
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Perhemiacetal formation and Cl/NO3-initiated chemistry of hydroperoxymethylthioformate (HPMTF) in atmospheric DMS oxidation†
L.
Vereecken
 *,
A.
Novelli
,
D.
Taraborrelli
and
A.
Wahner
*,
A.
Novelli
,
D.
Taraborrelli
and
A.
Wahner
Institute of Climate and Energy Systems, ICE-3: Troposphere, Forschungszentrum Jülich GmbH, Jülich, Germany. E-mail: l.vereecken@fz-juelich.de
First published on 12th December 2024
Abstract
The emission of dimethylsulfide (DMS) is an important source of sulfur in the atmosphere. Its oxidation leads to enhanced particle formation, where OCS is a critical reaction intermediate as it can reach the stratosphere and oxidize to low-volatility H2SO4 acting as a condensation nucleus. The mechanism for OCS formation from DMS is currently understood to proceed through the hydroperoxymethylthioformate intermediate (HOOCH2SCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, HPMTF), and experimental data indicate that the OH-initiated HPMTF oxidation generates high yields of OCS. The total atmospheric OCS formation is assumed to remain limited due to competition by phase transfer of the soluble HPMTF to water droplets, but the fate of HPMTF, once it transitions to the aqueous phase, remains unclear. In this work, we theoretically study the formation of cyclic thioperhemiacetal isomers of HPMTF both in the gas phase and in acidic aqueous phase, finding that formation of thioperhemiacetal can be rapid when catalyzed by acids. The subsequent oxidation of thioperhemiacetal is shown not to form OCS, but rather lead to formic and thioformic acid, HCOOH + HCOSH. Based on these theoretical predictions we propose that thioperhemiacetal formation is the main loss process blocking OCS formation from HPMTF in the aqueous phase. To complement the models incorporating the OH-initiated HPMTF oxidation, we also theoretically predict the rate coefficients of HPMTF with Cl atoms and NO3 radicals. The reaction with Cl is found to be fast and leads primarily to OCS, while the reaction with NO3 is slow and does not contribute appreciably to HPMTF loss.
O, HPMTF), and experimental data indicate that the OH-initiated HPMTF oxidation generates high yields of OCS. The total atmospheric OCS formation is assumed to remain limited due to competition by phase transfer of the soluble HPMTF to water droplets, but the fate of HPMTF, once it transitions to the aqueous phase, remains unclear. In this work, we theoretically study the formation of cyclic thioperhemiacetal isomers of HPMTF both in the gas phase and in acidic aqueous phase, finding that formation of thioperhemiacetal can be rapid when catalyzed by acids. The subsequent oxidation of thioperhemiacetal is shown not to form OCS, but rather lead to formic and thioformic acid, HCOOH + HCOSH. Based on these theoretical predictions we propose that thioperhemiacetal formation is the main loss process blocking OCS formation from HPMTF in the aqueous phase. To complement the models incorporating the OH-initiated HPMTF oxidation, we also theoretically predict the rate coefficients of HPMTF with Cl atoms and NO3 radicals. The reaction with Cl is found to be fast and leads primarily to OCS, while the reaction with NO3 is slow and does not contribute appreciably to HPMTF loss.
Environmental significanceAtmospheric sulfur compounds have an important environmental impact. One of the key sulfuric species is H2SO4, which leads to aerosol formation, affecting the Earth's radiative budget and air quality. OCS, a sulfur equivalent of CO2, can be transported to the stratosphere and enhance formation of stratospheric clouds. Hydroperoxymethylthioformate (HPMTF), formed from dimethyl sulfide in marine emissions, has been shown to decompose forming over 9% of OCS, which leads to OCS overestimations in models. We show that HPMTF can convert to a thioperhemiacetal molecule in the aqueous phase, blocking formation of OCS and providing a rationale how HPMTF uptake in aqueous phase can help close the atmospheric OCS budget. |
1. Introduction
Atmospheric sulfur has an important impact on climate and air quality.1–3 The formation of low-volatility H2SO4 is of particular importance, as it has a large influence on the formation and growth of organic particulate matter, where formation of high-albedo atmospheric particles has a distinct influence on the Earth's radiative budget while particles in the lower troposphere have important health and air quality effects. Another sulfur compound of significant interest is the OCS molecule formed from organo-sulfur molecules, as it is the primary compound transporting sulfur to the stratosphere, ultimately forming H2SO4 as a nucleus for formation of stratospheric clouds.4 In the troposphere, a wide range of organo-sulfur compounds is present, originating either from direct emissions or from in situ transformation of primary emitted sulfur-bearing compounds. Particulate matter also often contains sulfonated compounds. Stratospheric sulfur injection is also proposed as a viable way of geo-engineering global warming e.g. ref. 5.The sources, sinks, and transformation chemistry of these sulfur compounds are not well known, with regular updates to estimated emission inventories and reports of novel chemical pathways for transformation.6–13 Among the directly emitted organic sulfur-sources, dimethyl sulfide (DMS, CH3SCH3) is the most extensively studied molecule, but even for this compound its atmospheric oxidation mechanism is not clearly characterized.2,12,14–31 Recent literature showed that the dominant H-abstraction channel in the OH-initiated oxidation of DMS leads to the formation of hydroperoxymethylthioformate (HPMTF, HOOCH2SCHO). Subsequent oxidation of this compound by OH radicals leads to the formation of SO2 and OCS, where the yield of the latter is found to be significantly larger (≥9%) than used in earlier atmospheric kinetic models (∼0.7%).2,25,27 Data on rates and product yields of other loss processes of HPMTF, such as reaction with NO3, Cl-atoms, O3, SO3, or its photolysis or deposition, remains very limited.
Recently, Jacob et al.32 proposed a generalized oxidation mechanism for atmospheric DMS oxidation, based on the available literature data. For the critical HPMTF oxidation, the study draws on our extensive mechanism for the OH-based degradation of HPMTF recently proposed in Jernigan et al.25 incorporating experimental observations and theoretical calculations (see Fig. 1). Many uncertainties regarding the fate of HPMTF remain. The direct gas-phase yield of OCS from HPMTF obtained in current experiments is incompatible with the measured OCS atmospheric concentration. However, in addition to chemical losses, HPMTF was found to be highly soluble and is expected to readily transition to the aqueous phase (surface water and cloud droplets) in the marine environment where DMS is predominantly emitted.19,22 It is assumed that this phase transition is irreversible and thus prevents further formation of OCS; this has been confirmed by recent experiments by Jernigan et al.,30 but the chemical mechanism preventing OCS formation has not been elucidated. Formation of a perhemiacetal was proposed,30,33,34 but such an isomerisation is reversible, and furthermore the aqueous phase decomposition pathways of thioperhemiacetal (TPHAC) have not been characterized.
