
Structural diversity of nucleotide coordination polymers of cytidine mono-, di-, and tri-phosphates and their selective recognition of tryptophan and tyrosine†
Yaqoot
Khan
 a,
Yunyun
Du
a,
Li
Yan
b,
Niu
Zhang
a,
Menglei
Zhang
a,
Hongwei
Ma
*b and
Hui
Li
*a
a,
Yunyun
Du
a,
Li
Yan
b,
Niu
Zhang
a,
Menglei
Zhang
a,
Hongwei
Ma
*b and
Hui
Li
*a
aKey Laboratory of Cluster Science of Ministry of Education, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: lihui@bit.edu.cn; Tel: +86-10-68912667-602
bAnalysis & Testing Center of Beijing Institute of Technology, Beijing 102488, P. R. China. E-mail: hwma@bit.edu.cn
First published on 16th May 2025
Abstract
Understanding the coordination geometry of nucleotide mono-, di-, and triphosphates is pivotal for unraveling the intricate relationships between molecular structure and biological function, particularly in metal–ligand interactions and their role in biomolecular recognition. This study investigates the structure of nucleotide–metal polymers and their selective interactions with amino acids, specifically tryptophan (Trp) and tyrosine (Tyr). We synthesized and comprehensively analyzed five coordination polymers of cytidine nucleotides: cytidine monophosphate (CMP), deoxycytidine monophosphate (dCMP), cytidine diphosphate (CDP), and cytidine triphosphate (CTP), which are {[Cu(CMP)(bpa)(H2O)3](CMP)·3H2O}n (1), {[Cu2(dCMP)(4,4′-bipy)2(H2O)2]·4H2O}n (2), {[Cu2(CDP)2(azpy)(H2O)]·3H2O}n (3), {[Cd2(CDP)2(bpa)2(H2O)2]·8H2O}n (4), and {[Cu(CTP)(2,2′-bipy)]·2H2O}n (5), where (3), (4) and (5) mark the first report of CDP and CTP coordination complexes. Single-crystal X-ray diffraction unveiled the structure of the polymers to be one-dimensional (1, 5) or two-dimensional (2, 3, 4). Circular dichroism (CD) spectroscopy in both the solid and solution states elucidates the chirality-driven assembly in these nucleotide–metal polymers. The selective interactions of coordination polymers with tryptophan (Trp) and tyrosine (Tyr) were studied using spectroscopic titrations. Experimental and computational analyses reveal distinct interactions between all five coordination polymers and the amino acids, highlighting their biosensing potential. Binding affinity variations across the polymers offer insights into nucleotide–metal coordination chemistry and suggest applications in molecular recognition and functional materials.
Introduction
Nucleotides, the primary components of nucleic acids, are essential for various biological functions, including the storage and transfer of genetic information, energy metabolism, and cellular signaling.1–3 They consist of a nitrogenous base, a pentose sugar, and one or more phosphate groups, which can be classified into mono-, di-, and triphosphates based on the number of phosphate groups present.4–7 The structural diversity of nucleotides, particularly their chirality and metal-binding sites,8 makes them highly versatile ligands in coordination chemistry.9,10 The ability of nucleotides to self-assemble into well-ordered structures through hydrogen bonding and stacking interactions is crucial for their biological roles and has implications for the design of biomimetic materials.11 Research into nucleotide mono-, di-, and triphosphates12 has significantly advanced our understanding of their roles in biochemical processes. These compounds are not only integral to the synthesis of nucleic acids but also serve as substrates in various enzymatic reactions.9,13–15 The coordination chemistry of such nucleotides with metal ions has garnered attention due to the impact of metal ions on the structural integrity and reactivity of nucleotide complexes.12The exploration of nucleotide–metal coordination complexes has revealed their potential as tools for studying the conformation of nucleotides,16 molecular recognition,17 self-assembly processes18 and interactions with biomolecules.19,20 Moreover, the ability of coordination complexes to selectively bind to amino acids,21 particularly tryptophan and tyrosine,22 underscores their significance in biochemical applications.23 Recent investigations have highlighted the role of metal ions in modulating the interactions between nucleotides and amino acids.24 Computational analysis of binding energies provides insights into the stability and reactivity of nucleotide–metal complexes, enhancing their selective interactions with amino acids.25,26 Studies show that metal ion coordination boosts the binding affinity of nucleotide complexes towards specific amino acids, critical for understanding biochemical processes.27–29 These analyses reveal that the coordination environment affects both the stability and selectivity of interactions.30 Combining computational studies with spectroscopic techniques has clarified the interplay between metal ions, nucleotides, and amino acids,28 offering new possibilities in materials science and biomedicine.31
In this study, we focus on the structure and selective binding properties of nucleotide–metal coordination polymers towards the L- and D-forms of amino acids, specifically tryptophan and tyrosine. By employing spectroscopic techniques and computational analyses, including binding energy calculations, we aim to elucidate the binding affinities and mechanisms underlying these interactions. We designed and studied five coordination polymers of cytidine nucleotides: cytidine monophosphate (CMP), deoxycytidine monophosphate (dCMP), cytidine diphosphate (CDP), and cytidine triphosphate (CTP), which are {[Cu(CMP)(bpa)(H2O)3](CMP)·3H2O}(1), {[Cu2(dCMP)(4,4′-bipy)2(H2O)2]·4H2O}n (2), {[Cu2(CDP)2(azpy)(H2O)]·3H2O}n (3), {[Cd2(CDP)2(bpa)2(H2O)2]·8H2O}n (4), and {[Cu(CTP)(2,2′-bipy)]·2H2O}n (5) (bpa = 1,2′-bis(4-pyridyl)ethane, azpy = 4,4′-azopyridine, 4,4′-bipy = 4,4′-bipyridine, 2,2′-bipy = 2,2′-bipyridine) (Schemes 1, S2, and S3†).
 | ||
| Scheme 1 Schematic overview of the synthesis and crystallization of nucleotide–metal coordination complexes, along with amino acid recognition studies using experimental and computational approaches. | ||
Experimental section
Materials and methods
Reagents and organic solvents were sourced from reputable commercial suppliers and utilized without additional purification. Ligands 1,2′-bis(4-pyridyl)ethane (bpa), 2,2′-bipyridine (2,2′-bipy), 4,4′-azopyridine (azpy), and 4,4′-bipyridine (4,4-bipy) were obtained from Adamas, and metal nitrates (Cu2+ or Cd2+) were purchased from Aladdin. Deoxycytidine 5′-monophosphate, cytidine 5′-monophosphate, cytidine 5′-diphosphate, and cytidine 5′-triphosphate disodium salts were acquired from Alfa Aesar. L- and D-forms of tryptophan and tyrosine were purchased from Macklin.FT-IR spectra were recorded using a Thermo IS5 FT-IR spectrometer, employing KBr samples over a range of 400 to 4000 cm−1. Powder X-ray diffraction (PXRD) patterns were obtained using a Bruker D8 Advance X-ray diffractometer, with a scan speed of 5° min−1 and a 2θ measurement range of 5° to 50°. Single-crystal analysis was conducted using X-ray diffraction equipment fitted with graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å). Elemental composition (C, H, and N) was measured with an EA3000 elemental analyzer. The pH of the solution was determined with a PHB-1PH meter. UV-visible absorption spectra were collected using a Shimadzu UV-2600 spectrophotometer. Fluorescence spectra were measured using a model F-7000 FL spectrophotometer. Circular dichroism (CD) spectra of solid samples were measured at room temperature using a JASCO J-1500 spectropolarimeter, employing KBr pellets at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 under a constant nitrogen flow. For solution measurements, CD spectra were obtained at a concentration of 0.025 mmol L−1. Thermogravimetric analyses (TGA) were carried out with a DTG-60H thermal analyzer. All TGA analyses were carried out under a nitrogen atmosphere, covering a temperature span of 50 °C to 700 °C, with a heating rate of 10 °C min−1.
