Global-scale analysis of the effect of gas-phase Criegee intermediates (CIs) on sulphate aerosol formation: general trend and the importance of hydroxy radicals decomposed from vibrationally excited CIs†
Received
5th October 2024
, Accepted 25th February 2025
First published on 25th February 2025
Abstract
Stabilised Criegee intermediates (sCIs), which are formed in the atmosphere through the ozonolysis of alkenes, are known precursors of sulphate aerosols (SO42−(p)). Several previous studies have focused on the kinetics of sCI-related chemistry using both experimental and theoretical methods. Nonetheless, detailed evaluations of how the sCI affects global-scale SO42−(p) formation using chemical transport models (CTMs) have rarely been conducted. In this study, the impact of sCIs on SO42−(p) and other particulate matter was estimated using a global CTM by implementing approximately 100 chemical reactions associated with CI chemistry. The results suggest that sCIs contribute maximally less than 0.5% in remote areas, such as Amazon rainforests, Central Africa, and Australia. This value is lower than the previously estimated value, despite certain kinetic parameters related to CI chemistry being provisional due to insufficient data. Future work should focus on obtaining these kinetic parameters through experimental studies or theoretical calculations. The sCI that contributed the most to SO42−(p) formation was E-methyl glyoxal-1-oxide, which was generated by the ozonolysis of methyl vinyl ketone owing to its low-rate coefficient for the loss reaction of unimolecular decomposition and water vapour. The change in SO42−(p) enhanced the formation of secondary organic aerosols, whereas the reactions of the sCIs with NO2 decreased the formation of nitrate radicals. The results of the sensitivity analyses showed that in highly industrialised sites in China and India, OH radicals formed by the unimolecular decomposition of vibrationally excited CIs (vCIs) contributed to SO42−(p) formation, which maximally accounted for nearly ten times more than that of sCIs, whereas the contribution of vCIs and sCIs to SO42−(p) formation was estimated to be almost equal in rural and remote sites. The estimated sCI loss by HNO3 and organic acids was comparable to that of the unimolecular decomposition of sCIs and scavenging by water. This study provides full insight into the impact of gas-phase CI chemistry on a global scale.
Environmental significance
Atmospheric aerosols pose health risks to humans and animals and are also recognised as climate forcers. Therefore, understanding the fundamental chemistry of aerosol formation is crucial for the scientific community to address these issues. Criegee intermediates (CIs) are known precursors of sulphate and other atmospheric aerosols. While numerous studies have clarified the role of stabilised Criegee intermediates (sCIs) in aerosol formation, the impact of vibrationally excited Criegee intermediates (vCIs), which produce OH radicals through unimolecular decomposition, has been less explored. This study quantifies the significance of vCIs in aerosol formation and provides insights into Criegee chemistry using a global chemical transport model.
|
Introduction
Sulphate aerosols (SO42−(p)) are known as the nuclei of fine particulate matter (PM2.5), which has a negative impact on human health1–3 and contributes to the positive and negative radiative forcing.4–6 As described in Fig. 1, the ozonolysis of alkenes forms primary molozonide (POZ) and decomposes to vibrationally excited Criegee intermediates (vCIs) and carbonyl species.7–11 The vCIs then decompose to form hydroxy radicals (OH) or release energy through quenching in ambient air to form stabilised Criegee intermediates (sCIs).7–13 The sCIs are known to be strong oxidisers in the atmosphere, reacting with several air pollutants such as sulphur dioxide (SO2), nitrogen oxides (NOx), and organic acids.7–11 In particular, since the rate coefficient of the reaction between sCIs and SO2, which finally transforms into SO42−(p), was estimated to be high (approximately 10−11 cm3 molecule−1 s−1),7–11 sCIs have been considered as one of the main contributors of atmospheric SO42−(p) formation, and several studies analysed the impact of sCIs on SO42−(p) formation on both regional and global scales using chemical transport models (CTMs). Sarwer et al. included sCI-related chemical reactions to the CTM and estimated the contribution of sCIs to SO42−(p) formation, concluding that sCIs contributed 20% to 75% of total SO42−(p) formation in the U.S.14 Percival et al. conducted regional and global analyses of the effect of sCIs on SO42−(p) formation, and they concluded that the contribution of sCIs is as high as that of gas-phase oxidation of OH, especially in urban areas, although the global CTM could not resolve the impact of sCIs on SO42−(p) formation in the urban area due to the averaged emissions in each calculated grid.15 On the other hand, Nakamura et al. reported negligible contributions of sCIs to SO42−(p) formation in the Greater Tokyo Area (GTA), the metropolitan area of Japan.16 The estimated amount of sCIs in GTA was consistent with a previous study that estimated the amount of sCIs in the urban area of the U.K.17 Our previous study suggested that isoprene-derived sCIs enhance SO42−(p) formation in the forestry area of the Asian region, which was comparable to the rate of aqueous-S(IV) chemistries,18 although Newland et al. suggested the less importance of isoprene-derived sCIs to SO2 oxidation on a global scale.19 Vereecken et al. estimated several rate coefficients related to sCIs using quantum chemical calculations combined with transition state theory and showed the importance of unimolecular decompositions of sCIs to the total evaluation of sCI-related pollution by the global CTM.20 Other several studies also evaluated the impact of sCIs on atmospheric pollutants, such as SO2, organic acids, and peroxy radicals, by the CTM, sometimes combined with reaction rate coefficients derived from experiments and calculations using transition state theory.21–25
 |
| | Fig. 1 Reactions of alkene ozonolysis and formation of CI. | |
The impact of the CI chemistry on the atmosphere has not been analysed in detail. Most previous studies have investigated the atmospheric implications of the newly estimated rate coefficients or the impact of specific kinetics by applying the global CTM, especially focusing on the impact of sCIs on SO42−(p) formation. However, their impact on other aerosols has rarely been investigated. In addition, vCIs may have a more significant impact on SO42−(p) formation than sCIs. Khan et al. suggested that the OH formation through the unimolecular decomposition of vCIs contributes maximally 13% of total OH formation in the terrestrial rainforest and boreal forest regions.10 Also, Nakamura et al.16 implied that the formation of SO42−(p) due to incorporation of sCIs was mainly due to the increase in OH concentration caused by the unimolecular decomposition of vCIs, not the direct oxidation of SO2 by sCIs. Further studies should be conducted to investigate the role of vCIs in SO42−(p) formation through a comparative analysis of the contributions of sCIs and OH derived from vCIs.
This study aimed to reveal the impact of CI chemistry on SO42−(p) and other aerosol formation on a global scale and to compare the impact of sCIs and vCIs on SO42−(p) formation. This study comprised three steps: first, we applied the detailed sCI-related chemical mechanisms proposed by Nakamura et al.,16 most of which were originally based on the reactions of Vereecken et al.20 to the global CTM to evaluate the effect of sCIs on SO42−(p) formation on a global scale. Second, we investigated sCI impact on ionic and SOA formation in addition to SO42−(p) to show the cross-effect of sCIs on other particulate species. Third, we conducted a sensitivity analysis to compare the contribution of SO42−(p) formation between sCIs and OH derived from the unimolecular decomposition of vCIs and the contribution of other sCI loss reactions. Although there have been many previous studies related to the impact of gas-phase reactions of sCIs on the atmosphere using the CTM, this study contributes to providing new insights into the atmospheric contribution of sCIs and vCIs and the fundamental information of the whole CI chemistry.
Methodology
Global chemical transport modelling
The Goddard Earth Observing System Chemistry model (GEOS-Chem v14.2.3)26 was used as the global CTM. The modern era retrospective analysis for research and applications version 2 (MERRA2) was used as the meteorological input with a grid resolution of 2° × 2.5° (latitude × longitude) holding 72 vertical layers from the troposphere to the stratosphere.27 Emission inventories provided by the harmonized emission component (HEMCO) system were applied in this study.28 Briefly, the emission database for the community emission data system (CEDSv2) was chosen for anthropogenic emissions,29 hemispheric transport of air pollution (HTAPv3) was chosen for ship emissions,30 monthly mean data of the Aviation Emissions Inventory (AEIC2019) was chosen for aircraft emissions,31 global fire emission database (GFED) was chosen for biomass burning,32 Aerosol Comparisons between Observations and Models (AeroCom) was selected for the SO2 emissions from volcanoes,33 and the off-line model of emissions of gases and aerosols from nature (MEGAN) was chosen for BVOC emissions.34 The targeted period calculated in this study spanned from 1 July 2018 to 31 December 2019. The first six months (1 July 2018 to 31 December 2018) were treated as the spin-up period, and the entire year of 2019 was treated as the analysis term. The reactions of the Criegee intermediates (CIs) were added to the full chemistry module of the kinetic pre-processor (KPP). The added CI reactions were based on Nakamura et al., who analysed the impact of sCIs on SO42−(p) formation in the GTA of Japan.16 Nakamura et al. cited several reaction kinetics from the findings of Vereecken et al., who determined the rate coefficients of sCI atmospheric reactions using quantum chemical calculations coupled with transition state theory.20 The default KPP has already implemented the chemical reactions of CIs generated by the ozonolysis of ethylene and propene. In this study, those CI chemistries were excluded to unify the reactions applied in Nakamura et al.16 The details of the added reactions are described in a later section.
