
The influence of charge transfer on the emissive properties of pyridine dipyrrolide bismuth complexes†
Katharina L.
Deuter
 *,
Katharina
Vollmar
,
Jan
Rieser
,
Michael
Linseis
and
Rainer F.
Winter
*
*,
Katharina
Vollmar
,
Jan
Rieser
,
Michael
Linseis
and
Rainer F.
Winter
*
Faculty for Chemistry, University of Konstanz, Universitätsstrasse 10, 78457, Konstanz, Germany. E-mail: rainer.winter@uni-konstanz.de
First published on 5th June 2025
Abstract
We report on the emissive properties of four new pyridine dipyrrolide bismuth complexes Bi(R1PDPR2)Br, modified with electron-donating (–OMe) or electron-withdrawing (–CF3) substituents at the ligand. These complexes exhibit red phosphorescence at cryogenic temperatures with emission wavelengths ranging from 602 to 642 nm as a response to the electronic character of the substituents R1 and R2. A profound similarity between the pairs Bi(CF3PDPCF3)Br and Bi(CF3PDPOMe)Br, and Bi(OMePDPCF3)Br and Bi(OMePDPOMe)Br is attributed to improved conjugation between the respective phenyl ring and the PDP core, as based on TD-DFT calculations and crystallographic studies. The larger quantum yield of Bi(CF3PDPCF3)Br is attributed to the enhancement of the 3LMCT character of the electronic transition through the introduction of the electron-withdrawing –CF3 substituents.
Introduction
Many metal complexes are bright luminophores and have thus found application as emitters in bioimaging,8,9 OLEDs,10,11 in photodynamic therapy,12 photo(redox) catalysis,3,13,14 solar energy harvesting in the form of dye-sensitized solar cells,15 or energy storage.16 Currently, these applications are often based on rare, expensive and sometimes toxic late transition row elements such as rhenium, ruthenium, iridium, platinum, or osmium, utilizing the high spin-orbit coupling (SOC) constants17 of these elements to induce intersystem crossing (ISC) and thus populate the triplet states, which are a requirement for the aforementioned applications.18 This propensity of heavy elements to induce the population of triplet states through ISC is known as the heavy atom effect (HAE).19 Luminescent molecular complexes of main group metals have received far less attention in this respect, and of these limited reports, most focus on either the boron group or luminescent complexes of the group 14 elements.20–22 Comparatively little attention has been paid to the heavier analogues of nitrogen such as bismuth, although bismuth is particularly promising as it exhibits low toxicity,23,24 good affordability, and the largest SOC constant of the non-radioactive elements within the periodic table.17 These favourable properties are explored primarily for solid-state phosphorescent materials,20,25–27 often coupled with balanced energy transfer to other co-dopants,28–32 or as a dopant in perovskites.33,34 A number of reports (refer to the top panel of Fig. 1A for a selection) have shown that the incorporation of bismuth into molecular complexes may provide phosphorescence emission in solution at r.t.1–4,35 We and others have recently demonstrated the high potential of bismuth for generating emissive materials in combination with tridentate pincer ligands.1,5,36,37 In accordance with El Sayed's rule,19 which states that ISC is rendered particularly efficient if differing orbital types are involved in the electronic transition, those complexes displaying CT involving the bismuth ion as contributor to either the donor or the acceptor molecular orbitals (MOs) possess significantly shorter lifetimes and higher quantum yields than those which lack such CT contributions.1,3 | ||
| Fig. 1 (A) Compilation of the structures of select phosphorescent bismuth complexes that emit at r.t. in solution.1–4 (B) Select main group element and Zr complexes with ligands containing the pyridine dipyrrolide structural motif.5–7 (C) Concept for the design of the bismuth complexes reported in this manuscript. | ||
The tridentate pyridine-dipyrrolide (PDP2−) imine/amide ligand has been a popular structural motif in recent years, and complexes utilizing these ligands have been studied for C–H bond activation chemistry,38 as photosensitizers for photoredox catalysis,6 and as catalysts for Heck-type cyclization reactions.39 The high versatility of these systems stems from their high rigidity, the incorporation of particularly electron-rich pyrrolide units in concert with the electron accepting pyridine unit, and the potential of these ligands to strongly bind varying metal ions in their doubly deprotonated form. Complexes bearing ligands with the PDP2− motif have been investigated regarding their luminescent properties in combination with various d-40,41 and lanthanide f-block,42 or main group elements.7 Thus, complexes of Si, Ge and Sn (cf.Fig. 1B) exhibiting dually emissive properties with temperature-dependent equilibria of thermally activated delayed fluorescence and phosphorescence have recently been reported.7 In addition, substituent effects on the emissive and electrochemical properties of complexes of zirconium and of the proligands H2PDP were investigated (cf.Fig. 1B).43 In combination with bismuth, these ligands were used to access complexes that can be used to polymerize lactides44 and ring-open cyclic ethers45 when reduced to the Bi(II) form.
We have recently reported on complexes (R1PDPR2)BiX (X = Cl, Br, I) with regard to their photophysical behaviour as well as their solid-state structures and electrochemical properties.5 These compounds were found to be emissive at cryogenic temperatures with phosphorescence emission maxima λphos ≈ 640 nm. The optical properties of these compounds were studied with the aid of time-dependent density functional theory (TD-DFT) calculations. We found a significant charge transfer (CT) character of the highest molecular orbital (HOMO) to lowest unoccupied molecular orbital (LUMO) transition, where the former is entirely ligand-based whereas the latter receives contributions from the Bi3+ ion, resulting in a LMCT character of the underlying transition. These findings were further supported by the solvatochromism of the underlying absorption and emission. We mused that the introduction of electron withdrawing (σp-CF3 = +0.53)46 or electron donating (σp-MeO = −0.27)46para substituents onto the previously established diphenyl-substituted PDP core might support (–CF3) or counteract (–OMe) the underlying CT character of the phosphorescent emission, as the emissive excited state has Bi contributions, while the ground state is localized on the ligand, endowing the emissive decay with MLCT character. This conceptualization is illustrated within the bottom panel of Fig. 1.