:200 under a constant nitrogen flow. For solution measurements, CD spectra were obtained at a concentration of 0.025 mmol L−1. Thermogravimetric analyses (TGA) were carried out with a DTG-60H thermal analyzer. All TGA analyses were carried out under a nitrogen atmosphere, covering a temperature span of 50 °C to 700 °C, with a heating rate of 10 °C min−1.
Synthesis and structural characterization
Crystallographic data collection and refinement
Coordination polymers with appropriate dimensions were selected for single-crystal X-ray diffraction analysis. Data collection was performed at room temperature (296.15 K) utilizing a Bruker DUO APEX II-CCD diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Peak identification and unit cell parameter determination were performed using XSCAN software at 45 kV and 30 mA. Diffraction data were obtained in the ω − 2θ scanning mode, and all scanned data underwent empirical absorption correction. The crystal structure was determined through the SHELXT program,32 implemented via Olex2 software.33 Refinement was conducted using full matrix least squares techniques with the SHELXL program34 based on F2 values. The coordinates of the non-hydrogen atoms in the complexes were determined from Fourier electron density maps and refined anisotropically. The hydrogen atom positions were assigned based on Fourier maps combined with geometric constraints, followed by isotropic refinement. Crystallographic data and CCDC numbers for the coordination complexes are provided in Table 1.| Complex | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. | |||||
| CCDC no. | 2365439 | 2365440 | 2365441 | 2365442 | 2365443 |
| Chemical formula | C30H50CuN8O22P2 | C19H31CuN5O12.5P | C28H43Cu2N10O27P4 | C42H67.11Cd2N10O31.55P4 | C19H24CuN5O16P3 |
| Formula weight (g mol−1) | 1000.26 | 624.00 | 1202.68 | 1565.64 | 734.88 |
| Colour | Green | Green | Green | Colorless | Green |
| T (K) | 284.07 | 284.07 | 298.00 | 297.61 | 298.00 |
| Crystal system | Triclinic | Orthorhombic | Monoclinic | Triclinic | Triclinic |
| Space group | P1 | C2221 | P21 | P1 | P1 |
| a (Å) | 7.5466(4) | 11.0705(4) | 11.8834(5) | 11.0430(5) | 7.0931(4) |
| b (Å) | 11.1792(6) | 22.3081(10) | 15.5226(8) | 11.1177(5) | 9.8028(5) |
| c (Å) | 13.3465(7) | 20.3416(9) | 12.9286(6) | 14.0646(6) | 10.6048(5) |
| α (°) | 107.060(2) | 90.00 | 90 | 88.3730(10) | 104.396(2) |
| β (°) | 92.186(2) | 90.00 | 101.7890(10) | 68.5750(10) | 106.502(2) |
| γ (°) | 106.080(2) | 90.00 | 90 | 70.7720(10) | 93.529(2) |
| V (Å3) | 1025.54(10) | 5023.6(4) | 2334.52(19) | 1509.44(12) | 677.83(6) |
| Z | 1 | 8 | 2 | 1 | 1 |
| ρ cald (g cm−3) | 1.620 | 1.650 | 1.711 | 1.722 | 1.800 |
| μ (Mo Kα) (mm−1) | 0.707 | 1.008 | 1.150 | 0.911 | 1.071 |
| F(000) | 521.0 | 2592.0 | 1230.0 | 798.0 | 375.0 |
| θ range/° | 5.666–56.87 | 4.57–56.73 | 4.376–56.832 | 4.662–56.68 | 6.052–56.68 |
| Range of h, k, l | −10 < h < 10 | −14 < h < 14 | −15 < h < 15 | −13 < h < 14 | −9 < h < 8 |
| −14 < k < 14 | −28 < k < 29 | −20 < k < 20 | −14 < k < 14 | −13 < k < 12 | |
| −17 < l < 17 | −27 < l < 27 | −16 < l < 17 | −18 < l < 18 | −14 < l < 14 | |
| Reflections collected | 27068 |
43457 |
38641 |
23256 |
10034 |
| Independent reflections | 10132 |
6253 | 11266 |
12322 |
5435 |
| Unique data (Rint) | 0.0349 | 0.0512 | 0.0438 | 0.0220 | 0.0205 |
| R 1 /wR2b [I > 2(Iσ)] | 0.0292/0.0566 | 0.0323/0.0726 | 0.0410/0.0988 | 0.0338/0.0840 | 0.0262/0.0586 |
| R 1 /wR2b [all data] | 0.0385/0.0589 | 0.0475/0.0780 | 0.0588/0.1064 | 0.0382/0.0858 | 0.0300/0.0606 |
| GOF | 0.981 | 1.022 | 1.053 | 1.056 | 0.950 |
| H atom treatment | Constr | Constr | Constr | Constr | Constr |
| Residuals (e Å−3) | 0.22/−0.33 | 0.58/−0.46 | 1.32/−0.65 | 0.78/−0.66 | 0.38/−0.32 |
| Flack parameter | 0.007(4) | 0.012(15) | 0.015(12) | 0.018(7) | 0.001(19) |
Results and discussion
Design and syntheses
The central objective of this research was to synthesize and explore the coordination polymers of cytidine mono-, di-, and tri-phosphates with copper(II) and cadmium(II), focusing on their structural diversity and selective binding to amino acids, specifically tryptophan (Trp) and tyrosine (Tyr). The polymers were synthesized through slow evaporation crystallization at room temperature with continuous stirring in aqueous–organic media, yielding five distinct coordination polymers. Single crystals of the products were analyzed via single-crystal X-ray diffraction (SCXRD), confirming the formation of one-dimensional (1, 5) and two-dimensional (2, 3, 4) coordination polymers.The binding affinities of the synthesized coordination polymers for various amino acids were assessed through UV-visible and fluorescence titrations.35 To evaluate the selectivity of the coordination complexes, a broad screening was conducted using glycine, alanine, valine, leucine, isoleucine, serine, aspartic acid, glutamic acid, proline, methionine, tryptophan, and tyrosine. The results revealed that only tryptophan and tyrosine exhibited significant interactions, while the other amino acids showed minimal or no response, highlighting the specificity of the complexes (Fig. S36†). Additionally, both L- and D-forms of tryptophan and tyrosine exhibited identical spectroscopic titration spectra, indicating similar binding interactions. Given the indistinguishable spectral responses, we focused on the L-forms of the amino acids for further analysis. The polymers exhibited distinct binding behaviors with the two amino acids, reflecting their potential for selective molecular recognition. Binding energy calculations supported the experimental observations, revealing that solution-phase dynamics36 significantly influenced the binding trends.
Structural description
{[Cu(CMP)(bpa)(H2O)3](CMP)·3H2O}n (1). In complex 1, the CMP ligands coordinate with Cu2+ ions through the phosphate terminal in a bridging mode. Additionally, a second CMP2− anion forms a connection to the linear cationic sequence through a strong hydrogen bond between the oxygen atom (O5) of the phosphate group in the coordinated CMP and the oxygen atom (O12) of the non-coordinated CMP ligand (O12–H12A⋯O5, with bond lengths of 1.72 Å and 2.45 Å, and an angle of 148°). The bpa ligand serves as a bridge between metal centers, leading to the classification of complex 1 as a 1D coordination polymer with a triclinic system and a chiral P1 space group. In the asymmetric unit of 1, each Cu2+ ion exhibits a six-coordinate geometry, involving three oxygen atoms (O1, O2, and O3) from coordinated water molecules, two nitrogen atoms from the bpa bridging ligands (N1 and N2), and one oxygen atom (O4) from the phosphate group of CMP (Fig. 1a and S1†). The spacing between Cu2+ ions connected through bridging by the phosphate and bpa ligands is 7.546(5) Å and 13.346(8) Å, respectively (Fig. 1b). The nucleotides are organized along the linear chain derived from π–π stacking interactions, with intrachain distances of 3.793 Å and 4.363 Å, and interchain distances of 3.664 Å and 4.053 Å (Fig. S2a and c†). Moreover, the layers are connected through hydrogen bonding interactions. Complex 1 forms an i-motif structure through strong hydrogen bonding interactions (N5–H5A⋯O19, 1.92 Å, 2.78 Å, 176°; N4–H4⋯N7, 1.97 Å, 2.82 Å, 172°; N8–H8A⋯O11, 2.09 Å, 2.90 Å, 157°). The bpa ligand has a dihedral angle of 12.28(13)°, while the angle between the two cytosine bases participating in i-motif is 10.21(8)° (Fig. 1d, S2, and S3†).
