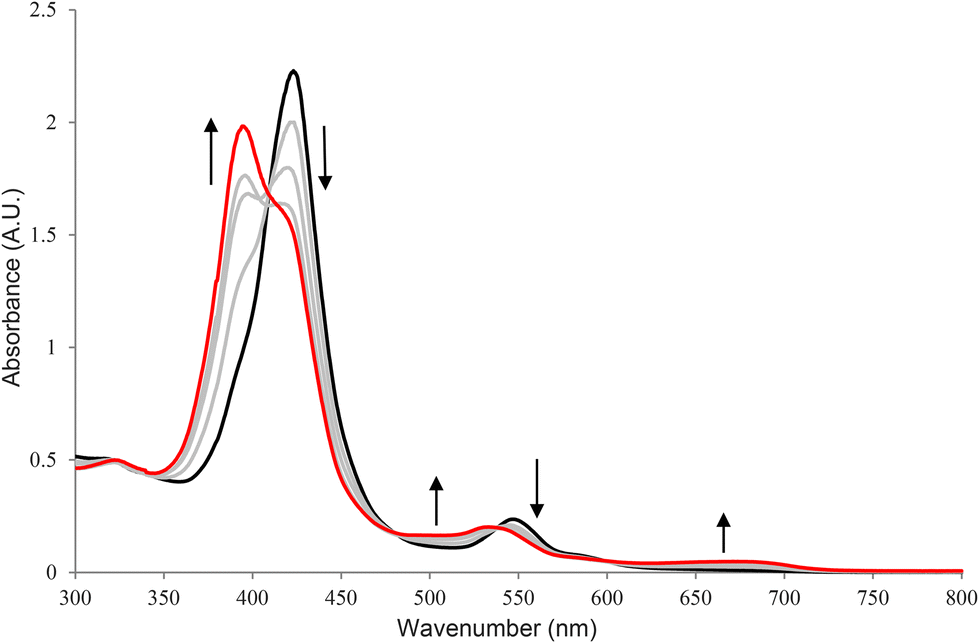
The CV of Na4[1] in 0.1 M HCl (pH 1) displayed two reversible oxidation couples at +0.66 and +0.94 V versus the saturated calomel electrode (SCE) (Fig. 3). To gain insight into the nature of the first couple, the oxidation of Na4[1] with a 1-electron oxidant, cerium(IV) ammonium nitrate ([NH4]2[Ce(NO3)6]) (CAN), which has a redox potential of +0.88 V versus SCE,41 was studied. Treatment of Na4[1] with 1 equivalent of CAN in water resulted in a blue-shift of the Soret band from 425 to 398 nm in the UV/visible spectrum (Fig. 4). A similar spectral change was found for the oxidation of Na4[1] with CAN in 0.1 M HCl. No absorption at ∼800 nm that is characteristic of a porphyrin radical cation was found, suggesting that the oxidation at +0.66 V is a metal-centred event, either Fe(V/IV), Ru(V/IV) or a combination of both. No further spectral change was found when excess (>3 equivalents) CAN was added. The nature of the second redox couple is unclear, but it is very likely that it resulted from a porphyrin-based oxidation. When the pH was increased to 3, similar half-wave potentials were found for the two redox couples, but they were less reversible (Fig. 5). At pH 6, only irreversible oxidation waves were found (Fig. 6). In contrast, the CV of (Ph4P)4[1] in CH2Cl2 displayed an irreversible oxidation wave with Epa = +0.51 V versus Fc+/0 (Fc = ferrocene) (Fig. S11, ESI†), whereas the reported compound [(py)(TPP)Fe(μ-N)Ru(LOEt)Cl2] exhibited two oxidation events at +0.29 and +0.77 V in CH2Cl2.16
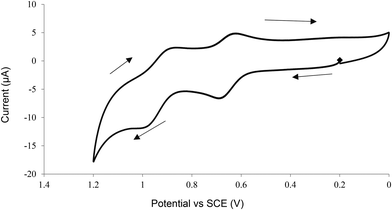 |
| | Fig. 3 CV of Na4[1] in 0.1 M HCl (pH 1). Working electrode: glassy carbon, reference electrode: standard calomel electrode, scan rate = 100 mV s−1. | |
 |
| | Fig. 4 UV/visible spectral change for the reaction of Na4[1] with 1 equivalent of CAN in water at room temperature. 1/4 equiv. of CAN was added in each scan. | |
 |
| | Fig. 5 CV of Na4[1] at pH 3. Working electrode: glassy carbon, reference electrode: standard calomel electrode, scan rate = 100 mV s−1. | |
 |
| | Fig. 6 CV of Na4[1] at pH 6. Working electrode: glassy carbon, reference electrode: standard calomel electrode, scan rate = 100 mV s−1. | |
Reaction of Na4[1] with oxidizing agents
Attempts have been made to synthesize high-valent heterometallic nitrido complexes by chemical oxidation of Na4[1]. Treatment of Na4[1] with 1 equivalent of AgNO3 in water resulted in a red-shift of the Soret band from 425 to ca. 440 nm (see the ESI†). No AgCl precipitate was found, indicating that the Ag(I) ion did not abstract the chloride ion at the Ru center. Attempts to isolate the product by adding excess acetone to the aqueous reaction mixture failed. To increase