
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-release of acetonitrile in ruthenium(II) complexes with various substituted terpyridine ligands†
Vladyslav
Mudrak
 ,
Pascal G.
Lacroix
*,
Pablo
Labra-Vázquez
,
Marine
Tassé
,
Sonia
Mallet-Ladeira
and
Isabelle
Malfant
*
,
Pascal G.
Lacroix
*,
Pablo
Labra-Vázquez
,
Marine
Tassé
,
Sonia
Mallet-Ladeira
and
Isabelle
Malfant
*
CNRS, LCC (Laboratoire de Chimie de Coordination), 205, route de Narbonne, F-31077 Toulouse, France. E-mail: pascal.lacroix@lcc-toulouse.fr; isabelle.malfant@lcc-toulouse.fr
First published on 6th May 2025
Abstract
The photo-release of acetonitrile is investigated in a series of ruthenium(II) complexes of the general formula [Ru(R-phtpy)(acac)(MeCN)](PF6) (phtpy stands for 4′-phenyl-2,2′:6′,2′′-terpyridine, and R = Et2N, Me2N, MeO, Me, H, NO2). The experimental quantum yields of photo-release (ϕMeCN = MeCN released/photons absorbed) increases with the donating capability of R, with values ranging from ϕMeCN = 0 (NO2) to ϕMeCN = 0.05 (Et2N). The origin of this effect is investigated computationally using the density functional theory and compared to those reported recently by our group on related [Ru(R-phtpy)(acac)(NO)](PF6) species capable of causing photo-release of NO. In the present case, the capability for MeCN release appears related to the relative energies of the metal-centered (3MC) vs. metal–ligand-charge-transfer (3MLCT) triplet states. The 3MC state, in which the Ru–NC distance is elongated to 4.2 Å, is expected to be responsible for the release. Additionally, four crystal structures are reported for the compounds in which R = Et2N, MeO, H, and NO2.
Introduction
In the general context of medicinal coordination chemistry, ruthenium complexes have seen widespread use as therapeutic agents and drug carriers, and numerous comprehensive reviews have summarized their applications.1–6 The biological action of ruthenium complexes was first pointed out in the 1950s,7 and they gained substantial attention due to the report of their anticancer activity8 and the recognition of their usually low toxicity, compared to the well-known cisplatin.9,10Using ruthenium derivatives offers appealing perspectives for the photorelease of bioactive ligands from metal-based prodrug reagents, which could reduce undesirable side effects due to the highly focused and noninvasive character of light. In this context, numerous ruthenium-nitrosyl Ru(NO) complexes have been reported for their photo-release properties, in relation to the recognized biological role of NO.11–18 Similarly, nitrile-containing species are currently in use for their biological activities.19 For instance, compounds 1–3 (Chart 1) are widely prescribed pharmaceuticals used in the treatment of breast cancer (1),20,21 and heart diseases (2 and 3).22–24 The key role of the nitriles in the therapeutic efficiency of these drugs has been evidenced.25–28
 | ||
| Chart 1 Examples of nitrile-containing drugs: anastrazole (1), verapamil (2) and gallopamil (3) currently prescribed as pharmaceuticals. | ||
There are only a few reports on ruthenium complexes with bioactive ligands linked by a nitrile group, which can be released under irradiation.29–35 In these species, it is widely believed that the ability for ligand release is related to the interplay between two key triplet excited states,36,37 as illustrated in Fig. 1. After the initial photon absorption from the singlet (1GS) ground state, the populated metal-to-ligand charge transfer 1MLCT excited singlet state undergoes a fast intersystem crossing to the lowest-energy 3MLCT triplet within a few hundreds of fs.38 Although Turro et al. have recently provided evidence for ligand released capabilities in the 3MLCT state,39,40 it has been usually accepted that the release arises after internal conversion to a thermally populated metal-centered 3MC state.41–43 The Ru–NC bond length is significantly increased in 3MC, involving a Ru–NC σ* antibonding character, which may favor the ligand release. This situation contrasts with a recent report pointing out that, contrary to 3MC, the 3MLCT state having the strongest Ru–NC bonds is that exhibiting the highest quantum yields of photo-release within a series of 15 complexes.44
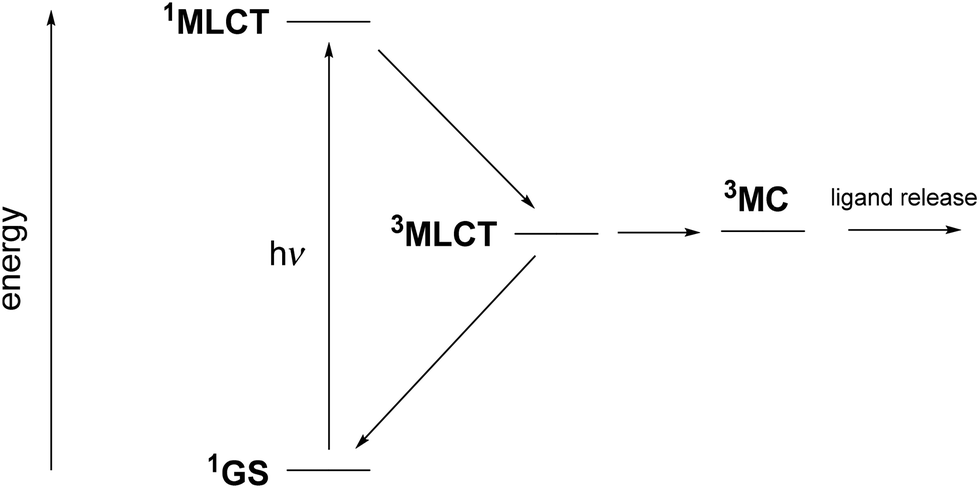 | ||
| Fig. 1 Jablonsky diagram showing the evolution of ruthenium complexes containing nitrile-based ligands after 1GS to 1MLCT light absorption. | ||
The above features suggest that for the complete understanding of the mechanism of a nitrile-containing ligand it still deserves further investigations. Along this line, investigating the MeCN release from Ru(MeCN) complexes offers a simplified and fruitful benchmark to conduct mechanistic investigations of the photo-release of nitrile-containing pharmaceuticals. Numerous reports have been published by Turro et al. to elucidate the mechanism underlying the efficient photo-release of acetonitrile from Ru(MeCN) complexes.30,32,33,37,45–48 The most recent works of this group have focused on identifying this pattern in complexes of the type [Ru(tpy)(L)(MeCN)]n+, where tpy = 2,2′:6′,2′′-terpyridine and L is a bidentate ligand.29,39,40,49,50 According to these studies, acetylacetonate (acac) appears to be a promising candidate for the L bidentate ligand, ensuring good stability and solubility in water. In addition, the resulting Ru(MeCN) complexes exhibit absorptions at higher wavelengths, which allow a release in the therapeutic window of transparency of biological tissues (λ = 600 nm to 1300 nm).51
Following these reports, and to further investigate the role of the terpyridine ligands in Ru(MeCN) compounds, we wish to report on a series of [Ru(R-phtpy)(acac)(MeCN)]+ complexes in which phtpy stands for 4′-phenyl-2,2′:6′,2′′-terpyridine, and R stands for substituents of various donor/acceptor characters. The different R-phtpy ligands under investigation are shown in Chart 2. In the present contribution, the X-ray crystal structures of four Ru(MeCN) complexes, [Ru(Et2N-phtpy)(acac)(MeCN)](PF6), [Ru(MeO-phtpy)(acac)(MeCN)](PF6), [Ru(H-phtpy)(acac)(MeCN)](PF6), and [Ru(NO2-phtpy)(acac)(MeCN)](PF6), are reported. The geometry of the coordination spheres around the ruthenium atoms is compared to those of related ruthenium-nitrosyl Ru(NO) complexes widely investigated for their capability to photo-release nitric oxide (NO).11,12,16–18,52 The experimental electronic spectra and capabilities for photo-release of MeCN are discussed within the framework of the density functional theory (DFT) to find a rationale for the origin of the properties.