Results and discussion
Synthesis and characterization
The synthesis of the ligands was conducted analogously to previously reported procedures for 2,4-disubstituted PDP ligands.5,6,47 The bismuth complexes were then prepared by twofold deprotonation of the respective proligand with potassium hydride under inert conditions, followed by treatment with BiBr3. Detailed synthetic procedures and the ESI-MS mass spectra as well as the 1H-, 13C- and 19F-NMR spectra of all proligands H2R1PDPR2 and of the complexes Bi(R1PDPR2)Br are available for review in the ESI (refer to Fig. S1–S52†). Fig. 2 depicts the relevant regions of the 1H-NMR spectra of the four complexes, while Tables S1 and S2 within the ESI† summarize select 1H and 13C-NMR resonance shifts for the proligands and the complexes. | ||
| Fig. 2 Relevant regions of the 1H-NMR spectra of the complexes Bi(R1PDPR2)Br in THF-d8 at r.t., 400 MHz. | ||
Chemical shifts, multiplicities, and coupling constants within the 1H, 13C and 19F NMR spectra agree with expectations and match the reports on other PDP bismuth complexes. The influence of the electron withdrawing –CF3 or the electron donating –OMe substituents are apparent in the NMR shifts of all compounds. Exemplarily, the pyrrolic H-6 resonance (underlaid in purple in Fig. 2) shifts downfield from 6.35 ppm for Bi(OMePDPOMe)Br to 6.68 ppm for Bi(CF3PDPCF3)Br. Additionally, the downfield shift of the central pyridine proton H-1 of Bi(CF3PDPCF3)Br against Bi(OMePDPOMe)Br by Δδ = 0.14 ppm, underlaid in green in Fig. 2, implies electronic delocalization across the entire PDP2− ligand, as even spatially remote substituents exert an influence on the location of the H-1 resonance. Notably, the heterosubstituted complexes with R1 ≠ R2 possess different resonance shifts with larger downfield shifts of the H-6 resonance for Bi(OMePDPCF3)Br compared to Bi(CF3PDPOMe)Br. We note in this vein a larger similarity between the pairs with the same R1 substituent. Our findings are thus at odds with those of Zhang et al., who had reported that the R2 substituent exerts a larger influence on the electronic properties of homoleptic Zr4+ complexes Zr(R1PDPR2)2 than the substituent R1.43 Compared to our previously reported reference system Bi(HPDPH)Br with R1 = R2 = H,5 the differences in resonance shifts are similarly pronounced for the bis(trifluoromethyl)-substituted congener Bi(CF3PDPCF3)Br (Δδ(H-6) = −0.17 ppm) and the di –OMe substituted system Bi(OMePDPOMe)Br (Δδ(H-6) = +0.15 ppm), despite the larger electron-withdrawing capabilities of the –CF3 substituent (σp-CF3 = +0.53) when compared to the electron donating effect of the –OMe substituent (σp-MeO = −0.27).46
UV-Vis absorption studies
The UV-Vis absorption spectra of the present complexes in THF solution are illustrated in Fig. 3, with pertinent data compiled in Table 1. For clarity, the spectra have been normalized to the HOMO–LUMO transition at approximately 500 nm, which is responsible for the deep red colour of these compounds. | ||
| Fig. 3 Top: UV-Vis spectra of the four complexes, normalized to their HOMO–LUMO transition in comparison. Bottom: shift of the HOMO–LUMO band of the complex Bi(OMePDPCF3)Br in differing solvents. Note that the region between 650 and 658 nm is excluded due to the presence of an artefact of the experimental setup. | ||
| Compound | λ max [nm] (ελ [×10−3 M−1 cm−1]) |
|---|---|
| H2CF3PDPCF3 | 328 (49.2), 370 (28.2) |
| H2CF3PDPOMe | 339 (40.9), 377 (26.6) |
| H2OMePDPCF3 | 308 (45.1), 381 (26.6) |
| H2OMePDPOMe | 329 (32.9), 370 (26.7) |
| Bi(CF3PDPCF3)Br | 329 (26.8), 371 (15.0), 501 (6.0) |
| Bi(CF3PDPOMe)Br | 320 (23.7), 369 (14.9), 511 (8.6) |
| Bi(OMePDPCF3)Br | 312 (25.6), 387 (9.7), 526 (8.4) |
| Bi(OMePDPOMe)Br | 300 (23.5), 358 (15.7), 427 (5.6), 535 (7.4) |
Qualitatively, the UV-Vis absorption spectra for the bismuth complexes are similar and exhibit several bands in the UV region of the spectrum with absorption maxima ranging from 300 to 427 nm. These bands are assigned to ligand-centred transitions (vide infra), except for the band at ca. 500 nm, corresponding to the HOMO–LUMO transition. Supportive of this assignment are the UV-Vis spectra of the ligands, which exhibit two transitions in the UV ranging from 308 to 339 nm for the high-energy transition, and 337 to 382 nm for the transition at lower energy, and lack the prominent LMCT band at >500 nm that the complexes display. The wavelength of this lowest-energy band shows a systematic shift from 501 to 535 nm, from electron-withdrawing to electron-donating substituents, in the order Bi(CF3PDPCF3)Br < Bi(CF3PDPOMe)Br < Bi(HPDPH)Br < Bi(OMePDPCF3)Br < Bi(OMePDPOMe)Br, with our previously reported reference compound situated in between the heterosubstituted systems.5 As is the case for the shifts in the NMR resonance signals, a greater similarity is observed for those systems bearing the same R1 substituent.
Extinction coefficients ε reflect the intense red colour of these compounds with values between 6.0–8.6 × 103 M−1 cm−1 for the HOMO–LUMO transitions and values up to 26.8 × 103 M−1 cm−1 for the UV transitions. Noteworthy are the slightly larger ελ values for the HOMO–LUMO transition of the heterosubstitued systems compared to the congeners carrying the same substituents R1 = R2 (refer to Table 1). The ligands absorb intensely in the UV with ελ values ranging from 32.9 × 103 M−1 cm−1 for H2OMePDPOMe to 49.2 × 103 M−1 cm−1 for H2CF3PDPCF3.
