
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of precipitation on gaseous oxidized and elemental mercury concentrations as quantified by two types of atmospheric mercury measurement systems
Peter S.
Weiss-Penzias
 *a,
Seth N.
Lyman
bc,
Tyler
Elgiar
b,
Lynne E.
Gratz
d,
Winston T.
Luke
e,
Gabriel
Quevedo
f,
Nicole
Choma
g and
Mae Sexauer
Gustin
g
*a,
Seth N.
Lyman
bc,
Tyler
Elgiar
b,
Lynne E.
Gratz
d,
Winston T.
Luke
e,
Gabriel
Quevedo
f,
Nicole
Choma
g and
Mae Sexauer
Gustin
g
aUniversity of California, Santa Cruz, Santa Cruz, CA 95064, USA. E-mail: pweiss@ucsc.edu
bBingham Research Center, Utah State University, Vernal, UT, USA
cDepartment of Chemistry and Biochemistry, Utah State University, Logan, UT, USA
dDepartment of Chemistry and Environmental Studies Program, Reed College, Portland, OR 97202, USA
eAir Resources Laboratory, National Oceanic and Atmospheric Administration, College Park, Maryland 20740, USA
fUniversity of California, Los Angeles, Los Angeles, CA, USA
gUniversity of Nevada, Reno, Reno, NV, USA
First published on 23rd December 2024
Abstract
Gaseous and particulate-bound oxidized mercury (Hg) compounds (HgII) have high solubility in precipitation compared to gaseous elemental Hg (Hg0). Wet and dry deposition are the primary routes of entry for atmospheric HgII into aquatic ecosystems. Information on how much HgII is removed from the atmosphere to the landscape during precipitation is lacking. In this study, oxidized HgII concentrations were measured with a dual-channel system (DCS) at two sites in the United States, Storm Peak Laboratory (SPL), in Colorado (2021–2022), and Beltsville (MD99) in Maryland (2022–2024), and compared with data from 16 co-located Atmospheric Mercury Network (AMNet) and Mercury Deposition Network (MDN) sites that used a KCl denuder method. At the two DCS sites, gaseous oxidized Hg concentrations were segregated by wet and dry periods from the nearest precipitation gauge to determine values for median dry HgII and median wet HgII concentrations (dry-wet = “HgII washout”) for each site. SPL had higher median ambient HgII and higher median HgII washout (90 pg m−3 and 22 pg m−3, respectively) compared to that for MD99 (43 pg m−3 and 7 pg m−3). This difference could be due to site elevation (3161 vs. 77 m) as there is generally more HgII higher in the atmosphere. In contrast, the ambient HgII/washout HgII ratios were more similar, 4.1 for SPL and 5.8 at MD99. The mean ambient HgII/washout HgII ratio for the 16 AMNet sites was 1.8 ± 0.1. The AMNet HgII data are known to be biased low due to issues with the KCl-denuder method, and this low bias appears to result in lower ambient HgII/washout HgII ratio observed for the AMNet sites. Correction factors for AMNet data using HgII measurements from DCS instruments were found to range from 2–3 and could be used to improve the accuracy of older data.