the solubility of the product in organic solvents, [1]4− was reacted with Ag(I) in the presence of a ligand that can bind to the Fe center. Thus, treatment of Na4[1] with Ag(CF3SO3) in methanol, followed by the addition of PPh3, led to the isolation of red crystals that were characterized as Na3[(H2O)(TPPS)Fe(μ-N)Ru(LOEt)Cl2{Ag(PPh3)}] (2) (Scheme 4). The 1H NMR spectrum of 2 showed resonances that can be attributed to the TPPS4−, LOEt− and PPh3 ligands, indicative of the diamagnetic properties. The 31P{1H} NMR spectrum showed a resonance at δ 15.99 ppm due to the Ag-bound PPh3 ligand. We have not been able to crystallize 2 for structure determination. However, we have structurally characterized the TPP analogue of 2, [(TPP)Fe(μ-N)Ru(LOEt)Cl2{Ag(PPh3)}](CF3SO3) (2-TPP), which was synthesized from [(TPP)Fe(μ-N)Ru(LOEt)Cl2], Ag(CF3SO3) and PPh3 (Scheme 4). A preliminary X-ray diffraction study revealed that the [Ag(PPh3)]+ unit binds to the two chloride ligands of the [Ru(LOEt)Cl2] moiety in 2-TPP (Fig. S3, ESI†). We believe that 2 has a core structure similar to 2-TPP featuring Ru(μ-Cl)Ag bridges. Thus, instead of oxidizing the metal centers, the Ag(I) ion binds to the chloride ligands in [1]4−.
 |
| | Scheme 4 Reactions of Fe–N–Ru porphyrin complexes with Ag(I) and PPh3. | |
As mentioned earlier, the oxidation of Na4[1] with CAN in water gave a high-valent species that displayed absorption bands at 398 and 507 nm in the UV/visible spectrum. Attempts to precipitate this species by the addition of acetone to the aqueous reaction mixture failed.
When Na4[1] in water at room temperature was treated with 1 equivalent of H2O2, no significant change was found in the UV/visible spectrum, but some gas bubbles, presumably oxygen, were observed. When excess H2O2 (>10 equivalents) was added, new absorption bands centred at 398 and 515 nm that are similar to those found for the CAN oxidation of [1]4− appeared, suggesting that a high-valent species, possibly an Fe(V) or Ru(V) complex, was generated. When Na4[1] was reacted with H2O2 in methanol, the same spectral change was found. Upon addition of excess methyl p-tolyl sulfide (20 equivalents) to the methanol solution, methyl p-tolyl sulfoxide that was identified by GC-MS was formed, but there was no change in the Soret band at 398 nm, indicating that this high-valent species was not involved in the sulfide oxidation. However, when a large excess of methyl p-tolyl sulfide (200 equivalents) was added to the mixture, the absorption peak at 398 nm dropped slowly and the Soret band of [1]4− (425 nm) appeared (Fig. S9, ESI†). Therefore, in the presence of a large excess of thioether, this high-valent species will also be slowly reduced to [1]4−.