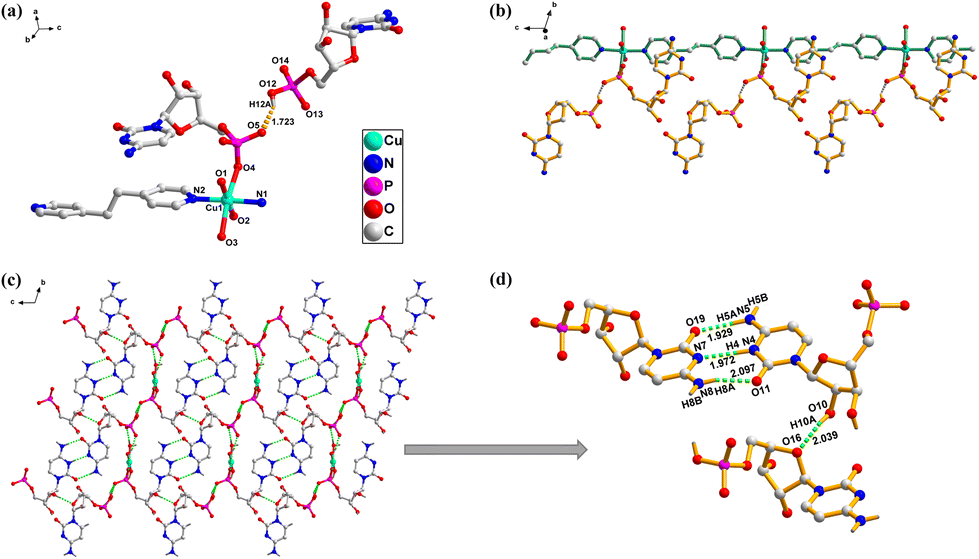 | ||
| Fig. 1 (a) Representation of the coordination geometry of Cu2+ in 1; H atoms are omitted for clarity. (b) 1D coordination network of 1, viewed along the a-axis, uncoordinated water molecules and some H atoms are omitted for clarity. (c) 3D structure based on H-bonding interactions, viewed along the a-axis. (d) Detailed illustration of i-motif formation and ribose–ribose H-bonding interactions. | ||
A novel hydrogen bond is formed between the oxygen atom of the sugar in the coordinated CMP and the hydroxyl group of the sugar in the uncoordinated CMP (O10–H10A⋯O16, 2.03 Å, 2.83 Å, 164°) (Fig. 1d and S2d†). This type of (sugar–sugar) hydrogen bonding represents a new class of supramolecular interactions, which may have applications in molecular recognition and supramolecular assembly in future studies. Additionally, there exists a hydrogen bond between the phosphate group and a coordinated water molecule (O2–H2A⋯O6, 1.78 Å, 2.61 Å, 162°). The structural arrangement of the auxiliary ligand bpa has been confirmed to be conducive for the formation of the i-motif, facilitating the alteration of the orientation of the cytosine base through intra- and inter-chain π–π stacking interactions between the pyridine ring and the cytosine base. The i-motif is situated between adjacent chains, and interactions between phosphate oxygen atoms of different nucleotides create hydrogen bonds that lead to the construction of a one-dimensional supramolecular assembly (O12–H12A⋯O5, 1.72 Å, 2.45 Å, 148°) (Fig. 1c and S4†). Consequently, a homochiral supramolecular framework is produced, which transmits the chirality of the CMP ligands into a three-dimensional supramolecular assembly (Fig. S3 and S4†).
{[Cu2(dCMP)(4,4′-bipy)2(H2O)2]·4H2O}n (2). In contrast to complex 1, complex 2 utilizes the dCMP ligand in place of the CMP ligand, and this leads to the construction of a 2D coordination polymer defined by an orthorhombic system and a chiral space group C2221. The coordination geometry of the Cu2+ ions in 2 resembles that in 1, with the exception of the orientation of the dCMP ligands. Notably, this complex contains two crystallographically distinct central metal ions. Each individual Cu2+ ion is six-coordinated, involving two phosphate oxygen atoms (O2, O2#), two bridging 4,4′-bipy ligands (N1, N4), and two coordinated solvent molecules (O1, O1#), which together create a distorted octahedral geometry (Fig. 2a and S5†). Furthermore, the extended axial chirality (EAC) is integrated into the two-dimensional sheet using coordination bonding among Cu ions and the phosphate bridge, resulting in alternating Cu⋯Cu distances along the two-dimensional chain of 11.1340(8) Å and 11.1741(8) Å (Fig. 2b and S6†). The 4,4′-bipy molecules exhibit two distinct dihedral angles of 51.21(19)° and 34.44(17)°. The orientation of the plane corresponds to the vectors of the crystal cell, resulting in a dihedral angle of 51.2° between Cu2 and Cu1 in the 4,4′-bipy ligands, rather than the supplementary angle. The configuration of the 4,4′-bipy in the EAC of complex 2 is designated as M, as all surrounding dCMP ligands are arranged anticlockwise around the axis (Fig. S7†).
 | ||
| Fig. 2 (a) Representation of the coordination geometry of Cu2+ in 2; H atoms are omitted for clarity. (b) 1D coordination chain of 2, viewed along the a-axis, uncoordinated water molecules and H atoms are omitted for clarity. (c) 3D supramolecular framework. (d) 2D assembly based on π–π stacking interactions. | ||
Strong π–π stacking interactions occur between the pyridine rings and the pyrimidine bases of the nucleotide ligands within the chain, with a separation distance of 3.732 Å. These interactions, combined with intralayer hydrogen bonding, orient the nucleotide ligands above and below the layer in a uniform direction (Fig. S6c†). The chirality of the two-dimensional coordination layers is conveyed to the three-dimensional supramolecular structure through weak interlayer hydrogen bonding. As a result, a single unit of the EAC in complex 2 comprises two adjacent 4,4′-bipy ligands (Fig. 2c and d). Although the EAC retains an M-conformation, it consists of 4,4′-bipy components with reverse chirality, as confirmed by circular dichroism spectroscopy. The chirality of dCMP is transmitted across the two-dimensional coordination chain via the central metal and the EAC of the 4,4′-bipy ligands, which are crucial for “locking” the axial chirality of the auxiliary ligand. The dCMP nucleotides in 2 are positioned along the chiral axial chain with reduced versatility, as the cytosine bases are constrained by intrachain hydrogen bonding and π–π stacking. Notably, two types of chiral sources are present in 2, and there is no supramolecular helix present. It is evident that no i-motif hydrogen bonds are found in 2 due to the absence of an OH group in the dCMP ligand.