Environmental significanceOxidized mercury (Hg) compounds in the atmosphere, both in gaseous and particulate forms, are the main sources of Hg in precipitation, which contributes Hg to aquatic ecosystems where it is transformed into methylmercury. Understanding the connection between emissions of Hg to the atmosphere and their contribution to Hg in precipitation is limited by several factors including uncertainties associated with the measurement methods for oxidized Hg. In this study, an improved measurement method for oxidized Hg was utilized at a mountaintop site in the Rocky Mountains and at a suburban site in Maryland, where high-resolution datasets were obtained at each site for two-year periods. The data were separated by precipitation depth as measured by the closest rain gauge (>0 (wet) and =0 (dry)). The medians of each of these categories were calculated and median wet-median dry was then termed oxidized Hg “washout”. We then calculated the ratio of ambient oxidized Hg to washout oxidized Hg and found the ratios to be relatively consistent at the two sites that used the improved instrumental method. The mean ratio for these two sites was then used to correct older measurements of oxidized Hg across a network of 16 sites in the U.S. and Canada, which had a mean ratio of ambient oxidized Hg to washout oxidized Hg that was 2.7 times lower. This analysis revealed that the older oxidized Hg data could be adjusted upward by factors of 2–3 depending on the site. We suggest that assessments of the impact of Hg emissions on Hg in precipitation could be underestimated if only uncorrected data were used. |
1 Introduction
Mercury (Hg) is a global toxicant that affects humans and wildlife through the dietary intake of methylmercury,1,2 as evidenced by recent Hg concentration surveys of high trophic level and sentinel species that showed exceedances of benchmarks for fish and wildlife.3 Most Hg pollution is emitted to the atmosphere, where it is transported and transformed from the relatively inert elemental form (Hg0) to the divalent form (HgII) that is more reactive and water-soluble.4 Experimental, theoretical, and modeling advances have suggested that Hg0 is initially oxidized by Br and OH, and subsequently by ozone and other atmospheric constituents,5 and much of the resultant gas-phase HgII subsequently becomes part of dissolved organic compound complexes in aerosols and cloud droplets.6 Gas- and aerosol-phase HgII can then either be photochemically reduced back to Hg0 or wet and dry deposited to the Earth's surface.7–10 Other atmospheric Hg redox pathways may also exist; however, understanding of atmospheric Hg chemistry is currently hindered by an inability to identify specific HgII compounds in the atmosphere. Hg0 can also be dry deposited to terrestrial vegetation through stomatal uptake, and by some estimates this is the dominant removal pathway,11 although bidirectional exchange releases Hg back to the atmosphere from plants and soil surfaces.12,13Wet deposition of Hg is measured by collection of precipitation that contains particulate and soluble forms of HgII. Wet deposition is an important source of Hg to ecosystems where HgII undergoes methylation by microbes and enters the food web.14 However, controls on methylmercury formation and concentrations in the environment are numerous, and there is not a simple relationship between wet deposition of inorganic Hg and methylmercury in fish.15 Although atmospheric washout as a removal phenomenon for Hg was identified years ago through observations of the decrease in Hg concentrations in sequential rain samples,16,17 the origins of HgII in precipitation are not well known. One complicating factor is that most measurements of ambient HgII concentrations have been collected using methods that are now known to be biased low due to interferences on the KCl-denuder and the quartz fiber filter.18,19 A second factor is that most wet deposition measurements made by the Mercury Deposition Network (MDN) that is part of the National Atmospheric Deposition Program (NADP) are weekly samples limiting the ability to quantitatively determine HgII sources using individual precipitation events.
There is considerable evidence that rainfall has a diluting effect on Hg concentrations in precipitation ([Hg]aq) and that a log–log relationship exists between [Hg]aq and precipitation depth both in weekly integrated and event-based samples.20–27 Scavenging coefficients of Hg in precipitation are lower than for other airborne metals and ions due to a continuous supply of soluble forms of HgII at least in the vicinity anthropogenic emissions.21 Gaseous oxidized Hg (GOM) is generally more water soluble than particulate bound Hg (PBM) and is thought to contribute more to [Hg]aq than PBM,21 although the opposite could occur in polluted urban atmospheres with heavy burdens of particulate matter.27 Some studies have suggested that HgII is incorporated into precipitation via gas-phase inclusion or particulate nucleation within the cloud, with below-cloud scavenging in the boundary layer probably being less important.21,28 Indeed, Shah and Jaeglé,29 in a modeling study, found that HgII produced in the upper and middle troposphere constitutes 91% of the annual HgII wet deposition flux. This may be specifically true in the western U.S. where the free troposphere is an important source of surface GOM and PBM, and wet deposition of Hg increases with altitude.30,31
Because of the importance of Hg wet deposition to ecosystems, monitoring networks have been established to make regular, standardized measurements of Hg loads to surfaces. The MDN is currently composed of 76 active sites that collect weekly precipitation samples to be analyzed for [Hg]aq.32 At 16 MDN sites, there were measurements of atmospheric speciated Hg concentrations (PBM, GOM, and Hg0) carried out for multiyear periods by AMNet during the years 2008–2020. Although these atmospheric Hg data sets are large and potentially valuable for understanding the origins of Hg in wet deposition, the quantitative accuracy of these data is in question because PBM and GOM measurements are subject to low-bias and interferences, due to the use of the KCl denuder and quartz filter, respectively.10,33,34 In response, alternative techniques have been developed that quantify HgII compounds using cation-exchange membranes in dual-channel systems that can measure HgII by difference (total Hg–Hg0),35–39 but datasets are from fewer locations and do not span as much time as the AMNet data.