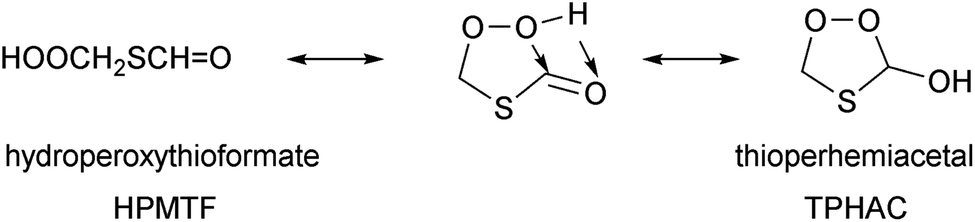
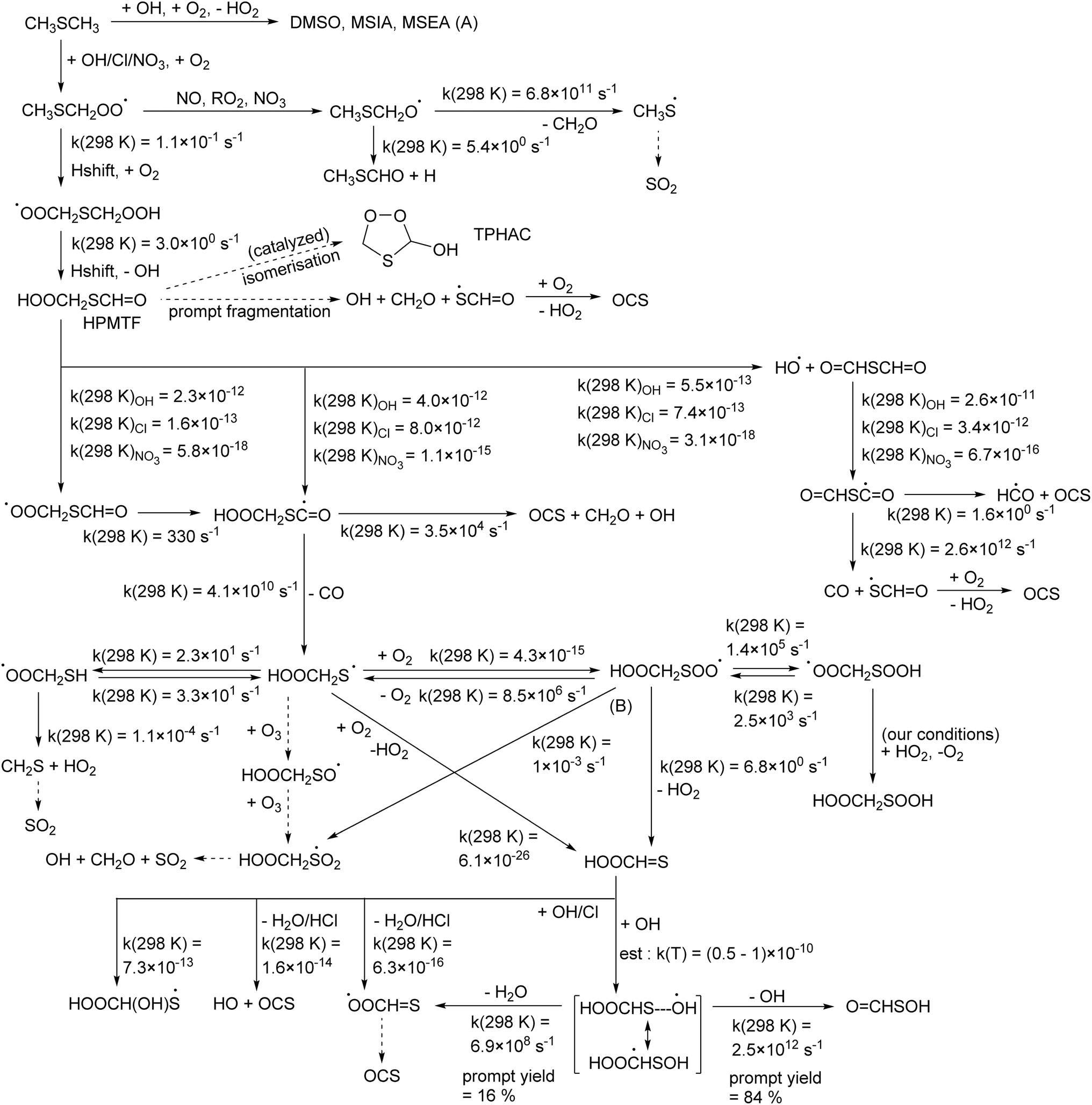 | ||
| Fig. 1 Hydroperoxymethylthioformate (HPMTF) formation from DMS, and subsequent OH-, Cl-, and NO3-initiated oxidation steps based on the theoretical work by Jernigan et al.25 and this work. (A) For a discussion of chemistry forming dimethylsulfoxide (DMSO), methanesulfinic acid (MSIA), and methane sulfenic acid (MSEA) we refer to Cala et al.,35 Chen et al.,36 Jacob et al.,32 and references therein. (B) Theoretical work by Chen et al.20 suggests that formation of HOOCH2SO2 is dominant instead of HOOCHS formation. | ||
In addition, it should be noted that TPHAC may be released back to the gas phase, e.g. by phase-transfer, or by evaporation of the containing droplets as proposed recently for methanediol.37 Its subsequent gas phase chemistry, i.e. kinetics and product yields, may thus also be of importance.
In this work, we further explore the fate of HPMTF in the atmosphere using theoretical methodologies, examining its reaction with NO3 and Cl oxidants of importance in the atmosphere and used in experimental studies. We also examine the interaction of HPMTF with the aqueous phase, where we focus on the formation and degradation pathways of the thioperhemiacetal TPHAC, an isomer of HPMTF.
2. Methodologies
The theoretical analysis extends the potential energy surfaces reported earlier by Jernigan et al.,25 using the same methodologies. Briefly, for all intermediates and transition states (TSs) an exhaustive characterisation of all conformers was done first at the M06-2X/cc-pVDZ level of theory,38,39 where TSs were verified by running IRC calculations at this level. A further optimization was then done for all conformers at the M06-2X-D3/aug-cc-pV(T+d)Z level of theory, suitable for compounds containing the 3rd row sulfur atom.40–42 The vibrational wavenumbers were likewise characterized at this level, using a scaling factor of 0.971.43,44 The relative energies between the lowest energy reactant and transition state conformers was then refined at the CCSD(T)/aug-cc-pV(Q+d)Z level of theory.40,45 The expected uncertainty on the reaction barrier heights at this level of theory is ±0.5 kcal mol−1. All T1 diagnostics are below the 0.044 cut-off suggested by Rienstra-Kiracofe et al.,46 except those for catalyzed TPHAC unimolecular dissociation. In those calculations, formation of singlet biradicals from a closed shell species uses the broken symmetry approach along the pathway,47 but future work should consider multi-reference calculations to improve the characterization of these channels. All calculations were performed using the Gaussian-16 software suite.48The high-pressure thermal rate coefficients were calculated using multi-conformer transition state theory, MC-TST, incorporating all conformers characterized as above using a rigid rotor harmonic oscillator (RRHO) model for each conformer.49 Tunneling is included using asymmetric Eckart tunneling rate corrections,50,51 while for bimolecular reactions with the OH and Cl radicals the spin orbit splitting of 27.95 cm−1 and 882.4 cm−1, respectively,52,53 is accounted for, assuming negligible spin orbit splitting in the TS. The reactions characterized in this work are not expected to be pressure-dependent under atmospheric conditions, and the estimated accuracy of the rate coefficients is a factor 2 to 3, except for the bimolecular reactions involving NO3, which are less reliable due to the internal rotations of the NO3 moiety in the TS, which have low barriers and high moments of inertia, and are less accurately represented in the RRHO multi-conformer paradigm (estimated uncertainty factor 3 to 4).
The updated mechanism was implemented in a 0D kinetic box model (available in EASY format in Section ESI-C†) where rate coefficients for the reactions of species added in this work and in Jernigan et al.25 were used as calculated by theory. For the new RO2 radicals in the scheme, bimolecular reactions with HO2, RO2, NO, and NO3 were added following the rate coefficients in the Master Chemical Mechanism MCMv3.3.1,54,55i.e. analogous to CH3SCH2OO˙. The subsequent fate of the alkoxy radicals derived from those RO2 intermediates is not implemented as they constitute only a small fraction of the sulfur budget (less than 5%). For S-based radicals where no theoretical data is available, we applied analogies to the chemistry of S-radicals described in the literature.2,56,57 The DMSO chemistry following OH-addition on DMS was implemented as in the available literature,2,32 but has little bearing on the chemistry studied here, and it is not shown in Section ESI-C.† The model there likewise does not include photochemistry or heterogeneous chemistry of the stable intermediates, nor bimolecular reactions of the longer-lived radical intermediates with potential reaction partners such as O3, NO2 or other reactive species that might be present in higher concentrations in experiments.