{[Cu2(CDP)2(azpy)(H2O)]·3H2O}n (3). Complex 3 is a two-dimensional Cu(II)-CDP coordination polymer characterized by a monoclinic system and chiral space group P21. In the asymmetrical unit, there are two Cu(II) ions, with square pyramidal coordination geometry. The Cu1 ion is coordinated by two CDP ligands in axial positions and one coordinated water molecule, while the Cu2 ion is bonded to two azpy ligands in equatorial positions and one coordinated water molecule. Notably, in the 2D grid topology, the unit Cu1 with two CDP ligands acts as the linker, whereas Cu2 with two azpy ligands acts as the node (Fig S11†). This observation demonstrates that Cu2 is coordinated to only one phosphate oxygen, in contrast to Cu1, which achieves a square planar coordination geometry through the oxygen atoms of the phosphate groups (Fig. 3a, b and S9†). The hexagonal chelate ring within the pyrophosphate backbone adopts a conformation that can be characterized as a deformed boat shape (Fig. S10a and b†). The distances between Cu⋯Cu ions linked through the bridging phosphate are measured at 11.883(5) Å, while the Cu⋯Cu distance built upon the length of the bridging ligand azpy is 12.928(6) Å (Fig. 3c and S11†). In contrast to the nucleotide monophosphate complexes, the pyrimidine cores of the CDP nucleotide form aromatic stacking interactions with the azpy chelator on the neighboring chain in complex 3, with distances of 4.043 Å and 4.301 Å (Fig. S11†). This behavior is markedly different from that observed in CMP complexes and is attributed to the greater flexibility of the CDP ligand. The π–π stacking constrains the CDP molecules to orient uniformly along the one-dimensional chain, limiting rotational freedom around the double-bonded nitrogen atoms of the pyridine rings within the azpy ligands. Consequently, supramolecular chirality is generated, defined by P-EAC and a dihedral angle of 24.33(19)° (Fig. S12†).
 | ||
| Fig. 3 (a) Representation of the coordination geometry of Cu2+ in 3; H atoms are omitted for clarity. (b) The coordination model of Cu1. (c) 1D coordination network of 3, viewed along the b-axis, uncoordinated water molecules and H atoms are omitted for clarity. (d) 2D framework based on ribose–ribose H-bonding interaction, viewed along the b-axis. | ||
The pentose ring adopts an envelope (E) conformation (Fig. S10c and d†). The two-dimensional supramolecular layers are positioned in a parallel configuration, facilitated by H-bonding between the hydroxyl group of the pentoses (O(12)–H(12)) and another oxygen atom (O(21)) of the CDP pentose (Fig. 3d). The inherent chiral symmetry of the CDP ligands is transmitted to the three-dimensional supramolecular assembly of 3via metal ion coordination and noncovalent interactions, including hydrogen bonding and π–π stacking, leading to the formation of a chiral-based supramolecular architecture. In addition, the H-bonds involving the OH groups of the ribose rings enhance the stability of the 3E conformation.
{[Cd2(CDP)2(bpa)2(H2O)2]·8H2O}n (4). Complex 4 is a 2D coordination polymer characterized by a triclinic system and a chiral space group P1. Within the one-dimensional belt layer, the Cd2+ ion shows a six-coordination mode, with the equatorial positions coordinated by two distinct CDP molecules in a closed-type (II) coordination geometry (Fig. 4a, S14a and b†). The alternating coordination of bpa ligands, coupled with the linking coordination of diphosphate oxygen atoms, leads to the construction of a two-dimensional grid network within the b–c plane (Fig. 4b). Strong aromatic stacking interactions between the pyridine rings of the bpa molecules induce adjacent bpa ligands to adopt distinct axial chirality conformations, with dihedral angles of 9.73(9)° and 17.62(12)°. Notably, a single unit of the EAC along the Cd(II)-bpa-Cd(II) chain in 4 consists of two bpa ligands, which differs from the configuration observed in complex 3. The orientation of the CDP ligands surrounding the bpa axis in 4 is opposite, resulting in an equal distribution of right-handed and left-handed chiral inducers. Consequently, the chiral configuration of the M-EAC in 4 is primarily determined by the right-handed metal center (Fig. S16†). Moreover, several π–π stacking interactions compel the two CDP ligands to extend outside the two-dimensional belt layer structure. The Cd2+ ions, linked via bridging phosphate and bpa ligands, exhibit distances of 11.117(5) Å and 14.064(6) Å, respectively (Fig. 4c and d). The CDP ligands are stably integrated within the two-dimensional belt layer through hydrogen bonds among the phosphate groups and coordinated water molecules, indicating that the additional β-phosphate group of CDP contributes to its greater flexibility compared to the CMP nucleotide (Fig. S17†).
 | ||
| Fig. 4 (a) Representation of the coordination geometry of Cd2+ in 4; H atoms are omitted for clarity. (b) 1D coordination chain of 4, viewed along the a-axis, uncoordinated water molecules and H atoms are omitted for clarity. (c and d) 3D supramolecular framework based on i-motif H-bonding and π–π-stacking interactions, viewed along the a-axis. | ||
The detailed structure reveals that the pentose ring conformations are distinct, with pentose-1 adopting a twisted conformation and pentose-2 exhibiting an envelope form (Fig. S14e and f†). This observation is consistent with the hydrophobic microenvironment established by the bpa ligands, which offers significant flexibility for the CDP nucleotide. Ultimately, the neighboring two-dimensional planar sheets can be further constructed into a three-dimensional supramolecular structure, facilitated by π–π stacking interactions and i-motif H-bonding. The i-motif in 4 is constructed through strong H-bonding (N6–H6A⋯O11, 1.92 Å, 2.78 Å, 174°; N5–H5⋯N2, 1.97 Å, 2.83 Å, 172°; N3–H3A⋯O21, 2.17 Å, 2.90 Å, 142°), which is slightly distorted compared to that in complex 1 (Fig. 4c and d). Nucleotides are aligned along the one-dimensional belt of bpa ligands, forming a one-dimensional chain based on π–π stacking interactions with spacings of 3.646 Å and 4.168 Å in layer A, and 3.984 Å and 3.698 Å in layer B, respectively. The countercation is stably maintained within the three-dimensional structure of 4 through hydrogen bonds between the coordinated water molecules and the phosphate groups. Additionally, these H-bonds play a role in the stabilization of the envelope and twisted conformations in the precise coordination of the two different CDP ligands in 4 (Fig. S17†).
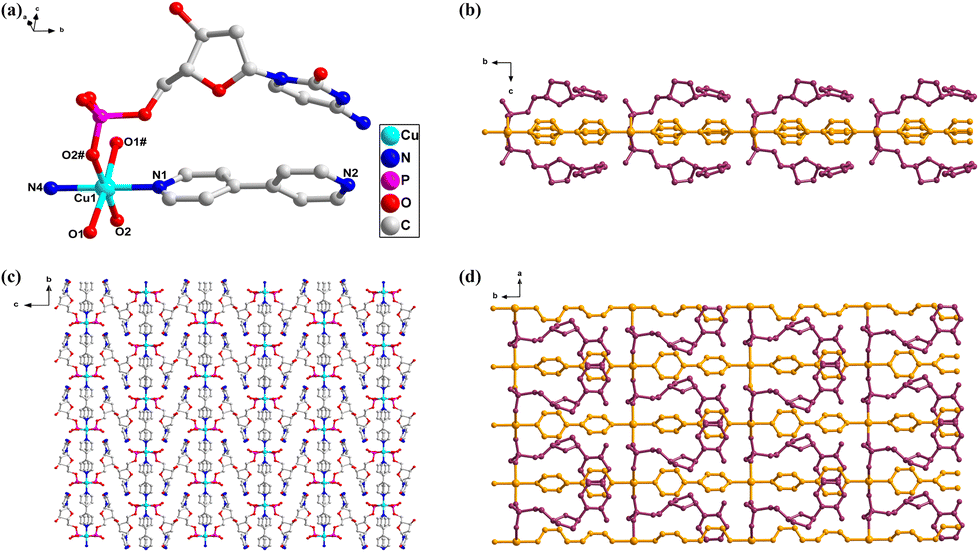
{[Cu(CTP)(2,2′-bipy)]·2H2O}n (5). Complex 5 crystallizes in the triclinic system and adopts the chiral space group P1. It features a one-dimensional structure centered around a Cu(II) ion. Crystallographic analysis reveals that the asymmetric unit comprises one Cu(II) ion center, one CTP ligand, one 2,2′-bipy ligand, and two free water molecules. In comparison with complexes 1–4, the Cu2+ ion in complex 5 exhibits a five-coordinate distorted trigonal bipyramidal geometry. Notably, complex 5 does not contain any coordinated water molecules; all bonds are formed by the triphosphate group and the 2,2′-bipy ligand. The Cu2+ ion is bonded to three oxygen atoms (O1, O2, and O9) from the α-, β- and γ-phosphate groups of the CTP ligand and two nitrogen atoms (N1, N2) from one 2,2′-bipy ligand (Fig. 5a and S18†). The 2,2′-bipy ligand does not exhibit any chiral conformation, with a dihedral angle of 1.56(14)°. Despite this small dihedral angle, no supramolecular chirality is observed in complex 5, as the cis arrangement of nitrogen atoms in the 2,2′-bipy ligand, combined with the coordination mode of the Cu2+ ion, prevents the extension of binding that would lead to chirality (Fig. S20†). The auxiliary ligand 2,2′-bipy plays an essential role in enhancing the chelate effect, where it coordinates in a bidentate fashion with the Cu2+ ion, stabilizing the metal center. This chelating interaction aids in the crystallization process of the CTP complex, creating a more ordered and stable structure. The presence of the 2,2′-bipy ligand is crucial for the formation of a stable one-dimensional chain structure along the a-axis, facilitating crystallization compared to the simpler secondary ligands used in complexes 1–4, such as bpa, azpy, and 4,4′-bpy. The axial position of the Cu-CTP complex extends infinitely along the a-axis through alternating connections with coordinated triphosphate groups, resulting in a one-dimensional vertical chain. This arrangement is distinctly different from the extended axial chirality (EAC) found in the monophosphate nucleotide complexes, such as 1 and 2, and the diphosphate nucleotide complexes, like 3 and 4. The Cu2+ ions, linked by bridging triphosphate groups, exhibit distances of 7.093(6) Å (Fig. 5b). Adjacent two-dimensional layers are formed into a three-dimensional supramolecular network through π–π stacking interactions (3.864 Å) and interlayer H-bonds between the hydroxyl group and phosphate groups (O12–H12⋯O3, 1.91 Å, 2.67 Å, 153°) as well as intra-molecular H-bonds between phosphate and OH groups (O13–H13A⋯O6, 1.94 Å, 2.74 Å, 162°) (Fig. 5c, d and S19†).
