
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaboratory development and validation of vapor phase PFAS methods for soil gas, sewer gas, and indoor air†
Heidi
Hayes
 *a,
Chris
Lutes
b,
Nicola
Watson
a,
Diane
Benton
a,
David J.
Hanigan
c,
Seth
McCoy
c,
Chase
Holton
d,
Katherine E.
Bronstein
e,
Brian
Schumacher
f,
John
Zimmerman
f and
Alan
Williams
f
*a,
Chris
Lutes
b,
Nicola
Watson
a,
Diane
Benton
a,
David J.
Hanigan
c,
Seth
McCoy
c,
Chase
Holton
d,
Katherine E.
Bronstein
e,
Brian
Schumacher
f,
John
Zimmerman
f and
Alan
Williams
f
aEurofins Environment Testing Northern California, LLC, Folsom, CA, USA. E-mail: heidi.hayes@et.eurofinsus.com
bJacobs Engineering, Cary, NC, USA
cDepartment of Civil and Environmental Engineering, University of Nevada Reno, Reno, NV, USA
dGSI Environmental, Inc, Lakewood, CO, USA
eRTI International, Research Triangle Park, NC, USA
fU.S. Environmental Protection Agency, Office of Research and Development Center for Environmental Measurement and Modeling, Watershed and Ecosystem Characterization Division, Research Triangle Park, NC, USA
First published on 20th November 2024
Abstract
There is no standard sampling and analysis method for vapor phase per- and polyfluoroalkyl substances (PFAS) that can be routinely applied to soil gas, sewer/conduit gas, and indoor air samples. We have validated a thermal desorption GC/MS/MS method for the measurement of a set of fluorotelomer alcohols and perfluorooctanesulfonamides collected on multi-bed sorbent tubes. Applications to perfluorocarboxylic acids were also evaluated since there is debate regarding under what circumstances these compounds could be observed moving into gas phase. Perfluorooctanoic acid (PFOA) met Method TO-17 calibration requirements when calibrated using National Institute of Standards and Technology (NIST) traceable standard solutions introduced through the thermal desorption system and using multiple reaction monitoring (MRM) transitions based on precursor mass ions identified in the PFOA spectra. However, subsequent detailed studies suggested that PFOA was decomposing during the thermal desorption sample introduction step when comparing two alternative GC/MS sample introduction techniques. The primary peak resulting from the thermal desorption of PFOA standard had spectra consistent with perfluoro-1-heptene (PFHp-1), suggesting that a degradation reaction was occurring. Therefore, the identification of the PFCA compounds in this method is currently subject to a potential positive interference from the corresponding perfluoroalkene and other thermally labile PFAS. Thus, it may be beneficial to limit the application of the thermal desorption GC/MS/MS method to the fluorotelomer alcohols and perfluorooctanesulfonamides and use a parallel solvent extraction approach to quantify the PFCA-related compounds. Method validation including desorption efficiency, second source verification, storage stability and method detection limit tests were successfully completed for the fluorotelomer alcohols and perfluorooctanesulfonamides target analytes.
Environmental significanceThere is currently no standard sampling and analysis method for vapor phase per- and polyfluoroalkyl substances (PFAS) in soil gas, sewer gas, and indoor air. Vapor intrusion (VI) to occupied buildings occurs due to pressure differentials between indoor air and contaminated soil gas. Such a method is critically needed given the prevalence of PFAS as subsurface contaminants (e.g., manufacturing facilities and fire-fighting training locations). A substantial number of PFAS are expected to be sufficiently volatile and sufficiently toxic via inhalation to require VI consideration. We have validated a thermal desorption GC/MS/MS method for measurement of fluorotelomer alcohols and perfluorooctanesulfonamides collected on multi-bed sorbent tubes. We identified critical weaknesses when the method is applied to perfluoroalkyl carboxylic acids. |
Introduction
The prevalence of per- and polyfluoroalkyl substances (PFAS) in indoor air and as subsurface contaminants1 and the associated concern of subsurface vapor intrusion (VI) to indoor air2 is driving the development of a standard analytical method for the routine measurement of vapor phase PFAS in soil gas, sewer/conduit gas, and indoor air.Current practice for evaluating the VI pathway consists of a combination of direct measurements in groundwater, external soil gas, subslab soil gas, and indoor air. No single line of evidence is considered definitive, and direct measurements can be costly and have significant spatial and temporal variability, generally requiring repeated measurements at multiple locations to assess the chronic risks of long-term volatile organic compound (VOC) exposure.
VI occurs due to pressure and concentration differentials between indoor air and the subsurface environment in which subsurface gases become contaminated with volatile or semivolatile anthropogenic compounds. Indoor environments are often negatively pressurized with respect to outdoor air and soil gas, which allows subsurface vapors to migrate into indoor air through advection. In addition, established concentration gradients may cause VOCs to migrate from subsurface areas of higher concentration to indoor areas of lower concentration through diffusion.
At least four polyfluorinated organic substances (i.e., 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2-, 6:2-, 8:2- and 10:2-fluorotelomer alcohols; FTOHs) have sufficient vapor pressure to be designated as vapor-forming chemicals3 in accordance with the U.S. Environmental Protection Agency's (US EPA's) VI Guide.2 The FTOHs are known to be present in indoor air in residential and commercial environments attributed to their use in paints, adhesives, textiles, waxes, coatings.4–7 A recent study in US homes showed that FTOHs were dominant species in residential indoor air.8 The range of values for vapor pressure for many PFCA compounds range across the 1 mm of Hg (133 Pa) volatility criterion. For example, ITRC reports 16 values for PFHxA, all but one are between 0.1 and 4.2 mm Hg (13 and 560 Pa).1
:2-, 6:2-, 8:2- and 10:2-fluorotelomer alcohols; FTOHs) have sufficient vapor pressure to be designated as vapor-forming chemicals3 in accordance with the U.S. Environmental Protection Agency's (US EPA's) VI Guide.2 The FTOHs are known to be present in indoor air in residential and commercial environments attributed to their use in paints, adhesives, textiles, waxes, coatings.4–7 A recent study in US homes showed that FTOHs were dominant species in residential indoor air.8 The range of values for vapor pressure for many PFCA compounds range across the 1 mm of Hg (133 Pa) volatility criterion. For example, ITRC reports 16 values for PFHxA, all but one are between 0.1 and 4.2 mm Hg (13 and 560 Pa).1
At many VI sites, flow moves preferentially through utility conduits and can be referred to as “conduit vapor intrusion.”9 Sewers are a common conduit for VI. PFAS have been widely observed at wastewater treatment plants10 and are likely present in substantial concentrations in sewers from disposal and use of consumer products. Biotransformation of FTOHs to PFCAs has also been observed in municipal wastewater systems.5 Sewer system components include drop structures that may generate mists or droplets and various ventilation features including slope changes and ventilated manholes that influence the flow of sewer gas. Other components such as full inverted siphons and p-traps can limit the migration of VOCs through sewer systems.11,12 Therefore, it may be difficult to distinguish “conduit vapor intrusion” of PFAS released to soil and groundwater from direct releases in sewage. Sewer gas is often near saturated relative humidity and condensation occurs within them due to temperature differentials.13 Therefore, the presence of condensed water droplets in sampled sewer gas is expected.
Some PFAS have been observed to be volatile or readily aerosolized, resulting in global atmospheric distribution and the potential for workplace and household exposure14–16 including through consumer products.17,18 Several studies have measured PFAS in commercial aqueous firefighting foam (AFFF) and found various low molecular weight potentially volatile PFAS compounds,19,20 which a conservative model suggests could pose a VI concern.20
There are currently no US EPA standard methods available specifically for the measurement of PFAS compounds in ambient air. Sampling methods used in research for PFAS in ambient and indoor air are based on collection with polyurethane foam (PUF), XAD-2 resin, or glass or quartz fiber filters.21–23 Initial studies towards the development of a thermal desorption gas chromatography/mass spectrometry (GC/MS) method for PFAS, primarily targeting FTOHs and short-chain PFCAs, were reported by Thaxton et al.,24 and have recently been reflected in methods/results released by instrument manufacturers in the form of application notes.25–27 Robbins et al. reported the development of a thermal desorption (TD)-GC/MS/MS method for consumer product emissions of 4:2, 6:2, 8:2 and 10:2 FTOH into indoor air.7 This development served as the basis of the recently published ASTM standard test method D8591-24 for determining FTOHs in test chamber air using TD-GC/MS/MS.28 Thermal desorption methods are especially desirable for VI assessment of volatiles because they provide high sensitivity with small sample volumes, which are compatible with the typical low sampling volumes of soil gas samples. In contrast, the methods most used for semivolatile compounds require solvent extraction and thus require substantially larger air sampling volumes.
PFCAs are generally considered to be predominantly partitioned to the particulate phase in the atmosphere.1 Since PFCAs are primarily ionic at environmentally relevant pH in aqueous solution, their volatility was initially expected to be low.1 However, multiple mechanisms for the PFCAs to enter the gas phase have been suggested: foam bubble bursting;14 cosolvent effects on pKa;29 and direct sublimation from the solid phase to the gas phase.30 Details of this work are discussed in the ESI.†
Roth et al. investigated the occurrence of PFAS in the headspace above mildly agitated and dilute AFFF mixtures and found that 16 of the 30 measured PFAS were present: five FTOHs (0.5–38.1 μg m−3), 10 PFCAs (0.4–13670 μg m−3), and 1 perfluorotelomer sulfonic acid (72.1 μg m−3).31 The 10 PFCAs detected ranged in carbon chain length from C5 to C16. The dominant compounds were PFOA, PFHxA, and perfluorodecanoic acid (PFDA). In a subsequent comment on the Roth et al. publication,31 it was noted that PFOA appeared to thermally degrade to form perfluoro-1-heptene (PFHp-1) when PFOA was injected onto thermal desorption tubes and desorbed to a GC/MS, making it difficult to differentiate between PFHp-1 and other thermally labile PFAS such as PFOA.32 Upon closer inspection of the experimental and published spectra of PFOA and PFHp-1, Roth et al. noted several inconsistencies between the expected and observed spectra, including the lack/presence of a m/z 44 (loss of CO2) and lack/presence of a m/z 45 (loss of COOH) present in the NIST published standard mass spectrum but not in other published GC/MS spectra. Thus, it is possible that the “PFOA-like peak” observed by GC/MS was aerosolized or volatilized PFOA (which was known to be present in the AFFF tested), but it is also possible that the single GC peak represents PFHp-1 and/or the sum of various thermally labile PFAS.31,32 This uncertainty about the volatility of the PFCAs motivates the current paper.