Catalytic sulfide oxidation with H2O2
The above results indicate that the reaction of [1]4− with H2O2 generated a reactive species that is capable of OAT. The use of [1]4− as a catalyst for sulfide oxidation with H2O2 has been explored, and the results are summarized in Table 1. Treatment of methyl p-tolyl sulfide with H2O2 in MeCN/H2O (1:1, v/v) in the presence of 2 mol% of Na4[1] afforded methyl p-tolylsulfoxide selectively in 95% yield. Only a trace amount of sulfone was detected. A similar yield (92%) was obtained when methanol was used as the solvent. However, only 5% of sulfoxide was formed when the oxidation was carried out in CH2Cl2 using (Ph4P)4[1] as a catalyst, indicating that the aqueous medium favors the catalytic oxidation of the heterometallic nitrido complex. The reduced catalytic activity of the [PPh4]+ salt can be attributed to the high oxidation potential of [1]4− in CH2Cl2 (+0.51 V versus Fc+/0), which renders the generation of a high-valent active species difficult. Similarly, the binding of the cationic [Ag(PPh3)]+ moiety has a negative impact on the performance of the Fe/Ru catalyst; the yield of sulfoxide was decreased to 25% when trianionic 2 was used as the catalyst.
Table 1 Catalytic sulfide oxidation with H2O2a
MS monitoring of the reaction of Na4[1] with H2O2
The reaction of Na4[1] with H2O2 has been monitored by ESI-MS in the range of m/z 420–440 (Fig. 7). Upon addition of ca. 10 equivalents of H2O2, a new signal at m/z 434.2198, which can be assigned to [1 + 2O]4− (Fig. S14, ESI†), was observed. The intensity of the [1]4− signal decreased gradually as the [1 + 2O]4− peak rose. At about 16 min, a new signal at m/z 430.2228 that is attributed to [1 + O]4− (Fig. S15, ESI†) appeared. After 56 min, the [1 + 2O]4− peak became the predominant signal in the mass spectrum. It seems reasonable to assume that the reaction of [1]4− with H2O2 afforded [1 + 2O]4−, which was subsequently converted to [1 + O]4−.
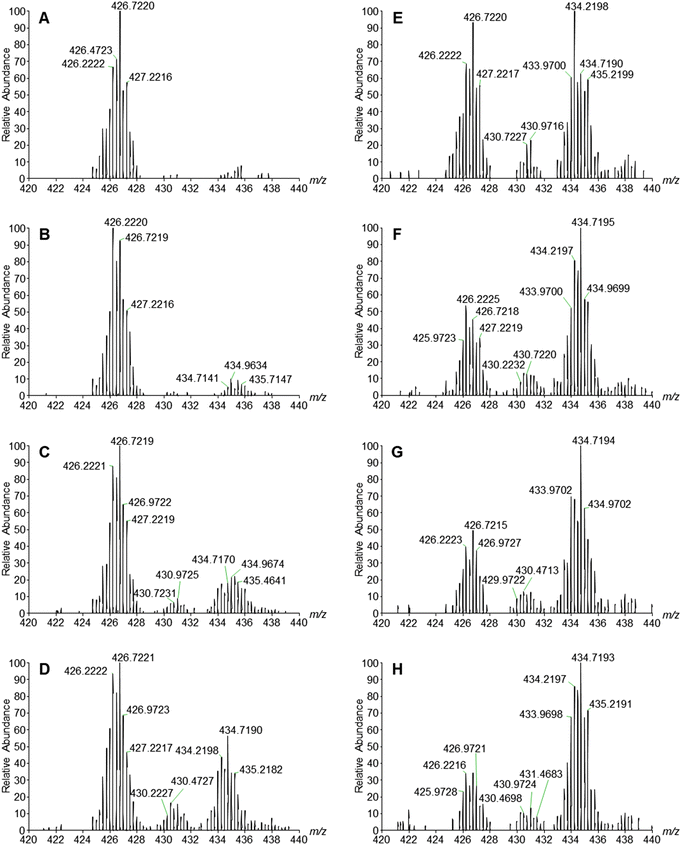 |
| | Fig. 7 ESI-MS monitoring of the formation of [1 + O]4− (m/z 430.2228) and [1 + 2O]4− (m/z 434.2198) species via the reaction of [1]4− with H2O2 (10 equivalents) in water at room temperature at t = 0 (A), 8 (B), 16 (C), 22 (D), 30 (E), 41 (F), 47 (G), and 55 (H) min. | |
A previous work has shown that the active oxidants of diiron-catalyzed oxidations with H2O2 are high-valent diiron-oxo species, which are formed via heterolytic O–O cleavage of (hydro)peroxo intermediates.8 We therefore assign the MS signals at m/z 434.2198 and 430.2228 as heterometallic Ru/Fe peroxo and oxo species, respectively. A possible mechanism for the catalytic sulfoxidation with [1]4− is shown in Scheme 5. Reaction of [1]4− with H2O2 generates a peroxo intermediate, I, which in the absence of a substrate decomposes to [1]4− readily. If excess H2O2 is used, a sufficient amount of I is accumulated, and it can be observed in ESI-MS. Subsequently, O–O cleavage of I affords a reactive dimetallic oxo species, II, which is the active oxidant for the sulfoxidation. The reaction of [1]4− with excess H2O2 also generates a high-valent species, possibly an Fe(V) or Ru(V) complex, via another pathway that is not clear at this stage. This high-valent species is, however, unable to undergo OAT with the thioether.