 | ||
| Fig. 5 (a) Representation of the coordination geometry of Cu2+ in 5; H atoms are omitted for clarity. (b) 1D coordination vertical chain of 5, viewed along the b-axis; uncoordinated water molecules and H atoms are omitted for clarity. (c and d) 2D framework created by strong π–π stacking interactions between chains, viewed along the a- and b-axes. | ||
In previous work by Orioli et al.37 the Zn-ATP complex with 2,2′-bipyridyl exhibited a dimeric structure where two Zn2+ ions were bridged by γ-phosphate groups, forming an eight-membered ring. This configuration primarily involved β- and γ-phosphates, with stability achieved through strong purine–bipyridyl stacking interactions, which enhanced resistance to hydrolysis. In contrast, our study of the Cu(II)-CTP complex presents a unique five-coordinate, distorted trigonal bipyramidal geometry centered around a single Cu2+ ion. Unlike the dimeric arrangement in the Zn complex, our structure forms a three-dimensional framework, controlled by π–π interactions between the nucleotide and 2,2′-bipyridyl. These interactions, along with hydrogen bonds, result in a more extended 3D network. Additionally, the Cu coordination involves the α-, β-, and γ-phosphates, indicating a broader interaction with the phosphate groups compared to the Zn complex. This comparison underscores how the choice of metal and nucleotide influences dimensionality and stabilization mechanisms. It is noteworthy that, although the flexibility of the CTP ligand is significantly reduced compared to that of the CDP ligand, the furanose ring of the CTP ligand adopts an E-conformation (Fig. S18e†). This is attributed to the hybrid-type (III) coordination modes38 of the triphosphate group (Fig. S21†), where the anti-gauche conformations of the triphosphate groups are arranged with the same positive or negative signs. This arrangement may be the primary reason why the furanose ring conformation of the CTP ligand tends to adopt the E-conformation.
Functional properties of the coordination polymers
In the solution state, the CD spectra of the ligands CMP, dCMP, CDP, and CTP exhibit characteristic Cotton effects that provide insights into their chiral environments. Negative Cotton effects observed near 218–224 nm are attributed to n–π* transitions, while positive Cotton effects in the range of 265–270 nm confirm the D-ribonucleotide configuration of these compounds. This interpretation is further supported by the corresponding UV/vis spectra. The similarity between the CD spectra of the nucleotides and their metal complexes suggests that the nucleotide ligands are the primary contributors to chirality in solution (Fig. S26†). For instance, the CD spectrum of CMP displays a distinct signal at approximately 220 nm, which arises from the additional hydroxyl group in its structure. This functional group influences the n–σ* electronic transition, leading to a unique spectral feature.
For the solution CD spectra of complexes 1–5, the patterns are similar to those of the corresponding nucleotide ligands, which indicates that the chirality of the complexes in solution are mainly contributed by the nucleotide molecules. The minor variations in the wavelength and intensity of the Cotton effects reflect the influence of metal coordination on the chiral environment of the nucleotides. In addition, supramolecular interactions such as non-classical base pairing (i-motif). However, the distinct features in the CD spectrum of complex 2 compared to complex 1, as well as those of complex 5, can be explained by pre-organization in solution, as evidenced by single-crystal structural analyses (Fig. S26†).
In the solid state, the CD spectra of the nucleotide ligands (CMP, dCMP, CDP, and CTP) and their corresponding complexes (1–5) exhibit clearly distinguishable trends. For example, CMP exhibits four broad Cotton effects centered at 217, 235, 275, and 315 nm, while dCMP shows effects at 218, 268, and 317 nm. CDP exhibits Cotton effects at 210, 226, and 265 nm, and CTP presents four effects at 210, 230, 270, and 326 nm. The presence of a prominent signal in the 310–410 nm range demonstrates the formation of EAC, which is a supramolecular chirality involving the nucleotide and twisted bipyridyl ligands (Fig. 6).41 Crystallographic analysis reveals that the sugar ring conformations of CMP, CDP, and CTP adopt T-conformations, whereas dCMP adopts an E-conformation.42,43 The Cotton effects observed between 210–240 nm are likely due to the different conformations of the ribose or deoxyribose moieties in the nucleotide ligands (Fig. 6).
 | ||
| Fig. 6 Solid-state circular dichroism CD spectra of CMP, dCMP, CDP, CTP, and their corresponding complexes 1–5 (KBr:[sample] = 200:1). | ||
In complex 1, the broad Cotton effects between 320–385 nm of complex compared to CMP indicates the formation of EAC (Fig. 6a). In complex 2, the EAC attributes the positive Cotton effect around 325 nm and the negative Cotton effect around 350 nm, respectively (Fig. 6b). The two positive Cotton effects at 275 and 285 nm regions are assigned to π–π transitions, which appear at longer wavelengths compared to the dCMP ligand due to strong π–π stacking interactions (Fig. 6b). In complexes 3 and 4, their EACs are confirmed by Cotton effects in the range of 310–400 nm (Fig. 6c). The supramolecular chirality has been supported by crystal structural analysis. Complexes 1, 2, and 4 exhibit M-chirality (Fig. S3, S7, and S16†), while complexes 3 and 5 exhibit P-chirality (Fig. S12, and S20†). The observed patterns are distinct and original compared to bpa and azpy, and they also vary due to differences in crystal structures (Fig. 6c). The Cotton effects in the range of 212–280 nm correspond to n–π* and strong π–π* transitions of the nucleobases in the CDP molecules, where the aromatic chromophores are incorporated into the framework, involving both intra- and intermolecular interactions of cytosine chromophores (Fig. 6c).44 Based on crystal structural analysis, complex 5 has no EAC, which has been confirmed by its CD spectrum (Fig. 6d). The features of complex 5 are similar to CTP with red shifts arising from intense π–π stacking interactions between the nucleobases and the 2,2′-bipy ligand.45 Upon polymer formation, the Cotton effects in the CD spectra shift to longer wavelengths, reflecting the alignment of nitrogen heterocycles and the promotion of electronic transitions.