Prior to the understanding of the limitations of the KCl-denuder based measurements, comparisons were made using the 2–3 h GOM and PBM concentrations with weekly [Hg]aq at co-located sites in AMNet/MDN40 to make predictions of GOM + PBM concentrations at MDN-only sites. Later, Cheng et al.41 made use of PBM measurements from AMNet sites and scavenging ratios of major ions in wet deposition from measurements at MDN sites to estimate the proportion of GOM, and coarse and fine PBM in precipitation. These scavenging ratios42 were then combined with weekly GOM concentrations from AMNet to estimate measurement biases of the KCl-denuder measurement system which ranged from 1.3–14.3 (mean = 2.3) times below predicted values. This range is similar to that suggested by Gustin et al.43 (1.6–12) and Gustin et al.44 (1.3–13), both of which were based on comparison between measurements by different methods.
Using uncorrected HgII concentration data taken with the KCl-denuder method could therefore be leading to misinterpretations regarding the relationship between HgII concentrations in air and [Hg]aq that is measured at an MDN site, resulting in uncertainties about the source of HgII in precipitation. For example, Lynam et al.45 saw Hg rainfall concentrations 2–4 times higher for concurrent events with similar precipitation depth at an urban site compared to a background site suggesting that scavenging of local Hg emissions was occurring. On the other hand, away from location emission sources, there may be a weak relationship between ambient HgII and [Hg]aq. A model evaluation study at the global level showed a variation of only ±10% in wet deposition for three different Hg emissions scenarios.46 Similarly, simulated Hg wet deposition flux in New York State changed only by 2% despite a near doubling of emissions in the Northeastern U.S.25 Considering the importance of the middle and upper troposphere as a source region for HgII in wet deposition,29 changes in surface concentrations of HgII may not be correlated with [Hg]aq in many locations.
In the current study, the effects of precipitation on air concentrations of HgII and Hg0 as measured with the dual-channel systems, which have been shown to quantitatively recover HgBr2 and HgCl2, at a remote high elevation site in the Rocky Mountains and in suburban Maryland were studied. The magnitudes of HgII washout (median [HgII]dry–median [HgII]wet) and Hg0 enhancement (median [Hg0]wet–median [Hg0]dry) were quantified at these sites over multiple years of data and seasonal patterns, and air mass histories were examined. It was then considered whether the information learned about precipitation-driven changes in HgII and Hg0 species from the dual-channel systems could be used to correct for biases in the AMNet GOM and PBM data (obtained with KCl-denuder/quartz filter measurement system) from 16 co-located AMNet-MDN sites. The mean DCS-derived ambient HgII/washout HgII ratio was determined and the subsequent analyses explored whether this ratio could be used as a correction of older measurement data from 16 co-located sites.