In more recent research, the University of Nevada measured the Henry's law constants (HLCs) of 15 PFAS, but PFOA was not volatile enough to be measured until the pH was reduced substantially, suggesting PFOA is not volatile when dissolved in water at circumneutral pH; however cosolvent and air/water interface effects were not evaluated.33–35 See the full discussion in the ESI.† Together, these studies suggest that in the experiments conducted by Roth et al. in which PFOA was measured in the gas phase, PFOA was aerosolized during the mild agitation of the AFFF or ejected as aerosols during popping of the AFFF bubbles immediately before the gas phase was collected, rather than present as a vapor.31
Substantial concentrations of FTOHs have been observed in soil and soil gas at a chemical manufacturing facility with known soil and groundwater PFAS impacts.36 However, the observation of several PFCA compounds in the soil gas samples was unexpected. Three PFCA compounds, PFHxA, PFBA, and PFOA, were observed in the subslab sample directly beneath the presumed source building at concentrations ranging from 59 to 650 μg m−3. Those three PFCAs as well as two additional compounds, PFPeA and PFHpA, were also observed at a downgradient location at soil gas concentrations ranging from 0.7 to 180 μg m−3. This is consistent with known degradation pathways under certain conditions.37 The soil pH at this site is between 5 and 7. Given that the water table and capillary fringe at the field site are shallow, the formation and bursting of bubbles by the PFAS surfactants during soil gas sampling even at <200 mL min−1 cannot be excluded. Given the complex history of industrial production at the field site reported by Schumacher et al.,36 the presence of cosolvents that could favor the presence of neutral forms of PFOA29 is probable.
The objective of the work presented was to develop a method for sampling vapor phase PFAS compounds that is suitable and accepted for widespread use in US laboratories for PFAS compounds in soil gas, sewer/conduit gas, and indoor air. We primarily conducted experiments in a California commercial laboratory aimed at developing and testing the robustness of a thermal desorption GC/MS/MS method for volatile and semivolatile PFAS. Given the questions surrounding the presence of PFCAs in the gas phase, particular attention was paid to the stability of PFCAs when measured by TD-GC/MS/MS. Limited experiments to evaluate transformations of PFCA compounds under thermal desorption conditions were independently conducted with different instrumentation and methods at the University of Nevada, Reno.
Methods
PFAS analytical approach and target compounds
Seventeen PFAS compounds listed in Table S1 in the ESI (ESI)† were targeted by a California commercial laboratory to develop and validate a method applicable to soil gas, sewer gas, and indoor air samples utilizing TD-GC/MS/MS. This validation effort was built on previous work conducted by the laboratory covering a subset of Table S1† compounds in support of soil gas measurements for previous field studies discussed by US EPA38 and Schumacher et al.36 The effort also incorporated subsequent method refinements by the laboratory to accommodate larger sample volumes required for indoor air measurements of fluorotelomer alcohols (10–300 L).The PFAS method utilized multi-bed TD sorbent tubes for vapor collection with analysis based on procedures described in EPA Method TO-17.39 The sorbent tube used for the commercial laboratory method evaluation was the CAMSCO PFAS/PFCA tube, which is a multi-bed tube packed with hydrophobic sorbents. These tubes were selected based on their performance in a previous field study and in the laboratory's subsequent indoor air recovery studies conducted for fluorotelomer alcohols. Alternative multi-bed TD tubes have also been used to collect vapor phase PFAS for a range of applications.25,31 The method, as currently proposed, directly samples onto sorbent media without a preliminary stage, which might be designed to remove particulates but would also potentially cause losses of volatiles.40,41 The exclusion of PFAS analytes not mobile in the gas phase can be achieved by limitation of flow rates to those not expected to cause artifactual particulate suspension from soil gas (<200 mL min−1)42 and small total air sample volumes (10 mL to 1 L of soil or sewer gas).
The thermal desorption system was configured with a GC/MS/MS, which is a modification to the single quadrupole (SQ) MS described in TO-17.39 The tandem MS and multiple reaction monitoring (MRM) mode was selected for the PFAS method over the conventional SQ MS in selected ion monitoring (SIM) mode based on its improved sensitivity and selectivity when measuring target compounds in complex environmental matrices (Fig. S1†). MRM relies on a series of ionization and mass filtering steps optimized for each target compound to minimize or eliminate background noise and matrix interference.43,44 Specifically, the target compound is ionized in the first quadrupole and compound-specific precursor ion(s) are selectively passed through a collision cell where collisions with an inert gas fragment the precursor ion(s) to generate product ion(s). Selected product ion(s) are then detected using the second quadrupole. The GC separation column selected was a DB-624 column, a mid-polarity column generally used for VOCs which demonstrated acceptable peak shape and resolution for the targeted PFAS compounds.24 Alternative mid-polarity columns suitable for VOC analysis have also been applied to similar PFAS applications on TD-GC/MS systems.25,31
The initial analytical method development conducted prior to US EPA38 and Schumacher et al.36 was based on successful completion of steps 1 through 5 outlined in Table 1. Detailed method parameters and analytical performance for this initial method and subsequent soil gas analysis can be found in the ESI (Tables S2 and S3).† The reporting limits and quality control criteria in Table S4† were incorporated into the quality assurance plan applied to the soil gas samples collected in the associated field study.
| (1) Determine target retention time, spectra |
| (2) Establish parent and product ions (using MRM) |
| (3) Generate initial calibration curve (ICC) to establish linearity (method criterion < 30%RSD) |
| (4) Assess desorption efficiency (method criterion >95%) |
| (5) Verify accuracy (second source verification) (70–130%) |
| (6) Complete method detection limit study (∼0.05 ng) |
| (7) Determine initial demonstration of capability – accuracy and precision based on replicate spikes |
| (8) Confirm storage stability on tubes |
Additional development, testing, and validation were undertaken to expand and optimize the targeted PFAS list, evaluate potential interferences for PFCAs, and extend the applicability of the method to include indoor air, sewer gas, and soil gas. This included adding compounds not included in US EPA38 and Schumacher et al.36 (9:2s FTOH, 11:2s FTOH, 12:2 FTOH, and 13:2s FTOH) as well 2-perfluorooctyl (1,2-13C2) ethanol (8:2 FTOH-C13) to serve as a field surrogate that underwent initial analytical method evaluation as outlined in Table 1. Evaluation of the stability of PFCAs through the analytical system was undertaken to determine subsequent impacts to compound identification. Tests included direct injection of a PFOA solution into a GC/MS over a range of injection port temperatures with follow-up tests conducted by investigating PFOA analyzed on TD-GC/MS across a span of desorption and cold trap temperatures.
Analytical method set up and validation
The analytical method was set up using two commercially available TD introduction systems, each with a different secondary trap design used to focus the thermally desorbed compounds:• Gerstel TD 3.5+ system with a cryogenically cooled trap with quartz wool and forward flush of compounds to the GC column, which is a non-selective approach useful for method development.
• Markes TD100-xr system configured with a back flush multi-bed sorbent trap in which the sample is focused in one direction and desorbed and injected onto the GC in the opposite direction to allow for sample split and re-collection for re-analysis.
Initial calibration, desorption efficiency, second source verification, initial demonstration of capability, and method detection limit studies were conducted for the FTOHs, N-MeFOSA, and N-EtFOSA using both systems. In both cases, the TD system was coupled to an Agilent 8890 GC and 7000D MS/MS using an Agilent DB-624 UI 60 m × 0.25 mm ID × 1.4 μm column for separation (Table S5†).
Prior to initial calibration, individual stock methanolic standards for the target analytes were prepared and spiked onto separate sorbent tubes and analyzed on the TD-GC/MS, collecting full scan data to determine retention time and spectra. Standards were then evaluated in MRM mode to determine primary and confirmation transitions with the goal of incorporating unique transitions for definitive compound identification. Preparation of the calibration standards and calibration procedures are detailed in the ESI.† Commercial PFCA stock methanolic mixes were received with added base (NaOH) to minimize conversion to their corresponding methyl esters. Due to the unavailability of calibration standards and the absence of EI spectra in the NIST library for 9:2s, 11:2s, and 13:2s FTOH, the laboratory developed an approach to tentatively identify these compounds based on expected MRM transitions and expected retention time windows. Details are provided in the ESI.†
Desorption efficiency was evaluated to verify that the thermal desorption step released more than 95% of the analyte mass from the sorbent tube. Desorption efficiency (DE) was measured by spiking a sorbent tube with the highest concentration analyzed for the initial calibration and measuring the mass from the analytical run. The tube was then spiked with internal standard and reanalyzed to measure residual mass on the tube. The %DE was calculated using the following equation:
| %DE = [mi (mi + mr)] × 100% |
Procedures for Method Detection Limit (MDL) studies conducted following CFR Appendix B Part 136 Revision 2 are discussed in the ESI.† An initial demonstration of capability (IDOC) was conducted to comply with a general requirement for environmental laboratory accreditation as outlined in The NELAC Institute (TNI) Standard, 2016 and referenced in the US Department of Defense Quality Systems Manual (Version 5.4).45 The IDOC was prepared by spiking four sorbent tubes using a second source methanolic working standard containing all target PFAS at concentration of 0.4 ng, a concentration one to four times the limit of quantitation as specified in the TNI Standard. The spikes were analyzed in the same analytical batch and the average recovery and RSD were calculated for the four replicate spikes to evaluate both accuracy and precision against the method or laboratory criteria.
The TD tube storage stability evaluation was conducted by spiking a set of 12 conditioned sorbent tubes with a methanolic working standard containing the target PFAS compounds and the field surrogate generating a final mass loading on each tube of 5 ng for each target compound and 1 ng for the field surrogate. Three of the spiked tubes were analyzed the day of preparation (Day 0) with another three tubes analyzed at Day 7, Day 14, and Day 28. Prior to analysis, storage of spiked tubes was at 4 ± 2 °C.
PFOA stability evaluation
The stability evaluation utilized two sample introduction techniques: direct injection of PFOA solution into the GC injection port and loading PFOA vapor or solution onto a sorbent tube and thermally desorbing the spiked tube onto the GC. The direct injection tests were used to evaluate impacts of the GC injection port temperature on its full scan electron ionization (EI) mass spectra and the presence of characteristic mass ion(s) that could be used to distinguish PFOA from potential co-eluting PFAS. The peak and spectra generated from the direct injection tests were compared to the thermal desorption tests to assess potential changes in PFOA behavior. After both the direct injection and thermal desorption experiments, the peak response and full scan spectra were evaluated at each temperature against the NIST spectrum with attention to the presence/absence of m/z 45 indicating a loss of –COOH.Three experiments were conducted in this PFOA stability evaluation. Experiment 1 evaluated PFOA performance through the GC/MS system over a range of GC inlet temperatures. Experiment 2 assessed PFOA peak response and EI spectra generated from vapor phase PFOA collected on sorbent tubes and analyzed by TD-GC/MS across a range of desorption temperatures. Experiment 3 further evaluated the behavior of PFOA during the thermal desorption step, examining perfluoro-1-heptene as a degradation product and potential interference.