3. Reactions of HPMTF with Cl and NO3
We calculated the rate coefficients of the reactions of HPMTF with Cl and NO3 oxidants; the site-specific and total rate coefficients are listed in Table 1 for a range of temperatures. For both reactants the dominant channel is abstraction of the thioformate H-atom, –SCHO (see Fig. 1), similar as for an OH co-reactant.25 However, the abstraction of the hydroperoxide, –OOH, or methylene hydrogen, –CH2–, by Cl and NO3 have minor contributions of less than 5% at 298 K, compared to ≥40% with OH, such that the HOOCH2SC˙O radical will be the sole product of importance. The theoretically predicted total rate coefficient with Cl atoms is similar to that with OH radicals at 298 K, while that with NO3 is a factor ∼6000 slower. For atmospherically relevant NO3 radical concentrations, loss of HPMTF to this radical is thus negligible. The rate coefficients for the OCHSCHO thioformic anhydride, likewise of importance in atmospheric HPMTF oxidation, present similar ratios against the loss with OH, with the NO3 reaction again being negligibly slow, k(298 K) ≤ 1 × 10−15 cm3 molecule−1 s−1.
| Reaction | Products | E b | k(298 K) | A | n | E a |
|---|---|---|---|---|---|---|
| a Corrected by 0.84 kcal mol−1 for spin orbit splitting. b Assumed to be similar to HOOCH2SOO˙. However, the CCSD(T)//M06-2X-D3 results shown here are likely unreliable, as discussed in ref. 20 and 25. c No distinct transition state geometry was localized. Geometry scans along the constrained O–O bond show a sudden collapse to the CO2 product at an O–O bond length just beyond 1.58 Å; the barrier height and estimated rate coefficient reported are based on the energetics at this point. | ||||||
| HOOCH2SCHO + ˙Cl |
→ HOOCH2SC˙O + HCl |
−0.1a | 8.0 × 10−12 | 1.22 × 10−14 | 1.09 | −88 |
| → HOOC˙HSCHO + HCl |
1.14a | 1.5 × 10−13 | 1.88 × 10−18 | 2.38 | 672 | |
| → ˙OOCH2SCHO + HCl |
2.4a | 7.4 × 10−13 | 2.37 × 10−21 | 3.19 | 277 | |
| → All products (total rate) | 8.2 × 10−12 | 7.58 × 10−16 | 1.52 | −182 | ||
| OCHSCHO + ˙Cl |
→ OCHSC˙O + HCl |
0.58 | 3.4 × 10−12 | 7.16 × 10−16 | 1.49 | 7 |
| HOOCH2SCHO + ˙NO3 |
→ HOOCH2SC˙O + HNO3 |
1.9 | 1.1 × 10−15 | 1.01 × 10−21 | 2.89 | 767 |
| → HOOC˙HSCHO + HNO3 |
3.4 | 3.1 × 10−18 | 4.72 × 10−34 | 6.93 | 920 | |
| → ˙OOCH2SCHO + HNO3 |
3.6 | 5.8 × 10−18 | 4.36 × 10−27 | 4.00 | 534 | |
| → All products (total rate) | 1.1 × 10−15 | 5.51 × 10−22 | 2.99 | 745 | ||
| OCHSCHO + ˙NO3 |
→ OCHSC˙O + HNO3 |
2.6 | 6.7 × 10−16 | 3.65 × 10−24 | 3.87 | 904 |
| HOOCH2SCHO + H2O |
→ TPHAC + H2O | 20.1 | 3.4 × 10−30 | 3.14 × 10−47 | 10.03 | 5340 |
| HOOCH2SCHO + HCOOH |
→ TPHAC + HCOOH | 7.2 | 5.1 × 10−22 | 5.22 × 10−48 | 9.99 | −873 |
| HOOCH2SCHO + HNO3 |
→ TPHAC + HNO3 | 10.6 | 6.2 × 10−25 | 1.13 × 10−20 | 1.06 | 4721 |
| HOOCH2SCHO |
→ TPHAC | 40.6 | 3.13 × 10−16 | 3.13 × 10−182 | 62.97 | −6788 |
| HOOCH2SCHO–H2O |
→ TPHAC–H2O | 26.2 | 2.9 × 10−8 | 3.47 × 10−18 | 9.20 | 8825 |
| TPHAC–H2O | → HOOCH2SCHO–H2O |
27.3 | 3.1 × 10−8 | 1.79 × 10−20 | 10.48 | 9401 |
| → HCOOH + ˙OCH2S˙–H2O (singlet) | 26.7 | 4.7 × 10−7 | 2.25 × 10−4 | 5.70 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 509 509 |
|
| HOOCH2SCHO–H3O+ |
→ TPHAC–H3O+ | 13.7 | 7.4 × 101 | 1.34 × 1011 | 0.24 | 6760 |
| TPHAC–H3O+ | → HOOCH2SCHO–H3O+ |
2.6 | 4.2 × 1011 | 3.72 × 108 | 1.97 | 1243 |
| → HCOOH + ˙OCH2S˙–H3O+ (singlet) | 16.5 | 6.2 × 101 | 1.13 × 100 | 4.81 | 6979 | |
| HOOCH2SCHO–H2O–H3O+ |
→ TPHAC–H2O–H3O+ | 9.7 | 2.6 × 104 | 3.75 × 1011 | −0.12 | 4711 |
| TPHAC–H2O–H3O+ | → HOOCH2SCHO–H2O–H3O+ |
9.4 | 2.7 × 106 | 2.15 × 109 | 1.57 | 4662 |
| HOOCH2SCHO–HCOOH |
→ TPHAC–HCOOH | 17.8 | 2.0 × 10−1 | 2.79 × 10−19 | 9.80 | 4397 |
| TPHAC–HCOOH | → HOOCH2SCHO–HCOOH |
17.0 | 1.6 × 100 | 4.85 × 10−22 | 11.03 | 3960 |
| HOOCH2SCHO–HNO3 |
→ TPHAC–HNO3 | 19.9 | 1.8 × 10−3 | 6.52 × 1011 | −0.19 | 9671 |
| TPHAC–HNO3 | → HOOCH2SCHO–HNO3 |
20.0 | 3.1 × 10−3 | 9.50 × 108 | 1.05 | 9662 |
| TPHAC + ˙OH | → OCHOOCH2S˙ + H2O |
−10.6 | ≥ 1 × 10−10 | ≥ 1 × 10−10 | ||
| → HCOOH + ˙SCHO + H2O |
−0.3 | 1.2 × 10−12 | 8.90 × 10−25 | 4.03 | −1475 | |
| → OC(OH)SCH2O˙ + H2O |
2.1 | 6.6 × 10−14 | 1.52 × 10−33 | 7.22 | −1215 | |
| → cyc-SCH2OOCH(O˙)– + H2O | 3.9 | 4.4 × 10−15 | 1.56 × 10−52 | 13.06 | −3520 | |
| TPHAC | → HOOCH2SCHO |
41.4 | 8.4 × 10−16 | 2.79 × 10−185 | 64.82 | −6269 |
| → HCOOH + ˙OCH2S˙ (singlet) | 31.7 | 1.4 × 10−10 | 2.10 × 109 | 1.66 | 15982 |
|
| → OCHOOCH2SH |
37.4 | 4.7 × 10−15 | 1.16 × 101 | 3.97 | 17311 |
|
| → HOCOOCH2SH (carbene) | 49.5 | 5.7 × 10−23 | 5.63 × 10−22 | 11.59 | 20370 |
|
| → HC(O)OOH + CH2S |
41.4 | 1.9 × 10−17 | 1.01 × 1011 | 1.12 | 20930 |
|
| OCHOOCH2S˙ |
→ OC˙OOCH2SH |
20.9 | 6.7 × 10−4 | 1.83 × 10−12 | 7.60 | 7025 |
| → OCHOO + CH2S |
31.2 | 1.4 × 10−10 | 4.62 × 107 | 1.97 | 15368 |
|
| → OCHO + ˙OCH2S˙ | 38.3 | 6.9 × 10−15 | 3.17 × 1010 | 1.43 | 19349 |
|
| → CO + ˙SCH2O(O)H (oxide) | 49.8 | 4.7 × 10−24 | 5.65 × 101 | 3.74 | 23553 |
|
| → ˙OOCH2SCHO |
34.5 | 2.1 × 10−13 | 2.76 × 10−84 | 31.52 | 4894 | |
| OC˙OOCH2SH |
→ OCHOOCH2S˙ |
10.4 | 2.0 × 105 | 1.03 × 1010 | 0.98 | 4908 |
| → CO2 + ˙OCH2SH | 5.3 | 1.3 × 1010 | 3.59 × 10−1 | 4.41 | 243 | |
| → CO + ˙OOCH2SH | 9.4 | 1.8 × 105 | 2.89 × 10−9 | 6.42 | 1442 | |
| OCHOOCH2S˙ + O2 |
→ OCHOOCH2SOO˙ |
1.7 | 2.5 × 10−15 | 1.14 × 10−22 | 3.20 | 393 |
| OCHOOCH2SOO˙ |
→ OCHOOCH2S˙ + O2 |
7.7 | 1.5 × 107 | 3.40 × 1010 | 0.89 | 3812 |
| → OC˙OOCH2SOOH |
21.6 | 1.0 × 10−2 | 6.98 × 10−35 | 14.05 | 1785 | |
| → OCHOOCHS + HO2 |
(16.5)b | (6.8 × 100)b | ||||
| → OCHOOCH2S(O)O˙ |
(21.2)b | (1.1 × 10−3)b | ||||
| OC˙OOCH2SOOH |
→ OCHOOCH2SOO˙ |
13.4 | 1.6 × 104 | 4.04 × 10−32 | 13.08 | −2230 |
| → CO2 + ˙OCH2SOOH | ∼5.8c | ∼1 × 109c | ||||
The theoretical work by Jernigan et al.25 extensively looked at the OCS formation channels, finding that the HOOCH2SC˙O intermediate from HPMTF, and the OCHSC˙O radical from thioformic anhydride, are the main pathways to OCS formation, either directly from prompt decomposition or after further radical chemistry. Given the low rate coefficient for NO3 radicals with HPMTF and the anhydride, it is expected that the OCS yield with this oxidant will be negligible. In contrast, the fast reaction of Cl radicals, combined with the higher yield of H-abstraction from DMS and higher yield of HOOCH2SC˙O radicals from HPMTF compared to the OH-based oxidation, suggests that Cl radicals may be more effective in producing OCS from DMS. The remainder of the HPMTF oxidation scheme as it was derived for OH radicals (Fig. 1) is not expected to require changes for the Cl and NO3 oxidants.