 |
| | Scheme 5 Proposed mechanism for the [1]4−-catalyzed sulfide oxidation. | |
Conclusions
We have synthesized a heterometallic Ru(IV)–Fe(IV) nitrido complex bearing a water-soluble porphyrin, [1]4−. (Ph4P)4[1] is the first Fe(IV) TPPS nitrido complex that has been characterized by X-ray diffraction. The binding of the Ru(VI) nitride moiety to [Fe(TPPS)]4− not only increases the solubility of the TPPS complex in organic solvents, facilitating its isolation and crystallization, but also inhibits its aggregation in aqueous solution. This strategy can be exploited for isolation and crystallization of other water-soluble metalloporphyrins in aqueous media. [1]4− undergoes 1-electron oxidation at a relatively low potential (E½ = +0.66 V versus SCE) in an acidic solution to yield a high-valent heterometallic nitrido complex that has an overall d7 configuration. [1]4− proved to be an efficient catalyst for sulfide oxidation with H2O2 in aqueous media. ESI-MS indicated that the oxidation of [1]4− with H2O2 produced peroxo and oxo species. It is believed that O–O cleavage of the peroxo intermediate generates an heterometallic oxo species, which is the active species for the sulfide oxidation. The results of this work demonstrate the potential application of water-soluble heterometallic nitrido complexes in catalytic OAT reactions in aqueous media.
Experimental
General information
All manipulations were performed under nitrogen using standard Schlenk techniques. NMR spectra were recorded on a Bruker ARX 400 spectrometer operating at 400, 376.5, and 162 MHz for 1H, 19F, and 31P, respectively. Chemical shifts (δ, ppm) were reported with reference to SiMe4 (1H), CF3C6H5 (19F), and H3PO4 (31P). UV-visible spectra were recorded on a Dynamica HALO DB-20 spectrometer. ESI mass spectrometry was performed on an UltiMate 3000 UHPLC system coupled with an Orbitrap Exploris™ 120 mass spectrometer (Thermo Fisher, Waltham, MA). Cyclic voltammetry was performed with a CH Instrument model 600D potentiostat. The reference electrodes for aqueous and non-aqueous studies were the standard calomel electrode and Ag–AgNO3 (0.1 M in acetonitrile) electrodes, respectively. Elemental analyses were performed by Medac Ltd, Surrey, UK. The compounds Na4[Fe(TPPS)(H2O)2]39 and [Ru(LOEt)(N)Cl2]42 were prepared according to literature methods. Other reagents were purchased from Aldrich Ltd and used as received.