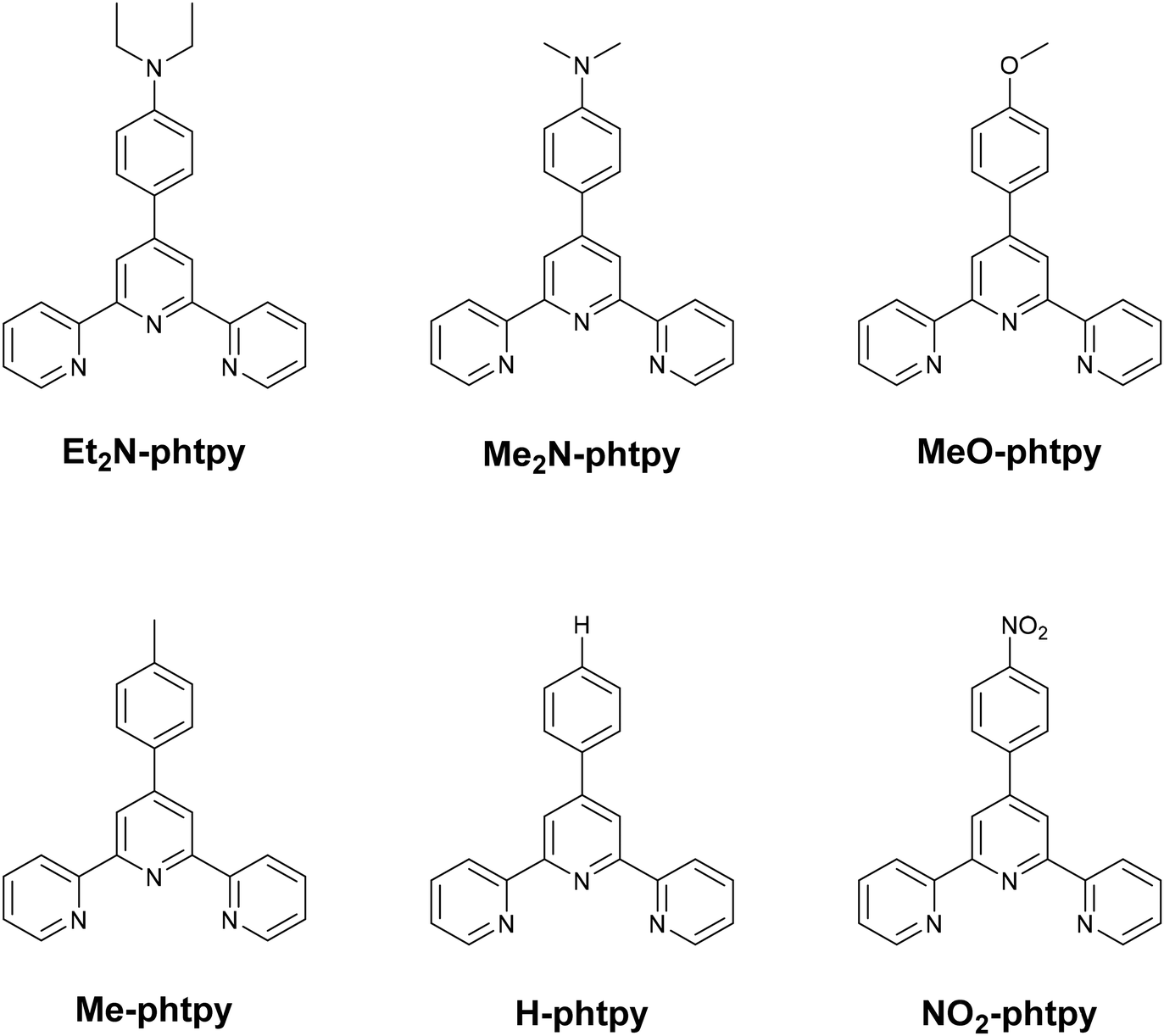 | ||
| Chart 2 R-tpy ligands used in [Ru(R-tpy)(acac)(MeCN)]+ complexes. | ||
Results and discussion
X-ray crystallographic studies
The X-ray crystal structures of [Ru(Et2N-phtpy)(acac)(MeCN)](PF6), [Ru(MeO-phtpy)(acac)(MeCN)](PF6), [Ru(H-phtpy)(acac)(MeCN)](PF6), and [Ru(NO2-phtpy)(acac)(MeCN)](PF6) have been investigated. Their asymmetric units are presented in Fig. 2. In any case, the presence of a single PF6− anion indicates that the oxidation state of the metal corresponds to RuII. The main crystal data are gathered in Table 1. Complete information on the X-ray studies including molecular geometries is provided in the ESI (Tables S1–16 and Fig. S3–S6†).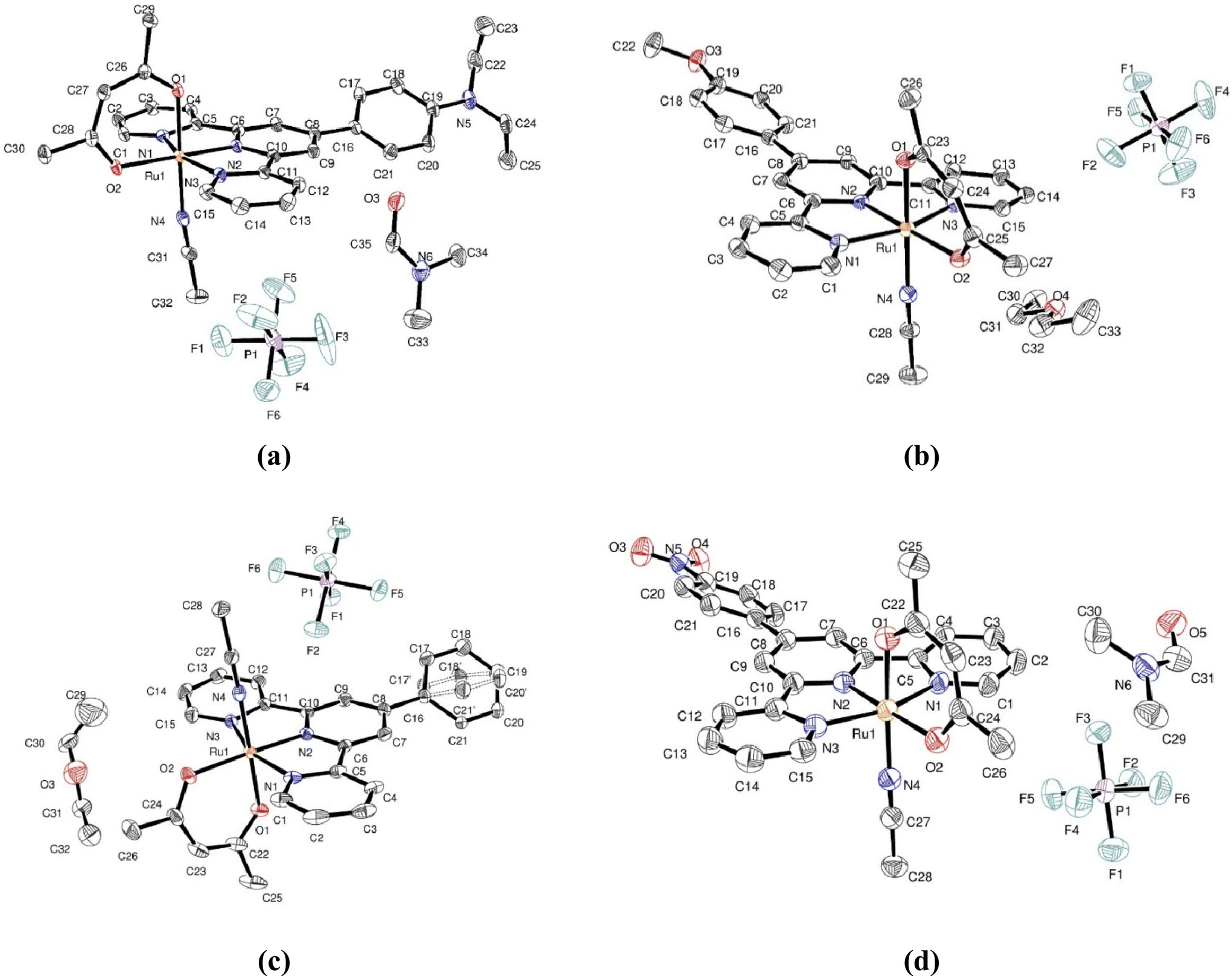 | ||
| Fig. 2 Asymmetric unit for [Ru(Et2N-phtpy)(acac)(MeCN)](PF6) (a), [Ru(MeO-phtpy)(acac)(MeCN)](PF6) (b), [Ru(H-phtpy)(acac)(MeCN)](PF6) (c), and [Ru(NO2-phtpy)(acac)(MeCN)](PF6) (d). | ||
| [Ru(Et2N-phtpy)(acac)(MeCN)](PF6) | [Ru(MeO-phtpy)(acac)(MeCN)](PF6) | [Ru(H-phtpy)(acac)(MeCN)](PF6) | [Ru(NO2-phtpy)(acac)(MeCN)](PF6) | |
|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc|∑|Fo|. b wR2 = [∑[w(Fo2 − Fc2)2]/∑[(wFo2)2]]1/2. | ||||
| Formula | C32H32N5O2Ru, F6P, C3H9N | C29H27N4O3Ru, F6P, 0.5(C4H10O) | C28H25N4O2Ru, F6P, 0.5(C4H10O) | C28H24N5O4Ru, F6P, 0.5(C4H10O), C3H7NO |
| Formula weight | 839.78 | 762.64 | 732.62 | 813.66 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c | P21/c |
| T (K) | 100(2) | 100(2) | 100(2) | 100(2) |
| Wavelength (MoKα) (Å) | 1.54184 | 1.54184 | 1.54184 | 1.54184 |
| a (Å) | 8.32390(10) | 11.0282(3) | 13.44010(10) | 8.41560(10) |
| b (Å) | 14.8029(2) | 11.3656(3) | 23.9107(2) | 19.4068(2) |
| c (Å) | 15.4530(2) | 13.5530(3) | 9.60110(10) | 21.2910(2) |
| α (°) | 77.6210(10) | 82.448(2) | 90 | 90 |
| β (°) | 82.4720(10) | 76.204(2) | 97.3060(10) | 92.7290(10) |
| γ (°) | 82.5980(10) | 75.467(2) | 90 | 90 |
| V (Å3) | 1833.78(4) | 1592.33(7) | 3060.38(5) | 3473.30(6) |
| Z | 2 | 2 | 4 | 4 |
| D calc. (g cm−3) | 1.521 | 1.591 | 1.590 | 1.556 |
| Abs. coef. (mm−1) | 4.525 | 5.141 | 5.298 | 4.806 |
| Unique reflections collected | 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 219 219 |
20459 |
56490 |
64597 |
| 7294 | 6267 | 6353 | 7089 | |
| R int | 0.0794 | 0.0590 | 0.0619 | 0.0655 |
|
R
1a |
0.0402 | 0.0473 | 0.0406 | 0.0524 |
| wR2b [I > 2σ(I)] [for all] |
0.1072 | 0.1253 | 0.1113 | 0.1414 |
| 0.1082 | 0.1281 | 0.1120 | 0.1432 | |
| GOF (F2) | 1.052 | 1.085 | 1.051 | 1.035 |
[Ru(Et2N-phtpy)(acac)(MeCN)](PF6) crystallizes in the triclinic P space group with one ruthenium complex and one molecule of DMF per asymmetric unit cell. The terpyridine skeleton (C15N3) is nearly planar with the largest distance to the mean plane of 0.108 Å, observed at C2. The terpyridine and acac unit (C5O2) are roughly orthogonal with a torsion angle between mean planes equal to 87.59°. The torsion angle between the terpyridine and its phenyl substituent (C6) is equal to 34.93°. The MeCN ligand has short contacts of 2.435 Å with one PF6− anion and 2.372 Å with an oxygen atom of a neighboring complex. Packing effects appear important in the structure with 17 short contacts involving the terpyridine unit.