The GC program used for the PFOA direct injection evaluation started at 50 °C for 1.0 min, ramping to 240 °C at a rate of 15 °C min−1. A 1.0 μL injection of each PFOA stock solution was conducted using an inlet split of 1:15 at inlet temperatures of 60, 100, 150, 200, and 280 °C. The peak area response and EI spectra for each injection temperature were evaluated, assessing changes as a function of injection temperature to identify potential evidence of thermal degradation of PFOA.
The PFOA vapor collected on the sorbent tube was thermally desorbed on the TD-GC/MS/MS system with the Markes TD100-xr configuration and parameters established during the PFAS method set up and validation. These parameters included a tube desorption temperature of 320 °C for 10 min and a secondary trap desorption maximum temperature of 290 °C. A follow-up test was conducted at lower desorption temperatures in the range of 100 to 250 °C to evaluate the impact of temperature on PFOA peak response and EI spectra. This subsequent test was similar to the initial test using a vial with PFOA crystals placed in the chamber at 60 °C. The tube desorption and secondary trap desorption temperatures were varied in tandem at temperature conditions of 100, 150, 200, and 250 °C.
Second laboratory confirmation of select observations using alternative equipment
The University of Nevada team conducted separate but complementary experiments using a Shimadzu TQ8040 GC/MS/MS equipped with a CTC Analytics Prep and Load Solution (PAL3 marketed as part number AOC6000 by Shimadzu) and an in-tube extraction dynamic headspace tool (ITEX-DHS). ITEX uses a syringe to draw sample headspace gases through a packed bed (Tenax TA for these experiments, Fig. S3†). The syringe was used to draw 100 μL of headspace gas over the sorbent from a sample vial containing 100 mg, 99% pure, solid phase PFOA agitated (orbital motion) and heated to 50 °C for 5 min. The headspace gas was backflushed into the vial and another 100 μL of headspace aspirated. The sorbent was then heated to desorb analytes back into the headspace gas and the 100 μL mixture was injected into the GC inlet. The loading and desorption temperatures were 50 and 300 °C. The analyses were performed on a Shimadzu Rxi-5Sil-MS 30 m × 0.25 mm ID × 0.25 μm film column, which was equivalent to the HP-5MS column used in experiment 1 (GC/MS direct injection PFOA evaluation). A splitless injection was used and inlet temperature varied from 250 to 325 °C. Column flow was constant 1.00 mL min−1, with an initial oven temperature of 35 °C held for 2 min, followed by a 30 °C min−1 ramp to a 320 °C GC oven temperature. The transfer line and ion source were 250 and 200 °C, respectively.Results and discussion
Instrument validation
| Analyte | Gerstel 3.5+ | Markes TD100-xr | ||
|---|---|---|---|---|
| Linearity (%RSD) | Method detection limit (ng) | Linearity (%RSD) | Method detection limit (ng) | |
| a NA = not available, variable recovery observed. | ||||
| 4:2 FTOH |
8.2 | 0.029 | 5.3 | 0.011 |
| 5:2s FTOH |
13 | 0.045 | 5.9 | 0.028 |
| 6:2 FTOH |
6.4 | 0.047 | 9.2 | 0.12 |
| 7:2s FTOH |
6.7 | 0.045 | 6.1 | 0.022 |
| 8:2 FTOH |
13 | 0.032 | 8.9 | 0.051 |
| 10:2 FTOH |
13 | 0.062 | 5.4 | 0.027 |
| 12:2 FTOH |
13 | 0.041 | 3.8 | 0.030 |
| n-MeFOSA | 10 | NA | 12 | 0.043 |
| n-EtFOSA | 12 | NA | 12 | 0.035 |
The IDOC for each TD system is summarized in Table S9,† showing the average recovery and %RSD of the four spikes prepared from a second source standard at 0.4 ng. Both systems had an average recovery between 70% and 130% and %RSD < 30%.
It was noted that n-MeFOSA and n-EtFOSA demonstrated sporadic anomalous recoveries during the MDL study on the Gerstel system and during the initial set up on the Markes system. Upon further investigation, the inconsistent recoveries were attributed to a small subset of sorbent tubes that were subsequently removed from the inventory and replaced with other tubes with the same model number that performed well. The root cause of the low recovery exhibited by this set of tubes was not identified.
PFCAs were then added to the target compound list on the Markes TD100-xr system and compared to the performance of the Gerstel system used for the soil gas screening as part of the field studies described by US EPA38 (ESI Tables S2 and S3†). The PFCAs demonstrated good linearity across the range of 0.1 to 10 ng, meeting the TO-17 method criterion of <30%RSD and met the initial demonstration of capability accuracy and precision objectives (Table 3).
| Analyte | Linearity (%RSD) | Method detection limit (ng) | Initial demonstration of capability | |
|---|---|---|---|---|
| Average recovery | %RSD | |||
| PFBA | 16 | 0.11 | 87% | 7.2% |
| PFPeA | 2.2 | 0.027 | 101% | 3.4% |
| PFHxA | 2.3 | 0.019 | 106% | 3.5% |
| PFHpA | 2.9 | 0.021 | 101% | 5.4% |
| PFOA | 11 | 0.049 | 95% | 4.4% |
The comparable performance across both platforms using divergent trapping techniques demonstrates that thermal desorption is a reliable technique to achieve the analytical criteria established in Table S4.† Based on comparable performance of the Gerstel and Markes instruments, subsequent method development and validation was conducted on the Markes system to take advantage of the system's sample re-collection feature to allow for sample archival and follow-up analysis.
PFOA stability evaluation
Additionally, the spectra when PFOA was injected at inlet temperatures of 60 °C and 280 °C were consistent for both the dichloromethane and methanol PFOA solutions. The spectra generated at the peak apex under each of these conditions yielded PFOA as the top NIST20 library match factor score ranging between 919 and 922, indicating high identification confidence (Fig. S6a–d†).
Review of the peak spectra revealed that neither the PFOA molecular ion (m/z 414) nor the molecular ion minus the loss of H (m/z 413) was present at any of the injection port temperatures for either the dichloromethane or methanol PFOA high concentration solutions which is consistent with NIST reference spectra. However, fragments advantageous in the identification of the PFOA molecular structure (m/z 45 (COOH+) and m/z 395 (C8HF14O2+)) were measurable across the range of inlet temperatures yielding relatively consistent m/z responses (Table 4). Because the PFOA peak demonstrated tailing on the HP-5MS column, peak height is tabulated rather than peak area. These data suggest that PFOA can be measured by GC/MS techniques without thermal desorption.
| PFOA solution (μg mL−1) | Solvent | PFOA on-column (ng) | Inlet temp (°C) | Peak height | ||
|---|---|---|---|---|---|---|
| TIC | m/z = 45 | m/z = 395 | ||||
| 5700 | DCM | 380 | 60 | 36227996 |
3546402 |
175424 |
| 100 | 38473818 |
3740600 |
204096 |
|||
| 150 | 36300426 |
3631561 |
201088 |
|||
| 200 | 35982879 |
3515088 |
182044 |
|||
| 280 | 30255547 |
2898693 |
151232 |
|||
| 4300 | MeOH | 290 | 60 | 22225211 |
2411244 |
80112 |
| 100 | 24696738 |
2497784 |
66544 |
|||
| 150 | 23623208 |
2503154 |
91096 |
|||
| 280 | 24774637 |
2514684 |
84600 |
|||
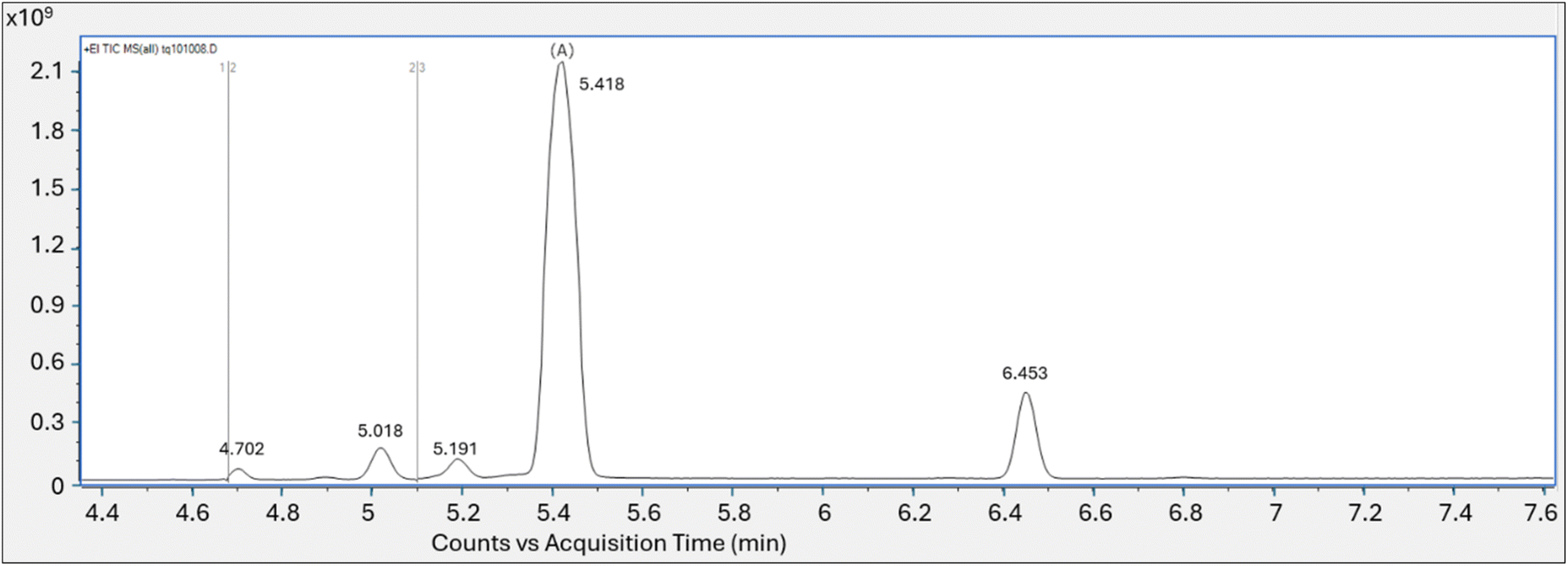 | ||
| Fig. 1 Experiment 2: total ion chromatogram of PFOA vapor collected on PFAS tube analyzed using routine TD-GC/MS parameters yields multiple peaks; Y axis is area counts, X axis is acquisition time (min). | ||
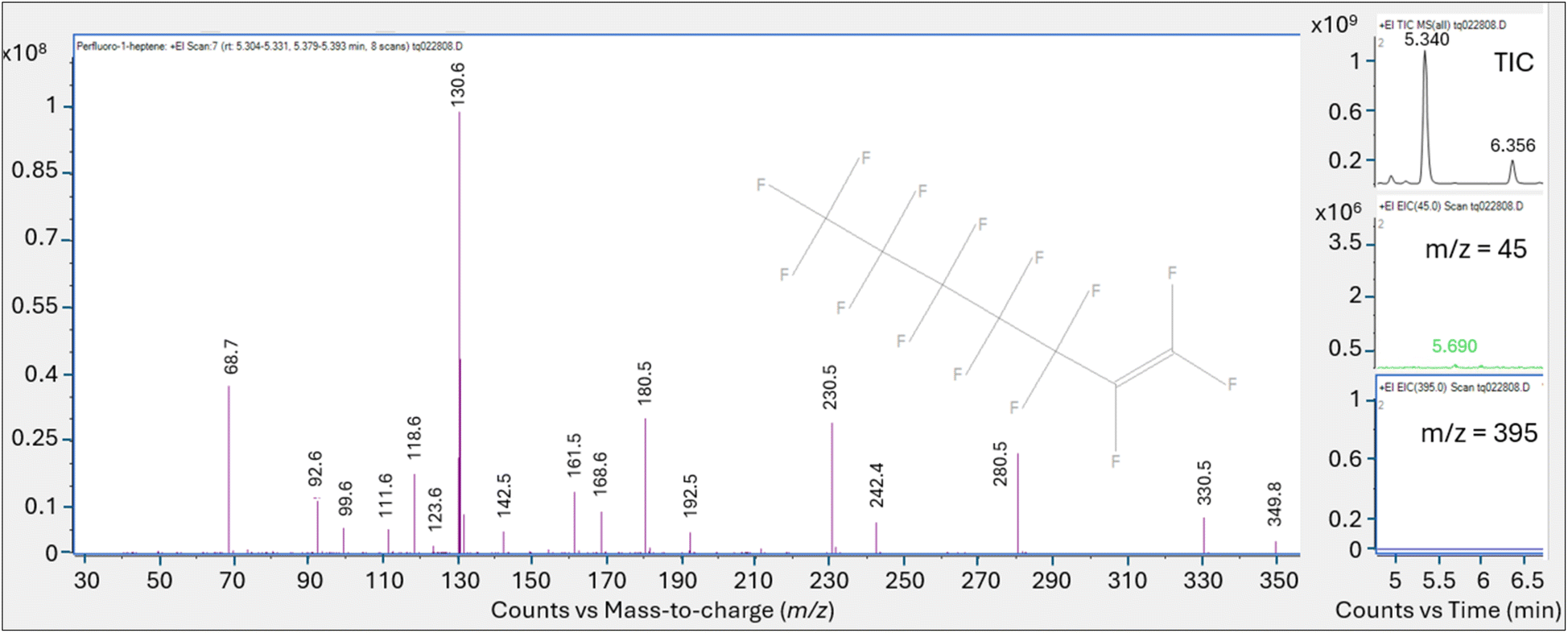
Despite the saturated peak (which would maximize the chance of observing weak fragments) there was no evidence of the oxygen containing m/z 395 or m/z 45 fragment ions (Fig. S7a†), which had been observed with the high concentration PFOA solutions directly injected into the GC/MS. Scans on each shoulder of the saturated peak were also evaluated for spectral match (5.368 min and 5.477 min, Fig. S7b†) yielding a top match with perfluoro-1-heptene at a match factor score of 839. The smaller peaks present in the analytical run were tentatively identified as perfluorinated compounds using the NIST20 library, although the match scores were quite low.