Preparation of Na4[(H2O)Fe(TPPS)(μ-N)RuCl2LOEt] (Na4[1]). To a suspension of Na4[Fe(TPPS)(H2O)2] (50 mg, 0.045 mmol) in MeOH (10 mL) was added 1 equivalent of [Ru(N)(LOEt)Cl2] (32 mg, 0.045 mmol), and the reaction mixture was stirred at room temperature overnight. The solvent was pumped off, and the residue was extracted with MeOH/MeCN (5 mL, 1:3 v/v). Recrystallization from MeOH/MeCN/acetone afforded red crystals. Yield: 39 mg, 50%. 1H NMR (400 MHz, D2O): δ 0.81 (t, J = 7.2 Hz, 6H, CH3), 0.89 (t, J = 7.2 Hz, 6H, CH3), 1.14 (t, J = 7.2 Hz, 6H, CH3), 2.44 (m, 4H, CH2), 3.34 (m, 4H, CH2), 4.66 (s, 5H, Cp), 8.22 (d, J = 8.4 Hz, 8H, Hm), 8.41 (m, 8H, Ho), 9.03 (s, 8H, Hpyrrole). 31P{1H} NMR (162 MHz, D2O): δ 117.79 (m). MS-ESI: m/z 426 [1 − H2O]4−. Anal. calc. for C61H61Cl2CoFeN5Na4O22P3RuS4·9H2O: C, 37.04; H, 4.03; N, 3.54. Found: C, 37.11; H, 3.86; N, 4.08.
Preparation of ((Ph4P)4[(H2O)(TPPS)Fe(μ-N)RuCl2LOEt] (PPh4)4[1]). To a suspension of Na4[1] (50 mg, 0.029 mmol) in MeOH (5 mL) were added 4 equivalents of Ph4PCl (44 mg, 0.12 mmol). The solvent was pumped off, and the residue was washed with water and dried in vacuo. Recrystallization from CH2Cl2/Et2O/hexane gave dark red single crystals. Yield: 43 mg, 48%. 1H NMR (400 MHz, CDCl3): δ 0.68 (t, J = 7.2 Hz, 6H, CH3), 0.91 (t, J = 7.2 Hz, 6H, CH3), 1.08 (t, J = 7.2 Hz, 6H, CH3), 2.27 (m, 3H, CH2), 2.41 (m, 3H, CH2), 3.28 (q, J = 7.2 Hz, 6H, CH2), 4.49 (s, 5H, Cp), 7.84 (m, 65H, Ph), 7.97 (m, 19H, Ph), 8.10 (m, 12H, Ph), 8.76 (s, 8H, Hpyrrole). 31P{1H} NMR (162 MHz, CDCl3): δ 22.91 (s, PPh4+), 117.14 (m, LOEt−). Anal. calc. for C157H160Cl2CoFeN5O32P7RuS4: C, 57.83; H, 4.95; N, 2.15. Found: C, 57.85; H, 4.87; N, 2.33.
Preparation of Na3[(H2O)(TPPS)Fe(μ-N)Ru(LOEt)Cl2{Ag(PPh3)}] (2). To a solution of Na4[1] (50 mg, 0.029 mmol) in MeOH (5 mL) were added AgCF3SO3 (7.5 mg, 0.029 mmol) and PPh3 (6.4 mg, 0.029 mmol), and the mixture was stirred overnight at room temperature and filtered. The solvent was pumped off, and the residue was extracted with MeOH/MeCN (5 mL, 1:3 v/v). Recrystallization from MeOH/MeCN/acetone afforded red crystals. Yield: 39 mg, 50%. 1H NMR (400 MHz, D2O): δ 0.58 (overlapping t, 6H, CH3), 0.83 (overlapping t, 6H, CH3), 1.01 (overlapping t, 6H, CH3), 2.43 (m, 2H, CH2), 2.55 (m, 2H, CH2), 3.25 (m, 8H, CH2), 4.66 (s, 5H, Cp), 7.26 (m, 5H, Ph), 7.40 (m, 5H, Ph), 7.51 (m, 3H, Ph), 8.18 (m, 10H, Ph and Hm), 8.37 (m, 8H, Ho), 9.01 (s, 8H, Hpyrrole). 31P{1H} NMR (162 MHz, D2O): δ 15.99 (dd, 1J(31P-107Ag) = 654 Hz, 1J(31P-109Ag) = 755 Hz, PPh3), 117.79 (m, LOEt−). Anal. calc. for C79H74AgCl2CoFeN5Na3O21P4RuS4·8H2O: C, 41.45; H, 3.96; N, 3.06. Found: C, 38.39; H, 3.79; N, 2.88.
Catalytic oxidation of p-tolyl methyl sulfide with H2O2. To a solution of p-tolyl methyl sulfide (0.1 mmol) and Na4[1] (1 μmol) in water (10 mL) was added H2O2 (50 mmol), and the mixture was stirred at room temperature in air for 3 h. The yield of p-tolyl methyl sulfoxide was determined by GLC using bromobenzene as the internal standard.
