
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of bulky hydride ligands: m-terphenylborohydride complexes with trivalent uranium and neodymium†
Peter A.
Zacher
III
 ,
Daniel K.
Unruh
and
Scott R.
Daly
*
,
Daniel K.
Unruh
and
Scott R.
Daly
*
The University of Iowa, Department of Chemistry, E331 Chemistry Building, Iowa City, IA 52242, USA. E-mail: scott-daly@uiowa.edu
First published on 28th May 2025
Abstract
Here we describe the first coordination complexes containing a bulky m-terphenyltrihydroborate ligand. Treating [UI3(thf)4] and NdCl3 with three equiv. of Li(H3BArtBu4)(Et2O) (where ArtBu4 = 2,6-(3,5-tBu2C6H3)2C6H3) yielded [M(H3BArtBu4)3(thf)2] (M = U and Nd). [U(H3BArtBu4)3(dme)2] is also described, and structural comparisons reveal the influence of the Lewis base on H3BArtBu4 positioning.
Actinide borohydride complexes have been known since the discovery of [U(BH4)4] during the Manhattan Project.1 The simplest borohydride, BH41−, for example, forms molecular complexes with the general formula [M(BH4)4] with M = Th–Pu,1a,2 and similar homoleptic complexes are known with methyltrihydroborate (MeBH31−).3
Despite the well-known examples of borohydride complexes with actinides in the +4 oxidation state,4 conventional borohydride ligands like BH41− and MeBH31− are too small to form neutral, mononuclear, and homoleptic complexes with trivalent actinides because of their larger ionic radius and reduced charge. The only homoleptic examples with borohydrides are those with chelating ligands like aminodiboranates and phosphinodiboranates.5 Even though these latter ligands saturate a larger percentage of the metal coordination sphere, their complexes with trivalent actinides are only known to exist as dimers or oligomers in the solid state. In this context, a mononuclear and homoleptic borohydride complex has yet to be isolated with a trivalent f-element.
We therefore set out to develop sterically bulky borohydride ligands for the isolation of homoleptic and mononuclear complexes with trivalent actinides. A structural scaffold that has proven effective at isolating low-coordinate transition metal, lanthanide, and actinide complexes is m-terphenyl. The demanding steric presence of m-terphenyl groups generally serve to shield open coordination sites and stabilize metals and main group elements with low coordination numbers.6 Notable and recent examples with uranium are provided in Fig. 1a. Arnold and coworkers demonstrated how the bulky terphenyl ligand (2,6-(4-tBu-C6H4)2C6H3)1– could be used to isolate the first structurally authenticated homoleptic tris(aryl)U(III) complex.7 Boncella and Odom used the m-terphenylamido ligand (NHAriPr6)1− where AriPr6 = 2,6-(2,4,6-iPr3C6H2)2C6H3 to isolate a rare example of a formally U(II) complex.8 Though not a homoleptic example, Goodwin and coworkers recently described how the m-terphenylthiolate ligand (SAriPr6)1− can be used to isolate low coordinate U(III) complexes like [UIII(SAriPr6)2(BH4)].9 These and other examples of coordinatively unsaturated m-terphenyl complexes led us to explore the development of m-terphenylborohydride ligands for low-valent actinide chemistry.10
 | ||
| Fig. 1 (a) Notable examples of bulky m-terphenyl ligand scaffolds used to isolate mononuclear uranium complexes. (b) Synthesis of [UIII(H3BArtBu4)3(thf)2] (1-U). | ||
Herein, we report the synthesis and characterization of the m-terphenyltrihydroborate ligand,11 (H3BArtBu4)1− where ArtBu4 = 2,6-(3,5-tBu2C6H3)C6H3, and its use for the preparation of trivalent uranium and neodymium complexes (Fig. 1b). This ligand was targeted for our initial attempt because we anticipated that the 3,5-positioning of the tert-butyl substituents would provide greater steric protection as compared to more common m-terphenyl platforms like AriPr6 shown in Fig. 1. As we will show, despite its size, (H3BArtBu4)1− is still not large enough to prevent etherates like thf from coordinating to U3+ and Nd3+.