Validation of the modelling results using satellite data
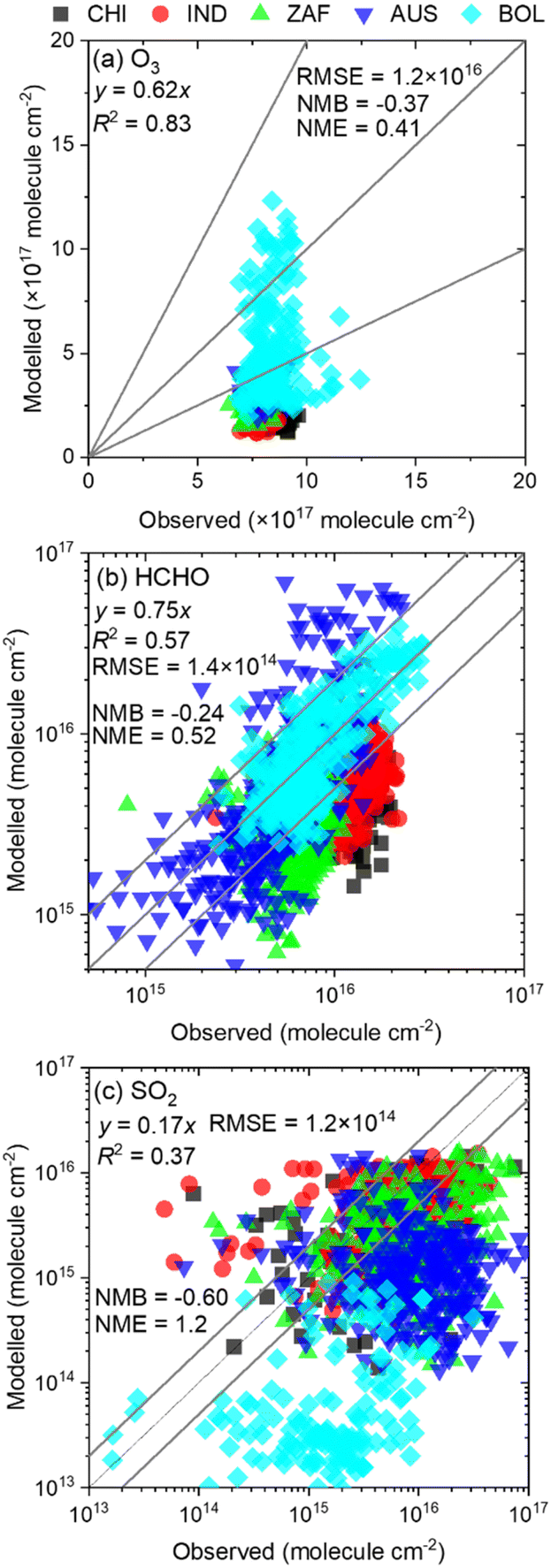
Validation of modelling results on a regional scale was typically conducted through comparison with ground observations. However, considering the coarse resolution of our analysis (2° × 2.5°), a direct comparison with ground observations is challenging. The results were validated by comparison with satellite observations from the tropospheric monitoring instrument (TROPOMI).35–37 Alkenes, O3, and SO2 are the main species forming sCIs and SO42−(p), and comparing these species would be ideal for model validation. Owing to the lack of satellite observations of alkenes, HCHO, which has been widely used as an indicator of VOC emissions (including alkenes),38 was used, along with tropospheric O3 and SO2. Level 2 data for HCHO, tropospheric O3, and SO2 were retrieved from COPERNICUS (O3; spatial resolution: 30 × 30 km2)39 and NASA Earth Observation Data (HCHO and SO2; special resolution: 7 × 3.5 km2).40 Datasets were screened based on the recommended quality flags (<0.5) to produce Level 3 data, solar zenith angle < 85°, and cloud fraction < 30%.39 The TROPOMI data were re-gridded to equalize the spatial resolution to that of GEOS-Chem (2° × 2.5°) via linear interpolation of the targeted grids. TROPOMI provides daytime data on the column densities of pollutants, which were compared with the daily average values calculated by GEOS-Chem. The determination factor (R2), root mean square error (RMSE), normalised mean bias (NMB), and normalised mean error (NME) were chosen as statistical indicators to verify the correlation between the observed and calculated results. Globally, five sites were selected for comparison: China (CHI), India (IND), South Africa (ZAF), Australia (AUS), and Bolivia (BOL: corresponding to the Amazon rainforests), where the impacts of sCIs were estimated to be high according to this study, the details of which are described in a later section. Detailed information on the five sites is provided in Table S1 of the ESI.†
Incorporated chemical reactions
The chemical mechanisms of the ozonolysis reactions of 8 alkenes (ethylene (C2H4), propene (PRPE), methyl vinyl ketone (MVK), methacrolein (MACR), isoprene (ISOP), pinene (MTPA), other monoterpenes (MTPO), and limonene (LIMO)), unimolecular reactions sCIs, and eight types of bimolecular reactions between sCIs and air pollutants (water monomer (H2O), water dimer ((H2O)2), nitrogen dioxide (NO2), sulphur dioxide (SO2), nitric acid (HNO3), formic acid (HCOOH), acetic acid (CH3COOH), and organic acids with more than C3-chains (RCOOH)) were incorporated into KPP. There were eight types of sCIs generated from alkene ozonolysis: formaldehyde oxide (SCI1), acetaldehyde oxide (SCI2), methyl vinyl ketone oxide (SCI3), methacrolein oxide (SCI4), two types of pinene oxides (SCI5 and SCI6), methyl glyoxal-1-oxide (SCI7), and methyl glyoxal-2-oxide (SCI8), of which all the sCIs aside from SCI1 are E- and Z-isomers. Detailed structures of the sCIs are shown in Fig. S1 of the ESI.† Most of the chemical mechanisms and rate coefficients are cited from Nakamura et al.16 Additionally, they analysed the effect of sCIs on SO42−(p) formation in the GTA using the community multiscale air quality modelling system (CMAQ)41 with the SAPRC-07 chemical mechanism,42 and several expressions of the chemical species were different from those of KPP. The corresponding species were chosen to substitute the SAPRC-07 components with KPP. The added reactions are summarised in Tables S2 to S10 of the ESI.†
Alkene ozonolysis
Table S11 of the ESI† lists the incorporated alkene ozonolysis reactions and rate coefficients whose information is originally cited from Master Chemical Mechanism (MCM).43 The default reactions of alkenes and O3 defined in KPP were substituted by the reactions listed in Table S11† because MCM has more detailed chemistries of sCI generation. The 15 sCI structures shown in Fig. S1 of the ESI,† including E- and Z-isomers of several sCIs, were generated by ozonolysis of the alkenes. The branching ratio of E- and Z-isomers was assumed to be 0.5.16,18 The alkene ozonolysis reactions leading to the generation of sCIs and OH are shown in Fig. S1.† For example, ethylene ozonolysis is defined as follows:| | | C2H4 + O3 → 0.37SCI1 + 0.13OH + … | (R1) |
The formation of OH by (R1) can be considered the end product of the unimolecular decomposition of vCIs.