4. Formation and destruction of thioperhemiacetal (TPHAC)
Quantum chemical calculations show that the energy barrier for TPHAC formation from HPMTF, 40.6 kcal mol−1, is a few kcal mol−1 lower than OH elimination, 42.8 kcal mol−1, both of which are lower than the energy release from formation of HPMTF (see Fig. 1S†). Jernigan et al.25 discuss the possibility of OH elimination from chemically activated HPMTF as a source of OCS. Updated RRKM master equation analysis analogous to those performed in that work indicate that the cyclisation required for TPHAC formation is entropically too unfavorable to contribute against prompt OH loss, and has negligible yield. Similarly, photoenergization of HPMTF could lead to TPHAC, where the required photon energy is only ∼700 nm (red light). Without enhanced energy content, however, the thermal rate of conversion of HPMTF to TPHAC is very slow, k(298 K) ∼10−16 s−1 (see Table 1). Perhemiacetal formation can be readily catalysed by molecules with a mobile H-atom, such as water and acids, where more acidic molecules are more effective. In the gas phase, this requires a collision with one such molecule, where Table 1 lists the catalyzed rate coefficients for conversion with water and the common volatile acids HCOOH and HNO3. With predicted bimolecular rate coefficients ≤10−21 cm3 molecule−1 s−1, atmospheric concentrations of these catalytic co-reactants are too low to make these reactions viable. Overall, thermal formation of TPHAC in the gas phase seems to be non-competitive against bimolecular or physical loss processes of HPMTF.In the aqueous phase, in contrast, HPMTF can be complexed with a co-reactant. Table 1 lists rate coefficients for isomerisation for HPMTF and TPHAC complexes with H2O, HCOOH, HNO3, H3O+ and H2O–H3O+. The results suggest that in an acidic aqueous phase where H2O and H3O+ are present, HPMTF and TPHAC could interconvert fast enough to instate a (near)equilibrium. The gas phase equilibrium constant of TPHAC over HPMTF was calculated as Keq(T) = 1.12 × 103T−1.85exp(519K/T), favoring the linear HPMTF over the cyclic TPHAC by a factor of 5.7 at 298 K.
Interconversion of HPMTF and TPHAC may thus be important in cloud droplets in the marine environment, which constitutes a major loss process for HPMTF.19,22,25,26 Franco et al.37 proposed that cloud droplets with dissolved carbonyl compounds may efficiently release their hydrated forms – typically only formed in the aqueous phase – to the gas phase. Similar to this, TPHAC could be released to the gas phase from the cloud droplets, by outgassing or droplet evaporation. Hence, atmospheric DMS oxidation in the marine environment might yield gas phase TPHAC despite the lack of viable gas phase formation pathways. All current observations of HPMTF, both in experiments and in the field, were performed by mass-spectrometric instruments, which may be incapable of distinguishing these isobaric compounds. At this time, we are thus unable to quantify TPHAC formation in such heterogeneous processes and, given that the TPHAC vapor pressure and Henry constant are also unknown, it is also not clear to what extent TPHAC will reside in the aqueous phase or gas phase, or even what the relative importance would be relative to HPMTF. Henry constants estimated using the Bond contribution method58 and VP/WSOL59 as implemented in the EPI Suite 4.11 (ref. 60) suggest that the accommodation of TPHAC (H = 4.4 × 104 (VP/WSOL method) to 3.5 × 106 (bond contribution method) M atm−1) is less favorable than for HPMTF (H = 1.3 × 104 (VP/WSOL) to 1.3 × 105 (bond) M atm−1), which could allow some TPHAC to be released to the gas phase.
Once formed, TPHAC can unimolecularly dissociate more easily than HPMTF (see Fig. 2), with a barrier of only 30.7 kcal mol−1 for breaking the O–O double bond (compared to ∼43 kcal mol−1 for HPMTF).