The synthesis of (H3BArtBu4)1− is summarized in Scheme 1 (see ESI† for full details), and it followed the standard double benzyne reaction and I2 quenching that is commonly used to prepare iodated m-terphenyl ligand precursors.12 Subsequent lithiation of I–ArtBu4 with nBuLi to form Li–ArtBu4 followed by treatment with B(OMe)3 in Et2O afforded (MeO)2BArtBu4 in moderate yield (58%). Reduction of the borate with LiAlH4 in Et2O yielded the final terphenyl trihydroborate salt Li(H3BArtBu4)(Et2O) in 75% yield.13 The 11B NMR spectrum revealed a diagnostic quartet at δB −29.1 ppm with 1JBH = 75 Hz due to coupling with the three hydrides.13 The 1H and 13C NMR spectra revealed the expected m-terphenyl resonances, as well as the presence of one Et2O molecule based on 1H integrations. X-ray diffraction (XRD) studies on transparent crystals isolated during a synthesis of 1-U (vide infra) revealed the structure of unsolvated [Li(H3BArtBu4)]2. The structure is dinuclear with the Li cations bridged by BH3 groups and coordinated in an η3 fashion to a flanking aryl from the m-terphenyl group (Fig. 2).
 | ||
| Scheme 1 Synthesis of Li(H3BArtBu4)(Et2O). | ||
 | ||
| Fig. 2 Truncated molecular structure of unsolvated [Li(H3BArtBu4)]2. Ellipsoids are drawn at 50% probability, except hydrogens which are drawn as arbitrary sized spheres. The flanking aryl group carbon atoms are shown as wires and hydrogen atoms on carbon have been removed for clarity. | ||
The reaction of [UI3(thf)4] with three equivalents of Li(H3BArtBu4)(Et2O) in chlorobenzene initially afforded a suspension of white and dark purple solids that slowly turned brown over the course of 16 hours. Evaporation of the reaction mixture, followed by extraction with benzene, afforded a deep red solution from which orange crystals of [UIII(H3BArtBu4)3(thf)2] (1-U) were isolated by slow evaporation in low yield (27%). Despite the use of chlorobenzene as the solvent, 1-U retains two thf ligands from the [UI3(thf)4] starting material. Attempts to remove the thf by heating under vacuum at 50–70 °C yielded visual evidence of decomposition. 1-U is appreciably soluble in aromatic solvents like benzene, toluene, fluorobenzene, and chlorobenzene, but is only sparingly soluble in Et2O and pentane.
The solid-state structure of 1-U from single-crystal XRD shows a monomeric trivalent uranium complex with three equatorial (H3BArtBu4)1− ligands and two axial thf molecules (Fig. 3). The three U–B distances ranged from 2.622(2)–2.646(3) Å, which is consistent with κ3 coordination with three hydrides bound to the metal. These U–B distances are shorter than the κ3-H3B distances reported previously for [UIII(H3BNMe2BH3)3] and [UIII(H3BPtBu2BH3)3]2 at U–B = 2.665(6) and 2.69(1) Å respectively.5b,d The U–O bond lengths of 2.4847(15) and 2.4873(15) Å are significantly shorter than those previously reported for [UIII(H3BNMe2BH3)3(thf)] (U–O = 2.549(4) Å) and trans U–O distances in the oligomeric structure of [UIII(BH4)3(thf)2] (2.519(5) Å).5a,14 The nine U–H and two U–O bonds indicate a total coordination number of 11 for the uranium center, which is lower than those observed for other U(III) borohydride complexes that range from 12–14.5,14 The B–U–B angles of 116.71(8)°, 119.33(8)°, and 123.93(8)° and the axial O–U–O bond angle of 178.96(6)° indicate a distorted trigonal bipyramidal coordination geometry (τ5 = 0.88).15
 | ||
| Fig. 3 Molecular structure of [UIII(H3BArtBu4)3(thf)2] (1-U). Ellipsoids are drawn at 50% probability, except hydrogens which are drawn as arbitrary sized spheres. The flanking aryl group carbon atoms are shown as wires and hydrogen atoms on carbon have been omitted for clarity. Selected distances (Å): U(1)–B(1) = 2.638(3), U(1)–B(2) = 2.646(3), U(1)–B(3) = 2.622(2), U(1)–O(1) = 2.4573(15), U(1)–O(2) = 2.4848(15). | ||
The IR spectrum of ground samples of 1-U revealed bridging U–H–B stretches at 2173 cm−1 and a shoulder at 2072 cm−1, consistent with the κ3-BH3 binding with uranium. The 11B NMR spectrum for 1-U in C6D6 showed a single resonance at δB 181.5 ppm, which is shifted more downfield compared to [UIII(BH4)3(thf)2] at δB 152.9 ppm.16 The 1H NMR spectrum of 1-U in C6D6 revealed a complex set of resonances between δH −8 and 18 ppm due to the paramagnetic UIII (5f3) metal center, with a characteristically downfield shifted resonance at δH 134.2 ppm assigned to BH3. The UV-vis/NIR electronic absorption spectrum of 1-U in thf (Fig. S26; ESI†) displays features at 408 nm, 471 nm, and 529 nm with ε = 940, 670, and 405 M−1 cm−1 respectively. These absorptions are consistent with the orange color of the complex, and similar colors spanning yellow to red have been observed for other UIII borohydride complexes.5a,16b,17 Between 700 and 1500 nm there are weak absorptions (ε = 25–90 M−1 cm−1) characteristic of f–f transitions.