[Ru(MeO-phtpy)(acac)(MeCN)](PF6) crystallizes in the same triclinic P space group, with one ruthenium complex per asymmetric unit cell. However, the structure appears significantly different from that of [Ru(Et2N-phtpy)(acac)(MeCN)](PF6) with 0.5 molecule of diethyl ether present in the asymmetric unit with some disorder. The terpyridine is nearly planar with the largest distance to the mean plane of 0.089 Å, observed at C15. The torsion angle with the acac ligand is equal to 73.96°, and that with the phenyl ring of the MeOphenyl substituent is equal to 17.49°. The MeCN ligand exhibits two short contacts of 2.554 Å and 2.622 Å with two different PF6− anions and a third one of 2.253 Å with the diethyl ether molecule. There are 13 short contacts with neighboring molecules which involve the terpyridine.
[Ru(H-phtpy)(acac)(MeCN)](PF6) crystallizes in the monoclinic P21/c space group with one ruthenium complex per asymmetric unit cell, and half a disordered molecule of diethyl ether. The largest deviation to planarity is observed at C2, and found to be equal to 0.096 Å in the terpyridine fragment. The torsion angle between terpyridine and acac is equal to 79.59°. The unsubstituted phenyl linked to the terpyridine is disordered with two conformations having torsion angles of 22.14° and 37.01° with the terpyridine. The MeCN ligand exhibits 10 short contacts with two PF6− anions, one molecule of diethyl ether and a neighboring complex, while 14 short contacts involve the terpyridine unit.
[Ru(NO2-phtpy)(acac)(MeCN)](PF6) crystallizes in the monoclinic P21/c space group with one ruthenium complex per asymmetric unit cell, and one molecule of DMF. The largest deviation to planarity in the terpyridine fragment is observed at C3, and found to be equal to 0.123 Å. The torsion angles between the terpyridine and acac are equal to 82.84° and 23.95° with the phenyl substituent. The MeCN ligand is involved in 6 short contacts with one PF6− anion and two neighboring complexes.
Details of the coordination spheres around the ruthenium atoms are provided in the ESI (Table S17†). The differences introduced by changing the substituents (Et2N, MeO, H, and NO2) with the series of crystals are weak and any discussion on their origin is somewhat irrelevant, as it appears difficult to separate intra- and inter-molecular effects in the X-ray crystal structures. Nevertheless, it is worth comparing the differences computed by DFT, together with those of the equivalent complexes in which the nitrosyl ligand (NO) was used instead of MeCN, in a previous investigation.53 It should be noted that although the structural parameters obtained by DFT are computed and not measured, they are independent of geometrical effects caused by the crystal packing, thus allowing a fair assessment of the geometrical effects caused by changes in the ligand framework. The data are provided in Table 2.
| R = NMe2 | R = OMe | R = H | R = NO2a |
Δ | |
|---|---|---|---|---|---|
| a Averaged values on the two complexes present in the asymmetric unit cell. | |||||
| Ru(R-phtpy)(acac)(MeCN)] + | |||||
| Ru-acac | |||||
| Ru–O(trans MeCN) | 2.0587 | 2.0577 | 2.0573 | 2.0554 | −0.0033 |
| Ru–O(cis MeCN) | 2.1053 | 2.1032 | 2.1023 | 2.1002 | −0.0051 |
| Ru-tpy | |||||
| Ru–N(lateral) | 2.0842 | 2.0830 | 2.0833 | 2.0823 | |
| Ru–N(central) | 1.9543 | 1.9530 | 1.9517 | 1.9487 | −0.0056 |
| Ru–N(lateral) | 2.0841 | 2.0842 | 2.0826 | 2.0824 | |
| Ru-MeCN | |||||
| Ru–N | 2.0061 | 2.0074 | 2.0083 | 2.0102 | +0.0041 |
| CN | 1.1603 | 1.1602 | 1.1601 | 1.1599 | −0.0004 |
| [Ru(R-phtpy)(acac)(NO)] 2+ | |||||
| Ru-acac | |||||
| Ru–O(trans NO) | 1.9936 | 1.9906 | 1.9893 | 1.9881 | −0.0055 |
| Ru–O(cis NO) | 2.0803 | 2.0759 | 2.0746 | 2.0721 | −0.0082 |
| Cd | |||||
| Ru-tpy | |||||
| Ru–N(lateral) | 2.0968 | 2.0973 | 2.0959 | 2.0952 | |
| Ru–N(central) | 1.9734 | 1.9789 | 1.9810 | 1.9834 | +0.0100 |
| Ru–N(lateral) | 2.0953 | 2.0957 | 2.0966 | 2.0952 | |
| Ru-NO | |||||
| Ru–N | 1.7598 | 1.7610 | 1.7617 | 1.7629 | +0.0031 |
| NO | 1.1404 | 1.1392 | 1.1387 | 1.1382 | −0.0022 |
The bond length computed by DFT (Table 2) and those obtained from the X-ray crystal structure analysis (Table S7†) are grossly similar. The examination of Table 2 reveals a few trends, which can be discussed as follows:
- The Ru–NO distances are significantly shorter than the related Ru–NC distances, in relation to the strong withdrawing character of NO, which is absent in MeCN (vide infra). Therefore, the well-known dRu–π*NO charge transfer effect observed in Ru(NO) complexes which results in a shortening of the Ru–NO distance does not have its counterpart in Ru(MeCN) complexes.
- The donor/acceptor strength of the R substituents modulates the charge transfer towards the π* orbitals of the nitrosyl ligand in Ru(NO) complexes, which significantly affects the N–O bond length. This effect is not present in CN; therefore the C–N bond lengths stay nearly constant (1.160 Å) along the series of Ru(MeCN) complexes.
- The examination of the Ru–acac bond lengths reveals a clear trend for reduced values in Ru(NO) species due to the possibility of acac → NO charge transfer, which is absent in Ru(MeCN) complexes.
- It seems especially interesting to point out that increasing the withdrawing character of R (NO2) reduces the tpy → NO charge transfer in Ru(NO) complexes, and then increases the Ru–tpy distance (Ru–N(central) = 1.9834 Å). The effect is the opposite in Ru(MeCN) complexes where the Ru → tpy charge transfer is dominant (vide infra). In this case, R = NO2 leads to a better charge transfer effect and hence a shortening of the Ru–N(central) value (1.9487 Å).
The above descriptions are in accordance with the expectation that, although NO acts as a strong withdrawing group in the reference Ru(NO) complexes with the outcome of significant electron densities transferred to the nitrosyl ligand upon irradiation, MeCN behaves as a relatively innocent ligand in terms of donor/acceptor capabilities in the present case. These features are thoroughly investigated in the next section.