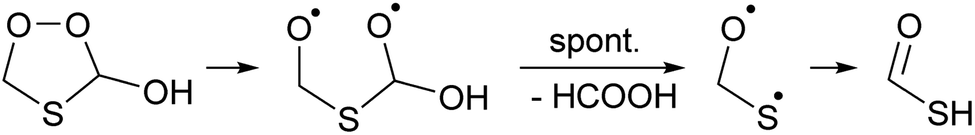
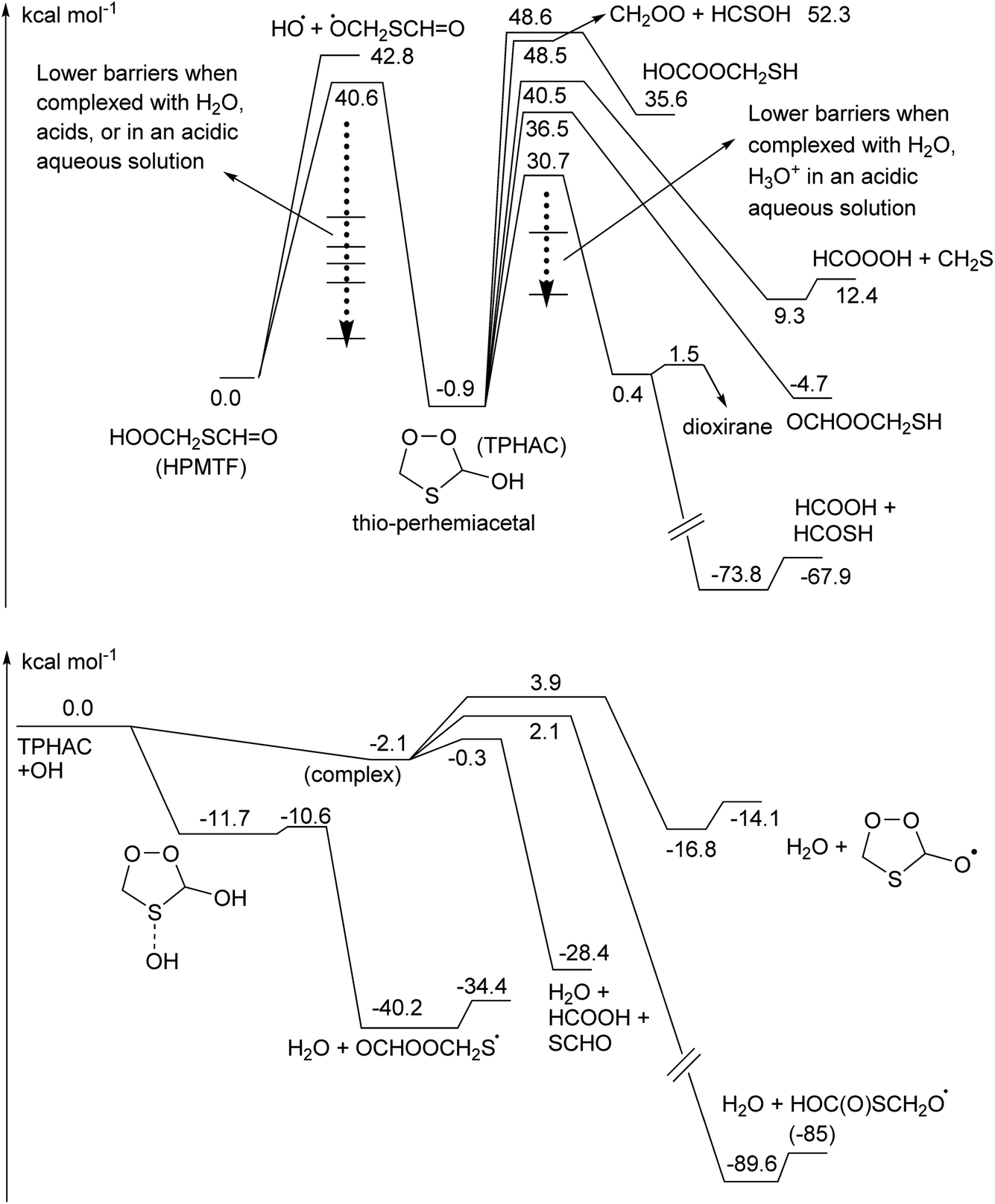 | ||
| Fig. 2 Potential energy surfaces for the chemistry of thioperhemiacetal (TPHAC), calculated at the CCSD(T)//M06-2X-D3 level of theory. (Top) Unimolecular decomposition channels. (Bottom) Reaction with OH radicals and subsequent reactions. | ||
The singlet bisoxy radical formed spontaneously eliminates formic acid, such that the products formed are HCOOH and the ˙OCH2S˙ singlet biradical, where the latter is expected to rearrange readily to thioformic acid similar to the singlet bisoxy to acid rearrangement known from ozonolysis reactions.61 As the HC(O)SH thioformic acid isomer is calculated to be 3.5 kcal mol−1 more stable than HC(S)OH, we assume here the former isomer will be formed predominantly. Formation of formic and thioformic acid was already proposed earlier e.g. ref. 30. Other decomposition and isomerisation pathways exist but have barriers that are higher by ≥6 kcal mol−1 and are unlikely to be competitive under atmospheric conditions (see Fig. 2). Complexation with H2O or H3O+ reduces the barrier height for dissociation by several kcal mol−1 (see Table 1), greatly enhancing the rate of reaction. In the aqueous phase, we thus expect the decomposition of TPHAC to HCOOH + HCOSH to be comparatively fast, with a 298 K lifetime of less than a second (see Table 1). We did not examine the further fate of the sulfur-bearing products, nor any competing reactions such as (catalyzed) concerted dissociation and H-migration leading directly to thioformic acid, or bimolecular reactions with other reaction partners or catalysts. The recent experiments by Jernigan et al.30 suggest that at least in some environments the products are converted near-exclusively to sulphates.
TPHAC, once formed in the aqueous phase, can be released to the gas phase. There, the reaction of TPHAC with OH radicals can proceed by either H-abstraction or addition (see Fig. 2 and Table 1). The H-abstraction reactions are a bit slower, k(298 K) ≅ 1 × 10−12 cm3 molecule−1 s−1, than for HPMTF, 7 × 10−12 cm3 molecule−1 s−1, as no suitable H-bonding is possible in the abstraction TS due to geometric constraints. In contrast, the addition process is barrierless, forming an >S–OH adduct with an energy −11.7 kcal mol−1 below the free reactants. From there, a rapid migration of the hydroxy-H-atom is possible with a deeply submerged barrier −10.6 kcal mol−1 below the reactants, forming OCHOOCH2S˙ radicals with an overall rate coefficient nearing the collision limit, ∼10−10 cm3 molecule−1 s−1. These theory-based rate estimates deviate from SAR predictions based on Kwok and Atkinson62 as implemented in the AOP program,63 where H-abstraction is estimated at k(298 K) = 4 × 10−11 cm3 molecule−1 s−1 with negligible addition reaction with the heteroatom. Fig. 3 shows an extensive theoretical characterization of the subsequent chemistry, which is analogous to that for HOOCH2S˙ characterized earlier,25 though the kinetics are rather different (see Table 1). In particular, the formate-H migrations in OCHOOCH2S˙ and OCHOOCH2SOO˙ are slower by many orders of magnitude compared to the hydroperoxide-H migrations in the HPMTF-derived HOOCH2S˙ and HOOCH2SOO˙ intermediates (see Fig. 1), making formation of OC˙OOCH2SH and OC˙OOCH2SOOH minor channels. For environments with O3 concentration similar to or above the global background concentration (≥10 ppb), the reaction of OCHOOCH2S˙ with O3 will be the dominant subsequent reaction, leading to SO2 formation. In conditions with less O3, the chemistry shifts to unimolecular reactions of the equilibrated HOOCH2SOO˙ intermediate. The work of Chen et al.20 for CH3SOO˙ suggests our level of theory may be less suited to calculate the ratio of HO2 elimination versus OCHOOCH2S(O)O˙ formation. As higher-level theoretical calculations are prohibitively expensive, we refrain from calculating these pathways and instead adopt the rearrangement to OCHOOCH2S(O)O˙ and subsequent SO2 formation as the dominant unimolecular loss process, by analogy with our earlier work and Chen et al. for HOOCH2SOO˙ and for CH3S˙.20,25
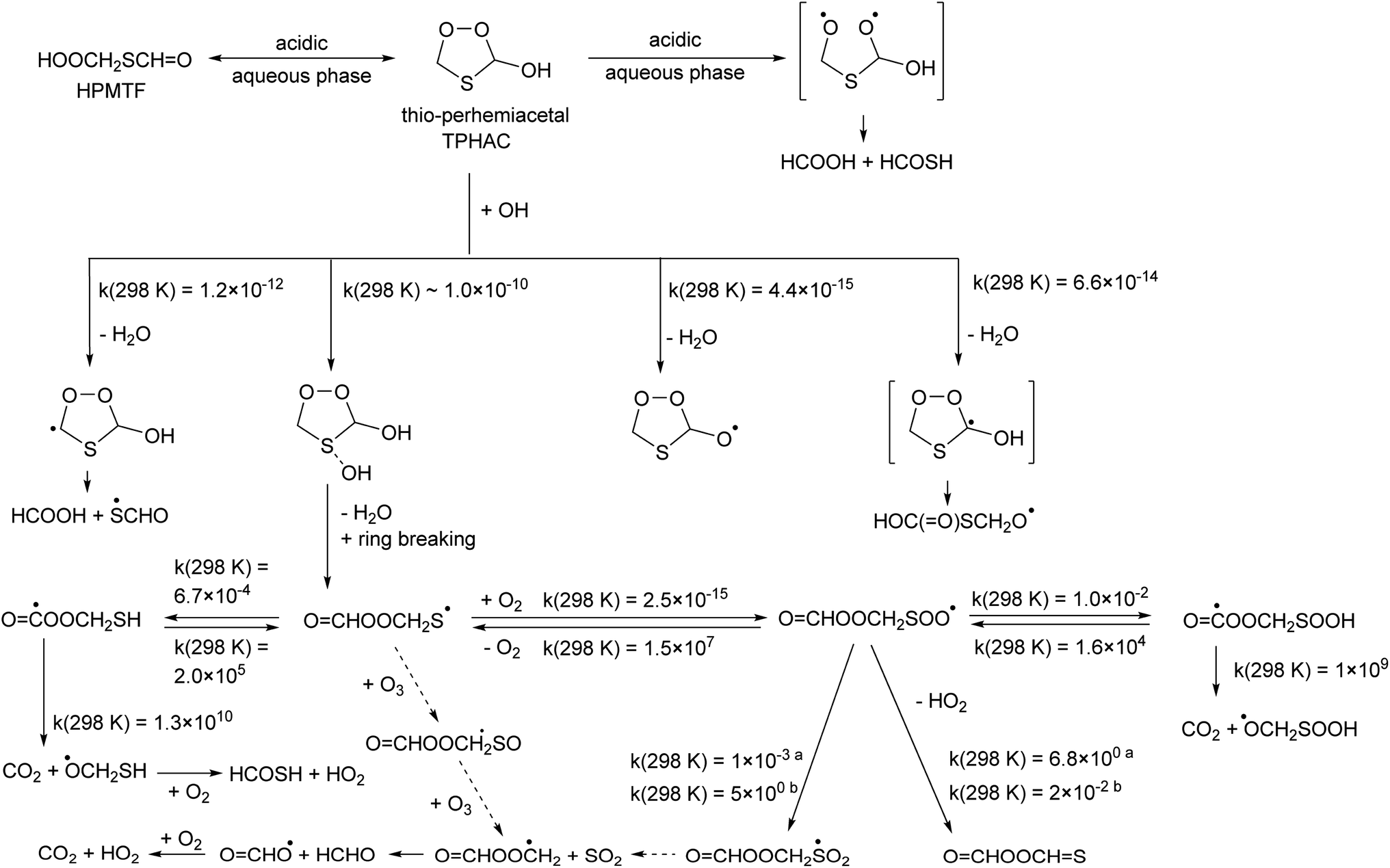 | ||
| Fig. 3 Thioperhemiacetal (TPHAC) formation from HPMTF, and subsequent OH-initiated oxidation steps based on the CCSD(T)//M06-2X-D3 theoretical calculations. Rate coefficients shown are for 298 K, in s−1 or cm3 molecule−1 s−1 as appropriate for the reaction molecularity. See text for more details on the OCHOOCH2SOO˙ chemistry; here both the rates from aJernigan et al.25 and bChen et al.20 are shown. | ||
TPHAC is not expected to react with O3, as it contains no double bonds, and the very fast addition channel is not accessible for Cl or NO3 co-reactants, making their reaction rates slower than with OH. Overall, the atmospheric gas phase lifetime of TPHAC is thus expected to be of the order of 3 hours, governed mostly by the reaction with OH, where none of the oxidation channels leads to OCS formation.
5. Chemical kinetic model