Stirring solid samples of 1-U in dimethoxyethane (dme) quickly leads to its dissolution and conversion to [UIII(H3BArtBu4)3(dme)2] (2-U). Evaporation of this solution, followed by crystallization from pentane afforded orange crystals suitable for single-crystal XRD studies. Unlike 1-U, 2-U is highly soluble in aromatic, as well as etherate and hydrocarbon solvents.
The molecular structure of 2-U reveals a distorted pentagonal bipyramidal geometry based on O and B positioning with two dme molecules and one borohydride in the equatorial plane (Fig. 4). The U–B distances of 2.695(3) Å, and 2.712(4) Å indicate the (H3BArtBu4)1− ligands retain the κ3 binding mode upon coordination of dme. The B1–U–B2 angle decreases to 102.03(7)°, while the B1–U–B1′ angle increases to 155.95(13)° breaking the pseudo trigonal arrangement observed in 1-U to create coordination sites for two dme molecules to bind (Fig. S2†). The O1–U–O1′ angle is also slightly decreased to 171.98(10)°. The two crystollographically unique U–O bond lengths of 2.596(2) Å and 2.788(2) Å reflect the steric crowding around the metal and are significantly longer than those observed in 1-U and the starting material [UI3(thf)4].16a,18 The nine U–H and four U–O bonds yield a total coordination number of 13 for 2-U. The 11B NMR spectrum of a crystalline sample of 2-U in C6D6 shows a single dominant feature at δB 271 ppm, as well as several minor species that may be attributed to different coordination isomers of [UIII(H3BArtBu4)3(dme)2] in solution.
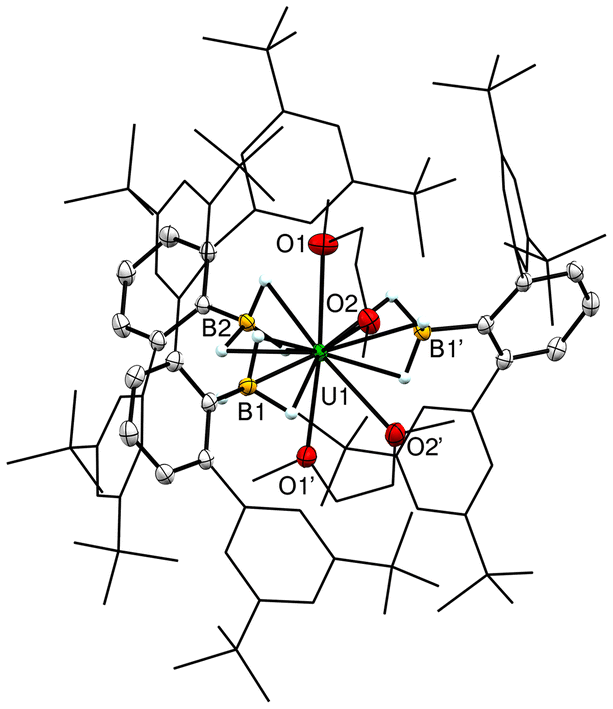 | ||
| Fig. 4 Molecular structure of [UIII(H3BArtBu4)3(dme)2] (2-U). Ellipsoids are drawn at 50% probability, except hydrogens which are drawn as arbitrary sized spheres. The flanking aryl groups and dme carbon atoms are shown as wires. Hydrogen atoms on carbon and co-crystallized pentane have been omitted for clarity. Selected distances (Å): U(1)–B(1) = 2.695(3), U(1)–B(2) = 2.712(4), U(1)–O(1) = 2.596(2), U(1)–O(2) = 2.788(2). | ||
In an attempt to prepare a lanthanide analog for a point of comparison to 1-U, we initially conducted salt metathesis reactions under the same conditions employed for 1-U. Interestingly, no evidence of reaction was observed after stirring NdI3 or NdI3(thf)3.5 with three equivalents of Li(H3BArtBu4)(Et2O) in chlorobenzene for 16 hours. The lack of reactivity with NdI3(thf)3.5, as compared to [UI3(thf)4], is possibly because this species can exist as the [NdI2(thf)5][NdI4(thf)2] ion pair, which tends to be less soluble.19 Metathesis