Spectroscopic properties
The experimental UV-visible spectra were recorded in water for the Ru(MeCN) complexes built with the 6 ligands presented in Chart 2. A representative example is provided for [Ru(Me2N-phtpy)(acac)(MeCN)](PF6) in Fig. 3. The spectrum arises from a dominant transition having an absorption maximum (λmax) around 500 nm, while additional and more intense transitions are present at higher energies (λ < 400 nm). The strong effect of the substituent in terpyridine ligands on the photophysical properties of the ruthenium complexes is well known in the literature.54 However, the UV-visible spectra for the 5 other complexes (Fig. S1 in the ESI†) seem closely related to that presented in Fig. 3. In particular, and in striking contrast to the Ru(NO) series where the nature of R deeply affects the λmax value (e.g. 510 nm for R = Me2N and ∼300 nm for R = NO2),53 the λmax values recorded in the Ru(MeCN) series stay in the λ = 512–524 nm range. Indeed, the presence of the strongly withdrawing NO ligand in Ru(NO) complexes leads to the possibility of an intense push–pull effect, when R is a strong donor (e.g. Me2N) with the outcome of deeply red-shifted transitions. In contrast, this effect does not take place in the Ru(MeCN) series. The evolution of the λmax values is tentatively related to the σp Hammett constants,55 used to quantify the donor/acceptor character of R, shown in Fig. 4.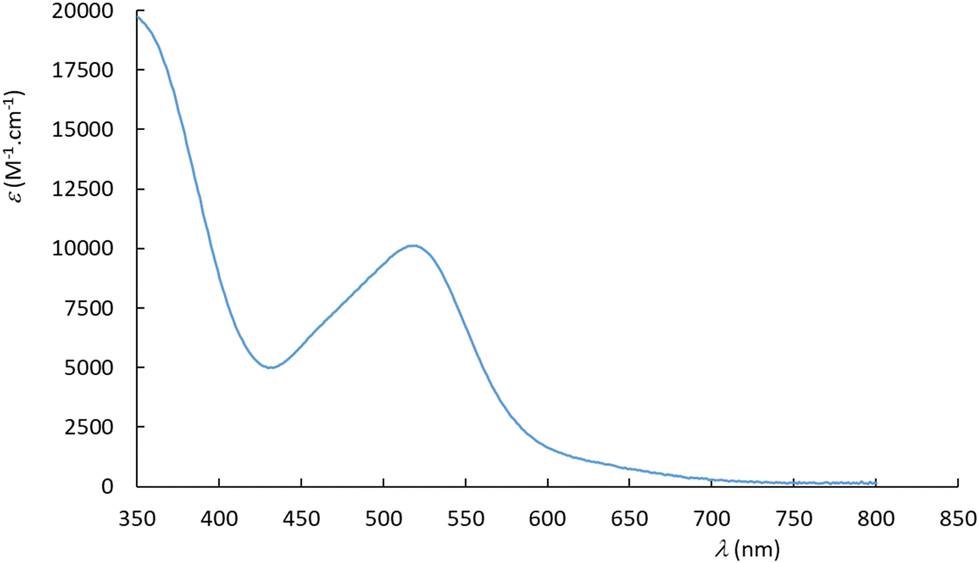 | ||
| Fig. 3 UV-visible spectrum recorded in water for [Ru(Me2N-phtpy)(acac)(MeCN)](PF6). | ||
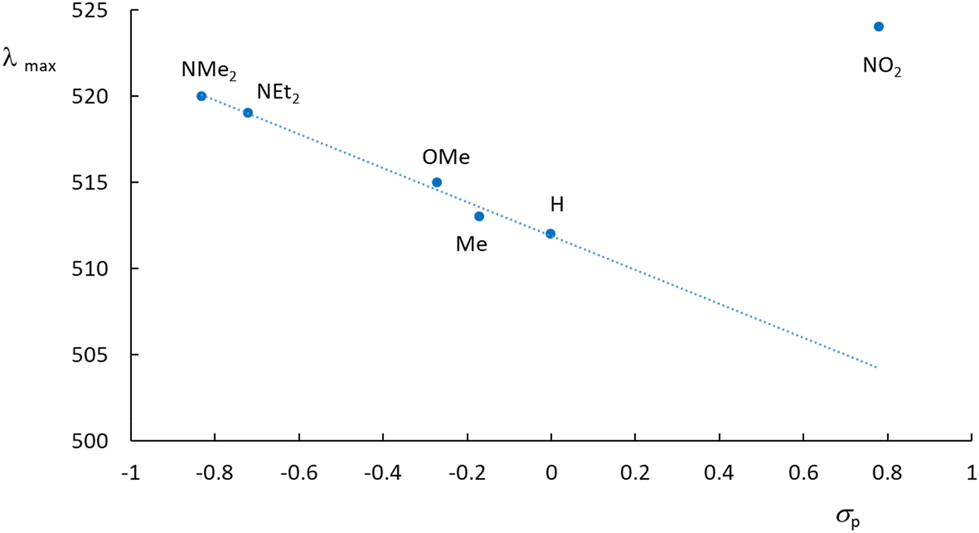 | ||
| Fig. 4 Absorption maxima drawn against the σp Hammett constants for the [Ru(R-phtpy)(acac)(MeCN)](PF6) series. The nature of the R substituent is shown. | ||
What immediately strikes in the examination of the figure is the good correlation linking λmax and σp as long as R is an electron donor (σp < 0). In contrast, the value of 524 nm recorded for NO2 looks at first somewhat irrelevant. To further clarify the origin of this behavior, the computed UV-visible spectra were examined by DFT for various R substituents. In any case, an intense transition is present at low energy, which readily accounts for the absorption maximum. The computed transitions are described in Table 3. Although no experimental data are available for R = CHO, the resulting complex was added to the table to introduce an intermediate withdrawing effect between H and NO2 (σp(CHO) = 0.42).55 Indeed, the resulting λmax = 421 nm value lies between those of R = H (416 nm) and R = NO2 (424 nm). Note that the energies computed by the CAM-B3LYP method are overestimated in these systems, as is well known (see the Experimental section). The computed behavior of the hypothetical complex with R = CHO brings further support for the relevance of Fig. 4 extended in the σp > 0 domain. Fig. 5 gives the details of the main orbitals involved in the dominant transition of these complexes.
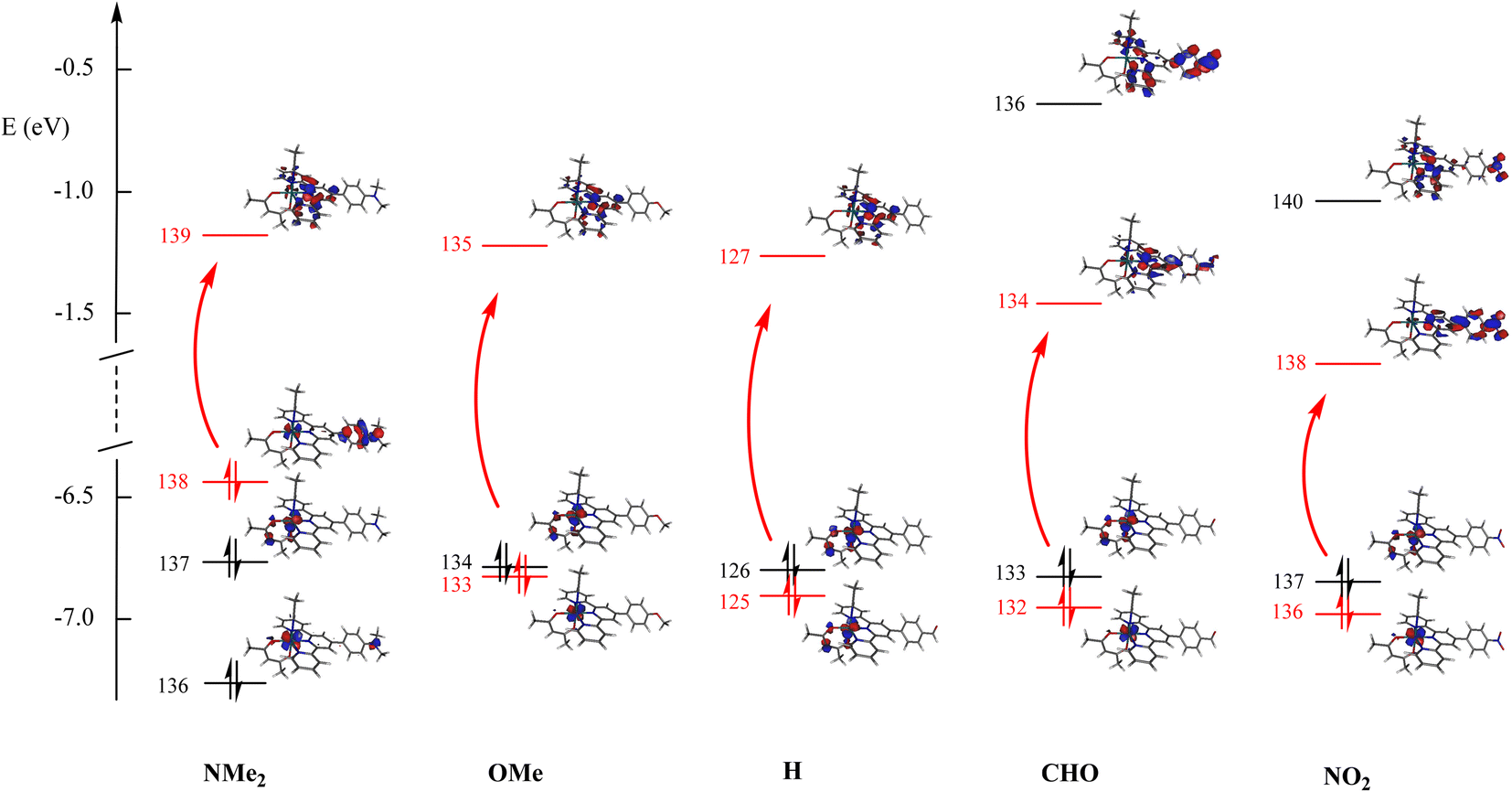 | ||
| Fig. 5 Energies of the main orbitals for the 5 complexes investigated in Table 3. The main contribution to the dominant transitions is shown in red. | ||
| R | Transition | λ max | f | Dominant compositiona | Character |
|---|---|---|---|---|---|
| a Orbital 138(139) is the HOMO(LUMO) for R = Me2N, 133(135) the HOMO−1(LUMO) for R = MeO, 125(127) the HOMO−1(LUMO) for R = H, and 136(138) the HOMO−1(LUMO) for R = NO2. b No experimental data are available for R = CHO. | |||||
| Me2N | 1 → 3 | 420 | 0.225 | 55% χ138→139 + 35% χ136→139 | Ru + Me2Nph → tpy |
| MeO | 1 → 4 | 416 | 0.114 | 80% χ133→135 | Ru + ε acac → tpy |
| H | 1 → 4 | 416 | 0.089 | 86% χ125→127 | Ru + ε acac → tpy |
| CHOb | 1 → 4 | 421 | 0.089 | 77% χ132→134 | Ru → CHO-phtpy |
| NO2 | 1 → 4 | 424 | 0.134 | 52% χ136→138 + 35% χ136→140 | Ru → NO2-phtpy |
The effect of donor/acceptor substituents appears from the examination of the figure. According to the DFT-computed data, for strong donors (NMe2), the energy of the occupied level increases, while that of the empty level remains roughly unaffected, thus decreasing the energy gap (increased λmax value). For a strong acceptor (NO2), the energy of the empty level decreases, while that of the occupied level remains roughly unaffected, thus decreasing the energy gap (increased λmax value). Finally, both effects are similar, and provide a V-shaped character for the λmax curve drawn in Fig. 4. The consequence of donor/acceptor substitutions will be investigated in the next section.