Follow-up tests lowering both the tube and secondary trap desorption temperatures in tandem showed no peak response when each was set at 100 °C or at 150 °C. Peak (A), eluting at retention time of the PFOA methanolic standard introduced by thermal desorption, was observed when the tube/trap desorption temperatures were at 200 °C and at 250 °C with a significant increase in peak response at the 250 °C tube/trap desorption temperature (Fig. 2). Similar to the initial test conducted at the tube and secondary trap desorption temperatures of 320 °C and 290 °C respectively, five non-targeted perfluorinated peaks eluting at similar retention times were identified in the 200 and 250 °C analyses.
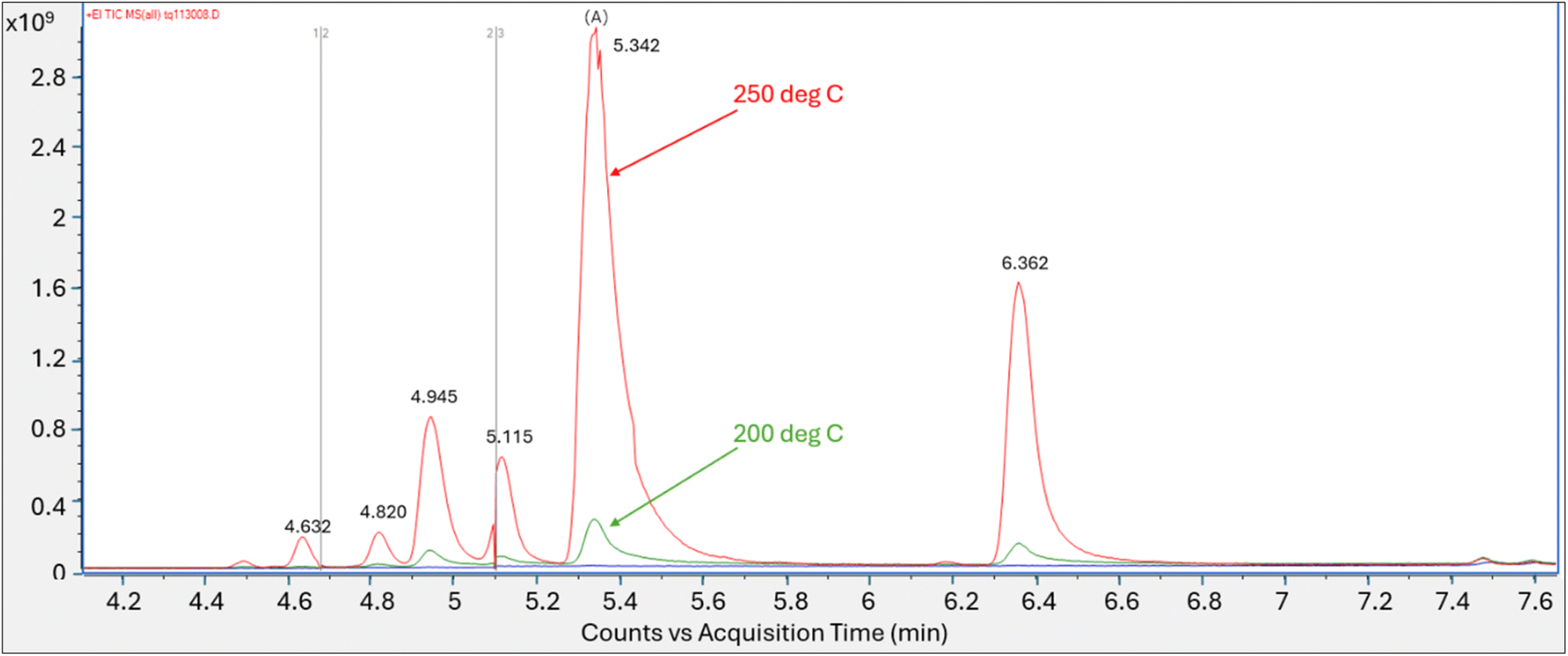 | ||
| Fig. 2 Total ion chromatogram of PFOA vapor collected on tube analyzed at 200 °C and 250 °C tube and trap desorption temperatures; Y axis is area counts, X axis is acquisition time (min). | ||
The spectra for peak (A) at the retention time of 5.34 min analyzed at the 200 °C tube and trap desorption temperatures showed a best library match with perfluoro-1-heptene at a match score of 861. The oxygen containing m/z 45 and 395 fragments diagnostic of PFOA were not present.
The analytical run conducted at the tube and trap desorption temperatures of 250 °C resulted in an increase in peak area and a saturated peak (A) eluting at the initially identified PFOA retention time. The saturated peak spectral information was reviewed for the presence of lower intensity mass ion fragments m/z 45 and 395, which were not present in the spectra (Fig. S8†). The scans evaluated on the left and right of the apex on the peak shoulder generated a match score of 832 for perfluoro-1-heptene.
These results indicate that the thermal desorption conditions required to extract PFOA from the CAMSCO multi-bed tube and subsequently release it from the secondary trap generated a peak that was identified as perfluoro-1-heptene based on the NIST library match. Despite the high PFOA mass loaded on the sorbent tubes, m/z 45 and 395 were not present in any of the thermally desorbed samples. These results suggest that PFOA appears to break down primarily to a peak tentatively identified as perfluoro-1-heptene (PFHp-1) in the thermal desorption introduction step making the identification and quantification of PFOA subject to error due to potential positive interference.
In the separate experiment conducted by the University of Nevada team using the ITEX introduction system, the PFOA headspace vapor collected using Tenax TA sorbent with subsequent thermal desorption and injection of the headspace into the GC/MS resulted in a peak eluting at 5.9 minutes with a spectra which exhibited m/z 45, consistent with the direct injection of PFOA solutions analyzed using an equivalent HP-5MS column. The team did note an increase in the common PFAS ions m/z 69, 119, and 131 at the holdup/dead time compared to blanks. The relative abundance increased with increasing inlet temperature from 250 to 325 °C, suggesting generation of unretained perfluorinated compounds as PFOA degrades in the heated inlet. Their abundance, however, was low compared to the peak identified as PFOA, and it did not appear that appreciable degradation of PFOA was occurring on the Tenax TA sorbent during ITEX thermal desorption.