2 Methodology
2.1 Dual-channel measurement sites
Elemental and total gaseous Hg were measured in ambient air at SPL using the dual-channel system.49–51 The system sampled air at 9 L min−1 through a PTFE-coated, heated (120 °C) elutriator and impactor that removed particles larger than 2.5 μm, through 0.5 m of heated perfluoroalkoxy (PFA) Teflon line, and then into one of two channels. One channel included a 650 °C thermolyzer to convert all atmospheric Hg to Hg0, measuring total atmospheric Hg, and the other a series of two cation-exchange membranes that retain HgII, to measure only Hg0. A Tekran 2537X analyzer customized to reduce the sample line volume and to increase sensitivity quantified the Hg0 output from both channels via the peak height method (cf.ref. 52), and HgII was calculated as the difference between the two channels. Poly(tetrafluoroethylene) (PTFE) Teflon valves were used to switch air sampled between the two channels at 5 min intervals. The instrument had a 1 h detection limit for HgII of less than 12 pg m−3 (3× standard deviation of ambient air measurements) and an expanded measurement uncertainty of 16%. HgII concentrations in this paper are reported to the ones place with no digit after the decimal point. The dual-channel system has been shown to quantitatively collect gas-phase HgII compounds and Hg0 injected into ambient air from an SI-traceable calibration source.42,50 Cation-exchange membranes used in the system to capture HgII have been shown to collect a wide variety of HgII compounds and do not collect a significant amount of Hg0.53 While no calibration method exists for particle-phase Hg, it is assumed that the dual-channel system captures all gas-phase HgII and HgII bound to particles smaller than 2.5 μm. Information about earlier versions of the dual-channel system and calibrator is available in Lyman et al.37 and Dunham-Cheatham et al.50 Soda lime traps were upstream of the 2537X to scrub reactive gases that can passivate the gold traps in the instrument. Soda lime traps and cation-exchange membranes were replaced biweekly. Periodic injections of Hg0 from a temperature controlled saturated Hg0 vapor source resulted in recovery of 101 ± 6%.
Oxidized mercury measured at MD99 as the difference between total Hg (THg) and gaseous elemental Hg. A Tekran® 1135 Particulate Unit module was modified to serve as the NOAA difference system. A custom pyrolyzer (URG Corp., Chapel Hill, NC) was based on a modified design of the Tekran® Corp. Regenerable Particle Filter (RPF). A porous quartz frit was fused approximately 5 cm from the threaded inlet of the pyrolyzer to minimize inlet losses of Reactive Mercury (RM) species and supported a 22 mm diameter quartz fiber filter and quartz wool plug. Quartz chips were packed behind the quartz frit to pyrolyze all mercury species to Hg0 at 800 °C. The pyrolyzer inlet was typically filtered with a PTFE membrane (25 mm diameter, 1 μm pore size) that was assumed to capture particle-phase HgII and allow gas-phase HgII to pass through.
Gaseous elemental Hg was measured by pulling air through two 47 mm diameter polyether sulfone (PES) membranes (0.22-micron pore size, MilliporeSigma™ Express™ PLUS Membrane Filters) to remove all PBM and GOM. We note that, while Dunham-Cheatham et al.55 found no significant difference between the cation-exchange membranes used at Storm Peak and the PES membranes used at Beltsville, Allen et al.56 found that PES membranes captured 14–23% less HgII than cation-exchange membranes (which are PES that have been treated with a proprietary process). The filters were housed in a Sulfinert (Restek, Bellafonte, PA) coated open-faced stainless steel filter holder (Model FJ-42SS, F&J Specialty Products, Inc., Ocala FL). The filters and holder were thermostatically controlled at 40 °C and were situated along the bottom edge of the 1135 module. All inlet filters were changed monthly, and the pyrolyzer was cleaned and replaced quarterly. Soda lime traps upstream of each 2537X analyzer were changed weekly. Ambient air was sampled through each channel at 1.0 standard liters per minute (0 °C, 1 atm), controlled by the Tekran analyzers.
The Tekran 1135 inlet module was mounted atop the 10 m walk-up scaffold at MD99. Two 50-foot lengths of ¼′′ diameter PFA Teflon tubes, encased in an insulated umbilical (Thermon Manufacturing) maintained at 50 °C, routed the flows from the THg and Hg0 channels to a valve switching box containing a series of PFA Teflon valves in the instrument shelter below. The valve switching assembly was connected to two Tekran® 2537X analyzers, and a Labview program actuated the valves to allow both analyzers to independently measure both THg and Hg0. The analyzers operated asynchronously, so that when analyzer 1 (X1) measured THg, Analyzer 2 (X2) measured Hg0, and vice versa. Each channel was sampled for two 6 minutes cycles by each analyzer. HgII was calculated as the difference of the hourly-averaged THg and Hg0 concentrations for each analyzer. The limit of detection was estimated as ±2 standard deviations of the average difference when the pyrolyzer was operated at 50 °C. LODs of 10–15 pg m−3 were achieved.