ESI-MS monitoring of the reaction of Na4[1] with H2O2 in water. To a solution of Na4[1] in double deionized water (950 μL, 6.5 μM) was added aqueous H2O2 (50 μL, 1.1 mM). The resulting mixture was subjected to MS analysis at t = 0, 8, 16, 22, 30, 41, 47, and 55 min.
X-ray crystallography
Crystallographic data and experimental details for [Ph4P]4[1] are summarized in Table S1 (ESI†). Intensity data of the complexes were collected with a Rigaku SuperNova Atlas X-ray diffractometer using graphite-monochromated Cu-Kα radiation. The collected frames were processed using CrysAlisPro software. Using Olex2,43 the structure was solved with the SHELXT44 structure solution program using intrinsic phasing and refined with the SHELXL45 refinement package using least squares minimisation. Atomic positions of non-hydrogen atoms were refined anisotropically and with restraints applied to those disordered atoms. Hydrogen atoms were refined as riding on their respective parent atoms before the final cycle of least-squares refinement. Disorder is found in some solvent molecules in the structure. However, as the disorder does not affect the interpretation of the overall structure of the compound, no further modeling was made.
Data availability
The data supporting this article, including syntheses of Na4[1-Me] and 2-TPP, NMR, UV-visible and mass spectra, cyclic voltammograms and X-ray crystallographic data, have been included as part of the ESI.†. Crystallographic data for (PPh4)4[1] have been deposited at the CCDC under CCDC 2417666.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Hong Kong Research Grants Council (project numbers 16301120, 16301621 and 16301424) and the Hong Kong University of Science and Technology.
References
- K. Dehnicke and J. Strähle, Angew. Chem., Int. Ed. Engl., 1992, 31, 955–978 CrossRef.
- P. Afanasiev and A. B. Sorokin, Acc. Chem. Res., 2016, 49, 583–593 CrossRef CAS PubMed.
- A. B. Sorokin, Adv. Inorg. Chem., 2017, 70, 107–165 CrossRef CAS.
- A. B. Sorokin, Catal. Today, 2021, 373, 38–58 CrossRef CAS.
- A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- E. V. Kudrik and A. B. Sorokin, Chem. – Eur. J., 2008, 14, 7123–7126 CrossRef CAS PubMed.
- E. V. Kudrik, P. Afanasiev, D. Bouchu, J. M. M. Millet and A. B. Sorokin, J. Porphyrins Phthalocyanines, 2008, 12, 1078–1089 CrossRef CAS.
- E. V. Kudrik, P. Afanasiev, L. X. Alvarez, P. Dubourdeaux, M. Clémancey, M. J.-M. Latour, G. Blondin, D. Bouchu, F. Albrieux, S. E. Nefedov and A. B. Sorokin, Nat. Chem., 2012, 4, 1024–1029 CrossRef CAS PubMed.
- Y. Yamada, J. Kura, Y. Toyoda and K. Tanaka, New J. Chem., 2020, 44, 19179–19183 RSC.
- Y. Yamada, K. Morita, N. Mihara, K. Igawa, K. Tomooka and K. Tanaka, New J. Chem., 2019, 43, 11477–11482 RSC.
- Y. Yamada, Y. Miwa, Y. Toyoda, T. Yamaguchi, S. Akine and K. Tanaka, Dalton Trans., 2021, 50, 16775–16781 RSC.
- Y. Yamada, Y. Miwa, Y. Toyoda, Y. Uno, Q. M. Phung and K. Tanaka, Dalton Trans., 2024, 53, 6556–6567 RSC.
- M. G. Quesne, D. Senthilnathan, D. Singh, D. Kumar, P. Maldivi, A. B. Sorokin and S. P. de Visser, ACS Catal., 2016, 6, 2230–2243 CrossRef CAS.
- M. Q. E. Mubarak, A. B. Sorokin and S. P. de Visser, JBIC, J. Biol. Inorg. Chem., 2019, 24, 1127–1134 CrossRef CAS PubMed.
- S. A. Zdanovich, E. Y. Tyulyaeva, V. S. Sukharev, M. V. Zaitsev and S. V. Zaitseva, Polyhedron, 2025, 270, 117448 CrossRef CAS.