Although thermal degradation of PFOA was not evident in the direct injection of the high concentration PFOA solutions into the GC inlet over the range of 60 to 280 °C, we suspect the short residence time in the heated inlet in conjunction with the high PFOA concentrations injected may have mitigated potential thermal degradation, thereby preserving the key mass ions m/z 45 and 395. Additionally, PFOA did not appear to be degraded when sorbed to Tenax TA only (ITEX experiment), in contrast to the multi-bed CAMSCO PFAS/PFCA tubes and the Markes Universal TD tubes.32 While the proprietary sorbents used to pack these multi-bed tubes are not disclosed, it is possible that if one or more carbon-based sorbents are used, PFCA degradation may be enhanced by catalyzed thermal decomposition. This hypothesis aligns with other research demonstrating that PFAS sorbed to activated carbon are degraded at lower temperature than PFAS, which are not sorbed to activated carbon.47,48 Similarly, recent modeling studies have highlighted the role of active sites on alumina surfaces in catalyzing the degradation of PFOA at 300 °C49 and shown that PFHp-1 is the primary product in the presence of alumina at 400 °C.50
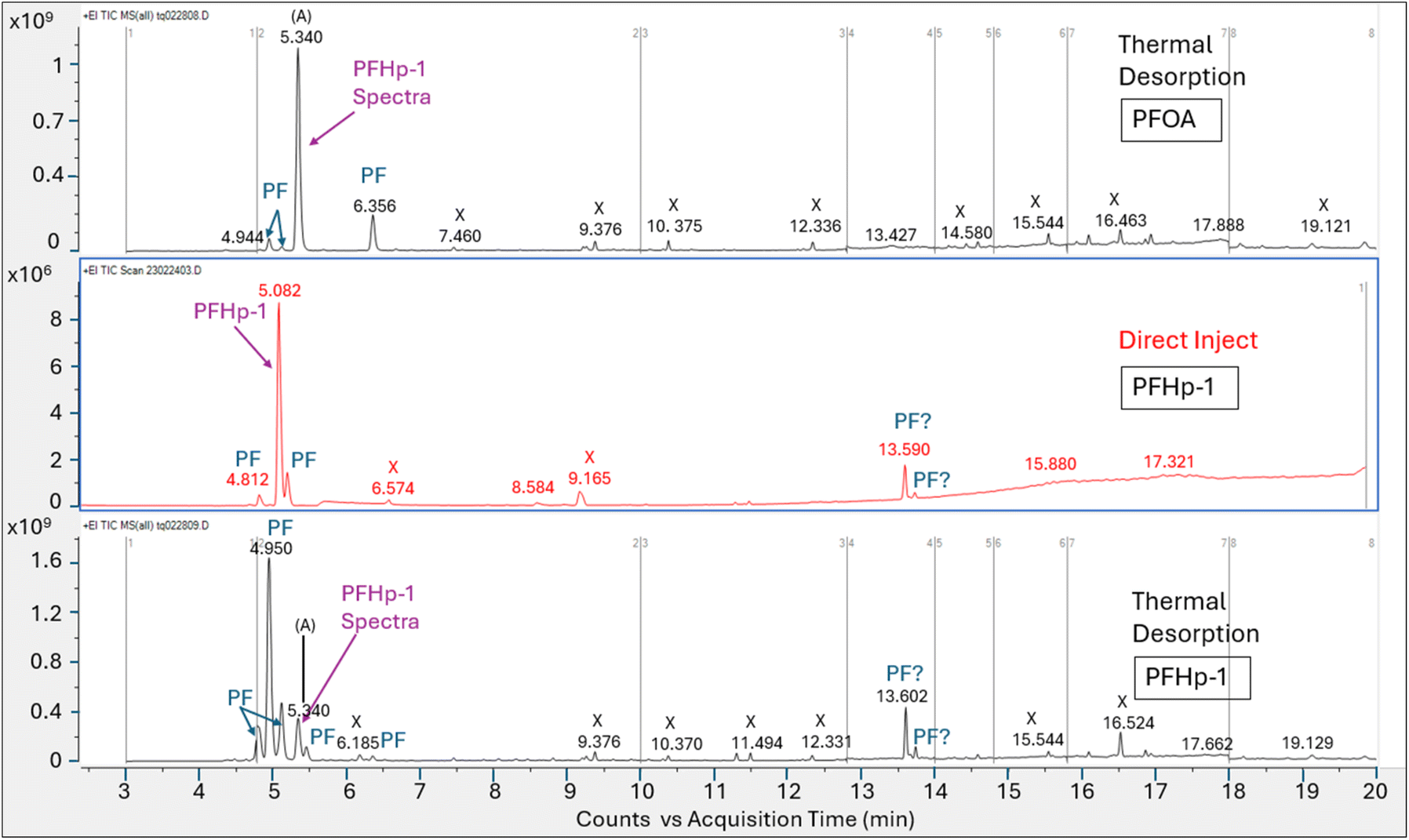
PFOA injection comparison tests. The total ion chromatograms and associated peak spectra generated by introduction of a 1.0 μL aliquot of a 1100 μg mL−1 PFOA methanolic standard using direct injection and thermal desorption onto comparable GC/MS systems configured with a HP-5MS column are compared in Fig. 3 and 4. Comparison data for the DB-624 column configuration are shown in Fig. 5 and 6.
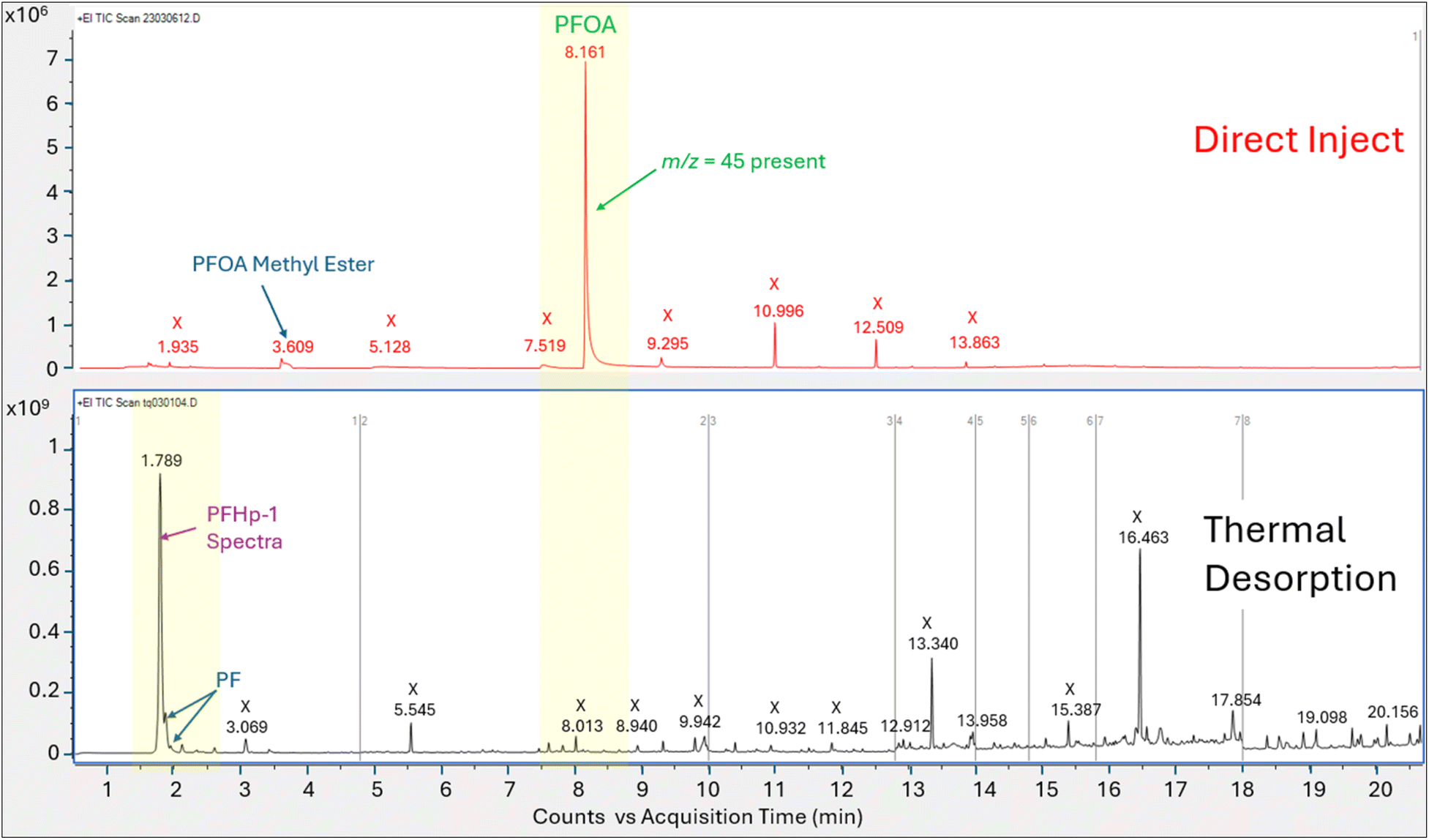 | ||
| Fig. 3 Total ion chromatograms for PFOA analysis on HP-5MS column with direct injection (top panel) vs. thermal desorption (bottom panel); area counts on Y axis, and retention time in minutes on the x axis, labels on figure are X = non-perfluorinated compound; PF = perfluorinated compound, identified structures labeled. | ||
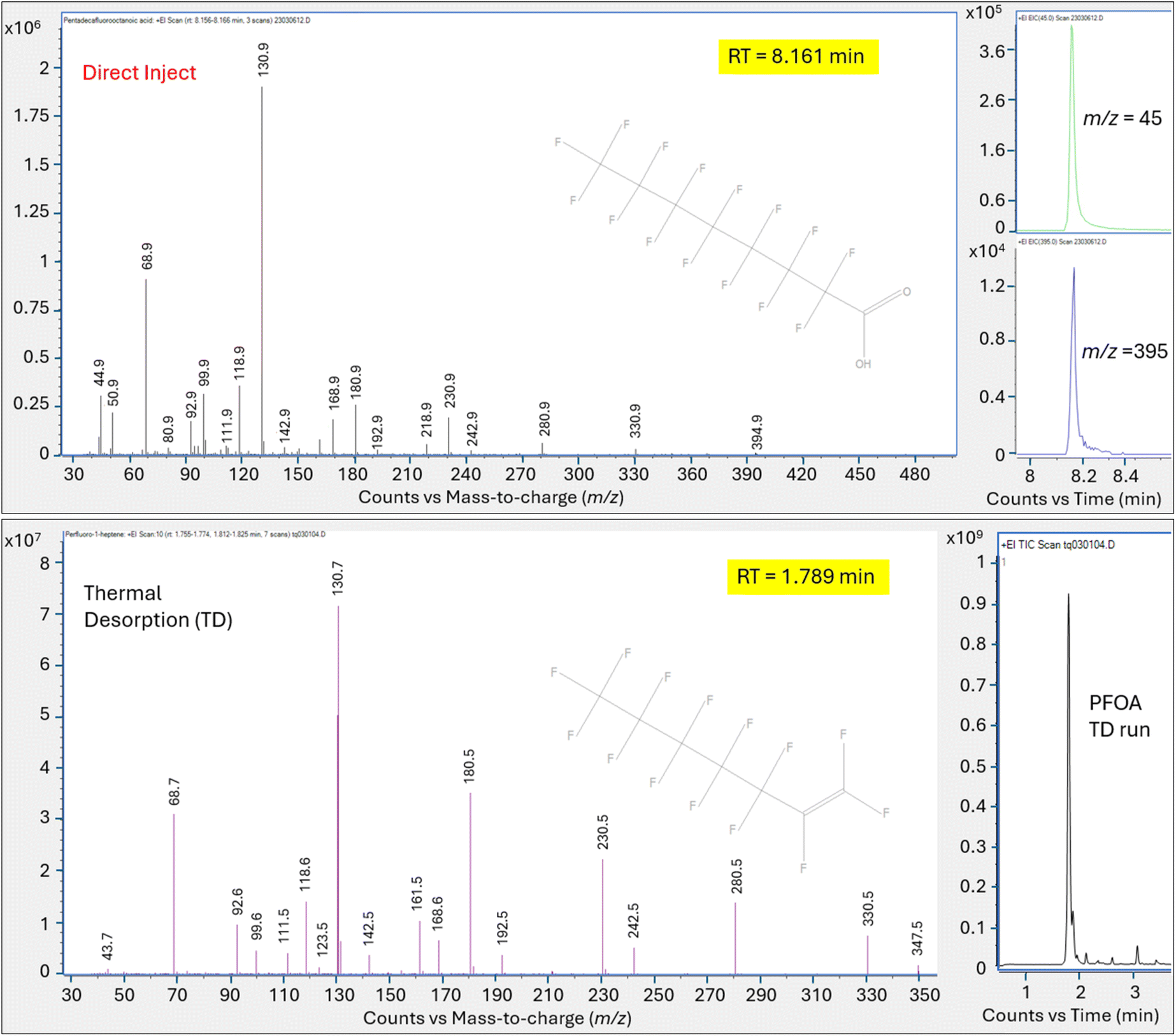 | ||
| Fig. 4 Mass spectra for primary peak measured from PFOA injection using direct injection and TD on HP-5MS column; X axis is mass to charge ratio, y axis is counts; Upper panel is direct injection method, bottom panel is thermal desorption, right inset shows chromatogram. | ||
 | ||
| Fig. 5 Total ion chromatograms for PFOA analysis on DB-624 column (Y-axis scaled to match Fig. 3) with direct injection (top panel) vs. thermal desorption (bottom panel); X axis is acquisition time in minutes, Y axis is area counts, labels on figure: X = non-perfluorinated compound; PF = perfluorinated compound not otherwise identified, identified compounds labeled. | ||
 | ||
| Fig. 6 Left and right mass spectral scans for peak A Measured from PFOA injection Using TD on DB-624 column. In main panel Y axis is area counts, X axis is mass to charge ratio; right inset is total ion chromatogram and selected ion chromatogram of peak of interest, with x axis retention time and y axis area counts. | ||
Direct injection of the PFOA methanolic mix on the HP-5MS column generated a primary peak eluting at 8.161 min with spectra that displayed m/z 45 and 395 and a NIST spectral match score factor to PFOA of 846, consistent with the direct injection results observed in experiment 1 (Fig. 4). However, when a 1.0 μL aliquot of the same PFOA methanolic standard was injected onto the sorbent tube and introduced onto the HP-5MS column using thermal desorption, the resulting primary peak eluted much earlier at 1.789 min. Additionally, the peak at 1.789 min did not show evidence of either of the oxygen containing ions m/z 45 or 395 despite saturation. Scans on the left and right shoulder of the 1.789 minutes peak yielded a best match with PFHp-1. Several unidentified perfluorinated compounds were evident in the thermal desorption run eluting near the primary peak.