In agreement with the distinct charge-transfer character of the HOMO–LUMO transition, the present bismuth complexes Bi(R1PDPR2)Br display a distinct negative solvatochromism, when measured in solutions of dry benzene, CH2Cl2, THF, Et2O, DMF, pyridine and propylene carbonate (PC), as exemplarily shown for Bi(OMePDPCF3)Br in the bottom panel of Fig. 3. The remaining absorption spectra are available for review in the ESI (Fig. S58†). The use of different solvents induces a dramatic shift of the HOMO–LUMO transition, comprising shifts of 2265 to 2600 cm−1 for the individual complexes, and invoking colour impressions ranging from orange (DMF, pyridine), over red (PC, THF, Et2O) and purple (CH2Cl2) to blue (benzene). This bathochromic shift in non-polar solvents indicates more polar electronic ground states, but coordination of donor solvents is assumed to play a role as well based on studies on similar systems.5
TD-DFT calculations
In order to directly interpret the UV-Vis absorption spectra and the emissive properties of these compounds, we performed TD-DFT calculations using the Gaussian 16 program package. Initial calculations using the PBE0 functional delivered qualitatively satisfactory UV-Vis absorption spectra, with the number of computed transitions matching the experimental spectra, but consistently underestimated the energy of the HOMO–LUMO transition by ca. 700 cm−1. We therefore speculated that the MN15 functional might provide better results, as this functional is known to provide broad accuracy even for partial CT character by adding an additional degree of Hartree–Fock exchange.48 Calculated and experimental spectra for all four complexes are compared in Fig. S76 of the ESI.†Fig. 4 provides rendered depictions and computed energies of the HOMO and LUMO for all four complexes and the reference system Bi(HPDPH)Br, including the electron density difference maps (EDDMs) that directly evidence the LMCT character of the HOMO–LUMO transition. | ||
| Fig. 4 Left: energy levels of the HOMO (purple shaded area) and the LUMO (green shaded area) of the complexes Bi(R1PDPR2)Br, as well as depictions of the respective MOs in light blue and white and the corresponding electron density difference maps corresponding to the underlying HOMO–LUMO transition, with electron density loss in blue and electron density gain in red. Right: contributions of various fragments of the complexes Bi(R1PDPR2)Br to the HOMO and LUMO as determined by TD-DFT calculations. R1 and R2 denote the contributions of the phenyl rings carrying the respective substituent. | ||
Based on the results of our TD-DFT calculations, we arrive at four conclusions: (1) the LUMO has substantial contributions from the metal ion and the halide ligand, resulting in a partial LMCT character in particular for the HOMO–LUMO transition. The CT character is however not wholly confined to the latter excitation, as remaining transitions also possess a small degree of bismuth involvement to the acceptor MO; (2) for most transitions involving differently substituted phenyl rings, electron density is shifted from the electron rich pyrrolide units and the anisyl rings to the bismuth, halide, and phenyl rings substituted with the –CF3 groups, so that each transition has additional intraligand CT (ILCT) character; (3) the HOMO and LUMO are both raised in energy by the incorporation of the electron-donating –OMe substituents, but the extent of destabilization is greater for the HOMO; (4) generally, the R1 substituents contribute more to the relevant molecular orbitals, in particular to the HOMO. These points are illustrated in Fig. 4, and a full overview of all relevant transitions is provided for each complex in Fig. S77–S80 of the ESI.†