Photo-release of acetonitrile
The photo-release experiments were carried out under irradiation at λirr = 455 nm for the 6 complexes [Ru(R-phtpy)(acac)(MeCN)](PF6) (R = Me2N, Et2N, OMe, Me, H, and NO2) in water solutions. The wavelength was selected to correspond to a domain where the 6 complexes exhibit a significant absorption. A representative example is illustrated in Fig. 6 for [Ru(MeO-phtpy)(acac)(MeCN)](PF6), which shows the evolution of the UV-visible spectrum under irradiation.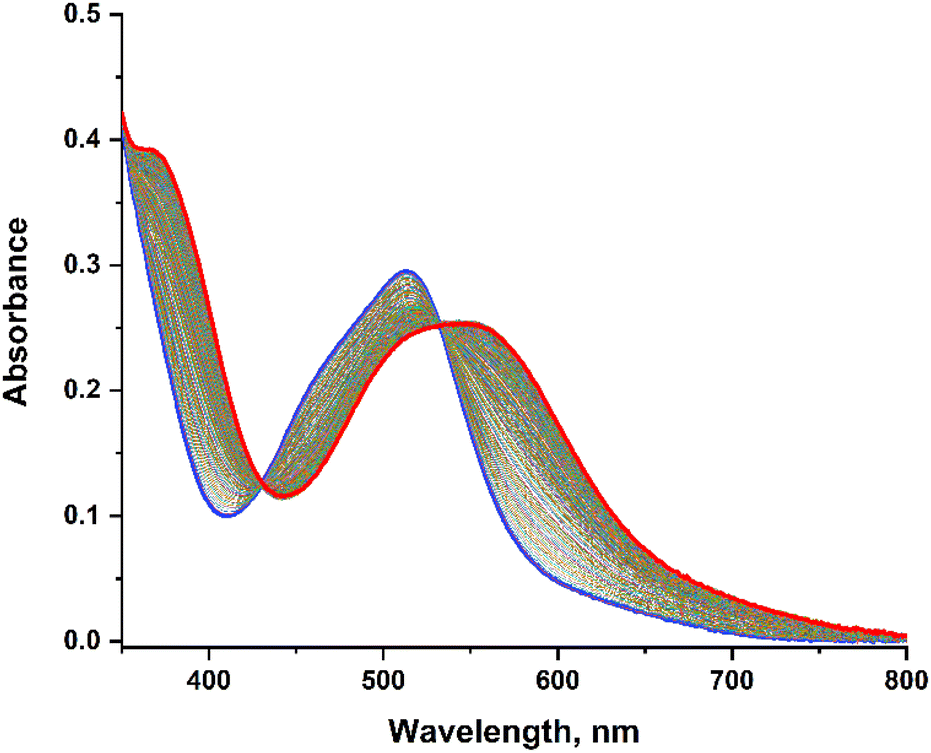 | ||
| Fig. 6 Evolution of the UV-visible spectrum of [Ru(MeO-phtpy)(acac)(MeCN)](PF6) in water, under irradiation at λirr = 455 nm, over a 2 hours period. The starting complex (t = 0) is in blue; the photoproduct (t = 2 hours) is in red. | ||
The experiments for the other complexes are provided in the ESI (Fig. S2†), except for [Ru(NO2-phtpy)(acac)(MeCN)](PF6) which undergoes no UV-visible changes. During the irradiation, a gradual evolution is observed with the appearance of an isosbestic point, which indicates that no other photo-active compound other than the starting Ru(MeCN) complex and the photoproduct is present. In any case, the dominant UV-visible band is shifted from ∼500 nm to ∼550 nm, which corresponds roughly to an energy shift of −1800 cm−1, and is in accordance with the observation made by Turro et al. on parent Ru(MeCN) complexes subjected to irradiation.50 To the best of our knowledge, the nature of the photoproduct was never addressed unambiguously in the previous investigation devoted to Ru(MeCN). However, the MeCN release achieved in water likely leads to [Ru(R-phtpy)(acac)(H2O)](PF6). This is supported by DFT computations which indicate an energy shift of about −1400 cm−1 on going from Ru(MeCN) to Ru(H2O) complexes (Table 4).
| R | [Ru(R-phtpy)(acac)(MeCN)]+ | [Ru(R-phtpy)(acac)(H2O)]+ | E shift occurring during the photo-release (red-shift) |
|---|---|---|---|
| Me2N | 420 nm | 447 nm | −1439 cm−1 |
| MeO | 416 nm | 443 nm | −1465 cm−1 |
| H | 416 nm | 443 nm | −1465 cm−1 |
| NO2 | 424 nm | 448 nm | −1264 cm−1 |
The quantum yields of the photo-release of acetonitrile defined as ϕMeCN = mol of MeCN released per mol of photons absorbed by the Ru(MeCN) complexes are shown in Fig. 7, with values presented in Table 5. It appears unambiguously that reducing the donating character of R reduces the ϕMeCN value, which totally vanishes when R becomes a strong acceptor (NO2). This observation substantially agrees with a previous report that better donor ligands lead to better MeCN release.50
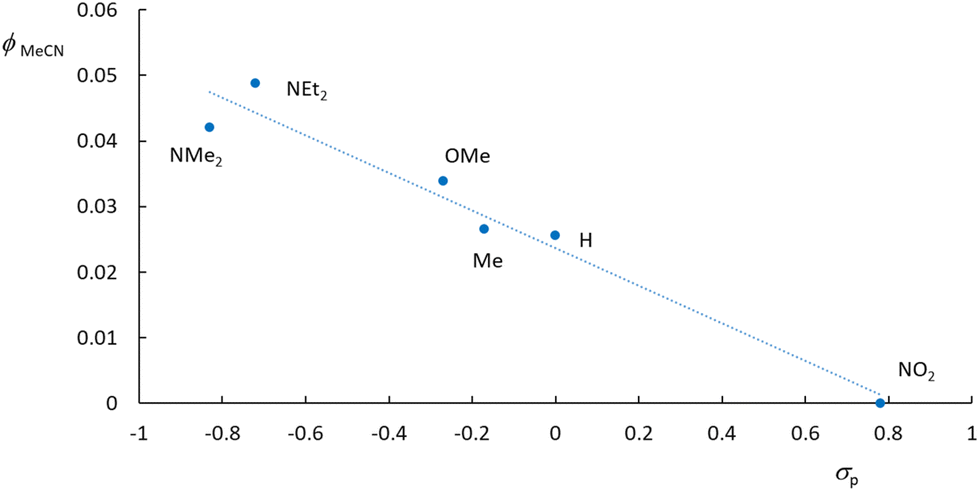 | ||
| Fig. 7 Quantum yield of MeCN photo-release in water in a series of [Ru(R-phtpy)(acac)(MeCN)](PF6) complexes drawn against the Hammett constant (σp) of the R substituents. | ||
| R | ϕ MeCN |
|---|---|
| NEt2 | 0.049 |
| NMe2 | 0.042 |
| OMe | 0.034 |
| Me | 0.027 |
| H | 0.026 |
| NO2 | 0.000 |
Following the generally accepted idea that the interplay between the 3MLCT and 3MC excited states of Ru(MeCN) complexes provides the rationale for understanding the photo-release capabilities of these species,36,37 the excited states of representative [Ru(R-phtpy)(acac)(MeCN)]+ complexes were computed by DFT to tentatively account for the data gathered in Fig. 7. The geometry of 3MC states was targeted at extended Ru–NC bonds by a scan procedure to reach final convergence. The energies are presented in Fig. 8. The point of interest in this figure is the relative position of the 3MLCT and 3MC states. It can be assumed from the examination of Fig. 5, and as previously reported by Turro's group,50 that the electron density is largely spread on the terpyridine fragment after the 1GS → 1MLCT initial transition. Therefore, it can be inferred that, after a fast intersystem crossing to 3MLCT, donating R substituents favor further evolution towards the 3MC state, where the spin density spread on the terpyridine is transferred to the ruthenium atom, as evidenced from the 3MLCT/3MC energy difference in Fig. 8. Interestingly, the Ru–N bond length in the Ru-MeCN fragment, which is around 2.0 Å in the complexes (Table 2), is significantly elongated to 4.2 Å in the 3MC states, due to the contribution of σ* overlaps in the Ru–N bonds, with the outcome of a strong weakening of the bond. This provides a dissociative character to 3MC. It is noteworthy that [Ru(NO2-phtpy)(acac)(MeCN)](PF6) is the only system where no MeCN release is observed (Fig. 7). It is also the only one where E(3MC) > E(3MLCT) (Fig. 8), thus prohibiting the thermal population of the potentially dissociative 3MC state.