The direct injection of the methanolic PFOA standard on the DB-624 column did not yield a prominent peak (Fig. 5). Several perfluorinated peaks were identified including a poor responding, tailing peak at 5.133 min which exhibited spectra most closely matching PFHp-1. The primary peak (A) in Fig. 5 identified on the thermal desorption system was consistent with the retention time during instrument calibration using the methanolic PFOA standards. As was observed on the thermal desorption HP-5MS run, peak (A) was saturated and characteristic oxygen containing PFOA m/z 45 and 395 were not present. The spectra from the left and right scans for peak (A) generated from the thermal desorption PFOA run is shown in Fig. 6. Several smaller unknown perfluorinated peaks were also identified in the thermal desorption run.
The PFOA direct injection and thermal desorption comparison tests conducted using the HP-5MS non-polar, general-purpose column and repeated using the DB-624 mid-polar volatiles column generated data supporting the occurrence of PFOA transformation during the thermal desorption step. When utilizing the HP-5MS column, the thermal desorption run yielded a chemical that eluted significantly earlier than the peak measured by direct inject and exhibited a spectral pattern distinct from the direct inject peak and missing the oxygen containing PFOA m/z 45 and 395. In contrast to the single prominent peak observed when injecting PFOA solution onto the HP-5MS column, no discernible peak was observed when directly injecting the same PFOA solution onto the volatiles DB-624 column. However, when the PFOA solution was introduced using thermal desorption onto the same DB-624 column a prominent Gaussian peak (A) was observed showing similar spectra as observed on HP-5MS thermal desorption injection. Consistent with the results of experiment 2, the peak generated from the thermal desorption sample introduction using either the HP-5MS and DB-624 was tentatively identified as PFHp-1 based on the NIST library search.
The absence of an observable PFOA peak on the direct injection run conducted using the DB-624 column is likely attributed to its acidic properties, since performance of GC analyses of underivatized acid analytes on DB-624 type phases are complex and include solvent interactions and irreversible sorption. Most methods using DB-624 for acid analytes require derivatization before analysis.51–53 The behavior exhibited by the acidic compound PFOA when analyzed on the DB-624 column using direct injection further supports the hypothesis that PFOA is breaking down during the thermal desorption step to a compound that exhibits significantly improved chromatographic performance.
Perfluoro-1-heptene injection comparison tests. To verify the tentative identification of the primary peak (A) in the thermal desorption injections as PFHp-1, a 1.0 μL aliquot of 3560 μg mL−1 authentic PFHp-1 standard in methanol was analyzed using direct liquid injection and thermal desorption introduction on each column configuration. The total ion chromatograms for the direct inject and thermal desorption runs of PFHp-1 on HP-5MS and DB-624 are shown in Fig. 7 and 8, respectively along with the thermal desorption total ion chromatogram generated from the PFOA injection.
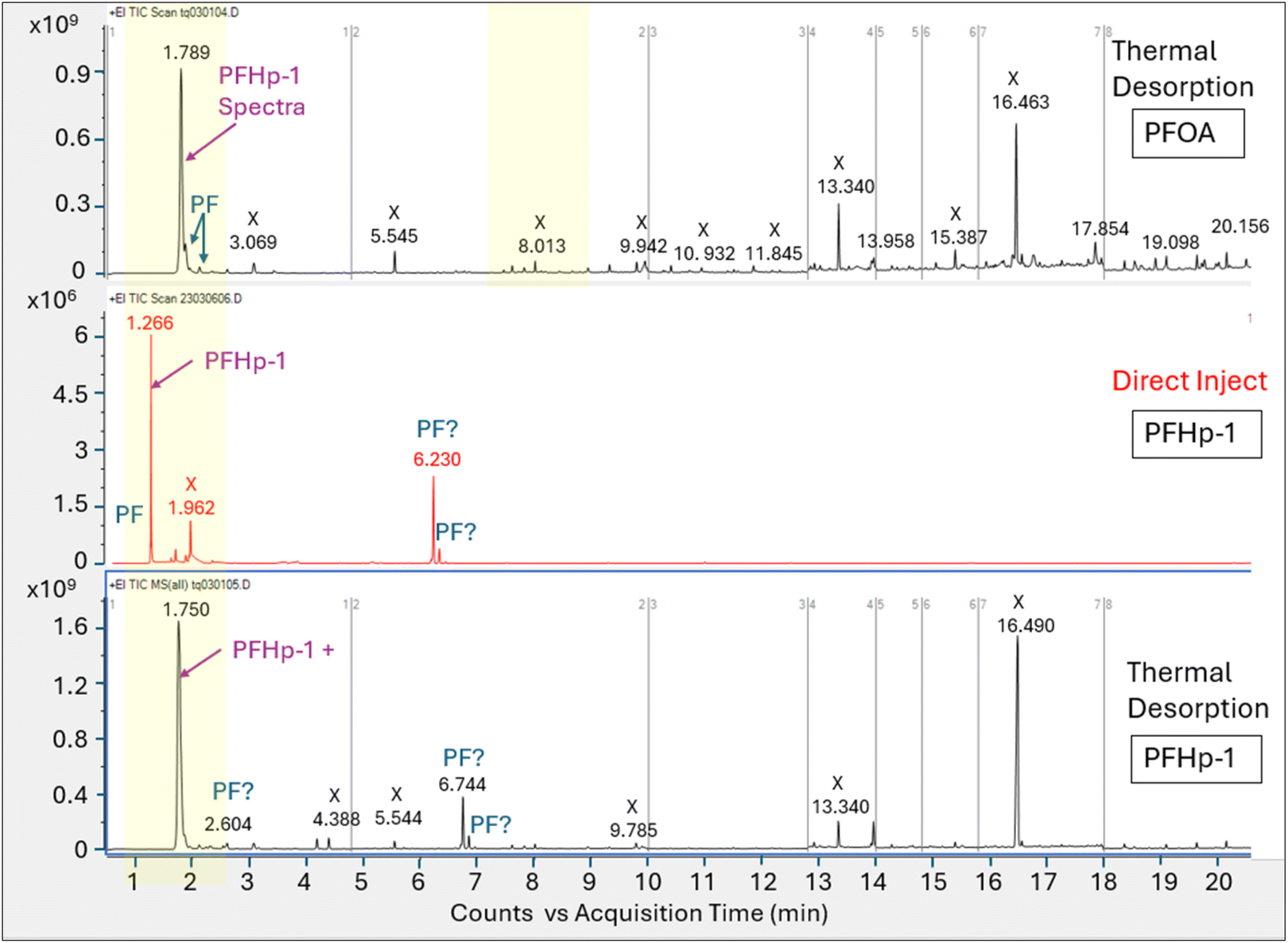 | ||
| Fig. 7 Total ion chromatograms for PFOA and PFHp-1 standards analyzed on HP-5MS column with thermal desorption (top and bottom panels) vs. direct injection (middle panel); X axis is acquisition time, Y axis is area counts, labels on figure: X = non-perfluorinated compound; PF = perfluorinated compound not otherwise identified, individual identified compounds labeled. | ||
 | ||
| Fig. 8 Total ion chromatograms for PFOA (TD) and PFHp-1 on DB-624 column; top panel is thermal desorption analysis of PFOA, middle panel is direct injection analysis of PFHp-1, bottom panel is thermal desorption analysis of PFHp-1; in each panel X axis is acquisition time (min) and Y axis area counts. Labels on peaks X = non-perfluorinated compound; PF = perfluorinated compound; specific identified compounds labeled. | ||
The introduction of the PFHp-1 standard using thermal desorption generated multiple perfluorinated peaks on both the HP-5MS and DB-624 columns, though this was most evident on the DB-624 column. While the HP-5MS column generated a prominent peak for the thermal desorption analysis of PFHp-1, review of the full scan spectra across the peak indicated that this single total ion peak represents the coelution of several unresolved perfluorinated compounds including PFHp-1.
The DB-624 column appeared to resolve the coeluting perfluorinated compounds noted on the HP-5MS thermal desorption run, and five perfluorinated peaks were identified on the DB-624 thermal desorption PFHp-1 run within ±1 minute of the retention time established using the PFOA standard. Interestingly, the largest peak identified in the PFHp-1 thermal desorption run did not match the NIST PFHp-1 spectra; however, one of the smaller peaks eluted at 5.340 min, consistent with peak (A), the prominent peak generated from the thermal desorption injection of PFOA. This smaller peak at 5.340 min also matched the PFHp-1 NIST spectra, generating a match factor of 876. The multiple perfluorinated peaks observed suggest that PFHp-1 is also not stable in the thermal desorption apparatus. A significant fraction of PFHp-1 injected appears to be thermally degraded, generating breakdown products that have not been completely identified.