2.2 AMNet and MDN data
Measurements of atmospheric PBM, GOM, and Hg0 have been made at 22 AMNet sites that also collect weekly precipitation samples analyzed for [Hg]aq concentration (MDN sites). Of these, 16 had sufficient data coverage to warrant detailed analysis (>1 year) (Fig. 1). Summary data for Hg concentrations measured at these sites is shown in Appendix A. Based on geography, and monthly and diel concentrations of Hg forms (Appendix A, Table 4) the AMNet sites were grouped into four categories: Local Emissions (AL19, IN21, NY06, NY43, OH02, UT97), Suburban North (MD08, MD99, NJ30, WI07), Suburban South (FL96, GA40, MS12, OK99), and Rural North (NS01, VT99). | ||
| Fig. 1 Map showing the co-located AMNet and MDN sites in the U.S. and Canada and the SPL site which was paired with MDN-only site CO97. | ||
Concentrations of the different forms of mercury in the AMNet network were quantified by automated Tekran® 1130/1135/2537 systems.57 All instrumentation across each site was operated in an identical manner according to standard operating procedures provided by AMNet. The Tekran® 1135 measures PBM <2.5 μm. GOM and PBM concentrations were determined every 3 h with 1–2 h sampling and 1 h desorption times. Hg0 was sampled and analyzed every 5 minutes and combined into hourly averages in the raw AMNet data set. AMNet GOM and PBM concentrations in this paper are reported to the ones place with no digits after the decimal point. Mercury concentrations in precipitation samples taken at the AMNet/MDN co-located sites were measured according to standardized protocols.32 Only validated data were used for this analysis. In this paper, HgII and Hg0 are used to refer to oxidized and elemental mercury measurements made at SPL and MD99 with the dual-channel system, whereas GOM, PBM, and Hg0 refer to the Hg forms measured using the Tekran® 1130/1135/2537 system, respectively.
2.3 Data analysis
Data tables from each site were used in their original downloaded formats to determine when there was precipitation at each site and to classify the Hg measurements accordingly. Hourly precipitation depth data reported by the electronic rain gauge associated with the MDN sites were used to classify individual Hg measurements at all sites as “wet” (precipitation depth >0) or “dry” (precipitation depth =0). Median concentrations for all Hg forms during “wet” or “dry” periods were calculated and used for subsequent comparisons between sites and between measurement systems. Using median wet and median dry concentrations, HgII washout for a given site over its record of measurement was defined as| [HgII]washout = [HgII]dry − [HgII]wet | (1) |
Medians were selected instead of means because of the large number of high concentrations of HgII in the AMNet, SPL, and MD99 datasets that greatly skewed the means but not the medians. When normally distributed datasets are grouped, means are reported. For the AMNet data, [GOM]washout and [PBM]washout were calculated separately and then combined to determine HgII washout:
| [HgII]washout = [GOM]washout + [PBM]washout | (2) |
The ratio of median ambient HgII concentration to the median HgII washout concentration was calculated for each site. A correction factor for ambient HgII concentrations measured at the AMNet sites was calculated using
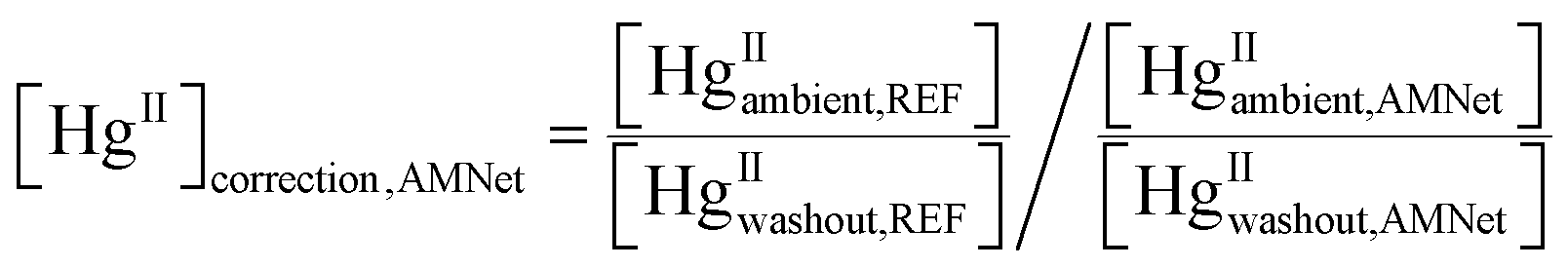 | (3) |
For Hg0 a precipitation enhancement was calculated
| [Hg0]enhancement = [Hg0]wet − [Hg0]dry | (4) |