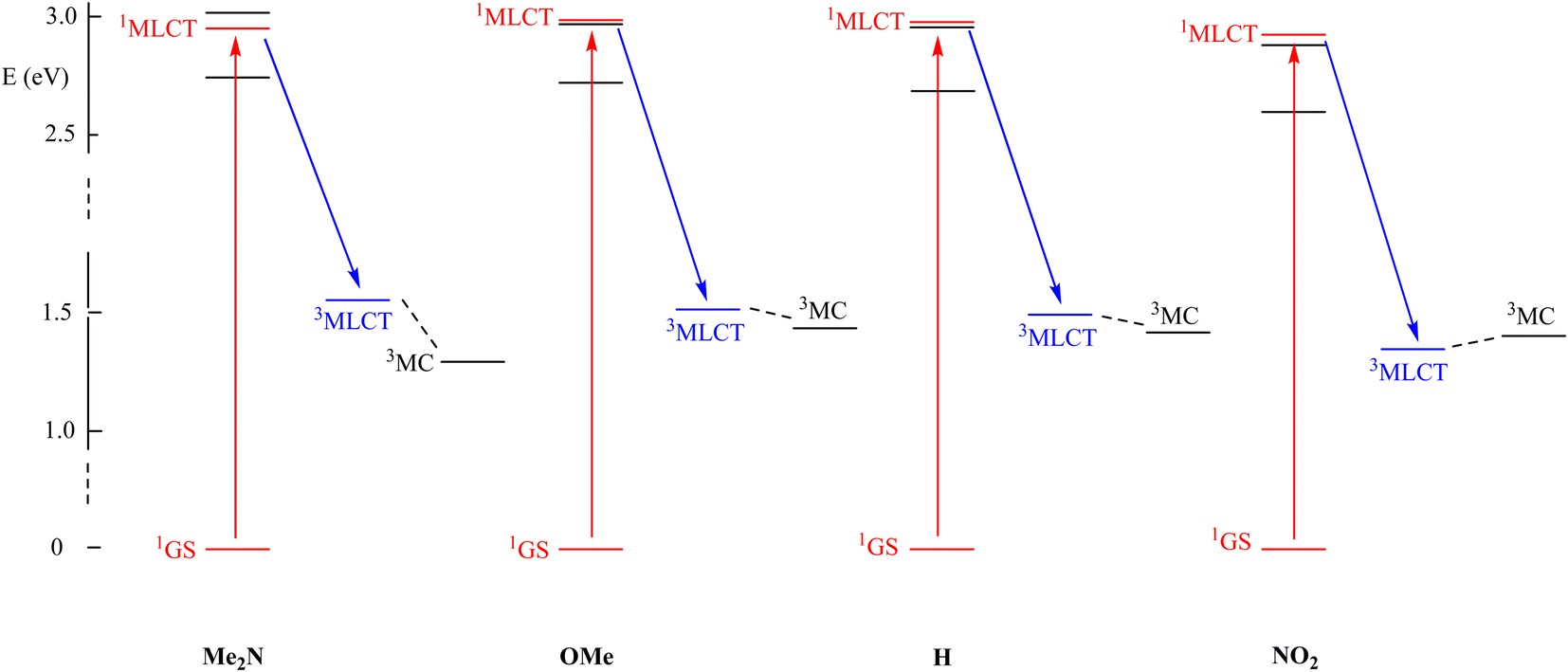 | ||
| Fig. 8 DFT computed energies of the main excited states involved during the absorption process of [Ru(R-phtpy)(acac)(MeCN)]+ for different R substituents, showing the initial absorption (in red) and the fast intersystem crossing (in blue). | ||
The observation that the gradual stabilization of the 3MC state (Fig. 7) grossly correlates with the evolution of ϕMeCN (Table 5) leads to the natural idea that this state with elongated Ru–NC bonds is indeed responsible for the MeCN release. However, great care must be taken before reaching this conclusion as no direct 3MLCT → 3MC internal conversion is evidenced at this stage, and the release may arise from still undiscovered reaction paths involving potential intersystem-crossings which are not considered here. Therefore, the search for the precise mechanism remains a challenge for theoretical chemists. In this regard, the comparison of the current study results with the already published results appears relevant.
Pioneering works by Ford et al. in the 1970s56,57 reported that the 3MLCT excited states in a series of fourteen Ru(NH3)5(py-R)2+ complexes bearing various substituents were relatively unreactive towards substitution. They proposed that the photoreactivity likely arises from a ligand field excited state rather than the 3MLCT state.
The work by Chen et al. sheds light on the effect of a ligand substituent's electron push/pull character on pyridine dissociation in a series of tetrahedral Ru(II) arene complexes,58 [(η6-p-cymene)Ru(dpb)(py-R)]2+ (dpb = 2,3-bis(2-pyridyl)benzoquinoxaline; py-R = 4-substituted pyridine, R = N(CH3)2, NH2, OCH3, H, COOCH3 and NO2). Interestingly, the authors describe the photorelease mechanism using the same excited-state model as in our study. However, in contrast to our observations, they found that electron-withdrawing substituents on the pyridine ligand enhance photorelease efficiency. This was rationalized by proposing that in these systems, the triplet excited states follow the energetic order 3MC > 3MLCT > 3IL (involving the dpb ligand), with small enough energy gaps to permit thermal activation from 3MLCT to 3MC and from 3IL to 3MLCT. In this case, electron-withdrawing groups stabilize the Ru-based t2g orbitals and weaken the ligand field, lowering the energy of the 3MC state and promoting more efficient photodissociation.
The ability to rationalize such seemingly opposing experimental results using the same sequential excited-state model (1MLCT → 3MLCT → 3MC) provides indirect support for the robustness and general applicability of this mechanistic framework.
In contrast to Ford's model, Turro et al. proposed that ligand release can occur directly from the 3MLCT excited state. Initially, they observed a linear correlation between ϕMeCN at 400 nm and the Hammett constants (σp) in a series of Ru(II) complexes (cis-[Ru(bpy)2(P(p-R-Ph)3)(CH3CN)]2+, where bpy = 2,2′-bipyridine and P(p-R-Ph)3 are para-substituted triphenylphosphines with R = –OCH3, –CH3, –H, –F, and –CF3).59 Complexes bearing stronger electron-donating substituents exhibited higher MeCN photorelease efficiencies, a trend that the authors were not able to explain with Ford's model. Extending their work to a broader series of 11 Ru(II)-MeCN complexes of the type [Ru(tpy)(L)(CH3CN)]n+ with various bidentate ligands L, they concluded that direct ligand photorelease from the 3MLCT state was the most consistent explanation.39
In our current contribution, we associate MeCN release with the capabilities of the complexes to populate a dissociative 3MC state. Although further studies are needed to confirm this mechanistic proposal, the theoretical investigations presented here show a correlation between the photorelease efficiency and the accessibility of the 3MC state. The scarcity of theoretical mechanistic studies60 on photoactivated chemotherapy agents underscores the significance of our current study, considering that discrete and relatively simple changes in the ligand framework modulated the accessibility of the 3MC state and consequently the photoreactivity of the complexes, thus providing a design strategy that could be exploited in other studies.