reactions conducted with the same Nd starting materials in Et2O, toluene, or thf also yielded no evidence of reaction. Given the divergence in reactivity between the U and Nd iodide starting materials, we instead tried metathesis reactions with NdCl3. Gratifyingly, stirring NdCl3 and three equivalents of Li(H3BArtBu4)(Et2O) in thf, followed by extraction with benzene and slow concentration of the solution, yielded very light blue blocky crystals of [NdIII(H3BArtBu4)3(thf)2] (1-Nd) in 31% yield.
XRD studies revealed a trigonal bipyramidal arrangement of (H3BArtBu4)1− and thf ligands identical to 1-U (Fig. S1; ESI†) The Nd–B distances of 2.628(2) Å, 2.623(2) Å, and 2.604(2) Å are consistent with a κ3 binding mode of the BH3− units and are on average ∼0.017 Å shorter than those observed for 1-U, consistent with the decrease in ionic radius from UIII (1.025 Å; CN = 6) to NdIII (0.983 Å; CN = 6).20 The B–Nd–B angles that comprise the trigonal arrangement of B atoms are 123.80(6)°, 116.85(6)°, and 119.32(7)°, which is also very similar to those observed in 1-U. Accounting for these angles and the axial O–Nd–O angle of 179.10(4)° yields a τ5 value of 0.92.15
The 11B NMR spectrum of 1-Nd in C6D6 depicts a single resonance at δB 197 ppm consistent with the equivalence of the boron atoms in the solid-state structure. The room temperature 1H NMR spectrum of 1-Nd in C6D6 unsurprisingly reveals a similarly complex set of paramagnetically shifted resonances to those in 1-U. The IR spectrum of 1-Nd (Nujol) features a B–H stretch at 2191 cm−1 with a shoulder at 2081 cm−1. The profile of these absorptions are nearly identical to those for 1-U, but they occur at slightly higher wavenumbers.
In summary, we have described the synthesis and characterization of the m-terphenyltrihydroborate salt Li(H3BArtBu4)(Et2O), and several complexes with trivalent U and Nd. The structures of [UIII(H3BArtBu4)3(thf)2] (1-U) and [NdIII(H3BArtBu4)3(thf)2] (1-Nd) demonstrate that despite the increased steric profile of the m-terphenyl scaffold, it is not sufficient to completely suppress coordination of Lewis bases like thf. Moreover, the formation of [UIII(H3BArtBu4)3(dme)2] (2-U) shows how these complexes can distort to increase their coordination numbers and accommodate binding of additional Lewis-base donors. We are continuing to investigate other m-terphenyl scaffolds to identify the steric criteria and experimental conditions that will allow mononuclear borohydride complexes to be isolated with trivalent actinides without Lewis bases.