In addition to conclusions regarding the energy levels of the MOs, the similarity between the pairs Bi(CF3PDPR2)Br and Bi(OMePDPR2)Br within the UV-Vis absorption data and the 1H- and 13C-NMR spectra becomes apparent from the larger calculated contribution of the R1 substituents owing to better conjugation of that particular ring with the PDP backbone. Indeed, in our TD-DFT calculations, the dihedral angle between the pyrrolide and the corresponding phenyl ring is smaller by ca. 15° than that between the pyrrolide and the phenyl rings bearing the R2 substituent (refer to Table S16 in the ESI† which summarizes the values for the dihedral angles as derived from DFT optimized structures). This distortion is also reflected in the solid-state structure of Bi(OMePDPCF3)Br, as well as in the solid-state structures of the ligands (see below and refer to Table S10 within the ESI†).
Photoluminescence measurements
Photoluminescence spectra were recorded at 77 K in glassy matrices of 2-MeTHF for the bismuth complexes Bi(R1PDPR2)Br and additionally, in THF solution at r.t. for the ligands. Select spectra are compiled in Fig. 5 and remaining data, including the biexponential fitting of the phosphorescence decay traces, is provided in Fig. S59–S62 of the ESI.†Table 2 summarizes extracted values for emission maxima, lifetimes and quantum yields of all complexes and their respective proligands.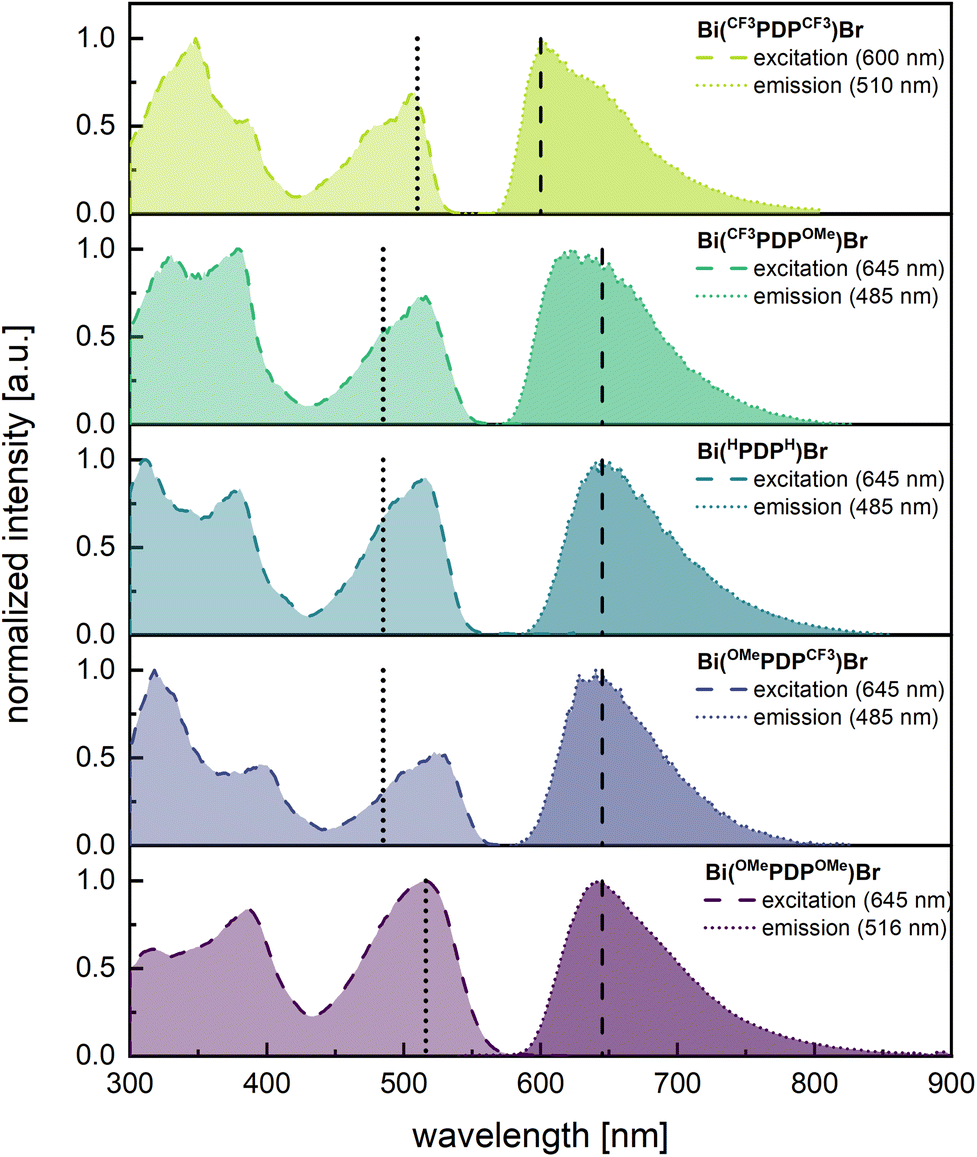 | ||
| Fig. 5 Emission (dotted line, darker shaded area) and excitation spectra (dashed line, lighter shaded area) for the phosphorescence emission of the complexes Bi(R1PDPR2)Br in 2-MeTHF at 77 K. The excitation and detection wavelengths are marked by vertical dotted or dashed lines, respectively. | ||
| λ exc, r.t/λexc, 77 K [nm] | λ flu, r.t/λflu, 77 K [nm] | λ phos, 77 K [nm] | τ flu, r.t/τflu, 77 K [ns] | τ phos, 77 K | Φ r.t./Φ77 K [%] | |
|---|---|---|---|---|---|---|
| H2CF3PDPCF3 | 330, 372/335, 375 | 416/406 | 536 | 2.3/2.5 | 0.7 s | 5.8/67.3 |
| H2CF3PDPOMe | 339, 378/345, 386 | 424/418 | 547 | 2.5/2.8 | 0.4 s | 33.5/73.0 |
| H2OMePDPCF3 | 314, 381/317, 393 | 433/420 | 548 | 1.3/2.8 | 0.4 s | 17.9/62.4 |
| H2OMePDPOMe | 330, 371/334, 376 | 426/418 | 541 | 1.4/2.9 | 0.9 s | 20.4/76.2 |
| Bi(CF3PDPCF3)Br | 348, 385, 509 | n.a. | 602 | n.a. | 65 (56%), 35 (44%) μs | n.a./18.5 |
| Bi(CF3PDPOMe)Br | 329, 380, 513 | n.a. | 621 | n.a. | 74 (31%), 33 (69%) μs | n.a./3.9 |
| Bi(OMePDPCF3)Br | 318, 398, 524 | n.a. | 640 | n.a. | 57 (24%), 25 (76%) μs | n.a./1.7 |
| Bi(OMePDPOMe)Br | 316, 386, 516 | n.a. | 641 | n.a. | 123 (13%), 51 (87%) μs | n.a./1.1 |
For the ligands H2R1PDPR2 at r.t. we observe blue fluorescence with maxima ranging from λflu, r.t. = 416 to 435 nm and fluorescence lifetimes in the nanosecond range (≤3 ns). Quantum yields range from Φflu, r.t. = 5.8% for H2CF3PDPCF3 to 33.5% for H2CF3PDPOMe. At 77 K, the ligands display primarily fluorescence, although a low-intensity phosphorescence may be found in the tail end of the fluorescent emission. Due to the partial overlap with the much more intense fluorescence, accurately determining exact maxima of this emission is difficult (cf. Fig. S68–S75†), but time-resolved emission spectroscopy (TRES) measurements allowed the separation of the fluorescence and phosphorescence components (cf. Fig. S74, S79, S84 and S89 of the ESI†). Colour impressions of the phosphorescence emission range from yellow for H2CF3PDPCF3 to orange for H2OMePDPOMe. Lifetimes τphos, 77 K lie in the range of several hundred milliseconds, and the combined quantum yields Φ77 K = Φflu, 77 K + Φphos, 77 K surpass those measured at r.t. with values between 62.4% and 76.2% (cf.Table 2).
In accordance with previous work on similar systems,5 the present bismuth complexes Bi(R1PDPR2)Br are non-emissive at r.t. in deaerated solutions of THF and in the solid state. When irradiated at cryogenic temperatures in glassy matrices of 2-MeTHF however, red phosphorescence with emission maxima >600 nm and phosphorescence decay rates in the microsecond range are observed. The appearance of phosphorescence emission upon coordination of the ligand to the bismuth ion is undoubtedly due to the large SOC constant of the bismuth ion, which accelerates ISC.