Among the five complexes, complexes 1 and 4 formed i-motif base pairing (Fig. S2 and S15†). Interestingly, the base-pairing interactions lead to the separation of adjacent nucleotides along the orientation of the secondary ligands, which may influence the EAC further. Therefore, complexes 1 and 4 have M-EAC, while complex 3 has P-EAC. Complex 2 has no i-motif since the bridge ligand 4,4′-bipy is short. To identify a positive Cotton effect in the CD spectra, it is essential to consider the P- and M-EAC configurations of the auxiliary ligands. The EAC formed by the nucleotides impacts the symmetry of the electronic transitions, thereby affecting the changes in the Cotton effects.46,47
Interaction of polymers with amino acids
Complex 1 demonstrated moderate selectivity for both tryptophan and tyrosine. For tryptophan, the binding constant for 1-Trp was 2.257 × 104 M−1, and for the Trp-1 system, the binding constant was 1.472 × 104 M−1, suggesting a moderate affinity for tryptophan. For tyrosine, 1-Tyr showed a binding constant of 1.710 × 104 M−1, and Tyr-1 exhibited a binding constant of 2.489 × 104 M−1, indicating a relatively stronger binding to tyrosine compared to tryptophan. Computational studies supported these experimental results. The binding energy for Trp-1 was calculated as −0.48877 eV, indicating a moderate interaction with tryptophan, while for tyrosine (Tyr-1), the binding energy was −0.15936 eV, suggesting a weaker interaction. The difference in binding constants observed in the UV-visible titration may be attributed to solvation effects or dynamic interactions not fully accounted in the computational models (Fig. S28a, c, S31a and c†).
Complex 2 exhibited similar selectivity. The binding constant for 2-Trp was 2.344 × 104 M−1, and for Trp-2, the binding constant was 1.587 × 104 M−1, indicating a moderate affinity for tryptophan. In contrast, 2-Tyr showed a binding constant of 1.696 × 104 M−1, and Tyr-2 exhibited a higher value of 2.752 × 104 M−1, suggesting a relatively stronger interaction with tyrosine. The corresponding computational binding energies for Trp-2 and Tyr-2 were −0.56412 eV and −0.47972 eV, respectively, indicating that the complex shows a higher affinity for tryptophan than for tyrosine, in agreement with the experimental results (Fig. S29a, c, S32a and c†).
Complex 3 demonstrated stronger selectivity for tryptophan in both UV-visible titration and computational calculations. The binding constant for 3-Trp was 2.761 × 104 M−1, and for Trp-3, the value was 1.700 × 104 M−1, showing a moderate affinity for tryptophan. For tyrosine, the binding constant for 3-Tyr was 2.596 × 104 M−1, and Tyr-3 showed 3.129 × 104 M−1, indicating a stronger interaction with tyrosine. Computational studies confirmed these findings, with a binding energy for Trp-3 of −1.0794 eV, indicating a stronger interaction with tryptophan compared to tyrosine, for which the binding energy was calculated to be −0.72156 eV. These results suggest that Complex 3 exhibits a preference for tryptophan over tyrosine, although the interaction with tyrosine is still significant (Fig. S30a, c, S33a and c†).
Complex 4 exhibited a similar trend, with binding constants of 2.421 × 104 M−1 for 4-Trp and 1.711 × 104 M−1 for Trp-4, indicating a strong affinity for tryptophan. For tyrosine, 4-Tyr exhibited a binding constant of 1.646 × 104 M−1, and Tyr-4 showed 2.486 × 104 M−1, indicating a stronger interaction with tyrosine. The computational binding energies for Trp-4 and Tyr-4 were −0.47703 eV and −0.5956 eV, respectively, indicating that the complex prefers tyrosine over tryptophan, but the interaction with both amino acids remains relatively significant (Fig. 7a, c, S34a and c†). Complex 5 showed moderate selectivity as well, with a binding constant of 2.318 × 104 M−1 for 5-Trp, and Trp-5 exhibited a binding constant of 1.521 × 104 M−1. For tyrosine, 5-Tyr showed a binding constant of 1.468 × 104 M−1, while Tyr-5 exhibited 2.331 × 104 M−1, indicating moderate interaction with both amino acids. The computational binding energies for Trp-5 and Tyr-5 were −0.54819 eV and −0.34112 eV, respectively, showing a stronger computational preference for tryptophan over tyrosine, which aligns with the experimental findings (Fig. 8a, c, S35a, and c†).
 | ||
| Fig. 7 (a) UV-visible absorption spectra of complex 4 with incremental additions of Trp; (b) fluorescence emission spectra (λex = 270 nm) of complex 4 with incremental additions of Trp; (c) UV-visible absorption spectra of complex 4 with incremental additions of Tyr; (d) fluorescence emission spectra (λex = 270 nm) of complex 4 with incremental additions of Tyr. All spectra were measured in methanol solution at a concentration of 2.5 × 10−5 M using a 1 cm cell. Binding isotherms for the interactions between complex 4 and Trp/Tyr are derived from both the UV-visible and fluorescence titration data. | ||
 | ||
| Fig. 8 (a) UV-visible absorption spectra of complex 5 with incremental additions of Trp; (b) fluorescence emission spectra (λex = 270 nm) of complex 5 with incremental additions of Trp; (c) UV-visible absorption spectra of complex 5 with incremental additions of Tyr; (d) fluorescence emission spectra (λex = 270 nm) of complex 5 with incremental additions of Tyr. All spectra were measured in methanol solution at a concentration of 2.5 × 10−5 M using a 1 cm cell. Binding isotherms for the interactions between complex 5 and Trp/Tyr are derived from both the UV-visible and fluorescence titration data. | ||
The UV-visible absorption spectra of the Cu-CTP-2,2′-bipy coordination complex, with a distorted trigonal bipyramidal geometry, exhibit four distinct isosbestic points within the range of 200–350 nm. These points highlight the dynamic interaction between the complex and guest molecules, such as tryptophan. The coordination complex consists of five bonds, two to 2,2′-bipy and three to the triphosphate group of the nucleotide, while the copper ion, in its +2-oxidation state, has the potential to form an additional bond with tryptophan, enhancing its coordination environment. This suggests that copper has the possibility to complete six bonds in total, influencing the binding interaction and stability of the complex. Such interactions highlight the potential of tryptophan as a selective ligand for metal coordination, which can be leveraged for amino acid detection.
In 1-Trp, the fluorescence emission at 289 nm with an intensity of 528 shifted to 292 nm with an intensity of 693, indicating a red shift and fluorescence enhancement upon titration. The second emission peak at 327 nm with an intensity of 318, and the third at 343 nm with an intensity of 267, merged into a broad peak at 339 nm with an intensity of 1595, signifying the formation of a stable complex and a marked increase in fluorescence intensity. In Trp-1, the fluorescence intensity at 339 nm initially increased from 1878 to 1925, before subsequently decreasing to 1240, reflecting fluorescence quenching as the complex bound to the amino acid (Fig. S28b and S31b†). For 1-Tyr, the emission at 290 nm with an intensity of 627 shifted to 301 nm with an intensity of 2514, revealing a red shift and considerable fluorescence enhancement, indicative of a strong interaction between the coordination polymer and tyrosine. In Tyr-1, the emission at 301 nm with an intensity of 2818 decreased to 1416 upon the addition of the complex, demonstrating fluorescence quenching. This suggests that the interaction between the coordination polymer and tyrosine results in competitive binding or displacement, leading to reduced emission intensity (Fig. S28d and S31d†).