Unimolecular decomposition of sCIs
Unimolecular decomposition of sCIs is one of the major pathways of sCI loss,20 and Table S2† lists the chemical mechanisms of the unimolecular reaction of sCIs and their kinetic parameters. Three types of unimolecular decompositions, 1,3-ring closure, 1,4-H-migration, and 1,5-ring closure, were considered, and the details of these reactions are described by Vereecken et al.20
Reactions of sCIs with H2O and (H2O)2
The reactions of sCIs with water monomers (H2O) and dimers ((H2O)2) are also one of the major paths for the loss of sCIs; Tables S3 and S4 of the ESI† list the chemical mechanisms and kinetic parameters of the reaction. Note that the kinetic parameters of the reactions of sCIs and (H2O)2 depend on the ratio of H2O to (H2O)2 in the atmosphere, which is described by the equilibrium constant Kw, expressed by eqn (1) according to a previous study.44| |  | (1) |
Further details of the derivation of the rate coefficients listed in Table S4† have been described in a previous study.16
Reactions of sCIs with SO2, NO2, HNO3, and organic acids
Tables S5–S11† show the incorporated reactions and kinetic parameters of the reactions between sCIs and SO2, NO2, HNO3, and organic acids. The main difference from Nakamura et al. is the definition of the reactions between sCIs and NO2. In a previous study,16 the products of sCIs + NO2 reactions were determined as NO3 and aldehydes (or ketones). Caravan et al.45 found out that the main product of the reaction was the sCIs–NO2 adduct, and only approximately 30% of sCIs–NO2 adduct decomposed to NO3 and aldehydes (or ketones), indicating that NO3 formation through the sCIs + NO2 reaction was overestimated by Nakamura et al.16 For this reason, in this study, we multiplied 0.3 with NO3 and aldehyde (or ketone) as the stoichiometric factor to incorporate the findings of Caravan et al.45
Sensitivity analysis to compare the contributions of sCIs and vCIs in SO42−(p) formation
Sensitivity analysis was conducted to reveal the important CI-related reaction scheme for SO42−(p) formation. Seven scenarios were used to evaluate sensitivity: unimolecular decomposition of vCIs (S1), reactions of sCIs with SO2 (S2), unimolecular decomposition of sCIs (S3), reactions of sCIs with water (S4), reactions of sCIs with NO2 (S5), reactions of sCIs with HNO3 (S6), and reactions of sCIs with organic acids (S7). The reactions evaluated in scenarios S1 and S2 contributed to SO42−(p) formation through SO2 oxidation by OH or sCIs. The other scenarios (S3–S7) were loss reactions of sCIs, which indirectly contributed to SO42−(p) formation. Note that although scenario S1 focuses on the contribution of vCIs to SO42−(p) formation, the exact species of vCIs are not defined by the chemical mechanism of this study. This is because vCIs are intermediate species in the ozonolysis reaction of alkenes and are either immediately decomposed into OH or stabilised through collisions with bath gas to form sCIs. Thus, scenario S1 only calculates the total sensitivity to ozonolysis minus the sensitivity to the reactions of sCIs. To evaluate the contribution of each process to SO42−(p) formation, the sensitivity, Si, of reaction scheme i was formulated as follows:| |  | (2) |
where ki is the rate coefficient of the targeted reaction scheme i and δki is the functional derivative of ki. [SO42−(p)] refers to the column density of SO42−(p) in the scenario with Criegee chemistry and δ[SO42−(p)] refers to the change in [SO42−(p)] after the reaction rates were multiplied by (1 + δki/ki). In this study, the value of δki/ki was set to 0.1. For example, the sensitivity of sCIs and SO2 reactions to SO42−(p) formation was calculated by multiplying the rate coefficients of all sCIs + SO2 reactions by 1.1 and investigating the change in the column density of SO42−(p). In terms of sensitivity scenario S1, the rate coefficients of the ozonolysis reactions of alkenes were multiplied by 1.1; however, this operation also led to the enhancement of later sCI-related reactions, which resulted in including both the effects of vCIs and sCIs on the sensitivities of SO42−(p) and other aerosol formation. To account for this problem, another scenario, S8, was calculated, which contained the operation of multiplying 1.1 by the rate coefficients of all sCI-related chemical reactions. The results of scenario S1 were then subtracted by the results of scenario S8 to ensure that S1 held only the sensitivities of the vCIs to aerosol formation. In the Results and discussion section, the sensitivity of S1 contains the subtracted result for scenario S8.
Results and discussion
Evaluation of the modelling performance in comparison with the observed data of TROPOMI
Fig. 2 shows the relationships between the observed and modelled column densities of O3, HCHO, and SO2. According to Fig. 2(a) and (b), O3 and HCHO showed high determination coefficients with R2 = 0.83 and 0.57, respectively, while the value of R2 for SO2 was 0.37, indicating a moderate linear correlation between the observed and modelled results. The slopes of all three chemicals were less than 1.0, with SO2 showing the lowest value, followed by O3 and HCHO. This suggests that the modelled results underestimated the observed results, especially in SO2. One of the emission sources of SO2 is anthropogenic activities, and thus the emission of SO2 is highly dependent on time. Fig. 2 shows a comparison between the 1 h observed data by TROPOMI during the daytime and the daily averaged modelled results by GEOS-Chem, and this is the reason why the modelled results underestimated the daytime observational results. Similarly, O3 exhibited higher concentrations during daytime compared to the nighttime, as it is generated by the photochemical reaction of NO2 and the related HOX cycle; thus, the modelled results underestimated the observed data due to the large concentration difference between daytime and nighttime. In contrast, the sources of HCHO are mostly secondary products of atmospheric oxidation of VOCs including nocturnal reactions. Therefore, HCHO is produced at all times of the day, leading to a relatively small underestimation of the modelled HCHO compared to the observed results. In addition, the results obtained using TROPOMI were linearly interpolated to fit the grid resolution of GEOS-Chem, which may have caused errors between the observed and calculated results. Comparisons between TROPOMI and GEOS-Chem were conducted using the column density rather than the surface mixing ratio, which may have contributed to error accumulation across vertical layers. These complex analyses resulted in relatively large discrepancies between TROPOMI and GEOS-Chem. Additionally, the accuracy of emission inventories for air pollutants is not perfect, as these inventories are estimated using statistical methods. Collectively, these factors influenced the observed differences between TROPOMI and GEOS-Chem. Overall, although discrepancies were observed between TROPOMI and GEOS-Chem for SO2, the calculated column densities of O3, HCHO, and SO2 correlated with the observed data in terms of determination coefficients, indicating an acceptable agreement between the datasets.
 |
| | Fig. 2 Comparison of observed and modelled results of the column density of (a) tropospheric O3, (b) HCHO, and (c) SO2. HCHO and SO2 are shown in the scale of common logarithm for readability. | |
Distribution of sCIs formed by alkene ozonolysis in 2019
Fig. 3 shows the annual average column density of the total sCIs in 2019 calculated using GEOS-Chem. The column density was high in the equatorial regions of the Amazon rainforests, Australia, Central Africa, Southeast Asia, and some countries of Central America and North America, including the U.S. and Mexico. The column densities of the precursors of the sCIs, O3, and total alkenes are shown in Fig. S2 of the ESI.† In a comparison between Fig. 3 and S2,† the abundance of sCIs was nearly correlated with the abundance of alkenes. Fig. S3–S8 of the ESI† show the column density of the detailed species of the sCIs, and Fig. S9† provides a detailed description of the alkene species. According to Fig. S3–S8,† SCI1 (formaldehyde oxide), ESCI3 (E-methyl vinyl ketone oxide), ESCI4 (E-methacrolein oxide), and ESCI7 (methyl glyoxal-1-oxide) were the main contributors of the total amount of sCIs. While SCI1 is generated from both anthropogenic and biogenic alkenes, ESCI3 and ESCI4 are generated from isoprene, a biogenic alkene, and ESCI7 is generated from methyl vinyl ketone, a byproduct of isoprene oxidation. The column density of ESCI7 was the highest among all sCIs. The high concentration of ESCI7 resulted from the low rates of unimolecular decay and ESCI7 and water reactions. Because of the geometrical structure of ESCI7, only 1,3-ring-closure was considered for the unimolecular isomerisation of ESCI7. For example, ESCI3, which is generated by isoprene ozonolysis, involves two unimolecular isomerisation processes: 1,3-ring closure and 1,4-H-migration. The rate coefficients of the 1,3-ring closure for ESCI7 and ESCI3 and of 1,4-H-migration for ESCI3 at 298 K were 0.058 s−1, 0.0040 s−1, and 51 s−1, respectively, calculated using the values listed in Table S2 of the ESI.† Similarly, assuming the concentration of water at 298 K as 8.3 × 1017 molecule cm−3,46 the reaction rates of SCI1, ESCI3, ESCI4, and ESCI7 with water were calculated to be 2866 s−1 (716 s−1 for H2O and 2150 s−1 for (H2O)2), 0.525 s−1 (0.065 s−1 for H2O and 0.460 s−1 for (H2O)2), 668 s−1 (158 s−1 for H2O and 510 s−1 for (H2O)2), and 22.0 s−1 (2.50 s−1 for H2O and 19.5 s−1 for (H2O)2), respectively, as calculated from the values listed in Tables S3 and S4 of the ESI.† Thus, because of the relatively high concentration of MVK in the atmosphere, which is shown in Fig. S9 of the ESI,† and because of the lower reaction rate in the main loss processes, which enabled a longer atmospheric lifetime, ESCI7 showed a high concentration in the atmosphere, as discussed by Vereecken et al.20 For similar reasons, E-isomers of sCIs are more dominant than Z-isomers because of the high reactivity of Z-isomers to unimolecular isomerization.20 Some of the previous studies suggested the importance of (E- and Z-)SCI3 and (E- and Z-)SCI4 as atmospheric oxidisers because SCI3 and SCI4 are directly generated from the ozonolysis of isoprene, which is the most relevant VOCs on a global scale.18,19 Nevertheless, this study showed the potential importance of ESCI7 as the most relevant atmospheric oxidiser among the sCIs owing to its long atmospheric lifetime. Therefore, further studies on the kinetics of ESCI7 in the atmosphere should be conducted in the future. Fig. S9† suggests that ethylene, isoprene, and methyl vinyl ketone are the relevant alkenes in the atmosphere and that ethylene is emitted from both anthropogenic and biogenic sources. Combining these results, most sCIs are formed from biogenic sources, particularly isoprene, on a global scale. The seasonal trend of the column density of sCIs in 2019 is shown in Fig. S10 of the ESI,† where the year is divided into four seasons: January–March (JFM), April–June (AMJ), July–September (JAS), and October–December (OND). In the Northern Hemisphere, the seasons of AMJ and JAS exhibit relatively high concentrations, particularly in the southeastern regions of the United States and the eastern part of Russia (Siberia). This is because AMJ and JAS correspond to warm to hot seasons in the Northern Hemisphere, during which high amounts of BVOCs are emitted. Conversely, except for the Amazon rainforest, the seasons of JFM and OND show relatively high concentrations of sCIs in the Southern Hemisphere, also due to high-temperature seasons in those regions. Although the Amazon rainforest is located in the equatorial region and the Southern Hemisphere, its rainy season during JFM, AMJ, and OND results in JAS having the highest concentration of sCIs.