The phosphorescence emission maxima λem within this series of bismuth complexes range from 602 nm for Bi(CF3PDPCF3)Br to 642 nm for Bi(OMePDPOMe)Br, reflecting the differing electron-withdrawing or -donating character of the substituents, and mirroring the trends in the absorption spectra. This stands in contrast to previous work on pyridine-dipyrrolide complexes of bismuth and zirconium,5,43 for which no clear trend between absorption and emission maxima could be observed upon introduction of differing substituents at either the R1 or R2 position. Despite the pronounced bathochromic shift of the emission maxima of 1010 cm−1 from Bi(CF3PDPCF3)Br to Bi(OMePDPOMe)Br, the phosphorescent emission invokes a very similar colour impression of bright red to the naked eye (cf. Fig. S63† for the chromaticity diagram). As can be derived from the excitation spectra, phosphorescence is triggered by irradiation into any absorption band of a complex (Fig. 4).
Slight discrepancies between the excitation and absorption spectra, not entirely attributable to the difference in solvent of THF vs. 2-MeTHF, are due to the rigidochromic effect. For the present compounds, the HOMO–LUMO transition at 77 K is shifted compared to r.t. by 313 to 688 cm−1. This is far less pronounced than in our previous report5 on similar bismuth complexes, for which the rigidochromic effect led to shifts of up to 1300 cm−1.5
In each instance, phosphorescence decay rates are biexponential with lifetimes τPhos ranging from 25 to 51 μs for their shorter-, and 57 to 123 μs for their longer-lived component. The contribution of the long-lived component decreases with increasing electron density within the PDP2− system, so that the ordering Bi(CF3PDPCF3)Br < Bi(CF3PDPOMe)Br < Bi(OMePDPCF3)Br < Bi(OMePDPOMe)Br applies. We note a consistent trend in the differences between absorption and emission maxima, the former derived from excitation spectra, with Δem-exc values ranging from 3035 cm−1 to 3779 cm−1 with an increase in the order Bi(CF3PDPCF3)Br < Bi(CF3PDPOMe)Br < Bi(OMePDPCF3)Br < Bi(OMePDPOMe)Br. These shifts are slightly lower than those of other literature-known bismuth complexes,5 but the long lifetimes in the microsecond range and the complete quenching at r.t. confirm the phosphorescent nature of this emission.
Intrigued by the emissive properties, and curious to see whether the anticipated enhancement of the CT character through the –CF3 substituents would reflect itself on the intensity of the emission, we measured the phosphorescence quantum yields of the four complexes at 77 K. Indeed, in those complexes in which the methoxy substituents oppose the MLCT character of the phosphorescence transition, the quantum yields are considerably lower with 1.1% and 1.7% for Bi(OMePDPOMe)Br and Bi(OMePDPCF3)Br. In the complexes in which the –CF3 substituents enhance the MLCT character, the quantum yields are higher with 3.9% for Bi(CF3PDPOMe)Br and 18.5% for Bi(CF3PDPCF3)Br.
In light of the insights provided by TD-DFT calculations (vide supra), the origin of the phosphorescent emission may be attributed to the MLCT character of the excited state, and bismuth's propensity at facilitating ISC through the HAE. In addition, it has been shown that incorporation of the bismuth central ion into the HOMO–LUMO transition promotes phosphorescence,1,3 and accordingly, the bismuth contribution to the LUMO was calculated to lie between 28–34% for the four complexes described herein.
Solid state structures
We succeeded in growing single crystals suitable for single-crystal X-ray diffraction of all ligands and the complexes Bi(OMePDPCF3)Br and Bi(CF3PDPCF3)Br. Fig. 6 shows the ORTEPs, and details pertaining to data collection and structure refinement parameters are provided in the ESI as Tables S3–S9.† | ||
| Fig. 6 Top: ORTEPs obtained for the solid-state structures of proligands H2R1PDPR2. Thermal displacement ellipsoids are at the 50% probability level, with the exception of H2CF3PDPCF3 × C6H6, which is set at 30% for clarity. Angles discussed in the text are highlighted using one molecule within the asymmetric unit of H2OMePDPCF3 as an example. The co-crystallized CH2Cl2 solvent molecule within the solid-state structure of H2OMePDPOMe × CH2Cl2 has been omitted for clarity reasons. Bottom: ORTEPs obtained for the solid-state structures of the two independent complex molecules of Bi(OMePDPCF3)Br present within the unit cell and ORTEPs for the solid-state structure of Bi(CF3PDPCF3)Br. Thermal displacement ellipsoids are displayed at the 50% probability level. Angles discussed in the text are highlighted using one molecule within the asymmetric unit of 2 Bi(OMePDPCF3)Br × THF as an example. | ||
H2CF3PDPCF3 × C6H6 crystallizes in the monoclinic space group C2/c with two halves of two ligands and half a benzene solvent molecule within the asymmetric unit, while 2 H2OMePDPCF3 and 2 H2CF3PDPOMe × 4 CHCl3 crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with the asymmetric unit comprising of two associated ligands and, in the latter case, four CHCl3 solvent molecules. H2OMePDPOMe × CH2Cl2 again crystallizes in the monoclinic space group C2/c with two halves of two ligands and one CH2Cl2 solvent molecule. With the exception of 2 H2OMePDPCF3, all ligands crystallize as mutually H-bonded dimers with H-bridges between the pyrrole protons and the pyridine nitrogen atoms. To enforce this interaction, the pyridine-dipyrrolide backbone distorts to some degree between the pyrrolide and the pyridine rings as quantified by the dihedral angles θPDP, highlighted exemplarily in blue in Fig. 6 for 2 H2OMePDPCF3. These values comprise θPDP(N1–C3–C4–N2) = −8.4(6)°, θPDP(N3–C24–C25–N4) = −14.7(5)° for H2CF3PDPCF3 × C6H6, θPDP(N2–C3–C4–N1) = −16.1(4)°, θPDP(N2–C23–C24–N3) = −23.6(4)°, θPDP(N5–C64–C65–N6) = −10.3(4)°, and θPDP(N5–C44–C45–N4) = −20.1(5)° for 2 H2CF3PDPOMe × 4 CHCl3, and θPDP(N1–C3–C4–N2) = 16.98(18)° and θPDP(N3–C20–C21–N4) = 12.05(19)° for H2OMePDPOMe × CH2Cl2. As an outlier, molecules are stacked on top of one another in the solid-state structure of H2OMePDPCF3, enabled by the comparative planarity of the pyridine-pyrrolide system and the ansiyl rings with θPDP(N2–C23–C24–N3) = −7.0(3)°, θPDP(N2–C3–C4–N1) = −8.3(3)°, θPDP(N5–C44–C45–N6) = −0.3(3)°, and θPDP(N5–63–C64–N4) = 1.0(3)°.