It is also worth pointing out an important difference observed between the MeCN release achieved from [Ru(R-phtpy)(acac)(MeCN)](PF6) (present study) and that of NO achieved from [Ru(R-phtpy)(acac)(NO)](PF6) complexes.53 While a clear correlation is evidenced between ϕMeCN and the Hammett constant of R (Fig. 6), this correlation is totally absent for ϕNO in the [Ru(R-phtpy)(acac)(NO)](PF6) series,53 as observed in a series of [Ru(R-phtpy)(Cl2)(NO)](PF6) compounds previously investigated in our group.61 It can be concluded that the mechanisms of MeCN and NO release are probably substantially different. Indeed, computational studies have led to the idea that after a first electronic transition, the linear Ru–NO fragment is subjected to an isomerization process to a bent and no dissociative geometry, followed by the absorption of a second photon required to populate the dissociative state.62–64 This mechanism does not operate here, where the photo-product appears dissociative, due to the weakness of the Ru–MeCN bond in the 3MC state. We would like to remark that elucidating the real ligand photorelease reaction mechanism in Ru(II) complexes requires further research efforts applied in this direction.
Conclusion
A series of six Ru(MeCN) complexes has been reported. All of them contain terpyridine ligands bearing substituents of various electron donor/acceptor capabilities. The MeCN release was measured under irradiation in water and rationalized. Contrary to the findings reported recently by our research group on closely related Ru(NO) complexes,53 a clear trend is evidenced between the quantum yield of photo-release and the donor/acceptor strengths of the substituents. This can be tentatively related to the position of the 3MC state, vs. that of the 3MLCT in these systems. Although no thorough theoretical analysis was carried out to elucidate this issue, 3MC, where the Ru–NC bond is elongated to ∼4.2 Å, appears to be a key state in the process of MeCN release. These investigations lead to the idea that donor ligands are required to increase the photo-release of acetonitrile in these Ru(MeCN) complexes. This could offer interesting perspectives, because the design of 4′-substituted terpyridine is well documented.65–67 Further studies should also delve into the photoluminescence of this class of complexes, as an appealing expectation considering the accessibility of low-lying 3MLCT states.Experimental section
Materials and equipment
The [Ru(R-phtpy)(acac)(MeCN)](PF6) complexes were synthesized as previously reported.53 All solvents used for optical characterization were purchased from commercial distributors and used without further purification. UV-Vis absorption spectroscopy and photokinetic studies were performed on an Agilent Cary 60 UV-Visible spectrophotometer with a cooling water regulator at 25 °C. All photokinetic measurements were performed under irradiation at λ = 455 nm. Light irradiation was provided using a chassis-wheeled wavelength switchable LED source from Mightex Company. The power of the LED was controlled via the Thorlabs PM130D Power Meter. All optical measurements were carried out in quartz cuvettes.X-Ray diffraction
Single crystals suitable for crystal structure determination were obtained by slow diffusion of diethyl ether into concentrated solutions of the substance in acetonitrile. Data were collected on a Rigaku XtaLAB Synergy Dualflex diffractometer using a PhotonJet X-ray source (CuKα, λ = 1.54184 Å). An Oxford Cryosystems Cryostream cooling device was used to collect data at low temperature (100(2) K). Omega scans were performed for data collection. An empirical absorption correction was applied and the structures were solved by an intrinsic phasing method (ShelXT).68 All non-hydrogen atoms were refined anisotropically by means of least-squares procedures on F2 with ShelXL.69 All the hydrogen atoms were refined isotropically at calculated positions using a riding model. For [Ru(MeO-phtpy)(acac)(MeCN)](PF6) and [Ru(H-phtpy)(acac)(MeCN)](PF6), the anisotropic displacement parameters of the disordered moieties were restrained using DELU and SIMU commands. The structure is deposited at the Cambridge Crystallographic Data Centre (CCDC 2433541–2433544†).Computational studies
The ruthenium complexes were fully optimized using the Gaussian-09 program package,70 within the framework of the density functional theory (DFT). The presence of solvent (water) was modelized by the polarizable continuum model (SCRF = PCM method).71 The double-ζ basis set 6-31G* was used for all atoms except the heavy ruthenium atom, for which the LANL2DZ basis set was applied to account for relativistic effects.72 The computations were performed using the B3PW91 method, for consistency with our previous investigations on substituted [Ru(R-phtpy)(acac)(NO)](PF6) compounds.53 Vibrational analysis was performed at the same level to verify that the stationary points correspond to minima on the potential energy surfaces. Several functionals B3PW91, PBE0 and CAM-B3LYP were tested for the computation of the UV-visible spectra by time-dependent (TD)-DFT. CAM-B3LYP73 was finally selected as it provides a satisfactory shape of the overall spectra and a simplified analysis restricted to a set of limited numbers of orbitals and is consistent with the previously reported investigation of [Ru(R-phtpy)(acac)(NO)](PF6) complexes frequently used for comparison in the present contribution. Contrary to [Ru(R-phtpy)(acac)(NO)](PF6), a noticeable discrepancy in the 4000–4500 cm−1 energy range was observed between computation and experimental data for the different complexes, the computed energies being higher than the observed ones. However, this trend is commonly observed in systems in which the path of π-delocalization is reduced, as it is the case in going from Ru(NO) to Ru(MeCN) complexes. It is explained by the fact that CAM-B3LYP was initially designed for the investigation of molecules having long range π-delocalized electronic structures. Nevertheless, we observed that, apart for this anticipated energy shift, the functional provides a qualitative description of the electronic spectra, with trends along the entire series of R-substituted ruthenium complexes satisfactorily reproduced. For each complex, the geometry of the triplet 3MLCT states was optimized at the same level of theory as the singlet states (B3PW91/631G*). Then, scans were performed on the Ru–NCMe distance to target the geometry of 3MC states, which were finally optimized. Both 3MLCT and 3MC states were identified from the value of the Mulliken spin density (MSD) parameters on the ruthenium centers which fell in the 0.7–0.9 range in 3MLCT and in the 1.7–1.8 range in 3MC.Photochemical studies
The methodology used for the photokinetics studies was that reported by Turro et al.29,49,50 To prepare solutions of Ru(MeCN) complexes for optical measurement, and due to their partial solubility in pure water, the following procedure was used: weighed amounts of complexes (5–10 mg) were dissolved in acetone. 0.1 mL of the obtained solutions was dissolved in 2.9 mL of water resulting in water solution of the Ru(MeCN) complex ([c] ∼10−5 mol L−1) with ∼3.33% acetone, suitable for further optical studies. The quantum yields of acetonitrile release (ϕMeCN) were determined using the program Sa3.3 written by D. Lavabre and V. Pimienta.74Data availability
The data supporting this article have been included as part of the ESI.†Crystallographic data for [Ru(Et2N-phtpy)(acac)(MeCN)](PF6), [Ru(MeO-phtpy)(acac)(MeCN)](PF6), [Ru(H-phtpy)(acac)(MeCN)](PF6), and [Ru(NO2-phtpy)(acac)(MeCN)](PF6) have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under [2433541–2433544†] numbers.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding through the MSCA4Ukraine project, which is funded by the European Union. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union. Neither the European Union nor the MSCA4Ukraine Consortium as a whole nor any individual member institutions of the MSCA4Ukraine Consortium can be held responsible for them. The authors thank Dr Martial Boggio-Pasqua (Laboratoire de Chimie et Physique Quantiques, UPS-Toulouse) for fruitful discussions.References
- L. Conti, E. Macedi, C. Giorgi, B. Valtancoli and V. Fusi, Coord. Chem. Rev., 2022, 469, 214656 CrossRef CAS.
- I. Romero-Canelón, in Metal-based Anticancer Agents, ed. A. Casini, A. Vessieres and S. M. Meier-Menches, The Royal Society of Chemistry, 2019, pp. 31–61 Search PubMed.
- A. Rilak Simović, R. Masnikosa, I. Bratsos and E. Alessio, Coord. Chem. Rev., 2019, 398, 113011 CrossRef.
- M. Mital and Z. Ziora, Coord. Chem. Rev., 2018, 375, 434–458 CrossRef CAS.
- V. Brabec and J. Kasparkova, Coord. Chem. Rev., 2018, 376, 75–94 CrossRef CAS.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190–191 CrossRef CAS PubMed.
- M. J. Clarke, in Metal Ions in Biological Systems, ed. H. Sigel, Marcel Dekker, 1980, pp. 231–283 Search PubMed.
- D. Pluim, R. C. A. M. van Waardenburg, J. H. Beijnen and J. H. M. Schellens, Cancer Chemother. Pharmacol., 2004, 54, 71–78 CrossRef CAS PubMed.
- T. V. Harris, R. K. Szilagyi and K. L. McFarlane Holman, J. Biol. Inorg. Chem., 2009, 14, 891–898 CrossRef CAS PubMed.
- I. Stepanenko, M. Zalibera, D. Schaniel, J. Tesler and V. V. Arion, Dalton Trans., 2022, 51, 5367–5393 RSC.
- P. G. Lacroix, I. Malfant, P. Labra-Vázquez, N. Farfan and G. Ramos-Ortiz, Dalton Trans., 2022, 51, 14833–14841 RSC.