In 2-Trp, the emission at 290 nm with an intensity of 2428 shifted to 292 nm with an intensity of 1093, showing a red shift and fluorescence quenching. The second peak at 327 nm with an intensity of 703, and the third peak at 341 nm with an intensity of 530, merged into a broad peak at 338 nm with an intensity of 1536, signifying fluorescence enhancement and stabilization of the complex. In Trp-2, the emission at 292 nm with an intensity of 693 shifted to 291 nm with an intensity of 1573, indicating a blue shift and significant fluorescence enhancement. The coordination of Cu2+ ions with dCMP ligands and 4,4′-bipy alters the electronic environment of Trp, stabilizing its excited state and reducing non-radiative decay, leading to the observed fluorescence enhancement. Conversely, the second peak at 339 nm with an intensity of 1947 decreased to 333 nm with an intensity of 1045, revealing fluorescence quenching due to competitive binding (Fig. S29b and S32b†). 2-Tyr exhibited an emission at 290 nm with an intensity of 2374, which shifted to 299 nm with an intensity of 2714, reflecting a red shift and fluorescence enhancement, suggesting a strong interaction between the coordination polymer and tyrosine. In Tyr-2, the emission at 302 nm with an intensity of 3282 shifted to 293 nm with an intensity of 2403, revealing a blue shift and fluorescence quenching. This quenching is likely due to the binding competition between the amino acid and the coordination polymer (Fig. S29d and S32d†).
In 3-Trp, the emission at 290 nm with an intensity of 2157 shifted to 292 nm with an intensity of 1145, indicating a red shift and fluorescence quenching. The second peak at 326 nm with an intensity of 750, and the third peak at 340 nm with an intensity of 581, merged into a broad peak at 337 nm with an intensity of 1555, highlighting fluorescence enhancement and further stabilization of the complex. In Trp-3, the emission at 293 nm with an intensity of 1065 increased to 291 nm with an intensity of 1684, showing a blue shift and fluorescence enhancement. The peak at 337 nm with an intensity of 1635 decreased to 332 nm with an intensity of 1123, indicating fluorescence quenching as a result of complex displacement or competition (Fig. S30b and S33b†). In 3-Tyr, the emission at 290 nm with an intensity of 1990 shifted to 295 nm with an intensity of 2755, showing a red shift and fluorescence enhancement, indicative of a strong interaction between the coordination polymer and tyrosine. In Tyr-3, the emission at 298 nm with an intensity of 3462 decreased to 293 nm with an intensity of 2441, reflecting a blue shift and fluorescence quenching, suggesting that the coordination polymer competes with the amino acid for binding, thereby reducing fluorescence emission (Fig. S30d and S33d†).
The 4-Trp demonstrated an emission at 290 nm with an intensity of 2514, which shifted to 292 nm with an intensity of 1335, exhibiting a red shift and fluorescence quenching. The second peak at 326 nm with an intensity of 756, and the third peak at 340 nm with an intensity of 548, merged into a broad peak at 338 nm with an intensity of 1492, representing fluorescence enhancement and stabilization of the coordination complex. In Trp-4, the emission at 293 nm with an intensity of 996 increased to 291 nm with an intensity of 1861, showing a blue shift and fluorescence enhancement. The second peak at 340 nm with an intensity of 1617 decreased to 333 nm with an intensity of 1356, indicating fluorescence quenching (Fig. 7b and S34b†). In 4-Tyr, the emission at 290 nm with an intensity of 2507 shifted to 296 nm with an intensity of 3219, indicating a red shift and a marked fluorescence enhancement. In Tyr-4, the emission at 300 nm with an intensity of 3757 shifted to 295 nm with an intensity of 2970, suggesting a blue shift and slight fluorescence quenching due to the interaction of the coordination polymer with tyrosine (Fig. 7d and S34d†).
In 5-Trp, the emission at 290 nm with an intensity of 2325 shifted to 292 nm with an intensity of 1442, revealing a red shift and fluorescence quenching. The second peak at 325 nm with an intensity of 770, and the third peak at 342 nm with an intensity of 546, merged into a broad peak at 337 nm with an intensity of 1373, signifying fluorescence enhancement. In Trp-5, the emission at 293 nm with an intensity of 1078 increased to 291 nm with an intensity of 1786, demonstrating a blue shift and fluorescence enhancement. Meanwhile, the second peak at 338 nm with an intensity of 1666 shifted to 329 nm with an intensity of 1032, indicating fluorescence quenching (Fig. 8b and S35b†). In 5-Tyr, the emission at 290 nm with an intensity of 2363 shifted to 295 nm with an intensity of 3093, showing a red shift and fluorescence enhancement, likely due to the strong interaction between the coordination polymer and tyrosine. In Tyr-5, the emission at 298 nm with an intensity of 3610 shifted to 293 nm with an intensity of 2601, indicating a blue shift and fluorescence quenching (Fig. 8d and S35d†).
Tryptophan (Trp) exhibits stronger and more stable fluorescence enhancement across all systems, making it the optimal target for fluorescence-based detection using coordination polymers. In contrast, tyrosine (Tyr) shows more variable responses, including fluorescence quenching, indicating lower sensitivity. UV-visible and fluorescence spectroscopy together provide a comprehensive view of the binding dynamics. UV-visible spectroscopy offers consistent binding constants, while fluorescence spectroscopy reveals subtle structural shifts and quenching effects.48 This dual approach is essential for advancing these polymers in selective amino acid recognition and biosensing. The strong selectivity for Trp and Tyr highlights their potential in biosensors and as functional materials for biomolecule capture, with diphosphate and triphosphate coordination polymers able to distinguish between the two amino acids for multifunctional applications in sensors.
 | ||
| Fig. 9 Computational binding energy representations of the interactions between the coordination complexes and the amino acid tryptophan. Tryptophan is highlighted in green, while hydrogen bonds between the complexes and tryptophan are indicated by yellow dotted lines. The figure illustrates the binding energies of all five complexes, highlighting the affinities and interactions of each complex with tryptophan. | ||
 | ||
| Fig. 10 Computational binding energy representations of the interactions between the coordination complexes and the amino acid tyrosine. Tyrosine is highlighted in yellow, while hydrogen bonds between the complexes and tyrosine are indicated by green dotted lines. The figure illustrates the binding energies of all five complexes, highlighting the affinities and interactions of each complex with tyrosine. | ||
Conclusions
This study presents a detailed exploration of five nucleotide–metal coordination polymers, emphasizing their structural diversity and functional properties. Single-crystal X-ray diffraction analysis unveiled distinct architectures, with coordination polymers 1 and 5 forming one-dimensional structures and polymers 2, 3, and 4 assembling into two-dimensional networks. Notably, this work marks the first report of CDP and CTP coordination polymers (complexes 3, 4, and 5), providing novel insights into nucleotide–metal interactions and expanding the understanding of nucleotide triphosphates in coordination chemistry. Furthermore, solid-state and solution-state circular dichroism (CD) spectroscopy provided insights into the chirality of these supramolecular assemblies, demonstrating the influence of metal coordination on the chiral properties of nucleotide frameworks. While complexes 1–4 exhibited supramolecular chirality due to their ligand architectures, complex 5, with its 2,2′-bipy ligand, does not exhibit extended axial chirality due to its small dihedral angle and cis nitrogen arrangement. Instead, complex 5 forms a distinctive one-dimensional chain along the a-axis through alternating triphosphate linkages, showcasing unique structural properties.The selectivity studies demonstrate that all five coordination polymers exhibit distinct and significant interactions with tryptophan and tyrosine, underscoring their potential for molecular recognition and biosensing. These polymers offer diverse binding profiles, making them promising candidates for selective detection in biosensing applications. Computational studies, conducted using a single-molecule model, provided further insight into the binding energies of the polymers. These findings largely align with the experimental results, offering a deeper understanding of the electronic interactions involved in amino acid binding. This integrated approach, combining experimental and computational techniques, advances our understanding of nucleotide–metal coordination polymers and paves the way for their application in biosensing, molecular diagnostics, and functional materials design.
Data availability
The experimental results and computational study data in the research paper titled “Structural diversity of nucleotide coordination polymers of cytidine mono-, di-, and tri-phosphates and their selective recognition of tryptophan and tyrosine” (DT-ART-04-2025-000786) are available in the paper, the ESI† and Cambridge Structural Database.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21071018, 21271026, and 21471017).References
- Y. Khan, I. Ismail, H. Ma, Z. Li and H. Li, CrystEngComm, 2024, 26, 2775–2783 RSC.
- H. Feng, S. Ma, Z. Chen, Y. Li, M. Wang and Y. Ding, Environ. Sci.:Nano, 2024, 11, 2655–2667 RSC.