with the asymmetric unit comprising of two associated ligands and, in the latter case, four CHCl3 solvent molecules. H2OMePDPOMe × CH2Cl2 again crystallizes in the monoclinic space group C2/c with two halves of two ligands and one CH2Cl2 solvent molecule. With the exception of 2 H2OMePDPCF3, all ligands crystallize as mutually H-bonded dimers with H-bridges between the pyrrole protons and the pyridine nitrogen atoms. To enforce this interaction, the pyridine-dipyrrolide backbone distorts to some degree between the pyrrolide and the pyridine rings as quantified by the dihedral angles θPDP, highlighted exemplarily in blue in Fig. 6 for 2 H2OMePDPCF3. These values comprise θPDP(N1–C3–C4–N2) = −8.4(6)°, θPDP(N3–C24–C25–N4) = −14.7(5)° for H2CF3PDPCF3 × C6H6, θPDP(N2–C3–C4–N1) = −16.1(4)°, θPDP(N2–C23–C24–N3) = −23.6(4)°, θPDP(N5–C64–C65–N6) = −10.3(4)°, and θPDP(N5–C44–C45–N4) = −20.1(5)° for 2 H2CF3PDPOMe × 4 CHCl3, and θPDP(N1–C3–C4–N2) = 16.98(18)° and θPDP(N3–C20–C21–N4) = 12.05(19)° for H2OMePDPOMe × CH2Cl2. As an outlier, molecules are stacked on top of one another in the solid-state structure of H2OMePDPCF3, enabled by the comparative planarity of the pyridine-pyrrolide system and the ansiyl rings with θPDP(N2–C23–C24–N3) = −7.0(3)°, θPDP(N2–C3–C4–N1) = −8.3(3)°, θPDP(N5–C44–C45–N6) = −0.3(3)°, and θPDP(N5–63–C64–N4) = 1.0(3)°.
For all ligands, the R1 substituents are tilted only slightly out of plane of the respective pyrrole unit to which they are attached, with angles ranging from θ1 = −23.8(6) to 0.0(4)° (exemplarily highlighted in red in Fig. 6). In contrast, R2 substituents are tilted considerably against the pyrrole units with torsion angles (green in Fig. 6) between θ2 = −44.1(4) to −79.9(4)° and 46.7(11) to 90.0(6)°. The larger angles for the R2 ring are presumably the source of the similarity between the pairs of compounds carrying the same phenyl ring R1, i.e.H2CF3PDPCF3, H2CF3PDPOMe on the one hand, and H2OMePDPCF3, H2OMePDPOMe on the other, with respect to their 1H- and 13C-NMR shifts and photophysical properties (see above). Table S10 of the ESI† summarizes all values for the dihedral angles between the two phenyl rings and the respective pyrrole to which they are bound as obtained from single-crystal X-ray diffraction studies.
Mirroring the behaviour of the corresponding ligand, 2 Bi(OMePDPCF3)Br × THF crystallizes in the triclinic space group P with two molecules within the asymmetric unit and one THF solvent molecule. The two molecules Bi1(OMePDPCF3)Br and Bi2(OMePDPCF3)Br in the asymmetric unit differ in the orientation of the methoxy substituents, which are one anti, one syn for molecule 1, or both anti for molecule 2 (see the bottom panel of Fig. 6). For both of the molecules of Bi(OMePDPCF3)Br, the bismuth central ion coordinates to the pyrrolide atoms N1, N3/N4, N6, as well as the pyridine nitrogen atoms N2/N5 and the bromido ligands Br1/Br2. The two independent molecules of Bi(OMePDPCF3)Br within the unit cell associate as dimers via short Bi⋯C contacts to pyrrolide or anisyl C atoms of the adjacent molecules (d(Bi1⋯C67) = 3.444(11) Å, d(Bi1⋯C68) = 3.102(12) Å, d(Bi1⋯C69) = 3.500(11) Å, d(Bi1⋯C74) = 3.578(11) Å; d(Bi2⋯C27) = 3.209(12) Å, d(Bi2⋯C28) = 3.288(10) Å, and d(Bi2⋯C29) = 3.364(10) Å), and, in the case of molecule 1, by an additional interaction with the pyrrolide N atom of molecule 2 of d(Bi1⋯N4) = 3.377(10) Å. These contacts are by 0.122 to 0.598 Å shorter than the sum of the van der Waals radii of the interacting atoms. The close proximity of the two molecules allows for additional C–H⋯π interactions of d(C80⋯H025) = 2.861(17) Å between the CF3 substituted aryl rings R2 as well as a C–H⋯π interaction of d(C1⋯H023) = 2.871(15) Å between the central pyridine ring and one of the R2 rings. The dimers associate with their neighbours by pairwise hydrogen bonds of the bromido ligands with a pyridine H-atom (d(Br1⋯H11) = 2.930(3) Å), which are mutually enforced by additional C–H⋯π interactions of d(C77⋯H01u) = 2.755(17) Å between C atom C77 and H01u as well as by π-stacking interactions between the roughly coplanar pyridine and pyrrolide rings of d(C42⋯C45) = 3.311(16) Å on the one side, or by weaker hydrogen bonds d(Br1⋯H02p) = 3.010(3) Å with a methoxy proton.