- J. M. Mir, B. A. Malik and R. C. Maurya, Rev. Inorg. Chem., 2019, 2, 91–112 Search PubMed.
- H. J. Xiang, M. Guo and J.-G. Liu, Eur. J. Inorg. Chem., 2017, 1586–1595 CrossRef CAS.
- E. Tfouni, D. R. Truzzi, A. Tavare, A. J. Gomes, L. E. Figueiredo and D. W. Franco, Nitric Oxide, 2012, 26, 38–53 CrossRef CAS PubMed.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 Search PubMed.
- M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093–2114 CrossRef CAS PubMed.
- P. C. Ford, Acc. Chem. Res., 2008, 41, 190–200 CrossRef CAS PubMed.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed.
- M. Milani, G. Jha and D. A. Potter, Clin. Med. Ther., 2009, 1, 141–156 CAS.
- S. Gangadhara and G. Bertelli, Ther. Clin. Risk Manage., 2009, 5, 291–300 CAS.
- R. M. Cooper-DeHoff, E. M. Handberg, G. Mancia, Q. Zhou, A. Champion, U. F. Legler and C. J. Pepine, Expert Rev. Cardiovasc. Ther., 2009, 7, 1329–1340 CrossRef CAS PubMed.
- D. A. Sica and L. M. Prisant, J. Clin. Hypertens., 2007, 9, 1–22 CrossRef CAS.
- R. N. Brogden, P. Benfield and A. Gallopamil, Drugs, 1994, 47, 93–115 CrossRef CAS PubMed.
- T. Jackson, L. W. L. Woo, M. N. Trusselle, A. Purohit, M. J. Reed and B. V. L. Potter, ChemMedChem, 2008,(no. 3), 603–618 CrossRef CAS PubMed.
- T. Jackson, L. W. L. Woo, M. N. Trusselle, S. K. Chander, A. Purohit, M. J. Reed and B. V. L. Potter, Org. Biomol. Chem., 2007,(no. 5), 2940–2952 RSC.
- R. Mannhold, H. D. Holtje and V. Koke, Arch. Pharm., 1986, 319, 990–998 CrossRef CAS PubMed.
- R. C. K. Cheng, D. B. Tikhonov and B. S. Zhorov, J. Biol. Chem., 2009, 284, 28332–28342 CrossRef CAS PubMed.
- T. N. Rohrabaugh, K. A. Collins, C. Xue, J. K. White, J. J. Kodanko and C. Turro, Dalton Trans., 2018, 47, 11851–11858 RSC.
- A. Li, C. Turro and J. J. Kodanko, Chem. Commun., 2018, 54, 1280–1290 RSC.
- T. Respondek, R. Sharma, M. K. Herroon, R. N. Garner, J. D. Knoll, E. Cueny, C. Turro, I. Podgorski and J. J. Kodanko, ChemMedChem, 2014, 9, 1306–1315 CrossRef CAS PubMed.
- M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS PubMed.
- T. Respondek, R. N. Garner, M. K. Herroon, I. Podgorski, C. Turro and J. Kodanko, J. Am. Chem. Soc., 2011, 133, 17164–17167 CrossRef CAS PubMed.
- R. N. Garner, J. C. Gallucci, K. R. Dunbar and C. Turro, Inorg. Chem., 2011, 50, 9213–9215 CrossRef CAS PubMed.
- R. J. Davidson, Y.-T. Hsu, D. S. Yufit and A. Beeby, ACS Omega, 2024, 9, 34098–34105 CrossRef CAS PubMed.
- A. Soupart, F. Alary, J. L. Heully, P. I. P. Elliot and I. M. Dixon, Coord. Chem. Rev., 2020, 408, 213184 CrossRef CAS.
- J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280–2287 CrossRef CAS PubMed.
- N. H. Damrauer, G. Cerullo, A. Yeh, T. R. Boussie, C. V. Shank and J. K. McCusker, Science, 1997, 275, 54–57 CrossRef CAS PubMed.
- S. J. Steinke, E. J. Piechota, L. M. Loftus and C. Turro, J. Am. Chem. Soc., 2022, 144, 20177–20182 CrossRef CAS PubMed.
- L. M. Loftus, J. J. Rack and C. Turro, Chem. Commun., 2020, 56, 4070–4073 RSC.
- E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133–142 RSC.
- E. Tfouni, Coord. Chem. Rev., 2000, 196, 281–305 CrossRef CAS.
- P. C. Ford, Coord. Chem. Rev., 1982, 44, 61–82 CrossRef CAS.
- M. McCutcheon, M. Freindorf and E. Kraka, J. Chem. Phys., 2022, 157, 014301 CrossRef CAS PubMed.
- Y. Liu, D. B. Turner, T. N. Singh, A. M. Angeles-Boza, A. Chouai, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2009, 131(1), 26–27 CrossRef CAS PubMed.
- Y.-J. Tu, S. Mazumder, J. F. Endicott, C. Turro, J. J. Kodanko and H. B. Schlegel, Inorg. Chem., 2015, 54(16), 8003–8011 CrossRef CAS PubMed.
- K. Arora, J. K. White, R. Sharma, S. Mazumder, P. D. Martin, H. B. Schlegel, C. Turro and J. J. Kodanko, Inorg. Chem., 2016, 55(14), 6968–6979 CrossRef CAS PubMed.
- L. M. Loftus, A. Li, K. L. Fillman, P. D. Martin, J. J. Kodanko and C. Turro, J. Am. Chem. Soc., 2017, 139(50), 18295–18306 CrossRef CAS PubMed.
- L. M. Loftus, K. F. Al-Afyouni and C. Turro, Chem. – Eur. J., 2018, 24, 11550–11553 CrossRef CAS PubMed.
- L. M. Loftus, K. F. Al-Afyouni, T. N. Rohrabaugh Jr., J. C. Gallucci, C. E. Moore, J. J. Rack and C. Turro, J. Phys. Chem. C, 2019, 123, 10291–10299 CrossRef CAS.
- M. R. Hamblin and T. N. Demidova, Proc. SPIE, Mech. Low-Light Ther., 2006, 6140, 614001–614012 Search PubMed.
- R. Santana da Silva, R. Galvao de Lima and S. P. Machado, Adv. Inorg. Chem., 2015, 67, 265–294 CrossRef CAS.
- P. Labra-Vázquez, V. Mudrak, M. Tassé, S. Mallet-Ladeira, A. Sournia-Saquet, J.-P. Malval, P. G. Lacroix and I. Malfant, Inorg. Chem., 2023, 62, 20349–20363 CrossRef PubMed.
- M. T. Rupp, N. Shevchenko, G. S. Hanan and D. G. Kurth, Coord. Chem. Rev., 2021, 446, 214127 CrossRef CAS.
- C. Hansch, A. Leo and R. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- G. Malouf and P. C. Ford, J. Am. Chem. Soc., 1974, 96(2), 601–603 CrossRef CAS.
- R. E. Hintze and P. C. Ford, J. Am. Chem. Soc., 1975, 97(10), 2664–2671 CrossRef CAS.
- Y. Chen, W. Lei, Y. Hou, C. Li, G. Jiang, B. Zhang, Q. Zhou and X. Wang, Dalton Trans., 2015, 44, 7347–7354 Search PubMed.
- A. P. Lanquist, S. Gupta, K. F. Al-Afyouni, M. Al-Afyouni, J. J. Kodanko and C. Turro, Chem. Sci., 2021, 12, 12056–12067 RSC.
- J. I. M. Dixon, S. Bonnet, F. Alary and J. Cuny, Phys. Chem. Lett., 2021, 12, 7278–7284 CrossRef PubMed.
- S. Amabilino, M. Tasse, P. G. Lacroix, S. Mallet-Ladeira, V. Pimienta, J. Akl, I. Sasaki and I. Malfant, New J. Chem., 2017, 41, 7371–7383 RSC.
- J. S. Garcia, F. Alary, M. Boggio-Pasqua, I. M. Dixon and J.-L. Heully, J. Mol. Model., 2016, 22, 284 CrossRef PubMed.
- L. Freitag and L. González, Inorg. Chem., 2014, 53, 6415–6426 CrossRef CAS PubMed.
- F. Talotta, L. González and M. Boggio-Pasqua, Molecules, 2020, 25, 2613 Search PubMed.
- R.-A. Fallahpour, Synthesis, 2003, 155–184 Search PubMed.
- W. Goodall, K. Wild, K. J. Arma and J. A. Gareth Williams, J. Chem. Soc., Perkin Trans. 2, 2002, 1669–1681 RSC.
- J. Wang and G. S. Hanan, Synlett, 2005,(08), 1251–1254 CAS.
- G. M. Sheldrick, ShelXT, University of Göttingen, 2015, Acta Crystallogr. Sect. A, 71, 3–8 Search PubMed.
- G. M. Sheldrick, ShelXL, University of Göttingen, 2015, Acta Crystallogr. Sect. C, 71, 3–8.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 Search PubMed.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (b) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- V. Pimienta, D. Lavabre and J. C. Micheau, https://cinet.chim.cnrs.fr/.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2433541–2433544. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00734h |
| This journal is © The Royal Society of Chemistry 2025 |