 |
| | Fig. 3 Annual mean total column density of sCIs in 2019. | |
Impact of Criegee chemistry on sulphate formation
Fig. 4 shows the calculated results of the column densities of SO42−(p) for (a) annual mean SO42−(p) (μg cm−2), (b) the change in SO42−(p) due to Criegee chemistry (μg cm−2), and (c) the ratio of the change in SO42−(p) (%). According to Fig. 4(a), SO42−(p) is distributed globally, but its concentration is high in the Northern Hemisphere, where countries such as China, India, and Middle East are located, which results in high SO2 emissions from the burning of fossil fuels, as shown in Fig. S2(b).† According to Fig. 4(b), the change in SO42−(p) was significant in China and India, both of which emit substantial amounts of SO2 due to the fossil fuel combustion, and in the Amazon rainforests, Central Africa to South Africa, and Australia, which are dominated by forests, causing high BVOC emissions and high sCI concentrations, as shown in Fig. 3. Thus, the effect of Criegee chemistry on SO42−(p) formation was high in high-SO2 and BVOC emissions. Fig. 4(c) demonstrates that the contribution of Criegee chemistries to the rate of change of SO42−(p) is maximum less than 0.5% in the Amazon rainforests, Australia, and Central Africa, the value of which is not drastic in terms of the air pollution issue. The percentage contribution of Criegee chemistry to SO42−(p) formation is high in those regions because the background concentration of SO42−(p) is low in remote areas, so the incorporation of the Criegee chemistry showed higher responsiveness in those regions. As mentioned above, SCI1, ESCI3, ESCI4, and ESCI7 were the dominant sCIs on a global scale, and these four sCIs mainly contributed to the formation of SO42−(p). Seasonal trends in the column density of SO42−(p) are shown in Fig. S11–S14 of the ESI,† with the seasons divided in the same manner as in Fig. S10:† January–March (JFM), April–June (AMJ), July–September (JAS), and October–December (OND). The absolute changes in SO42−(p) in the eastern part of China during JFM and OND, which correspond to the cool to cold seasons, show a significant increase due to high SO2 emissions driven by increased fossil fuel demand. In the Amazon rainforest, the changes in SO42−(p) are the highest during JAS, as JFM, AMJ, and OND coincide with the rainy seasons. The concentration of sCIs is also the highest during JAS, as discussed in the previous section.
 |
| | Fig. 4 (a) Annual mean column density of sulphate (g m−2), (b) the change in the column density of sulphate before and after the introduction of Criegee chemistry (mg m−2), and (c) the rate of change of sulphate before and after the introduction of Criegee chemistry (%). | |
The contributions of sCIs to the change in SO42−(p) shown in Fig. 4(c) are lower than the results of Newland et al., who evaluated the impact of monoterpene-derived sCIs on SO42−(p) formation, which suggested that sCIs from monoterpenes account for 1.2% of annual SO42−(p) formation in the terrestrial tropics.24 The difference might come from the included sCI chemical reactions. Newland et al. considered the reactions of sCIs with SO2, H2O (monomers), and the unimolecular decomposition of sCIs, whereas this study included additional reactions involving (H2O)2, NO2, HNO3, and three organic acids. The inclusion of these additional reactions would lower the concentration of sCIs, thereby leading to slower oxidation of SO2. Caravan et al. also suggested a 1–10% contribution of SO42−(p) formation in the terrestrial tropics from isoprene-derived Criegee intermediates (SCI3 in this study),23 but for the same reason as Newland et al.,24 the estimated contribution might be overestimated.
Effect of Criegee chemistry on NO3−(p), NH4+(p), and SOA formation
According to a previous study, the incorporation of Criegee chemistry led to not only an increase in SO42−(p) but also a change in other particulate species owing to the change in ionic balance and the enhancement of particle formation through nucleation around the surface of SO42−(p).18 These particulate species include nitrate aerosol (NO3−(p)), ammonium aerosol (NH4+(p)), and secondary organic aerosol (SOA), which are defined in ISORROPA II implemented in GEOS-Chem.47Fig. 5 shows the change in the composition of particulate matter at five sites: CHI, IND, ZAF, AUS, and BOL (Table S1†), which showed high SO42−(p) formation caused by Criegee chemistry incorporation, as shown in Fig. 4. According to Fig. 5(a), all sites showed an increase in SO42−(p) and SOA, the results of which are consistent with a previous study by Hata et al.18 Budisulistiorini et al. suggested that SOA formation is strongly correlated with the ambient concentration of SO42−(p), and a 25% decrease in SO42−(p) induces a 70% decrease in SOA.48 For this reason, the high increase in SOA shown in Fig. 5(a) is due to the increase in SO42−(p) induced by Criegee chemistry incorporation. On the other hand, a decrease in NO3−(p) was estimated for all the analysed sites, which is inconsistent with the results of a previous study by Hata et al.18 Hata et al. treated sCIs + NO2 reactions to completely form NO3 radicals. However, according to the findings of Caravan et al., only 30% of NO3 was estimated to be formed by sCIs + NO2 reactions, and the remaining NO2 was considered to form the adduct sCIs–NO2.45 This new finding was implemented in this study. Thus, the formation of sCIs–NO2 additives resulted in NO2 removal from the atmosphere, leading to a decrease in NO3−(p). As shown in Fig. 5(a), the decrease in NO3−(p) was large in CHI and IND, where highly anthropogenic air pollutants were likely to be emitted, as implied by the column density of SO2 (Fig. S2(b)†). These two sites emitted high amounts of NO, subsequently forming NO2via a reaction with O3, which caused a strong decline in NO3−(p) due to NO2 removal by sCIs. This suggests that in areas with high emission, the increases in SO42−(p) and SOA were cancelled by the reduction of NO3−(p), all of which were caused by Criegee chemistry incorporation. In contrast, the decrease in NO3−(p) was low at the remaining sites with relatively lower anthropogenic emissions. The behaviour of NH4+(p) differed in each site. Because NH4+ is the counter ion of SO42−, NO3−, and other anion species, NH4+(p) increased when the total cations increased (ZAF and AUS) and decreased when the total cations decreased (CHI and IND). According to Fig. 5(b), the change in column densities of SO42−(p), NO3−(p), NH4+(p), and SOA at the five sites ranged from −1.3% to 0.4% depending on the sites and species. The ratios were higher for AUS and BOL than for CHI and IND. This trend may have been due to the lower background concentrations of particulate species in AUS and BOL. The lower anthropogenic emissions led to lower particulate species in AUS and BOL, which have resulted in high responsiveness to concentration fluctuations derived from Criegee chemistry incorporation. In contrast, ZAF showed moderate anthropogenic emissions according to the column density of SO2 (Fig. S2(b)†), indicating an intermediate trend in the ratio (%) between the high- and low-emission sites.
 |
| | Fig. 5 Cross-effects of the change of SO42−(p) to NO3−(p), NH4+(p), and SOA by the introduction of Criegee chemistry for (a) absolute change and (b) ratio. | |
Sensitivity analysis of the rate coefficients of Criegee-induced chemical reactions for the formation of sulphate aerosol
vCIs vs. sCIs: Which CIs are important to SO42−(p) formation?