Bi(CF3PDPCF3)Br × THF crystallizes in the triclinic space group P. with one coordinating THF solvent molecule (cf. bottom of Fig. 6). The coordination geometry can be described as distorted octahedral with the bromide and coordinated solvent molecule and the pyrrolide atoms N1 and N3 in trans position to one another, respectively. The remaining positions are filled by the pyridine nitrogen atom N2 and a fluorine atom of a neighbouring molecule's CF3 group, which resides trans to the pyridine N2 with d(Bi1⋯F7) = 3.382(4) Å. The bismuth lone pair induces a slight variance from the ideal 180° angle between Br1 and O1, which is instead θ(O1–Bi1–Br1) = 164.25(7)°. The presence of the bismuth lone pair is further evidenced by a smaller angle N3–Bi–O1 = 80.04(10)° compared with N1–Bi–O1 = 95.5(1)°. Neighbouring molecules of Bi(CF3PDPCF3)Br within the solid state structure associate via π-stacking of one side of the PDP2− unit with short contacts d(C1⋯C4) = 3.311(5) Å, and d(C2⋯C3) = 3.329(5) Å. This association is supported by an additional C–H⋯π interaction of d(C36⋯H16) = 2.761(4) Å. Other interactions consist of arene–H⋯F and interactions between the hydrogen atoms belonging to the coordinated THF solvent molecule and the CF3 fluorine atoms.
As for the ligands, all bond lengths and angles compare favourably to those of other literature-known bismuth complexes bearing PDP2− ligands.5,44,45
Conclusions
Four new complexes Bi(R1PDPR2)Br with dianionic pyridine-2,6-bis(2′-pyrrolide) pincer ligands, modified with electron-donating (–OMe) or -withdrawing (–CF3) substituents at the pyrrolides, were successfully synthesized and characterized. At cryogenic temperatures, these complexes emit red phosphorescence with biexponential decay curves and lifetimes in the range of microseconds, while the respective proligands H2R1PDPR2 exhibit primarily fluorescence and very weak phosphorescence at 77 K with lifetimes in the range of several hundred milliseconds. The sole presence of phosphorescence in the complexes compared to the ligands attests to the capability of the Bi3+ ion to induce ISC and populate excited triplet states. In addition to shifting the emission wavelength from 602 nm to 641 nm by variation of substituents, we observed a profound increase in the phosphorescence quantum yield in the systems with R1 = CF3 which we attribute of the enhancement of the T1 → S0 MLCT character through the introduction of electron-withdrawing –CF3 groups. The experimentally observed negative solvatochromism and TD-DFT calculations indicate a substantial LMCT character to the HOMO–LUMO transition which is situated at ca. 500 nm in THF. Excitation of this band causes electron density to shift from the ligand-localized HOMO to the LUMO, which is situated predominantly on the pyridine ring and the Bi–Br antibonding orbitals. In addition to providing valuable insight into the UV-Vis absorption data, the TD-DFT calculations explain the profound similarities of the pairs with the same R1 substituents in NMR shifts, as well as the absorptive and emissive properties. From the optimized structures obtained in the calculations, it becomes apparent that the R2 rings are tilted considerably against the backbone of the PDP2− pincer ligand, thereby attenuating electronic participation of the substituents at this phenyl ring, while the R1 rings are better conjugated. These findings are mirrored in the solid state structures of the ligands and the complexes Bi(CF3PDPOMe)Br and Bi(CF3PDPCF3)Br.Author contributions
K. L. Deuter and R. F. Winter conceptualized the project. As part of a master course internship, J. Rieser prepared the complexes Bi(OMePDPOMe)Br and Bi(OMePDPCF3)Br from ligands synthesized by K. L. Deuter, and K. Vollmar prepared compounds Bi(CF3PDPCF3)Br and Bi(CF3PDPOMe)Br as part of her bachelor thesis. Data was aquired and evaluated by K. L. Deuter and in part by K. Vollmar and J. Rieser. Data visualization was conducted by K. L. Deuter and partially by K. Vollmar. M. Linseis supervised the acquisition and evaluation of crystallographic data. K. L. Deuter prepared the manuscript with input from all authors. R. F. Winter supervised the research and provided resources.Data availability
All pertinent synthesis and characterization data as well as the data from quantum chemical modelling are contained in the Electronic ESI.† X-ray structure data were deposited with the Cambridge Structure database.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Yurii Kolbediuk, Anneke Janshen, Gernot Kummer and Tristan Kölsch for the synthetic post-production of various ligands as part of their undergraduate studies. We thank Gernot Haug for glovebox maintenance and Anton Batalov from PicoQuant GmbH for his patient guidance during the repair of our spectrometer. K. L. Deuter would like to thank Kai Jellinek for proofreading early versions of this manuscript and Lars Vogelsang for his efforts at laying the foundations for 77 K quantum yield measurements. Special thanks is owed Alice Lulich for valuable discussions on the role of CT for luminescent bismuth complexes at the 45th ICCC in Ft. Collins. The authors gratefully acknowledge support by the state of Baden-Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575-1 FUGG (JUSTUS 2 cluster), and the NMR Core Facility at the University of Konstanz for experimental support and spectrometer upkeep.References
- K. L. Deuter, D. J.-J. Balaba, M. Linseis and R. F. Winter, Chem. Commun., 2025, 61, 3548–3551 RSC.
- W. Ma, L. Xu, S. Zhang, G. Li, T. Ma, B. Rao, M. Zhang and G. He, J. Am. Chem. Soc., 2021, 143, 1590–1597 CrossRef PubMed.
- L. A. Maurer, O. M. Pearce, F. D. R. Maharaj, N. L. Brown, C. K. Amador, N. H. Damrauer and M. P. Marshak, Inorg. Chem., 2021, 60, 10137–10146 CrossRef PubMed.
- J. Ohshita, S. Matsui, R. Yamamoto, T. Mizumo, Y. Ooyama, Y. Harima, T. Murafuji, K. Tao, Y. Kuramochi, T. Kaikoh and H. Higashimura, Organometallics, 2010, 29, 3239–3241 CrossRef.
- K. L. Deuter, F. Kather, M. Linseis, M. Bodensteiner and R. F. Winter, Chem. – Eur. J., 2024, 31, e202403761 CrossRef PubMed.
- Y. Zhang, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2016, 138, 13115–13118 CrossRef PubMed.
- A. S. Gowda, T. S. Lee, M. C. Rosko, J. L. Petersen, F. N. Castellano and C. Milsmann, Inorg. Chem., 2022, 61, 7338–7348 CrossRef PubMed.