- H. Li, Y. Zhu, P. Zhou, Q. Qiu, W. Song and L. Gu, Sci. Sin.: Chim., 2020, 50, 947–961 Search PubMed.
- X. Huang, S. Liu, Z. Zhou, H. Zhang, Z. Gao, G. Shen, H. Wang, Z. Wang, Q. Yao and D. Sun, Inorg. Chem. Front., 2023, 10, 1465–1474 RSC.
- I. Rozman Grinberg, O. Bimaï, S. Shahid, L. Philipp, M. Martínez-Carranza, I. Banerjee, D. Lundin, P. Stenmark, B.-M. Sjöberg and D. T. Logan, FEBS J., 2025 DOI:10.1111/febs.70037.
- G. Park, E. C. Wralstad, N. Faginas-Lago, K. Qian, R. T. Raines, G. Bistoni and C. C. Cummins, ACS Cent. Sci., 2024, 10, 1415–1422 CrossRef CAS PubMed.
- P. Banaszek and M. Podgórski, Sustainable Mater. Technol., 2025, 43, e01270 CrossRef CAS.
- K. Sheng, L.-M. Fan, X.-F. Tian, R. K. Gupta, L. Gao, C.-H. Tung and D. Sun, Sci. China: Chem., 2020, 63, 182–186 CrossRef CAS.
- C. Grauffel, T. Dudev and C. Lim, J. Chem. Theory Comput., 2019, 15, 6992–7003 CrossRef CAS PubMed.
- R. Ye, Y. Wang, Y. Liu, P. Cai and J. Song, Biomater. Sci., 2024, 12, 3712–3724 RSC.
- B. Roy, A. Depaix, C. Périgaud and S. Peyrottes, Chem. Rev., 2016, 116, 7854–7897 CrossRef CAS PubMed.
- T. Gollnest, T. D. de Oliveira, D. Schols, J. Balzarini and C. Meier, Nat. Commun., 2015, 6, 8716 CrossRef CAS PubMed.
- J. Gu, J. Liang, T. Tian and Y. Lin, JACS Au, 2025, 5, 486–520 CrossRef CAS PubMed.
- M. H. Shamsi and H.-B. Kraatz, J. Inorg. Organomet. Polym. Mater., 2013, 23, 4–23 CrossRef CAS.
- H. Sigel and R. Griesser, Chem. Soc. Rev., 2005, 34, 875–900 RSC.
- S. Roy, B. Wang, K. Roy, Y. Tian, M. Bhattacharya, S. Williams and Q. Yin, Commun. Biol., 2025, 8, 282 CrossRef CAS PubMed.
- J. Zhou, H. Han and J. Liu, Nano Res., 2022, 15, 71–84 CrossRef CAS.
- J. Feng, Y. Qiu, H. Gao and Y. Wu, Acc. Chem. Res., 2024, 57, 222–233 CrossRef CAS PubMed.
- A. Li, Y. Zhang, L. Wan, R. Peng, X. Zhang, Q. Guo, S. Xu, D. Qiao, P. Zheng, N. Li, W. Zhu and Q. Pan, ACS Appl. Mater. Interfaces, 2024, 16, 28041–28055 CrossRef CAS PubMed.
- M. Riaz, R. K. Gupta, D. Sun, M. Azam and P. Cui, Chin. J. Struct. Chem., 2024, 43, 100427 CrossRef CAS.
- S. Sarkar, P. Daga, S. K. Mondal and P. Mahata, Cryst. Growth Des., 2024, 24, 4748–4757 CrossRef CAS.
- M. I. El-Barghouthi, K. Bodoor, O. M. Abuhasan, K. I. Assaf, B. J. Al Hourani and A. M. M. Rawashdeh, ACS Omega, 2022, 7, 10729–10737 CrossRef CAS PubMed.
- J. Dong, X.-D. Zhang, X.-F. Xie, F. Guo and W.-Y. Sun, RSC Adv., 2020, 10, 37449–37455 RSC.
- A. Rimola, L. Rodríguez-Santiago and M. Sodupe, J. Phys. Chem. B, 2006, 110, 24189–24199 CrossRef CAS PubMed.
- J. Martinez, V. Truffault and M. Hothorn, J. Biol. Chem., 2015, 290, 23348–23360 CrossRef CAS PubMed.
- J. Purhonen, A. Hofer and J. Kallijärvi, Nucleic Acids Res., 2023, 52, e6–e6 CrossRef PubMed.
- A. Kumar, D. Lionetti, V. W. Day and J. D. Blakemore, J. Am. Chem. Soc., 2020, 142, 3032–3041 CrossRef CAS PubMed.
- R. Bruno, N. Marino, L. Bartella, L. Di Donna, G. De Munno, E. Pardo and D. Armentano, Chem. Commun., 2018, 54, 6356–6359 RSC.
- N. Kheeree, K. Kuptawach, S. Puthong, P. Sangtanoo, P. Srimongkol, P. Boonserm, O. Reamtong, K. Choowongkomon and A. Karnchanatat, Sci. Rep., 2022, 12, 4659 CrossRef CAS PubMed.
- I. Geronimo, P. Vidossich and M. De Vivo, ACS Catal., 2021, 11, 14110–14121 CrossRef CAS.
- L. Biancalana, I. Abdalghani, F. Chiellini, S. Zacchini, G. Pampaloni, M. Crucianelli and F. Marchetti, Eur. J. Inorg. Chem., 2018, 2018, 3041–3057 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- F. Feizi, M. Shamsipur, M.-B. Gholivand, A. Barati, F. Mousavi, F. Molaabasi, M. Mahlooji and M. Sedeghi, ACS Appl. Mater. Interfaces, 2024, 16, 48058–48072 CrossRef CAS PubMed.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, J. Chem. Theory Comput., 2020, 16, 528–552 CrossRef PubMed.
- P. Orioli, R. Cini, D. Donati and S. Mangani, Nature, 1980, 283, 691–693 CrossRef CAS PubMed.
- Y. Zhu, Z. Li, X. Zhong, X. Wu, Y. Lu, M. A. Khan and H. Li, Inorg. Chem., 2022, 61, 19425–19439 CrossRef CAS PubMed.
- Q. Zhang, R. Toyoda, L. Pfeifer and B. L. Feringa, J. Am. Chem. Soc., 2023, 145, 6976–6985 CrossRef CAS PubMed.
- Z. Chen, S. Qin, D. Liu, Y. Shen and F. Liang, Cryst. Growth Des., 2013, 13, 3389–3395 CrossRef CAS.
- P. Zhou, J.-F. Yao, C.-F. Sheng and H. Li, CrystEngComm, 2013, 15, 8430–8436 RSC.
- M. Xue, L. Zhang, M. Liu, Y. Bai, Y. Guo and Z. Zhang, J. Phys. Chem. B, 2020, 124, 8179–8187 CrossRef CAS PubMed.
- H. Ito, H. Tsukube and S. Shinoda, Chem. - Eur. J., 2013, 19, 3330–3339 CrossRef CAS PubMed.
- S. MacQuarrie, M. P. Thompson, A. Blanc, N. J. Mosey, R. P. Lemieux and C. M. Crudden, J. Am. Chem. Soc., 2008, 130, 14099–14101 CrossRef CAS PubMed.
- X. Feng, X. Wang, C. Redshaw and B. Z. Tang, Chem. Soc. Rev., 2023, 52, 6715–6753 RSC.
- H. Miyake, K. Terada and H. Tsukube, Chirality, 2014, 26, 293–299 CrossRef CAS PubMed.
- D. Baran, S. I. Ivlev and E. Meggers, Organometallics, 2022, 41, 52–59 CrossRef CAS.
- Y. Li, Z. Yang, M. Zhou and Y. Li, RSC Adv., 2017, 7, 49404–49422 RSC.
- C. Helene, J.-C. Maurizot and K. G. Wagner, Crit. Rev. Biochem., 1981, 10, 213–258 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2365439–2365443. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00786k |
| This journal is © The Royal Society of Chemistry 2025 |