Based on the current work, we propose an extension of the kinetic model derived by theoretical methodologies in Jernigan et al.25 (see ESI sections S8 and S9† in that work for a detailed description). Here, we extend the model with the Cl and NO3 initiated chemistry for HPMTF, with formation of TPHAC from HPMTF, either directly or after water/acid-based catalysis, as well as with the unimolecular chemistry and OH-initiated oxidation of TPHAC.To identify the dominant gas phase products following TPHAC formation, a chemical box model run was performed using fixed concentrations of reagents comparable to those in the atmosphere over the ocean (OH = 1.2 × 106 cm−3, O3 = 20 ppbv, NO = 50 pptv, NO2 = 70 pptv) and initiated with an arbitrary starting concentration of TPHAC of 1.5 × 1010 cm−3. In these reaction conditions TPHAC reacts with OH with the majority of the reaction flux evenly distributed between formation of OCHOOCHS (after HO2 elimination from OCHOOCH2SOO˙) and the sum of SO2, CO2 and HO2 (formed after O3 reaction of OCHOOCH2S˙). A test run performed using the slower HO2 elimination and faster isomerisation rates for OCHOOCH2SOO˙ as proposed by Chen et al.20 shows formation of ∼30% OCHOOCHS and 70% SO2 + CO2 + HO2. Neither scenario allows for direct OCS formation; photolysis of OCHOOCHS products might lead to ˙OCHS radicals that convert to secondary OCS. If a fast TPHAC unimolecular loss is added with a rate constant comparable to those found for H3O+-catalyzed TPHAC dissociation, k ≥ 1 s−1, this channel becomes the sole loss process and formic + thioformic acid are the sole products from TPHAC.
The aqueous chemistry of HPMTF is currently not known, but given that all of its H-abstraction channels as well as the breaking of the weak O–O hydroperoxide bond allow for chemistry that leads to OCS formation, the uptake of HPMTF from gas to aqueous phase is by itself likely not sufficient to prevent OCS formation. Considering that the TPHAC chemistry does not show viable direct routes to OCS formation, conversion of HPTMF to TPHAC could block OCS formation compared to direct HPMTF chemistry, even if TPHAC is released back to the gas phase. The reduction in OCS yield is then directly proportional to the fraction of HPMTF converted to TPHAC; note that a sizable fraction of HPMTF needs to be removed, as direct HPMTF oxidation leads to ≥9% OCS formation25,27 while atmospheric models only support about 1% of OCS formation to close the OCS budget. The gas phase equilibrium constant for TPHAC over HPMTF favors the linear HPMTF molecule, with a ∼1:6 ratio at 298 K, but the lifetime of TPHAC is shorter than that of HPMTF. Overall, this favors subsequent reactions through the TPHAC isomer. The chemical box model allows visiting scenarios of varying HPMTF to TPHAC conversion rates and ratios. For example, a modeling run where HPMTF and TPHAC are allowed to rapidly reach gas phase equilibrium ratios results in a 1.7 times higher loss reaction flux through TPHAC than through HPMTF, despite the higher prevalence of HPMTF. This in turn leads to an OCS yield reduction by 15% compared to a scenario without TPHAC formation. The limited OCS yield reduction is mostly due to prompt OCS formation from hot nascent HPMTF which occurs before isomerisation to TPHAC; our theoretical work suggests hot formation of TPHAC is not competitive (see above). If a fast loss process for TPHAC is added, e.g. with a rate ≥1 s−1 as for H3O+-catalyzed HPMTF dissociation, OCS formation is reduced by a factor 2, i.e. leaving only prompt OCS formation from nascent hot HPMTF. This suggests that HPMTF, once in the aqueous phase, will no longer yield OCS due to conversion to (thio)formic acid.
The theory-based model includes several OCS-forming reactions from HPMTF but each of these carries a significant uncertainty, leading to large uncertainties in the predicted reduction in OCS yield by TPHAC formation. In realistic atmospheric conditions the contribution of TPHAC formation and loss also strongly depends on the multi-phase chemistry, including the impact of the composition of the aqueous droplets on the TPHAC formation and destruction rates, and its equilibrium with HPMTF. While the current study probes this chemistry and suggest very fast channels converting HPMTF over TPHAC to thioformic acid, it does not constitute an in-depth study able to describe the ultimate fate of HPMTF once it reaches the aqueous phase. The atmospheric impact can only be quantified in a full earth system model including the multiphase chemistry, which is beyond the scope of the current work.
6. Conclusions
Based on our theoretical calculations, we find that hydroperoxymethylthioformate, HPMTF, and thioformic anhydride, OCHSCHO, react readily with Cl atoms, which can be a significant loss process especially in the marine environment where DMS processing is critical. The ultimate yield of OCS is expected to be higher than for OH-based chemistry. In contrast, the reaction of HPMTF and thioformic anhydride with NO3 is slow, making this a negligible loss process and not a gas phase source of OCS.