Author contributions
Peter A. Zacher III: writing – original draft, data curation, formal analysis. Daniel K. Unruh: data curation (crystal structures). Scott R. Daly: writing – original draft, formal analysis, supervision, funding acquisition, conceptualization.Data availability
Crystallographic data for 1-U, 2-U, 1-Nd, and [Li(H3BArtBu4)]2 have been deposited into the Cambridge Structural Database (CSD) and have been assigned the CCDC numbers 2439982–2439985.† All other data are available in the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Nuclear Security Administration (DE-NA0004151) for support of this research. We also thank Vincent Groner and Jim Boncella at Washington State University for assistance with the synthesis of the I–ArtBu4 precursor. XRD data were collected using the instrument supported by NSF CHE-1828117. Some of the NMR data were collected using the instrument supported by NSF CHE-2017828.References
- (a) H. I. Schlesinger and H. C. Brown, J. Am. Chem. Soc., 1953, 75, 219–221 CrossRef CAS; (b) H. I. Schlesinger, H. C. Brown, B. Abraham, A. C. Bond, N. Davidson, A. E. Finholt, J. R. Gilbreath, H. Hoekstra, L. Horvitz, E. K. Hyde, J. J. Katz, J. Knight, R. A. Lad, D. L. Mayfield, L. Rapp, D. M. Ritter, A. M. Schwartz, I. Sheft, L. D. Tuck and A. O. Walker, J. Am. Chem. Soc., 1953, 75, 186–190 CrossRef CAS.
- (a) R. H. Banks, N. M. Edelstein, R. R. Rietz, D. H. Templeton and A. Zalkin, J. Am. Chem. Soc., 1978, 100, 1957–1958 CrossRef CAS; (b) H. R. Hoekstra and J. J. Katz, J. Am. Chem. Soc., 1949, 71, 2488–2492 CrossRef CAS.
- (a) H. I. Schlesinger, H. C. Brown, L. Horvitz, A. C. Bond, L. D. Tuck and A. O. Walker, J. Am. Chem. Soc., 1953, 75, 222–224 CrossRef CAS; (b) R. Shinomoto, E. Gamp, N. M. Edelstein, D. H. Templeton and A. Zalkin, Inorg. Chem., 1983, 22, 2351–2355 CrossRef CAS.
- (a) S. R. Daly, Actinide Borohydrides, in The Heaviest Metals: Science and Technology of the Actinides and Beyond, ed. W. J. Evans and T. P. Hanusa, Major Reference Works, John Wiley & Sons, Ltd, 2018, pp. 319–334 Search PubMed; (b) M. Ephritikhine, Chem. Rev., 1997, 97, 2193–2242 CrossRef CAS PubMed; (c) T. J. Marks and J. R. Kolb, Chem. Rev., 1977, 77, 263–293 CrossRef CAS.
- (a) S. R. Daly and G. S. Girolami, Inorg. Chem., 2010, 49, 5157–5166 CrossRef CAS PubMed; (b) S. R. Daly and G. S. Girolami, Chem. Commun., 2010, 46, 407–408 RSC; (c) S. R. Daly, P. M. B. Piccoli, A. J. Schultz, T. K. Todorova, L. Gagliardi and G. S. Girolami, Angew. Chem., Int. Ed., 2010, 49, 3379–3381 CrossRef CAS PubMed; (d) A. V. Blake, T. V. Fetrow, Z. J. Theiler, B. Vlaisavljevich and S. R. Daly, Chem. Commun., 2018, 54, 5602–5605 RSC; (e) T. V. Fetrow, R. Bhowmick, A. J. Achazi, A. V. Blake, F. D. Eckstrom, B. Vlaisavljevich and S. R. Daly, Inorg. Chem., 2020, 59, 48–61 CrossRef CAS PubMed; (f) T. V. Fetrow and S. R. Daly, Dalton Trans., 2021, 50, 11472–11484 RSC; (g) T. V. Fetrow, J. Zgrabik, R. Bhowmick, F. D. Eckstrom, G. Crull, B. Vlaisavljevich and S. R. Daly, Angew. Chem., Int. Ed., 2022, 61, e202211145 CrossRef CAS PubMed; (h) S. R. Daly, B. J. Bellott, D. R. McAlister, E. P. Horwitz and G. S. Girolami, Inorg. Chem., 2022, 61, 7217–7221 CrossRef CAS PubMed; (i) J. C. Zgrabik, R. Bhowmick, F. D. Eckstrom, A. R. Harrison, T. V. Fetrow, A. V. Blake, B. Vlaisavljevich and S. R. Daly, Inorg. Chem., 2024, 63, 9451–9463 CrossRef CAS PubMed; (j) J. C. Zgrabik, D. J. Lussier, R. Bhowmick, N. Nguyen, P. A. Zacher III, T. Elkin, A. J. Gaunt, G. S. Goff, H. E. Mason, J. Murillo, B. L. Scott, B. Vlaisavljevich and S. R. Daly, J. Am. Chem. Soc., 2024, 146, 25943–25948 CrossRef CAS PubMed.