To estimate the magnitude of the potential artifact or bias to the PFOA concentration measured by TD-GC/MS/MS due to the presence of PFHp-1 in field vapor samples, four PFAS sorbent tubes were spiked with 100 ng of PFHp-1 methanolic solution. The spiked tubes were analyzed on the TD-GC/MS/MS system using the routine PFAS method. While the same peaks were present in each run, some variability in peak area was evident, specifically for the peak eluting at 5.347 min which aligns with the retention time (RT) previously established on the TD system for PFOA, identified as peak (A) (Fig. S9†). The peak eluting at 5.347 min most closely matches the PFHp-1 NIST spectra, consistent with the PFHp-1 thermal desorption results from experiment 3.
In addition to the full scan data evaluation, the MRM data from the four PFHp-1 spiked tubes were processed against the PFCA TD-GC/MS/MS initial calibration. A peak was present in each spike that met the laboratory's identification criteria for PFOA as demonstrated by (1) the quantitation and the two confirmation transition peaks eluting at the expected retention time (5.35 min) and (2) confirmation transition peak areas at the expected ratios (±30%) as determined by the calibration standards. Fig. S10† shows the MRM data and the peak identified as PFOA in a representative PFHp-1 spike.
Since internal standard was not injected onto the PFHp-1 tubes, the concentration measured as PFOA was estimated based on the response factor of the daily continuing calibration PFOA standard (1.0 ng). The apparent PFOA concentration generated from 100 ng of PFHp-1 analyzed by TD-GC/MS/MS ranged from approximately 1.8 to 3.2 ng, with an average PFOA concentration of 2.9 ng. The estimated PFOA mass measured in each of the 100 ng PFHp-1 injections are listed in Table 5. While the data suggests that PFHp-1 is a relatively weak interference for PFOA, the concentration range of PFHp-1 in the environment relative to PFOA is unknown.
| Run | Estimated concentrations (ng) |
|---|---|
| Spike 1 | 3.2 |
| Spike 2 | 1.8 |
| Spike 3 | 3.4 |
| Spike 4 | 3.2 |
| Average | 2.88 |
| %RSD | 26% |
Studies investigating thermal treatment of PFAS-impacted soils and spent water purification filters report PFHp-1 as a primary breakdown product of PFOA at temperatures of 200–400 °C as well as other perfluorinated products, specifically perfluoroalkenes, supporting our findings.54 Additionally, investigators have reported thermal degradation of PFHp-1 and the generation of multiple perfluorinated compounds at temperatures similar to temperatures used for tube desorption.55
Conclusion
A TD-GC/MS/MS method has been developed that exhibits good linearity, sensitivity, and sample stability for seven FTOH compounds for which at least one known standard is available, and FOSA compounds. Three additional FTOH compounds were incorporated based on retention time and structural similarities. Method evaluation was successfully conducted on two different manufacturer's instruments.For the five PFCA targeted, TO-17 calibration criteria can be met for TD-GC/MS/MS, but evidence suggested that the thermal desorption introduction step may cause inadvertent breakdown of these compounds. Perfluorooctanoic acid met TO-17 calibration requirements when calibrated using NIST traceable standard solutions introduced through the thermal desorption system followed by analysis using GC/MS/MS. However, subsequent detailed studies comparing direct injection of PFOA standards into the GC inlet to PFOA standards thermally desorbed from a multi-bed sorbent tube suggested that PFOA was breaking down during the thermal desorption step. But, when PFOA was thermally desorbed from the ITEX-Tenax TA sorbent bed as part of an in-tube dynamic headspace preparation technique, decomposition was minimally observed over a range of inlet temperatures. The primary peak resulting from the thermal desorption of PFOA standard loaded onto the multi-bed sorbent tube exhibited spectra consistent with perfluoro-1-heptene (PFHp-1).
PFHp-1 also exhibited degradation, which appeared to be enhanced during the thermal desorption step, raising further concerns as to extent that other PFAS can degrade to generate PFHp-1 or similar interference. Under the thermal desorption conditions used for these tests, a significant fraction of PFHp-1 injected into the analytical system appeared to generate breakdown products that have not been completely identified, which is a limitation of this study.
The implication of apparent breakdown of PFOA during the thermal desorption introduction step and potential interferences in its identification and quantification will likely extend to PFCAs in general since similar transformations are expected but were not directly studied here. The fluorotelomer alcohols and sulfonamindes maintain their molecular structure during the thermal desorption step and yield mass ions which are characteristic of their molecular structure and associated functional group. Alternate approaches using either a solvent extraction method for the PFCAs or an alternate thermally desorbed sorbent system should be explored.
Therefore, the identification of the PFCA compounds in this method is currently subject to a potential positive interference from the corresponding perfluoroalkene and potentially other thermally labile PFAS that may generate products with common spectra and retention time. Thus, it may be beneficial to limit the application of the thermal desorption GC/MS/MS method to the fluorotelomer alcohols and perfluorooctanesulfonamides and use a parallel solvent extraction approach to quantify the PFCA compounds.
Disclaimer
The views expressed in this paper are those of the author(s) and do not necessarily reflect the views or policies of the EPA. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.Data availability
Data supporting this article have been included as part of the ESI.† This file has been uploaded as part of the article submission.Author contributions
Heidi Hayes: methodology, supervision, writing – original draft; Chris Lutes: conceptualization, formal analysis, supervision, validation, writing – review & editing; Nicola Watson: investigation, writing – review & editing; Diane Benton: Investigation; David Hanigan: project administration, resources, writing – review and editing; Seth Mccoy: investigation, methodology, writing – review and editing; Chase Holton: writing – review & editing; Kate Bronstein: project administration, supervision, writing – review & editing; Brian Schumacher: conceptualization, funding acquisition, project administration, writing-review and editing; John Zimmerman: conceptualization, funding acquisition, project administration, writing-review and editing; Alan Williams: project administration, writing-review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research described in this article has been funded by the U.S. Environmental Protection Agency through contract number 68HERC21D0004 to Research Triangle Institute, International with partial support by the National Science Foundation under Grant No. 2128407.References
-
ITRC, Technical/Regulatory Guidance: Per- and Polyfluoralkyl Substances, https://pfas-1.itrcweb.org/ Search PubMed
.
-
US EPA, OSWER Technical Guide for Assessing and Mitigating the Vapor Intrusion Pathway from Subsurface Vapor Sources to Indoor Air. OSWER Publication 9200.2-154, U.S. Environmental Protection Agency (U.S. EPA), 2015 Search PubMed
-
C. Lutes, G. Buckley, V. Boyd, R. Truesdale, C. Holton, H. Dawson, H. Hayes, J. Zimmerman, A. Williams and B. Schumacher, presented in part at the AWMA Annual Conference, 2021 Search PubMed
- M. Shoeib, T. Harner, M. W. G and S. C. Lee, Indoor sources of poly- and perfluorinated compounds (PFCS) in Vancouver, Canada: implications for human exposure, Environ. Sci. Technol., 2011, 45, 7999–8005 CrossRef CAS
- H. Chen, H. Peng, M. Yang, J. Hu and Y. Zhang, Detection, Occurrence, and Fate of Fluorotelomer Alcohols in Municipal Wastewater Treatment Plants, Environ. Sci. Technol., 2017, 51, 8953–8961 CrossRef CAS PubMed
- M. E. Morales-McDevitt, J. Becanova, A. Blum, T. A. Bruton, S. Vojta, M. Woodward and R. Lohmann, The air that we breathe: Neutral and volatile PFAS in indoor air, Environ. Sci. Technol. Lett., 2021, 8, 897–902 CrossRef CAS PubMed
- Z. G. Robbins, X. Liu, B. A. Schumacher, M. G. Smeltz and H. K. Liberatore, Method development for thermal desorption-gas chromatography-tandem mass spectrometry (TD-GC-MS/MS) analysis of trace level fluorotelomer alcohols emitted from consumer products, J. Chromatogr. A, 2023, 1705, 464204 CrossRef CAS
- C. M. A. Eichler, N. Y. Chang, E. A. Cohen Hubal, D. E. Amparo, J. Zhou, J. D. Surratt, G. C. Morrison and B. J. Turpin, Cloth-air partitioning of neutral per- and polyfluoroalkyl substances (PFAS) in North Carolina homes during the Indoor PFAS Assessment (IPA) Campaign, Environ. Sci. Technol., 2023, 57, 15173–15183 CrossRef CAS
- R. Kapuscinski, Two Proposals Regarding Nomenclature About Vapor Intrusion, Groundwater Monit. Rem., 2021, 41, 7–9 CrossRef
- D. Gonzalez, K. Thompson, O. Quinones, E. Dickenson and C. Bott, Assessment of PFAS fate, transport, and treatment inhibition associated with a simulated AFFF release within a wastewater treatment plant, Chemosphere, 2021, 262, 127900 CrossRef CAS PubMed
-
B. Jordan, presented in part at the Water Enivronment Research Federation, 2014 Search PubMed
- M. Roghani, Y. Li, N. Rezaei, A. Robinson, E. Shirazi and K. G. Pennell, Modeling fate and transport of volatile organic compounds (VOCs) inside sewer systems, Groundwater Monit. Rem., 2021, 41, 112–121 CrossRef
- T. Wells and R. E. Melchers, Modelling concrete deterioration in sewers using theory and field observations, Cem. Concr. Res., 2015, 77, 82–96 CrossRef CAS
- J. H. Johansson, M. E. Salter, J. C. Acosta Navarro, C. Leck, E. D. Nilsson and I. T. Cousins, Global transport of perfluoroalkyl acids via sea spray aerosol, Environ. Sci.: Processes Impacts, 2019, 21, 635–649 RSC
- M. Shoeib, T. Harner and P. Vlahos, Perfluorinated chemicals in the arctic atmosphere, Environ. Sci. Technol., 2006, 40, 7577–7583 CrossRef CAS PubMed
- J. L. Barber, U. Berger, C. Chaemfa, S. Huber, A. Jahnke, C. Temme and K. C. Jones, Analysis of per- and polyfluorinated alkyl substances in air samples from Northwest Europe, J. Environ. Monit., 2007, 9, 530–541 RSC
- K. Winkens, J. Koponen, J. Schuster, M. Shoeib, R. Vestergren, U. Berger, A. M. Karvonen, J. Pekkanen, H. Kiviranta and I. T. Cousins, Perfluoroalkyl acids and their precursors in indoor air sampled in children's bedrooms, Environ. Pollut., 2017, 222, 423–432 CrossRef CAS PubMed
- X. Liu, Z. Guo, E. E. t. Folk and N. F. Roache, Determination of fluorotelomer alcohols in selected consumer products and preliminary investigation of their fate in the indoor environment, Chemosphere, 2015, 129, 81–86 CrossRef CAS PubMed
- T. P. Riedel, J. R. Lang, M. J. Strynar, A. B. Lindstrom and J. H. Offenberg, Gas-Phase Detection of Fluorotelomer Alcohols and Other Oxygenated Per- and Polyfluoroalkyl Substances by Chemical Ionization Mass Spectrometry, Environ. Sci. Technol. Lett., 2019, 6, 289–293 CrossRef CAS
- I. A. Titaley, J. Khattak, J. Dong, C. I. Olivares, B. DiGuiseppi, C. C. Lutes and J. A. Field, Neutral Per- and Polyfluoroalkyl Substances, Butyl Carbitol, and Organic Corrosion Inhibitors in Aqueous Film-Forming Foams: Implications for Vapor Intrusion and the Environment, Environ. Sci. Technol., 2022, 56, 10785–10797 CrossRef CAS PubMed
-
A. Bradman, R. Castorina, T. Pattabhiraman, A. Nirula, M. Wong and S. Calabretta, Assessment of Methods to Collect and Analyze Perfluoroalkyl and Polyfluoroalkyl Substances (PFASs) in Air, Dust and Soil, Center for Environmental Research and Children's Health University of California, Berkeley, p. 2021 Search PubMed
- M. A. G. Wallace, M. G. Smeltz, J. M. Mattila, H. K. Liberatore, S. R. Jackson, E. P. Shields, X. Xhani, E. Y. Li and J. H. Johansson, A review of sample collection and analytical methods for detecting per- and polyfluoroalkyl substances in indoor and outdoor air, Chemosphere, 2024, 358, 142129 CrossRef CAS PubMed
- N. Y. Chang, C. M. A. Eichler, D. E. Amparo, J. Zhou, K. Baumann, E. A. Cohen Hubal, J. D. Surratt, G. C. Morrison and B. J. Turpin, Indoor air concentrations of PM(2.5) quartz fiber filter-collected ionic PFAS and emissions to outdoor air: findings from the IPA campaign, Environ. Sci.: Processes Impacts, 2024 10.1039/d4em00359d
-
K. Thaxton, J. Stuff, J. A. Whitecavage, H. Hayes and J. Miller, Analysis of PFAS Compounds in Air using Solid Sorbent Tubes with Thermal Desorption Gas Chromatography Mass Spectrometry, presented at National Environmental Monitoring Conference, 2020 Search PubMed
-
Markes International, Application Note 158: Analysis of Trace Per- and Polyfluorinated Organic Vapours in Air Using Cryogen-Free Thermal Desorption and Gas Chromatography–Mass Spectrometry, 2022 Search PubMed
-
Markes International, Application Note 167 Measurement of PFAS in Indoor Air and Investigation of Source Materials, 2023 Search PubMed
- L. Miles, H. Calder, V. Nikiforov, D. Herzke, N. Warner, G. Riccardino, A. Ladak and D. Kutscher, High-Throughput Analysis of Both Neutral and Ionic PFAS in Ambient Air Using Thermal Desorption Coupled to Gas Chromatography – Mass Spectrometry (TD-GC-MS/MS), Thermo Fisher Scientific Application Note 001715, 2023 Search PubMed.