It was shown theoretically that HPMTF can be converted to thioperhemiacetal, TPHAC. In the gas phase, the thermal isomerisation reaction is slow, but can be aided by photo-absorption or by chemical activation from HPMTF formation processes. While the gas phase isomerisation can be catalyzed by water and acids, the concentration of these catalysts is too low in the atmosphere to allow efficient thermal TPHAC formation through these routes. In contrast, in acidic aqueous solutions, the isomerisation between HPMTF and TPHAC is expected to be fast. This might also affect chamber studies, where the walls often have a slight film of water and acids; photo-energization in the aqueous phase may further enhance isomerisation.
The unimolecular decomposition of TPHAC is slow in the gas phase, but becomes fast upon catalysis with water and H3O+ present in the acidic aqueous phase where TPHAC is likely formed. Importantly, this decomposition leads to formic and thioformic acid, neither of which are expected to be converted to OCS rapidly. If TPHAC were to be released to the gas phase, e.g. by droplet evaporation or outgassing, the reaction with OH radicals is expected to be very fast, with rate coefficients close to the collision limit, and proceeds predominantly by OH-addition and H2O elimination with spontaneous ring breaking. The dominant route proceeds over the OCHOOCH2S˙ radical, but extensive characterization of the subsequent radical chemistry does not reveal first generation pathways yielding OCS. The isomerisation of HPMTF to TPHAC, and the subsequent decomposition of TPHAC may thus explain the lack of OCS formation once HPMTF transitions to the aqueous phase. The main sulfur product is predicted to be thioformic acid, though in oxidative conditions other products may arise, such as the sulphates observed by Jernigan et al.30
The theoretical predictions in this work yield a mechanistic view on the HPMTF fate that is compatible with the current experimental data, but needs direct experimental validation as well as further theoretical work. In particular the gas phase/aqueous phase exchange of HPMTF and TPHAC, their aqueous phase chemistry, as well as the role of chemical activation, catalysis, and photolysis on the subsequent fate of these intermediates remains ill-characterized.
Data availability
Data for this article, including an additional potential energy surface and the kinetic model have been included as part of the ESI.† The replication data for the quantum chemical calculations are available at Jülich DATA at https://doi.org/10.26165/JUELICH-DATA/8MYZUY.Author contributions
All authors contributed to the conceptualization and writing of the manuscript. LV performed the theoretical calculations, while AN and DT implemented and applied the chemical model. AW provided funding and resources.Conflicts of interest
There are no conflicts of interest to declare.References
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, San Diego, 1999 Search PubMed.
- I. Barnes, J. Hjorth and N. Mihalopoulos, Chem. Rev., 2006, 106, 940–975 CrossRef CAS PubMed.
- H. Korhonen, K. S. Carslaw, D. V. Spracklen, G. W. Mann and M. T. Woodhouse, J. Geophys. Res. Atmos., 2008, 113, D15204 Search PubMed.
- S. Kremser, L. W. Thomason, M. Von Hobe, M. Hermann, T. Deshler, C. Timmreck, M. Toohey, A. Stenke, J. P. Schwarz, R. Weigel, S. Fueglistaler, F. J. Prata, J.-P. Vernier, H. Schlager, J. E. Barnes, J.-C. Antuña-Marrero, D. Fairlie, M. Palm, E. Mahieu, J. Notholt, M. Rex, C. Bingen, F. Vanhellemont, A. Bourassa, J. M. C. Plane, D. Klocke, S. A. Carn, L. Clarisse, T. Trickl, R. Neely, A. D. James, L. Rieger, J. C. Wilson and B. Meland, Rev. Geophys., 2016, 54, 278–335 CrossRef.
- V. Brovkin, V. Petoukhov, M. Claussen, E. Bauer, D. Archer and C. Jaeger, Clim. Change, 2009, 92, 243–259 CrossRef CAS.
- T. S. Bates, B. K. Lamb, A. Guenther, J. Dignon and R. E. Stoiber, J. Atmos. Chem., 1992, 14, 315–337 CrossRef CAS.
- A. Lana, T. G. Bell, R. Simo, S. M. Vallina, J. Ballabrera-Poy, A. J. Kettle, J. Dachs, L. Bopp, E. S. Saltzman, J. Stefels, J. E. Johnson and P. S. Liss, Global Biogeochem. Cycles, 2011, 25, GB1004 CrossRef.
- K. S. Carslaw, L. A. Lee, C. L. Reddington, K. J. Pringle, A. Rap, P. M. Forster, G. W. Mann, D. V. Spracklen, M. T. Woodhouse, L. A. Regayre and J. R. Pierce, Nature, 2013, 503, 67–71 CrossRef CAS PubMed.
- P. Van Rooy, K. L. Purvis-Roberts, P. J. Silva, M. J. Nee and D. Cocker, Atmos. Environ., 2021, 256, 118148 CrossRef CAS.
- P. Van Rooy, R. Drover, T. Cress, C. Michael, K. L. Purvis-Roberts, P. J. Silva, M. J. Nee and D. Cocker, Atmos. Environ., 2021, 261, 118504 CrossRef CAS.
- T. Berndt, E. H. Hoffmann, A. Tilgner, F. Stratmann and H. Herrmann, Nat. Commun., 2023, 14, 4849 CrossRef CAS.
- X. Zhang, J. Chen and B. Long, J. Chem. Theor. Comput., 2024, 1237, 114641 CrossRef CAS.
- M. B. Goss and J. H. Kroll, Atmos. Chem. Phys., 2024, 24, 1299–1314 CrossRef CAS PubMed.
- R. Wu, S. Wang and L. Wang, J. Phys. Chem. A, 2015, 119, 112–117 CrossRef CAS PubMed.
- A. Mardyukov and P. R. Schreiner, Acc. Chem. Res., 2018, 51, 475–483 CrossRef CAS PubMed.
- T. Berndt, W. Scholz, B. Mender, L. Fischer, E. H. Hoffmann, A. Tilgner, N. Hyttinen, N. L. Prisle, A. Hansel and H. Herrmann, J. Phys. Chem. Lett., 2019, 10, 6478–6483 CrossRef CAS.
- Z. Salta, J. Lupi, V. Barone and O. N. Ventura, ACS Earth Space Chem., 2020, 4, 403–419 CrossRef CAS.
- P. R. Veres, J. A. Neuman, T. H. Bertram, E. Assaf, G. M. Wolfe, C. J. Williamson, B. Weinzierl, S. Tilmes, C. R. Thompson, A. B. Thames, J. C. Schroder, A. Saiz-Lopez, A. W. Rollins, J. M. Roberts, D. Price, J. Peischl, B. A. Nault, K. H. Møller, D. O. Miller, S. Meinardi, Q. Li, J.-F. Lamarque, A. Kupc, H. G. Kjaergaard, D. Kinnison, J. L. Jimenez, C. M. Jernigan, R. S. Hornbrook, A. Hills, M. Dollner, D. A. Day, C. A. Cuevas, P. Campuzano-Jost, J. Burkholder, T. P. Bui, W. H. Brune, S. S. Brown, C. A. Brock, I. Bourgeois, D. R. Blake, E. C. Apel and T. B. Ryerson, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4505–4510 CrossRef CAS.
- M. P. Vermeuel, G. A. Novak, C. M. Jernigan and T. H. Bertram, Environ. Sci. Technol., 2020, 54, 12521–12529 CrossRef CAS.
- J. Chen, T. Berndt, K. H. Møller, J. R. Lane and H. G. Kjaergaard, J. Phys. Chem. A, 2021, 125, 8933–8941 CrossRef CAS PubMed.
- M. A. H. Khan, T. J. Bannan, R. Holland, D. E. Shallcross, A. T. Archibald, E. Matthews, A. Back, J. Allan, H. Coe, P. Artaxo and C. J. Percival, ACS Earth Space Chem., 2021, 5, 2577–2586 CrossRef CAS.
- G. A. Novak, C. H. Fite, C. D. Holmes, P. R. Veres, J. A. Neuman, I. Faloona, J. A. Thornton, G. M. Wolfe, M. P. Vermeuel, C. M. Jernigan, J. Peischl, T. B. Ryerson, C. R. Thompson, I. Bourgeois, C. Warneke, G. Gkatzelis, M. M. Coggon, K. Sekimoto, T. P. Bui, J. Dean-Day, G. S. Diskin, J. P. DiGangi, J. B. Nowak, R. H. Moore, E. B. Wiggins, E. L. Winstead, C. Robinson, K. L. Thornhill, K. J. Sanchez, S. R. Hall, K. Ullmann, M. Dollner, B. Weinzierl, D. R. Blake and T. H. Bertram, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110472118 CrossRef CAS PubMed.