- (a) B. Twamley, S. T. Haubrich and P. P. Power, Adv. Organomet. Chem., 1999, 44, 1–65 CrossRef CAS; (b) J. A. C. Clyburne and N. McMullen, Coord. Chem. Rev., 2000, 210, 73–99 CrossRef CAS; (c) E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CrossRef CAS PubMed; (d) D. L. Kays, Organomet. Chem., 2010, 36, 56–76 CAS; (e) C. Ni and P. P. Power, Struct. Bonding, 2010, 136, 59–112 CrossRef CAS PubMed; (f) D. P. Mills and S. T. Liddle, Ligand Design in Modern Lanthanide Chemistry, in Ligand Design in Metal Chemistry: Reactivity and Catalysis, ed. M. Stradiotto and R. J. Lundgren, John Wiley & Sons, 2016, pp. 330–363 Search PubMed; (g) D. L. Kays, Chem. Soc. Rev., 2016, 45, 1004–1018 RSC.
- M. A. Boreen, B. F. Parker, T. D. Lohrey and J. Arnold, J. Am. Chem. Soc., 2016, 138, 15865–15868 CrossRef CAS PubMed.
- B. S. Billow, B. N. Livesay, C. C. Mokhtarzadeh, J. McCracken, M. P. Shores, J. M. Boncella and A. L. Odom, J. Am. Chem. Soc., 2018, 140, 17369–17373 CrossRef CAS PubMed.
- B. L. L. Reant, J. A. Seed, G. F. S. Whitehead and C. A. P. Goodwin, Inorg. Chem., 2025, 64, 3161–3177 CrossRef CAS PubMed.
- (a) I. Korobkov, A. Arunachalampillai and S. Gambarotta, Organometallics, 2004, 23, 6248–6252 CrossRef CAS; (b) M. A. Boreen, B. F. Parker, S. Hohloch, B. A. Skeel and J. Arnold, Dalton Trans., 2018, 47, 96–104 RSC.
- A m-terphenyldihydroborate salt has been isolated previously and structurally characterized. R. J. Wehmschulte, A. A. Diaz and M. A. Khan, Organometallics, 2003, 22, 83–92 CrossRef CAS.
- A. J. Valentine, A. M. Geer, L. J. Taylor, A. M. Teale, K. E. Wood, H. E. L. Williams, W. Lewis, S. P. Argent, J. McMaster and D. L. Kays, Dalton Trans., 2021, 50, 722–728 RSC.
- B. Singaram, T. E. Cole and H. C. Brown, Organometallics, 1984, 3, 774–777 CrossRef CAS.
- J. T. Boronski, L. R. Doyle, J. A. Seed, A. J. Wooles and S. T. Liddle, Angew. Chem., Int. Ed., 2020, 59, 295–299 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- (a) T. V. Fetrow, J. P. Grabow, J. Leddy and S. R. Daly, Inorg. Chem., 2021, 60, 7593–7601 CrossRef CAS PubMed; (b) P. L. Arnold, C. J. Stevens, J. H. Farnaby, M. G. Gardiner, G. S. Nichol and J. B. Love, J. Am. Chem. Soc., 2014, 136, 10218–10221 CrossRef CAS PubMed.
- T. Arliguie, L. Belkhiri, S.-E. Bouaoud, P. Thuéry, C. Villiers, A. Boucekkine and M. Ephritikhine, Inorg. Chem., 2009, 48, 221–230 CrossRef CAS PubMed.
- L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 2248–2256 CrossRef CAS.
- G. V. Khoroshen'kov, A. A. Fag, M. N. Bochkarev, S. Dechert and H. Schumann, Russ. Chem. Bull., 2003, 52, 1715–1719 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and crystallographic details, structure of 1-Nd, spectroscopic data. CCDC 2439982–2439985. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5dt01076d |
| This journal is © The Royal Society of Chemistry 2025 |