Fig. 6 shows the results of the sensitivity analysis of the vCIs and sCIs (scenarios S1 and S2) for SO42−(p) formation at the five sites estimated by eqn (2). Fig. 6 suggests that vCIs contributed more to the formation of SO42−(p) than sCIs at all analysed sites. In areas with high emissions, CHI, IND, and ZAF, the contribution of vCIs was much higher than that of sCIs, whereas in sites with lower anthropogenic emissions, AUS and BOL, the contribution was equal. At sites with high to moderate anthropogenic emissions, such as CHI, IND, and ZAF, bimolecular reactions between sCIs and other pollutants, such as NO2 and HNO3, occurred frequently, which decreased the oxidation of SO2 by sCIs. Thus, a relatively high vCI contribution to SO42−(p) formation was estimated in these sites. In contrast, in AUS and BOL, low concentrations of air pollutants led to a relatively high contribution of sCIs to SO42−(p) formation in AUS and BOL, which was almost comparable to the contribution from vCIs. Previous studies have separately investigated the atmospheric roles of vCIs and sCIs. Khan et al. suggested an approximately 13% contribution of OH formation by the unimolecular reactions of vCIs in the terrestrial rainforests and boreal forest regions.10 Numerous studies have focused on the importance of sCIs in the formation of SO42−(p) in both remote and urban areas.14–18,20 However, the contribution of SO42−(p) formation in vCIs and sCIs has not been compared in previous studies. This study quantitatively evaluates their contributions through a detailed sensitivity analysis. The OH from the unimolecular decomposition of vCIs is a more dominant oxidiser of SO2 than the direct oxidation of SO2 by sCIs, especially in urban areas.
 |
| | Fig. 6 Sensitivities of sCI and vCI chemical reactions to SO42−(p) formation in the five analysed sites. | |
Sensitivities of the sCI-loss reactions via unimolecular decomposition and water
Fig. 7 shows the sensitivity of the unimolecular decomposition of the sCIs and the reaction of the sCIs with water for the formation of SO42−(p) (scenarios S3 and S4). Most of the results showed a negative impact on SO42−(p) formation because both the unimolecular decomposition of sCIs and the reaction of sCIs with water are sCI-scavenging reactions, leading to a decrease in the total amount of SO42−(p) formation. Only the sensitivity of the unimolecular decomposition of sCIs in CHI showed a positive value, possibly because the products of the unimolecular decomposition of sCIs included OH and the precursors of OH, such as aldehydes, which surpassed the negative effect of the consumption of sCIs. At all the analysed sites, the sensitivity of the reaction of sCIs and water was higher than that of the unimolecular decomposition of sCIs, indicating that the reaction of sCIs with water is more important for the consumption of sCIs and subsequent reduction of SO42−(p) formation. Only BOL showed high sensitivity to the unimolecular decomposition of sCIs, which was almost equal to the sensitivity of water. This was due to the abundant sCIs derived from BVOCs in BOL, which are in the terrestrial Amazon rainforests and have intense BVOC emissions (Fig. S9†). Vereecken et al. showed that sCIs from BVOCs tend to be lost faster by unimolecular decomposition than sCIs from AVOCs,20 leading to high sensitivity of the unimolecular reactions of sCIs.
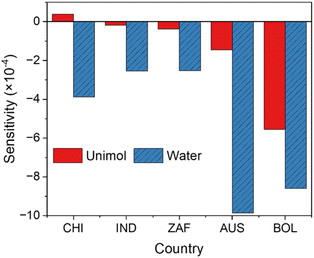 |
| | Fig. 7 Sensitivities of sCI-loss reactions by the unimolecular decomposition of sCIs (Unimol) and by the reaction of sCIs and water in the five analysed sites. | |
Cox et al.17 and Vereecken et al.20 estimated the contributions of sCI-loss reactions in urban and suburban areas and found that the contributions of unimolecular decomposition and water scavenging of sCIs are equal or that unimolecular decomposition is higher, and the results are opposite to the results shown in Fig. 7. This study investigated the temperature dependence of the ratio of H2O/(H2O)2, which differed from the results of previous studies. Furthermore, the contributions of the two sCI-loss processes were evaluated using the functional derivative of the sensitivity equation defined by eqn (2), while previous studies estimated the contributions by comparing the ratio of the rate coefficients between unimolecular decomposition and water weighed by the ambient concentrations.17,20 Therefore, it is difficult to completely compare the results between this study and previous studies due to the difference in the definition of the sensitivity and the sCI-chemical reactions included in this study.
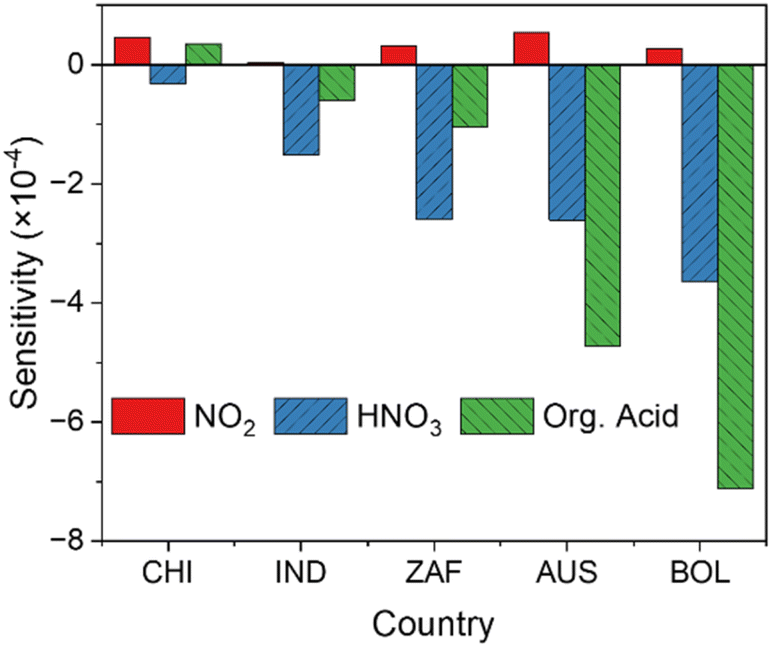
Sensitivities of the sCI-loss reactions via NO2, HNO3, and organic acids
Fig. 8 shows the sensitivities of the sCI-loss reactions by NO2, HNO3, and organic acids to SO42−(p) formation (scenarios S5, S6, and S7). At all analysed sites, the sensitivity of NO2 showed a positive contribution to SO42−(p) formation, whereas the sensitivities of HNO3 and organic acids showed negative contributions. We applied the reaction of sCIs and NO2 to form 30% of NO3, according to Caravan et al.45 NO3 is known as a nocturnal oxidiser and enhances the HOX cycle,49 and this scheme might contribute to the positive sensitivity of NO3 to SO42−(p) formation. However, the sensitivities of the sCI-loss reactions of HNO3 and organic acids were negative because these species consumed sCIs, which decreased the sCIs + SO2 reactions. It is important to note that the negative sensitivities of the reactions of HNO3 and organic acids with sCIs to SO42−(p) formation were of the same order of magnitude as those of unimolecular decomposition and water reactions, especially at remote sites, including AUS and BOL. Therefore, the presence of HNO3 and organic acids in the atmosphere led to the decrease of SO42−(p) formation through oxidation by sCIs, which are as significant as the unimolecular decomposition of sCIs and the reaction of sCIs and water.
 |
| | Fig. 8 Sensitivities of the reactions of sCIs with NO2, HNO3, and organic acids (denoted as Org. Acid in the figure). | |
Overall conclusions of the sensitivity analyses
This study shows the sensitivity of the sCI chemical reactions to the formation of SO42−(p) according to the detailed sensitivity analysis method defined by eqn (2). The results suggest that in all the analysed sites, the OH formed from vCIs more influenced the formation of SO42−(p) than sCIs, but in the remote sites, their difference became closer. Compared with the unimolecular reaction of sCIs and the reaction of sCIs with water, water was more relevant to the sCI-loss reactions at all analysed sites. The loss of sCIs by HNO3 and organic acids is as important as the loss by unimolecular decomposition and water. In terms of the comparison of vCIs and sCIs with SO42−(p) formation, this is the first study to clarify the quantitative relationship between them in highly industrialised and rural/remote sites, concluding that sCI chemistry should be considered in rural and remote areas.