Relationships between the Hg concentration in weekly integrated precipitation samples ([Hg]aq) and the air concentrations of Hg forms along with values of other ancillary meteorological and chemical parameters were also investigated. At SPL and MD99, a weekly [Hg]aq was compared with mean values of chemical and meteorological parameters by selecting those parameters' values that corresponded to when precipitation depth >0 and neglecting values when precipitation depth =0. For the MDN sites weekly [Hg]aq was compared against GOM + PBM = HgII concentrations averaged over the same weekly time step as the MDN sample, which included both dry and wet precipitation states.
AMNet data were further reduced to monthly and hourly means to characterize the seasonal and diel cycles (Appendix A, Fig. 8 and 9).
Statistical analyses included one-way ANOVA tests, linear regressions, multiple linear regressions, principal component analyses, and Shapiro–Wilk and Kolmogorov–Smirnov normality tests. OriginPro 2021 (OriginLab) was used for these tests. Statistical significance was met if the test statistic was <0.05. HgII concentrations were not normally distributed for any of the sites (except for SPL) therefore log transformation was carried out before further analysis.
2.4 Other chemical and meteorological measurements at SPL
Meteorological data were collected on the roof of SPL at a height of 10 m above ground level (AGL) at five-minute time resolution.51 Ozone (O3) was measured at a time resolution of 1 minute and calibrated daily using a Thermo Model 49i Analyzer with a precision of 1.0 ppb. As no precipitation data was collected at SPL, this study relied upon hourly precipitation measurements over the measurement period at SPL from the MDN site CO97 at Buffalo Pass, located 9 km to the northwest from SPL in the same mountain range at an elevation of 3234 m A.S.L. More details on the SPL ancillary measurements are given in Derry et al.51Back trajectory simulations for SPL were performed using the NOAA Air Resources Laboratory GDAS 1° data archive and Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) (http://ready.arl.noaa.gov/HYSPLIT.php) for the timespan March 12–September 30, 2021. Back trajectories were initialized every six hours for starting locations in a 0.5° × 0.5° grid centered on SPL. The initial trajectory heights were 100, 300, and 500 m above the ground, and the trajectories went backward 24 hours. Each trajectory contains information on the modeled boundary layer height, and this was used to calculate the mean height of the multiple trajectories above the boundary layer. All initial trajectory locations and heights were reduced to a 6 h average (mean of N = 675 trajectories). The mean pressure of the trajectories at time = 0 for a subset between the dates May 1–June 14, 2021, was 704 mbar (averaging all the 100, 300, and 500 m AGL starting heights) that is similar to the mean barometric pressure (698 mbar) during the previous study.47 This indicates that while the terrain at SPL is complex and meteorological fields are coarse, the back trajectory starting points reasonably represented the surface. For the comparison of [Hg]aq with mean back trajectory height, back trajectory data representing wet conditions were selected 12 h before the onset of precipitation at CO97 until the end time of the precipitation. This data selection decision was made to boost the number of 6 h observations used for averaging, since precipitation events were sometimes only a few hours per week, and to get a representation of the atmosphere just before the precipitation event. If two precipitation events were separated by 24 h or less in time, then an unbroken record of the 6 h data was selected until the end of the precipitation event.