-
ASTM, D8591-24, Standard Test Method for Determination of Fluorotelomer Alcohols in Test Chamber Air by Thermal Desorption-Gas Chromatography-Triple Quadrupole Tandem Mass Spectrometry (TD-GC-MS/MS), https://www.astm.org/d8591-24.html Search PubMed
- T. P. Bastow, G. B. Douglas and G. B. Davis, Volatilization potential of per- and poly-fluoroalkyl substances from airfield pavements and during recycling of asphalt, Environ. Toxicol. Chem., 2022, 41, 2202–2208 CrossRef CAS
- M. A. Kaiser, B. J. Dawson, C. A. Barton and M. A. Botelho, Understanding potential exposure sources of perfluorinated carboxylic acids in the workplace, Ann. Occup. Hyg., 2010, 54, 915–922 CAS
- J. Roth, I. Abusallout, T. Hill, C. Holton, U. Thapa and D. Hanigan, Release of Volatile Per- and Polyfluoroalkyl Substances from Aqueous Film-Forming Foam, Environ. Sci. Technol. Lett., 2020, 7, 164–170 CrossRef CAS
- I. A. Titaley, F. B. De la Cruz and J. A. Field, Comment on “Release of Volatile Per- and Polyfluoroalkyl Substances from Aqueous Film-Forming Foam”, Environ. Sci. Technol. Lett., 2020, 7, 866–868 CrossRef CAS
- T. Sakaki, P. E. Schulte, A. Cihan, J. A. Christ and T. H. Illangasekare, Airflow Pathway Development as Affected by Soil Moisture Variability in Heterogeneous Soils, Vadose Zone J., 2013, 12(1), 1–14 CrossRef
- S. Endo, J. Hammer and S. Matsuzawa, Experimental Determination of Air/Water Partition Coefficients for 21 Per- and Polyfluoroalkyl Substances Reveals Variable Performance of Property Prediction Models, Environ. Sci. Technol., 2023, 57, 8406–8413 CrossRef CAS
- I. Abusallout, C. Holton, J. Wang and D. Hanigan, Henry’s Law constants of 15 per- and polyfluoroalkyl substances determined by static headspace analysis, J. Hazard. Mater. Lett., 2022, 3, 100070 CrossRef CAS
- B. A. Schumacher, J. H. Zimmerman, A. C. Williams, C. C. Lutes, C. W. Holton, E. Escobar, H. Hayes and R. Warrier, Vapor intrusion potential of select per- and polyfluoroalkyl substances, J. Hazard. Mater., 2023, 464 Search PubMed
- R. R. Giri, H. Ozaki, T. Okada, S. Taniguchi and R. Takanami, Factors influencing UV photodecomposition of perfluorooctanoic acid in water, Chem. Eng. J., 2012, 180, 197–203 CrossRef CAS
-
US EPA, Subsurface Per- and Polyfluoroalkyl Substances (PFAS) Distribution at Two Contaminated Sites (EPA 600/R-23/294), 2023 Search PubMed
-
US EPA, Compendium of Methods for the Determination of Toxic Organic Compounds in Ambient Air Second Edition, Compendium Method TO-17: Determination of Volatile Organic Compounds in Ambient Air using Active Sampling onto Sorbent Tubes (EPA/625/R-96/010b), 1999 Search PubMed
- H. P. H. Arp and K.-U. Goss, Irreversible sorption of trace concentrations of perfluorocarboxylic acids to fiber filters used for air sampling, Atmos. Environ., 2008, 42, 6869–6872 CrossRef CAS
- J. H. Johansson, U. Berger and I. T. Cousins, Can the use of deactivated glass fibre filters eliminate sorption artefacts associated with active air sampling of perfluorooctanoic acid?, Environ. Pollut., 2017, 224, 779–786 CrossRef CAS PubMed
-
ITRC, Vapor Intrusion Pathway: A Practical Guideline, Interstate Technology Regulatory Council (ITRC), 2007 Search PubMed
- R. W. Kondrat, G. A. McClusky and R. G. Cooks, Multiple reaction monitoring in mass spectrometry/mass spectrometry for direct analysis of complex mixtures, Anal. Chem., 2002, 50, 2017–2021 CrossRef
- M. A. Mottaleb, Q. X. Ding, K. G. Pennell, E. N. Haynes and A. J. Morris, Direct injection analysis of per and polyfluoroalkyl substances in surface and drinking water by sample filtration and liquid chromatography-tandem mass spectrometry, J. Chromatogr. A, 2021, 1653, 462426 CrossRef CAS
-
DOD and DOE, Consolidated Quality Systems Manual (QSM) for Environmental Laboratories; Based on ISO/IEC 17025:2005(E), ISO/IEC 17025:2017(E) and the NELAC Institute (TNI) Standards, Volume 1, (September 2009); DoD Quality Systems Manual Versoin 5.4, 2021 Search PubMed
- N. Hanari, N. Itoh, K. Ishikawa, T. Yarita and M. Numata, Variation in concentration of perfluorooctanoic acid in methanol solutions during storage, Chemosphere, 2014, 94, 116–120 CrossRef CAS
- B. Sonmez Baghirzade, Y. Zhang, J. F. Reuther, N. B. Saleh, A. K. Venkatesan and O. G. Apul, Thermal regeneration of spent granular activated carbon presents an opportunity to break the forever PFAS cycle, Environ. Sci. Technol., 2021, 55, 5608–5619 CrossRef CAS
- P. C. Sasi, A. Alinezhad, B. Yao, A. Kubatova, S. A. Golovko, M. Y. Golovko and F. Xiao, Effect of granular activated carbon and other porous materials on thermal decomposition of per- and polyfluoroalkyl substances: Mechanisms and implications for water purification, Water Res., 2021, 200, 117271 CrossRef CAS
- S. Biswas and B. M. Wong, Degradation of Perfluorooctanoic Acid on Aluminum Oxide Surfaces: New Mechanisms from Ab Initio Molecular Dynamics Simulations, Environ. Sci. Technol., 2023, 57, 6695–6702 CrossRef CAS
- N. H. Weber, L. J. Dixon, S. P. Stockenhuber, C. C. Grimison, J. A. Lucas, J. C. Mackie, M. Stockenhuber and E. M. Kennedy, Thermal decomposition of PFOA: Influence of reactor and reaction conditions on product formation, Chem. Eng. Sci., 2023, 278, 118924 CrossRef CAS
- M. Pazda, P. Stepnowski, T. Sledzinski, M. Chmielewski and A. Mika, Suitability of selected chromatographic columns for analysis of fatty acids in dialyzed patients, Biomed. Chromatogr., 2017, 31(11), e4006 CrossRef PubMed
- G. Nyerges, J. Mátyási and J. Balla, Comparison of 624-type capillary columns, Period. Polytech., Chem.
Eng., 2023, 67, 427–434 CrossRef CAS
- M. Ren, N. Natsagdorj and N. Shun, Influence and mechanism of polar solvents on the retention time of short-chain fatty acids in gas chromatography, Separations, 2022, 9(5), 124 CrossRef CAS
- A. Alinezhad, P. C. Sasi, P. Zhang, B. Yao, A. Kubátová, S. A. Golovko, M. Y. Golovko and F. Xiao, An investigation of thermal air degradation and pyrolysis of per- and polyfluoroalkyl substances and aqueous film-forming foams in soil, Environ. Sci. Technol., 2022, 2, 198–209 CAS
- B. Yao, R. Sun, A. Alinezhad, A. Kubatova, M. F. Simcik, X. Guan and F. Xiao, The first quantitative investigation of compounds generated from PFAS, PFAS-containing aqueous film-forming foams and commercial fluorosurfactants in pyrolytic processes, J. Hazard. Mater., 2022, 436, 129313 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00084f |
| This journal is © The Royal Society of Chemistry 2025 |