Uncertainties of this work
Although several impacts of CIs on SO42−(p) formation were updated in this study, the uncertainties of the analysis must be addressed. There are two types of uncertainties in the analyses: those associated with the CTM itself and those related to the chemical reactions incorporated in this study. Regarding the uncertainties of the CTM, several factors contribute, including emission inventories, meteorological parameters, and gas, heterogeneous-, and aqueous-phase chemistries incorporated in the default CTM. The combinations of these uncertainties ultimately resulted in the outcomes shown in Fig. 2, which indicate a correlation between the observed and modelled results for the column density of O3, HCHO, and SO2, although the replicability is not perfect. As discussed above, the discrepancies may stem from the differences in grid resolution between the observed and calculated results, as well as the accumulation of errors in the vertical layer summation process calculated by the CTM. There is also a possibility of inaccuracies in the emission inventories. Nevertheless, we must accept the errors caused by the CTM itself, as all current CTMs have inherent uncertainties. It is important to note that the results obtained in this study should be treated as one of the conclusions with potential errors stemming from the CTM.
The uncertainties arising from the incorporated reactions of CIs should be treated more carefully, as the inclusion of CI chemistry is the main focus of this study. The results in Fig. 5 suggest that only 30% of NO3 formation by sCIs + NO2 reactions is considered to lead to NO2 removal,45 thus contributing to a reduction in tropospheric NO3, a phenomenon opposite to that observed in previous work.18 Furthermore, the sensitivity analyses shown in Fig. 6–8 indicate that the sensitivities of other CI-related chemical reactions are higher than those of the sCIs + NO2 reactions. This suggests that the definition and accuracy of the chemical mechanisms applied in the CTM affect the atmospheric impacts induced by CI chemistry. For example, Sarwer et al. conducted CTM calculations by adding the chemical mechanisms of sCIs for both low- and high-rate coefficients of sCIs + water reactions to clarify the potential influence of uncertainties in these reactions.50 The results suggested that the contribution of sCIs to SO42−(p) was more than an order of magnitude higher when lower rate coefficients for sCIs + water reactions were applied. Lade et al. conducted experimental and theoretical approaches to determine the rate coefficient of SCI1 (formaldehyde oxide) and water reactions, as well as CTM calculations to assess the atmospheric impact of the water monomer and dimer.51 They concluded that more than 98% of SCI1 is consumed by the water dimer, but the uncertainties in the product yields of SCI1 + water reactions limit the understanding of SCI1 chemistry. In terms of the rate coefficients for the unimolecular decomposition of sCIs, this study used the values obtained by Vereecken et al.,20 which were estimated based on classical TST calculations coupled with the semi-empirical method and structure–activity relationships. However, the experimental and theoretical studies for the determination of the rate coefficient for the unimolecular decomposition of dimethyl-substituted CIs, conducted by Lester and Klippenstein, suggested the importance of applying high-pressure limit calculations (master equation method) to determine the rate coefficient under ambient conditions and of adjusting tunnelling parameters.52 The rate coefficient estimated by Vereecken et al. was reported to be approximately 1.5 times higher than that of Lester and Klippenstein. The branching ratios of the reaction mechanisms of CI chemistry incorporated in this study also hold several uncertainties, as the products of nearly all reactions involve assumptions due to a lack of information (e.g. assuming 0.5 for the branching ratio of E- and Z-isomers of sCIs after alkene ozonolysis, assuming complete dissociation of unimolecular decay to form OH, assuming the same rate coefficients for all sCIs + HNO3 reactions, etc.).
Despite these uncertainties, one of the main topics of this study is to clarify the importance of vCI-initiated OH formation in SO42−(p) formation. According to the results in Fig. 6–8, the sensitivities of the reaction kinetics of vCIs and sCIs on SO42−(p) formation are an order of magnitude higher than those of the sCI-loss reactions. For these reasons, although several reaction mechanisms incorporated in this study involve uncertainties, the contribution of vCIs remains plausible. Further studies should be conducted to clarify the uncertainties in reaction kinetics, branching ratios, and other aspects of CI chemistry in future work.
Summary
The impact of chemical reactions related to CIs on SO42−(p) and other aerosol species was evaluated using a global CTM. The absolute amount of SO42−(p) formation was found to be high in China and India because of high SO2 pollution, while the percentage of contribution of Criegee chemistry was maximally approximately 0.5% in the Amazon rainforests. The most relevant sCIs on a global scale estimated in this study was ESCI7, the E-isomer of the sCIs formed by the ozonolysis of MVK, owing to the relatively low rate of atmospheric loss processes, which indicates the necessity for further kinetic studies related to ESCI7 chemistry. The results of the sensitivity analyses suggest that in all analysed sites, the OH formed by the unimolecular decomposition of vCIs can have more influence on SO42−(p) formation than on the direct oxidation of SO2 by sCIs, especially in industrialised areas. This indicates that, in terms of aerosol formation via Criegee chemistry, the unimolecular decomposition of vCIs is more important in aerosol formation, and sCIs has increasing importance in the remote area. The sCI loss reactions involving HNO3 and organic acids are as important for SO42−(p) formation as those involving the unimolecular decomposition of sCIs and water.
The results of this study showcase the overall global impact of CIs on the formation of tropospheric SO42−(p) and related aerosol species, but there are several assumptions for the rate coefficients set in this study (e.g., temperature dependence was not considered for the reactions of sCIs with SO2, NO2, etc., and the branching ratio of E- and Z-isomers was set to 0.5) because of the limited information available on the gas-phase kinetics of sCIs. Further kinetic studies on sCIs should be conducted in future. Furthermore, several studies have revealed the heterogeneous kinetics between the gas/liquid interface of CIs reacting with H2O, alcohol, etc., forming hydroxy hydroperoxide (HHP), alkoxy alkyl hydroperoxide (AAHP), liquid-phase OH, and RO2, which are important oxidisers in the liquid phase that contribute to the formation of particulate matter.53–55 Besides the detailed gas-phase chemistry of CI, the heterogeneous kinetics of CI should be evaluated by chemical transport modelling in future work to detail the effect of CI on the atmosphere.
Data availability
The data supporting this article are included as part of the main text and in the ESI.†
Author contributions
Hiroo Hata proposed the research concept, conducted the global chemical transport modelling, and wrote the manuscript draft. Yuya Nakamura determined the kinetic parameters of the Criegee intermediates and checked the manuscript for accuracy. Jairo Vazquez Santiago analysed satellite data obtained from TROPOMI and checked the manuscript for accuracy. Kenichi Tonokura supervised and checked the work for accuracy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS; Grant Number JP24K03088).
References
- A. C. Pope III, T. R. Burnett and J. M. Thun, Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution, JAMA, J. Am. Med. Assoc., 2002, 287(9), 1132–1141 CrossRef PubMed.
- K.-H. Kim, E. Kabir and S. Kabir, A review on the human health impact of airborne particulate matter, Environ. Int., 2015, 74, 136–143 CrossRef CAS PubMed.
- R. Burnett, H. Chen, M. Szyszkowicz and V. J. Spadaro, Global estimates of mortality associated with long-term exposure to outdoor fine particulate matter, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(38), 9592–9597 CrossRef CAS.
- R. Zhang, G. Wang, S. Guo, L. M. Zamora, Q. Ying, Y. Lin, W. Wang, M. Hu and Y. Wang, Formation of urban fine particulate matter, Chem. Rev., 2015, 115(10), 3803–3855 CrossRef CAS.
- S. Singh, K. Soni, T. Bano, S. R. Tanwar, S. Nath and C. B. Arya, Clear-sky direct aerosol radiative forcing variations over mega-city Delhi, Ann. Geophys., 2010, 28, 1157–1166 CrossRef.
- Q. Wang, J. D. Jacob, A. J. Fisher, J. Mao, M. E. Leibensperger, C. C. Carouge, P. Le Sager, Y. Kondo, L. J. Jimenez, J. M. Cubison and J. S. Doherty, Sources of carbonaceous aerosols and deposited black carbon in the Arctic in winter-spring: implications for radiative forcing, Atmos. Chem. Phys., 2011, 11, 12453–12473 CrossRef CAS.