- Q. Ye, M. B. Goss, G. Isaacman-VanWertz, A. Zaytsev, P. Massoli, C. Lim, P. Croteau, M. Canagaratna, D. A. Knopf, F. N. Keutsch, C. L. Heald and J. H. Kroll, ACS Earth Space Chem., 2021, 5, 2013–2020 CrossRef CAS.
- K. M. Fung, C. L. Heald, J. H. Kroll, S. Wang, D. S. Jo, A. Gettelman, Z. Lu, X. Liu, R. A. Zaveri, E. C. Apel, D. R. Blake, J.-L. Jimenez, P. Campuzano-Jost, P. R. Veres, T. S. Bates, J. E. Shilling and M. Zawadowicz, Atmos. Chem. Phys., 2022, 22, 1549–1573 CrossRef CAS.
- C. M. Jernigan, C. H. Fite, L. Vereecken, M. B. Berkelhammer, A. W. Rollins, P. S. Rickly, A. Novelli, D. Taraborrelli, C. D. Holmes and T. H. Bertram, Geophys. Res. Lett., 2022, 49, e2021GL096838 CrossRef CAS.
- C. M. Jernigan, C. D. Cappa and T. H. Bertram, J. Phys. Chem. A, 2022, 126, 4476–4481 CrossRef CAS PubMed.
- M. von Hobe, D. Taraborrelli, S. Alber, B. Bohn, H.-P. Dorn, H. Fuchs, Y. Li, C. Qiu, F. Rohrer, R. Sommariva, F. Stroh, Z. Tan, S. Wedel and A. Novelli, Atmos. Chem. Phys., 2023, 23, 10609–10623 CrossRef CAS.
- W. Scholz, J. Shen, D. Aliaga, C. Wu, S. Carbone, I. Moreno, Q. Zha, W. Huang, L. Heikkinen, J. L. Jaffrezo, G. Uzu, E. Partoll, M. Leiminger, F. Velarde, P. Laj, P. Ginot, P. Artaxo, A. Wiedensohler, M. Kulmala, C. Mohr, M. Andrade, V. Sinclair, F. Bianchi and A. Hansel, Atmos. Chem. Phys., 2023, 23, 895–920 CrossRef CAS.
- Q. Ye, M. B. B. Goss, J. E. E. Krechmer, F. Majluf, A. Zaytsev, Y. Li, J. R. R. Roscioli, M. Canagaratna, F. N. N. Keutsch, C. L. L. Heald and J. H. H. Kroll, Atmos. Chem. Phys., 2022, 22, 16003–16015 CrossRef CAS.
- C. M. Jernigan, M. J. Rivard, M. B. Berkelhammer and T. H. Bertram, ACS ES&T Air, 2024, 1, 397–404 Search PubMed.
- B. Long, Y. Q. Zhang, C. L. Xie, X. F. Tan and D. G. Truhlar, Reaction of Carbonyl Oxide with Hydroperoxymethyl Thioformate: Quantitative Kinetics and Atmospheric Implications, Research, 2024, 7, 0525 CrossRef.
- L. S. D. Jacob, C. Giorio and A. T. Archibald, Atmos. Chem. Phys., 2024, 24, 3329–3347 CrossRef CAS.
- P. J. Ziemann, J. Phys. Chem. A, 2003, 107, 2048–2060 CrossRef CAS.
- D. Pagonis and P. J. Ziemann, Aerosol Sci. Technol., 2018, 52, 1178–1193 CrossRef CAS.
- B. A. Cala, S. Archer-Nicholls, J. Weber, N. L. Abraham, P. T. Griffiths, L. Jacob, Y. M. Shin, L. E. Revell, M. Woodhouse and A. T. Archibald, Atmos. Chem. Phys., 2023, 23, 14735–14760 CrossRef CAS.
- J. Chen, J. R. Lane, K. H. Bates and H. G. Kjaergaard, Environ. Sci. Technol., 2023, 57, 21168–21177 CrossRef CAS PubMed.
- B. Franco, T. Blumenstock, C. Cho, L. Clarisse, C. Clerbaux, P.-F. Coheur, M. De Mazière, I. De Smedt, H.-P. Dorn, T. Emmerichs, H. Fuchs, G. Gkatzelis, D. W. T. Griffith, S. Gromov, J. W. Hannigan, F. Hase, T. Hohaus, N. Jones, A. Kerkweg, A. Kiendler-Scharr, E. Lutsch, E. Mahieu, A. Novelli, I. Ortega, C. Paton-Walsh, M. Pommier, A. Pozzer, D. Reimer, S. Rosanka, R. Sander, M. Schneider, K. Strong, R. Tillmann, M. Van Roozendael, L. Vereecken, C. Vigouroux, A. Wahner and D. Taraborrelli, Nature, 2021, 593, 233–237 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- T. H. Dunning, K. A. Peterson and A. K. Wilson, J. Chem. Phys., 2001, 114, 9244–9253 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi and S. Grimme, Phys. Chem. Chem. Phys., 2017, 19, 32184–32215 RSC.
- I. M. Alecu, J. Zheng, Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2010, 6, 2872–2887 CrossRef CAS PubMed.
- S. Kanchanakungwankul, J. L. Bao, J. Zheng, I. M. Alecu, B. J. Lynch, Y. Zhao and D. G. Truhlar, Database of Frequency Scale Factors for Electronic Model Chemistries (Version 5), http://comp.chem.umn.edu/freqscale/index.html.
- R. J. Bartlett and G. D. Purvis, Int. J. Quantum Chem., 1978, 14, 561–581 CrossRef CAS.
- J. C. Rienstra-Kiracofe, W. D. Allen and H. F. Schaefer, J. Phys. Chem. A, 2000, 104, 9823–9840 CrossRef CAS.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Eara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Mntgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian Inc., Wallington CT, 2016 Search PubMed.
- L. Vereecken and J. Peeters, J. Chem. Phys., 2003, 119, 5159–5170 CrossRef CAS.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- H. S. Johnston and J. Heicklen, J. Phys. Chem., 1962, 66, 532–533 CrossRef.
- E. Martínez, J. Albaladejo, E. Jiménez, A. Notario and Y. Díaz de Mera, Chem. Phys. Lett., 2000, 329, 191–199 CrossRef.
- M. Dagenais, J. W. C. Johns and A. R. W. Mckellar, Can. J. Phys., 1976, 54, 1438–1441 CrossRef CAS.
- M. E. Jenkin, S. M. Saunders and M. J. Pilling, Atmos. Environ., 1997, 31, 81–104 CrossRef CAS.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef CAS.
- P. Arathala, M. Katz and R. A. Musah, Phys. Chem. Chem. Phys., 2020, 22, 10027–10042 RSC.
- L. Zhu and J. W. Bozzelli, J. Phys. Chem. A, 2006, 110, 6923–6937 CrossRef CAS PubMed.
- W. M. Meylan and P. H. Howard, Environ. Toxicol. Chem., 1991, 10, 1283–1293 CrossRef CAS.
- W. B. Neely, in Environmental Exposure from Chemicals: Volume I, CRC Press, Boca Raton, FL, 1st edn, 2017, pp. 13–49 Search PubMed.
- US EPA, EPI Suite 4.11 Estimation Programs Interface Suite for Microsoft Windows, v.4.11 (version 4.11), 2024 Search PubMed.
- L. Vereecken and J. S. Francisco, Chem. Soc. Rev., 2012, 41, 6259–6293 RSC.
- E. Kwok and R. Atkinson, Atmos. Environ., 1995, 29, 1685–1695 CrossRef CAS.
- W. Meylan and P. Howard, Chemosphere, 1993, 26, 2293–2299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00134f |
| This journal is © The Royal Society of Chemistry 2025 |