- W.-M. Cheung, W.-H. Chiu, M. de Vere-Tucker, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, Inorg. Chem., 2017, 56, 5680–5687 CrossRef CAS PubMed.
- W.-M. Cheung, W.-M. Ng, W.-H. Wong, H. K. Lee, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, Inorg. Chem., 2018, 57, 9215–9222 CrossRef CAS PubMed.
- R. Ng, W.-M. Ng, W.-M. Cheung, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, J. Organomet. Chem., 2022, 967, 122354 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- C. Colombana, E. V. Kudrika, P. Afanasieva and A. B. Sorokin, Catal. Today, 2014, 235, 14–19 CrossRef.
- P. Hambright, in The Porphyrin Handbook, ed. K. Kaddish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 3, ch. 18, pp. 129–210 Search PubMed.
- P. K. Pandey and G. Zheng, in The Porphyrin Handbook, ed. K. Kaddish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2000, vol. 6, ch. 43, pp. 157–230 Search PubMed.
- P. Böhm and H. Gröger, ChemCatChem, 2015, 7, 22–28 CrossRef.
- J. W. Buchler, J. Porphyrins Phthalocyanines, 2004, 8, 43–66 CrossRef CAS.
- G. Lente and J. H. Espenson, Green Chem., 2005, 7, 28–34 RSC.
- M. Fukushima and K. Tatsumi, Environ. Sci. Technol., 2005, 39, 9337–9342 CrossRef CAS PubMed.
- K. Kano, Y. Itoh, H. Kitagishi, T. Hayashi and S. Hirota, J. Am. Chem. Soc., 2008, 130, 8006–8015 CrossRef CAS PubMed.
- H. Kitagishi, M. Tamaki, T. Ueda, S. Hirota, T. Ohta, Y. Naruta and K. Kano, J. Am. Chem. Soc., 2010, 132, 16730–16732 CrossRef CAS PubMed.
- T. Ueda, H. Kitagishi and K. Kano, Inorg. Chem., 2014, 53, 543–551 CrossRef CAS PubMed.
- H. Kitagishi, D. Shimoji, T. Ohta, R. Kamiya, Y. Kudo, A. Onoda, T. Hayashi, J. Weiss, J. A. Wytko and K. Kano, Chem. Sci., 2018, 9, 1989–1995 RSC.
- C. Khin, J. Heinecke and P. C. Ford, J. Am. Chem. Soc., 2008, 130, 13830–13831 CrossRef CAS PubMed.
- J. Heinecke and P. C. Ford, J. Am. Chem. Soc., 2010, 132, 9240–9243 CrossRef CAS PubMed.
- M. P. Jensen and D. P. Riley, Inorg. Chem., 2002, 41, 4788–4797 CrossRef CAS PubMed.
- H. Maid, P. Böhm, S. M. Huber, W. Bauer, W. Hummel, N. Jux and H. Gröger, Angew. Chem., Int. Ed., 2011, 50, 2397–2400 CrossRef CAS PubMed.
- E. B. Fleischer, J. M. Palmer, T. S. Srivastava and A. Chatterjee, J. Am. Chem. Soc., 1971, 93, 3162–3167 CrossRef CAS PubMed.
- D. A. Summerville and I. A. Cohen, J. Am. Chem. Soc., 1976, 98, 1747 CrossRef CAS PubMed.
- M. Li, M. Shang, N. Ehlinger, C. E. Schulz and W. R. Scheidt, Inorg. Chem., 2000, 39, 580–583 CrossRef CAS PubMed.
- Z. Dong and P. J. Scammells, J. Org. Chem., 2007, 72, 9881–9885 CrossRef CAS PubMed.
- A. Mazzeo, C. Gaviglio, J. Pellegrino and F. Doctorovich, Heliyon, 2022, 8, e09555 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- X.-Y. Yi, T. C. H. Lam, Y.-K. Sau, Q.-F. Zhang, I. D. Williams and W.-H. Leung, Inorg. Chem., 2007, 46, 7193–7198 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2005, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses of Na4[1-Me] and 2-TPP, NMR, UV-visible and mass spectra, cyclic voltammograms and X-ray crystallographic data. CCDC 2417666. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00712g |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Wa-Hung Leung
* and
Wa-Hung Leung