3 Results and discussion
3.1 Storm Peak Lab and CO97 Hg data
HgII and Hg0 concentrations measured at Storm Peak Lab with the Dual-Channel System during March–October 2021 and March–September in 2022 (ref. 51) were aligned with hourly precipitation measurements from Buffalo Pass (CO97). Table 1 shows the mean and median values for HgII and Hg0 grouped by precipitation state (dry or wet). HgII concentrations were statistically significantly lower during wet periods with a difference between the medians of each group of 22 pg m−3 (Fig. 2a). HgII measurements classified as wet constituted 9.4% of the total measurements. HgII was washed out of the atmosphere during precipitation, however Hg0 was released as shown by a wet-dry difference in median Hg0 concentrations of 0.052 ng m−3, equivalent to a 4.2% increase (Fig. 2b). The effect of season on HgII washout and Hg0 enhancement is shown in Fig. 2c and d. HgII washout can be seen in all months except March, and Hg0 enhancement occurred in all months except October. Both Hg species displayed little seasonality at SPL.51| Site | Parameter (unit) | N (hourly) | Mean | Median | Std dev. | SE |
|---|---|---|---|---|---|---|
| SPL | HgII all (pg m−3) | 7469 | 93 | 90 | 43 | 0.5 |
| SPL | HgII wet (pg m−3) | 703 | 71 | 70 | 34 | 1.3 |
| SPL | HgII dry (pg m−3) | 6766 | 96 | 92 | 43 | 0.5 |
| SPL | HgII dry-wet (pg m−3) | 25 | 22 | 9 | 0.3 | |
| SPL | Hg0 all (ng m−3) | 7574 | 1.256 | 1.270 | 0.118 | 0.001 |
| SPL | Hg0 wet (ng m−3) | 722 | 1.301 | 1.314 | 0.094 | 0.004 |
| SPL | Hg0 dry (ng m−3) | 6944 | 1.250 | 1.264 | 0.117 | 0.001 |
| SPL | Hg0 wet-dry (ng m−3) | 0.051 | 0.050 | 0.033 | 0.020 | |
| MD99 | HgII all (pg m−3) | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 465 465 |