- L. C.-C. Lee and K. K.-W. Lo, Chem. Rev., 2024, 124, 8825–9014 CrossRef PubMed.
- S.-S. Xue, Y. Pan, W. Pan, S. Liu, N. Li and B. Tang, Chem. Sci., 2022, 13, 9468–9484 RSC.
- C. Bizzarri, E. Spuling, D. M. Knoll, D. Volz and S. Bräse, Coord. Chem. Rev., 2018, 373, 49–82 CrossRef CAS.
- X. Li, Y. Xie and Z. Li, Chem. – Asian J., 2021, 16, 2817–2829 CrossRef CAS PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- A. S. Polo, M. K. Itokazu and N. Y. M. Iha, Coord. Chem. Rev., 2004, 248, 1343–1361 CrossRef CAS.
- A. Kumar, P. Daw and D. Milstein, Chem. Rev., 2022, 122, 385–441 CrossRef CAS PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 3rd edn, 2006 Search PubMed.
- A. J. S. Valentine and X. Li, J. Phys. Chem. Lett., 2022, 13, 3039–3046 CrossRef CAS PubMed.
- S. E. Braslavsky, Pure Appl. Chem., 2007, 79, 293–465 CrossRef CAS.
- S. M. Parke and E. Rivard, Isr. J. Chem., 2018, 58, 915–926 CrossRef CAS.
- S. Arunkumar, D. Ghosh and G. R. Kumar, Results Chem., 2022, 4, 100399 CrossRef CAS.
- J. C. Berrones-Reyes, C. C. Vidyasagar, B. M. Muñoz Flores and V. M. Jiménez-Pérez, J. Lumin., 2018, 195, 290–313 CrossRef CAS.
- Y. Sano, H. Satoh, M. Chiba, M. Okamoto, K. Serizawa, H. Nakashima and K. Omae, J. Occup. Health, 2005, 47, 293–298 CrossRef CAS PubMed.
- J. D. Rosário, F. H. Moreira, L. H. Rosa, W. Guerra and P. P. Silva-Caldeira, Molecules, 2023, 28, 5921 CrossRef PubMed.
- A. C. Marwitz, A. K. Dutta, R. L. Conner, L. A. Sanz, L. G. Jacobsohn and K. E. Knope, Inorg. Chem., 2024, 63, 11053–11062 CrossRef CAS PubMed.
- A. C. Marwitz, A. D. Nicholas, L. M. Breuer, J. A. Bertke and K. E. Knope, Inorg. Chem., 2021, 60, 16840–16851 CrossRef CAS PubMed.
- T. Oksana, A. Magali, M. Francesco, F. Alessandra, B. Chiara and M. Nicolas, Angew. Chem., Int. Ed., 2016, 55, 7998–8002 CrossRef PubMed.
- P. Xiong, Y. Li and M. Peng, iScience, 2020, 23, 101578 CrossRef CAS PubMed.
- P. Dang, D. Liu, G. Li, A. A. Al Kheraif and J. Lin, Adv. Opt. Mater., 2020, 8, 1901993 CrossRef CAS.
- P. Boutinaud, Phys. Chem. Chem. Phys., 2023, 25, 11027–11054 RSC.
- Y. Tang, M. Deng, M. Wang, X. Liu, Z. Zhou, J. Wang and Q. Liu, Adv. Opt. Mater., 2023, 11, 2201827 CrossRef CAS.
- S. Arunkumar and G. R. Kumar, J. Organomet. Chem., 2023, 1001, 122849 CrossRef CAS.
- L. Zhang, K. Wang and B. Zou, ChemSusChem, 2019, 12, 1612–1630 CrossRef CAS PubMed.
- Z. Jin, Z. Zhang, J. Xiu, H. Song, T. Gatti and Z. He, J. Mater. Chem., 2020, 8, 16166–16188 RSC.
- Y. Adachi, S. Terao, Y. Kanematsu and J. Ohshita, Chem. – Asian J., 2024, 19, e202301142 CrossRef CAS PubMed.
- M. Geppert, M. Müller, M. Linseis and R. F. Winter, Dalton Trans., 2025, 54, 1779–1783 RSC.
- A. Stamoulis, M. Mato, P. C. Bruzzese, M. Leutzsch, A. Cadranel, M. Gil-Sepulcre, F. Neese and J. Cornella, J. Am. Chem. Soc., 2025, 147, 6037–6048 CrossRef CAS PubMed.
- L. N. Grant, M. E. Carroll, P. J. Carroll and D. J. Mindiola, Inorg. Chem., 2016, 55, 7997–8002 CrossRef CAS PubMed.
- S. Yadav and C. Dash, Tetrahedron, 2020, 76, 131350 CrossRef CAS.
- Y. Zhang, T. S. Lee, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2018, 140, 5934–5947 CrossRef CAS PubMed.
- D. C. Leary, Y. Zhang, J. G. Rodriguez, N. G. Akhmedov, J. L. Petersen, B. S. Dolinar and C. Milsmann, Organometallics, 2023, 42, 1220–1231 CrossRef CAS PubMed.
- L. R. Valerio, B. M. Hakey, D. C. Leary, E. Stockdale, W. W. Brennessel, C. Milsmann and E. M. Matson, Inorg. Chem., 2024, 63, 9610–9623 CrossRef CAS PubMed.
- Y. Zhang, D. C. Leary, A. M. Belldina, J. L. Petersen and C. Milsmann, Inorg. Chem., 2020, 59, 14716–14730 CrossRef CAS PubMed.
- Z. R. Turner, J. T. Wilmore, N. H. Rees and J.-C. Buffet, Dalton Trans., 2022, 51, 3060–3074 RSC.
- Z. R. Turner, Inorg. Chem., 2019, 58, 14212–14227 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- M. Matsui, A. Oji, K. Hiramatsu, K. Shibata and H. Muramatsu, J. Chem. Soc., Perkin Trans. 2, 1992, 201–206 RSC.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2442540–2442545. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt00869g |
| This journal is © The Royal Society of Chemistry 2025 |