- Z. Hassan, M. Stahlberger, N. Rosenbaum and S. Bräse, Criegee intermediates beyond ozonolysis: synthetic and mechanistic insights, Angew. Chem., Int. Ed., 2021, 60, 15138–15152 CrossRef CAS PubMed.
- A. C. Taatjes, E. D. Shallcross and J. C. Percival, Research frontiers in the chemistry of Criegee intermediates and tropospheric ozonolysis, Phys. Chem. Chem. Phys., 2014, 16, 1704–1718 RSC.
- R. Chhantyal-Pun, H. A. M. Khan, A. C. Taatjes and J. C. Percival, Criegee intermediates: production, detection and reactivity, Int. Rev. Phys. Chem., 2020, 39(3), 385–424 Search PubMed.
- H. A. M. Khan, J. C. Percival, L. R. Caravan, A. C. Taatjes and E. D. Shallcross, Criegee intermediates and their impacts on the troposphere, Environ. Sci.: Processes Impacts, 2018, 20, 437–453 RSC.
- L. D. Osborn and A. C. Taatjes, The physical chemistry of Criegee intermediates in the gas phase, Int. Rev. Phys. Chem., 2015, 34(3), 309–360 Search PubMed.
- I. M. Lester and J. S. Klippenstein, Unimolecular decay of Criegee intermediates to OH radical product: prompt and thermal decay processes, Acc. Chem. Res., 2018, 51(4), 978–985 CrossRef PubMed.
- M. N. Kidwell, H. Li, X. Wang, M. J. Bowman and I. M. Lester, Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products, Nat. Chem., 2016, 8, 509–514 CrossRef PubMed.
- G. Sarwer, H. Simon, K. Fahey, R. Mathur, S. W. Goliff and R. W. Stockwell, Impact of sulfur dioxide oxidation by Stabilized Criegee Intermediate on sulfate, Atmos. Environ., 2014, 85, 204–214 CrossRef.
- J. C. Percival, O. Welz, J. A. Eskola, D. J. Savee, L. D. Osborn, O. D. Topping, D. Lowe, R. S. Utembe, A. Bacak, G. McFiggans, C. M. Cooke, P. Xiao, T. A. Archibald, E. M. Jenkin, G. R. Derwent, I. Riipinen, K. W. D. Mok, F. P. E. Lee, M. J. Dyke, A. C. Taatjes and E. D. Shallcross, Regional and global impacts of Criegee intermediates on atmospheric sulphuric acid concentrations and first steps of aerosol formation, Faraday Discuss., 2013, 165, 45–73 RSC.
- Y. Nakamura, H. Hata and K. Tonokura, Urban-scale analysis of the seasonal trend of stabilized-Criegee intermediates and their effect on sulphate formation in the Greater Tokyo Area, Environ. Sci.: Atmos., 2023, 3, 1758–1766 CAS.
- A. R. Cox, M. Ammann, N. J. Crowley, H. Herrmann, E. M. Jenkin, F. V. McNeill, A. Mellouki, J. Troe and J. T. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VII – Criegee intermediates, Atmos. Chem. Phys., 2020, 20(21), 13497–13519 CrossRef.
- H. Hata, S. Hoshino, M. Fujita and K. Tonokura, Atmospheric impact of isoprene-derived Criegee intermediates and isoprene hydroxy hydroperoxide on sulfate aerosol formation in the Asian region, Atmos. Environ.:X, 2023, 20, 100226 CAS.
- J. M. Newland, R. A. Rickard, L. Vereecken, A. Muñoz, M. Ródenas and J. W. Bloss, Atmospheric isoprene ozonolysis: impacts of stabilised Criegee intermediate reactions with SO2, H2O and dimethyl sulfide, Atmos. Chem. Phys., 2015, 15, 9251–9536 CrossRef.
- L. Vereecken, A. Novelli and D. Taraborrelli, Unimolecular decay strongly limits the atmospheric impact of Criegee intermediates, Phys. Chem. Chem. Phys., 2017, 19, 31599–31612 RSC.
- L. Sheps, B. Rotavera, J. A. Eskola, L. D. Osborn, A. C. Taatjes, K. Au, E. D. Shallcross, H. A. M. Khan and J. C. Percival, The reaction of Criegee intermediate CH2OO with water dimer: primary products and atmospheric impact, Phys. Chem. Chem. Phys., 2017, 19, 21970–21979 RSC.
- R. M. McGillen, E. F. B. Curchod, R. Chhantyal-Pun, M. J. Beames, N. Watson, H. A. M. Khan, L. McMahon, E. D. Shallcross and J. A. Orr-Ewing, Criegee intermediate-alcohol reactions, a potential source of functionalized hydroperoxides in the atmosphere, ACS Earth Space Chem., 2017, 1(10), 664–672 CrossRef.
- L. R. Caravan, F. M. Vansco, K. Au and I. M. Lester, Direct kinetic measurements and theoretical predictions of an isoprene-derived Criegee intermediate, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(18), 9733–9740 CrossRef PubMed.
- J. M. Newland, R. A. Rickard, T. Sherwen, J. M. Evans, L. Vereecken, A. Muñoz, M. Ródenas and J. W. Bloss, The atmospheric impacts of monoterpene ozonolysis on global stabilised Criegee intermediate budgets and SO2 oxidation: experiment, theory and modelling, Atmos. Chem. Phys., 2018, 18, 6095–6120 CrossRef.
- R. Chhantyal-Pun, H. A. M. Khan, N. Zachhuber, J. C. Percival, E. D. Shallcross and J. A. Orr-Ewing, Impact of Criegee intermediate reactions with peroxy radicals on tropospheric organic aerosol, ACS Earth Space Chem., 2020, 4(10), 1743–1755 CrossRef CAS.
-
GEOS-Chem v14.2.3, DOI:10.5281/zenodo.10246625.
- R. Gelaro, W. McCarty, M. J. Suárez, R. Todling, A. Molod, L. Takacs, C. A. Randles, A. Darmenov, M. G. Bosilovich, R. Reichle, K. Wargan, L. Coy, R. Cullather, C. Draper, S. Akella, V. Buchard, A. Conaty, A. M. da Silva, W. Gu, G.-K. Kim, R. Koster, R. Lucchesi, D. Merkova, J. E. Nielsen, G. Partyka, S. Pawson, W. Putman, M. Rienecker, S. D. Schubert, M. Sienkiewicz and B. Zhao, Modern era retrospective analysis for research and applications version 2 (MERRA-2), J. Clim., 2017, 30(14), 5419–5454 CrossRef PubMed.
- H. Lin, J. D. Jacob, W. E. Lundgren, P. M. Sulprizio, A. C. Keller, M. T. Fritz, D. S. Eastham, K. L. Emmons, C. P. Campbell, B. Baker, D. R. Saylor and R. Montuoro, Harmonized emissions component (HEMCO) 3.0 as a versatile emissions component for atmospheric models: application in the GEOS-Chem, NASA GEOS, WRF-GC, CESM2, NOAA GEFS-Aerosol, and NOAA UFS models, Geosci. Model Dev., 2021, 14, 5487–5506 CrossRef CAS.
- E. E. McDuffie, S. J. Smith, P. O'Rourke, K. Tibrewal, C. Venkataraman, E. A. Marais, B. Zheng, M. Crippa, M. Brauer and R. V. Martin, A global anthropogenic emission inventory of atmospheric pollutants from sector- and fuel-specific sources (1970-2017): an application of the Community Emissions Data System (CEDS), Earth Syst. Sci. Data, 2020, 12, 3413–3442 CrossRef.
- G. Janssens-Maenhout, M. Crippa, D. Guizzardi, F. Dentener, M. Muntean, G. Pouliot, T. Keating, Q. Zhang, J. Kurokawa, R. Wankmüller, H. Denier van der Gon, J. J. P. Kuenen, Z. Klimont, G. Frost, S. Darras, B. Koffi and M. Li, HTAP_v2.2: A mosaic of regional and global emission grid maps for 2008 and 2010 to study the hemispheric transport of air pollution, Atmos. Chem. Phys., 2015, 15, 11411–11432 CrossRef CAS.
- V. V. Pham, J. Tang, S. Alam, C. Lokan and H. A. Abbass, Aviation emission inventory development and analysis, Environ. Model. Software, 2010, 25(12), 1738–1753 CrossRef.
- G. R. van der Werf, J. T. Randerson, L. Giglio, T. T. van Leeuwen, Y. Chen, B. M. Rogers, M. Mu, M. J. E. van Marle, D. C. Morton, G. J. Collatz, R. J. Yokelson and P. S. Kasibhatla, Global fire emissions estimates during 1997-2016, Earth Syst. Sci. Data, 2017, 9(2), 697–720 CrossRef.
- F. Dentener, S. Kinne, T. Bond, O. Boucher, J. Cofala, S. Generoso, P. Ginoux, S. Gong, J. J. Hoelzemann, A. Ito, L. Marelli, J. E. Penner, J.-P. Putaud, C. Textor, M. Schulz, G. R. van der Werf and J. Wilson, Emissions of primary aerosol and precursor gases in the years 2000 and 1750 prescribed data-sets for AeroCom, Atmos. Chem. Phys., 2006, 6, 4321–4344 CrossRef CAS.
- A. B. Guenther, X. Jiang, C. L. Heald, T. Sakulyanontvittaya, T. Duhl, L. K. Emmons and X. Wang, The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 2012, 5, 1471–1492 CrossRef.
- K. Garane, M.-E. Koukouli, C. V. T. Lerot, K.-P. Heue, V. Fioletov, D. Balis, A. Bais, A. Bazureau, A. Dehn, F. Goutail, J. Granville, D. Griffin, D. Hubert, A. Keppens, J.-C. Lambert, D. Loyola, C. McLinden, A. Pazmino, J.-P. Pommereau, A. Redondas, F. Romahn, P. Valks, M. Van Roozendael, J. Xu, C. Zehner, C. Zerefos and W. Zimmer, TROPOMI/S5P total ozone column data: global ground-based validation and consistency with other satellite missions, Atmos. Meas. Tech., 2019, 12, 5263–5287 CrossRef.
- J. van Geffen, F. K. Boersma, H. Eskes, M. Sneep, M. ter Linden, M. Zara and P. J. Veefkind, S5P TROPOMI NO2 slant column retrieval: method, stability, uncertainties and comparisons with OMI, Atmos. Meas. Tech., 2020, 13(3), 1315–1335 CrossRef CAS.
- C. Vigouroux, B. Langerock, B. A. C. Aquino, T. Blumenstock, Z. Cheng, M. De Mazière, I. De Smedt, M. Grutter, W. J. Hannigan, N. Jones, R. Kivi, D. Loyola, E. Lutsch, E. Mahieu, M. Makarova, J.-M. Metzger, I. Morino, I. Murata, T. Nagahama, J. Notholt, I. Ortega, M. Palm, G. Pinardi, A. Röhling, D. Smale, W. Stremme, K. Strong, R. Sussmann, Y. Té, M. van Roozendael, P. Wang and H. Winkler, TROPOMI–Sentinel-5 Precursor formaldehyde validation using an extensive network of ground-based Fourier-transform infrared stations, Atmos. Meas. Tech., 2020, 13(7), 3751–3767 CrossRef CAS.
- Q. Hong, C. Liu, Q. Hu, Y. Zhang, C. Xing, W. Su, X. Ji and S. Xiao, Evaluating the feasibility of formaldehyde derived from hyperspectral remote sensing as a proxy for volatile organic compounds, Atmos. Res., 2021, 264, 105777 CrossRef CAS.
-
Data archive of COPERNICUS, https://sentinels.copernicus.eu/web/sentinel/home, accessed June 4, 2024 Search PubMed.
-
NASA EarthData Archive, https://www.earthdata.nasa.gov/, accessed June 4, 2024 Search PubMed.
- B. N. Murphy, G. C. Nolt, F. Sidi, O. J. Bash, W. K. Appel, C. Jang, D. Kang, J. Kelly, R. Mathur, S. Napelenok, G. Pouliot and T. O. H. Pye, The detailed emissions scaling, isolation, and diagnostic (DESID) module in the community multiscale air quality (CMAQ) modeling system version 5.3.2, Geosci. Model Dev., 2021, 14, 3407–3420 CrossRef CAS PubMed.
- W. P. L. Carter, Development of the SAPRC-07 chemical mechanism, Atmos. Environ., 2010, 44(40), 5324–5335 CrossRef CAS.
- M. S. Saunders, E. M. Jenkin, G. R. Derwent and J. M. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 2003, 3, 161–180 CrossRef.
- C. M. Smith, C.-H. Chang, W. Chao, L.-C. Lin, K. Takahashi, A. K. Boering and J. Jr-Min Lin, Strong negative temperature dependence of the simplest Criegee intermediate CH2OO reaction with water dimer, J. Phys. Chem. Lett., 2015, 6(14), 2708–2713 CrossRef PubMed.
- L. R. Caravan, H. A. M. Khan, F. Rotavera, E. Papajak, O. I. Antonov, M.-W. Chen, K. Au, W. Chao, L. D. Osborn, J. Jr-Min Lin, J. C. Percival, E. D. Shallcross and A. C. Taatjes, Products of Criegee intermediate reactions with NO2: experimental measurements and tropospheric implications, Faraday Discuss., 2017, 200, 313–330 RSC.
- H.-L. Huang, W. Chao and Jr. M. J. Lin, Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 10857–10862 CrossRef CAS PubMed.
- C. Fountoukis and A. Nenes, ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+–NH+4–Na+–SO42–NO3−–Cl−–H2O aerosols, Atmos. Chem. Phys., 2007, 7, 4639–4659 CrossRef CAS.
- S. H. Budisulistiorini, A. Nenes, A. G. Cariton, J. D. Surratt, V. F. McNeill and H. O. T. Pye, Simulating aqueous-phase isoprene-epoxydiol (IEPOX) secondary organic aerosol production during the 2013 Southern Oxidant and Aerosol Study (SOAS), Environ. Sci. Technol., 2017, 51(9), 5026–5034 CrossRef CAS PubMed.
- L. N. Ng, S. S. Brown, T. A. Archibald, E. Atlas, C. R. Cohen, N. J. Crowley, A. D. Day, M. N. Donahue, L. J. Fry, H. Fuchs, J. R. Griffin, I. M. Guzman, H. Herrmann, A. Hodzic, Y. Iinuma, L. J. Jimenez, A. Kiendler-Scharr, H. B. Lee, J. D. Luecken, J. Mao, R. McLaren, A. Mutzel, D. H. Osthoff, B. Ouyang, B. Picquet-Varrault, U. Platt, T. O. H. Pye, Y. Yinon Rudich, H. R. Schwantes, M. Shiraiwa, J. Stutz, A. J. Thornton, A. Tilgner, J. B. Williams and A. R. Zaveri, Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 2017, 17, 2103–2162 CrossRef PubMed.
- G. Sarwar, K. Fahey, R. Kwok, R. C. Gilliam, J. S. Roselle, R. Mathur, J. Xue, J. Yu and W. P. L. Carter, Potential impacts of two SO2 oxidation pathways on regional sulfate concentrations: Aqueous-phase oxidation by NO2 and gas-phase oxidation by Stabilized Criegee Intermediates, Atmos. Environ., 2013, 68, 186–197 CrossRef CAS.
- R. E. Lade, M. A. Blitz, M. Rowlinson, M. J. Evans, P. W. Seakins and D. Stone, Kinetics of the reactions of the Criegee intermediate CH2OO with water vapour: experimental measurements as a function of temperature and global atmospheric modelling, Environ. Sci.: Atmos., 2024, 4, 1294–1308 CAS.
- M. I. Lester and S. J. Klippenstein, Unimolecular decay of Criegee intermediates to OH radical products: prompt and thermal decay processes, Acc. Chem. Res., 2018, 51(4), 978–985 CrossRef CAS PubMed.
- J. Qiu, S. Ishizuka, K. Tonokura, J. A. Colussi and S. Enami, Reactivity of monoterpene Criegee intermediates at gas-liquid interfaces, J. Phys. Chem. A, 2018, 122(39), 7910–7917 CrossRef CAS PubMed.
- M. Zeng and R. K. Wilson, Efficient coupling of reaction pathways of Criegee intermediates and free radicals in the heterogeneous ozonolysis of alkenes, J. Phys. Chem. Lett., 2020, 11(16), 6580–6585 CrossRef CAS PubMed.
- Z. Zhao, Q. Xu, X. Yang and H. Zhang, Heterogeneous ozonolysis of endocyclic unsaturated organic aerosol proxies: implications for Criegee intermediate dynamics and later-generation reactions, ACS Earth Space Chem., 2019, 3(3), 344–356 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Yuya
Nakamura
*a,
Yuya
Nakamura