44 | 43 | 23 | 0.2 |
| MD99 | HgII wet (pg m−3) | 699 | 37 | 36.6 | 16 | 0.2 |
| MD99 | HgII dry (pg m−3) | 9766 | 45 | 44.0 | 23 | 0.6 |
| MD99 | HgII dry-wet (pg m−3) | 8 | 7 | 7 | 0.3 | |
| MD99 | Hg0 all (ng m−3) | 14186 |
1.288 | 1.289 | 0.150 | 0.001 |
| MD99 | Hg0 wet (ng m−3) | 846 | 1.341 | 1.347 | 0.135 | 0.001 |
| MD99 | Hg0 dry (ng m−3) | 12414 |
1.289 | 1.297 | 0.143 | 0.005 |
| MD99 | Hg0 wet-dry (ng m−3) | 0.052 | 0.050 | 0.008 | 0.004 |
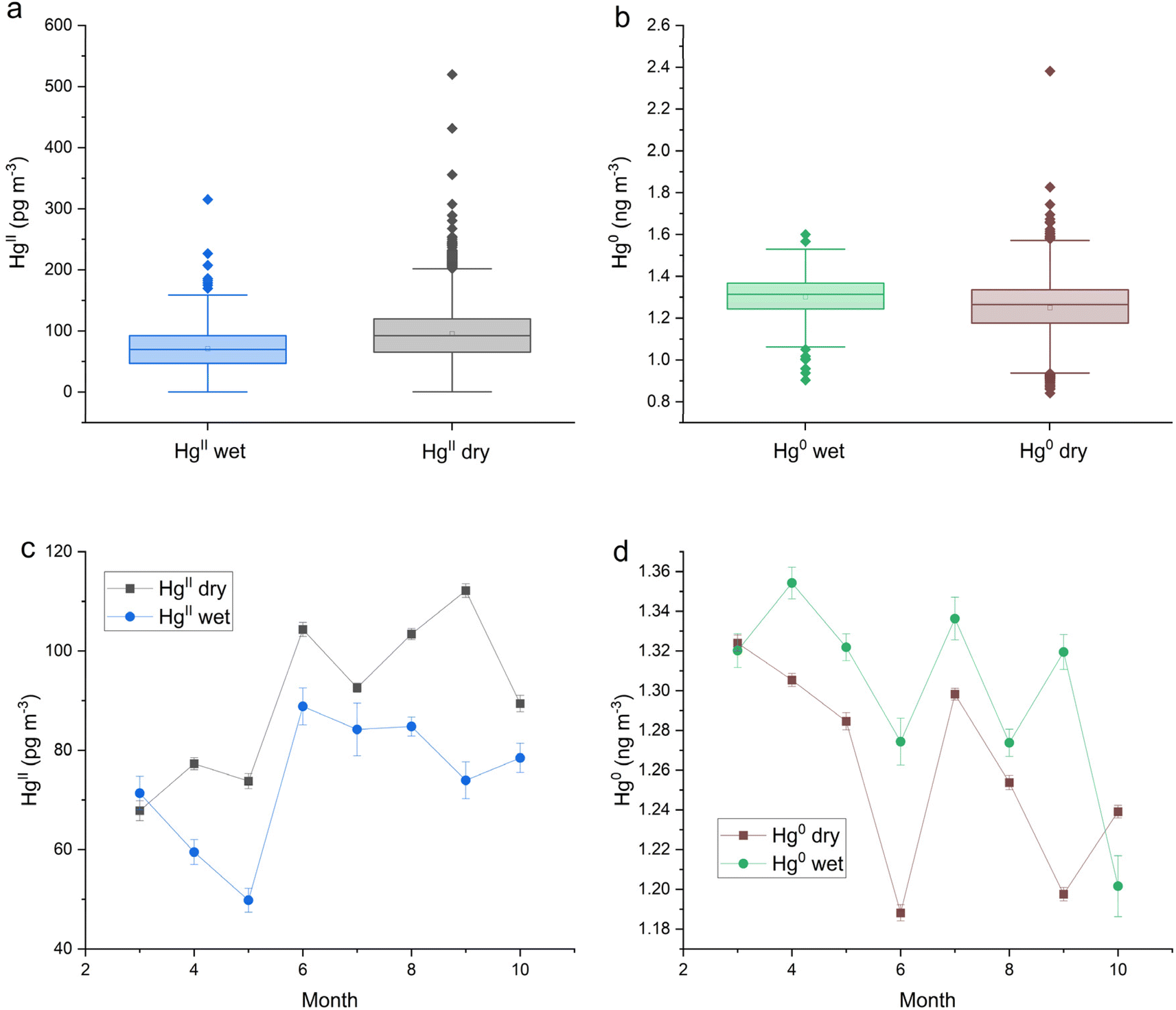 | ||
| Fig. 2 Storm Peak Laboratory (SPL) box and whisker plots of (a) HgII and (b) Hg0 concentrations by precipitation state, and (c) HgII and (d) Hg0 monthly medians by precipitation state. These data were measured with a dual-channel measurement system. Error bars in (c) and (d) represent the standard error. | ||
HgII and Hg0 concentrations plus ancillary parameters at SPL were compared with [Hg]aq concentrations in the weekly precipitation sample at CO97 (Table 2). Log[Hg]aq was significantly positively correlated with the following parameters: log HgII, log sample volume, relative humidity, ozone concentrations, PM10 aerosol scattering, and the mean altitude of 24 h back trajectory heights arriving at SPL (Table 2). Principal component analysis (Appendix A, Fig. 7) showed that the first component was consistent with a relatively strong anticorrelation between [Hg]aq and %RH. Taken together, these relationships suggest that precipitation that originates higher in the troposphere, as indicated by the trajectory heights, higher ozone, and lower relative humidity, generally has higher [Hg]aq, which is consistent with the recent analysis by Derry et al.,51 Shah and Jaeglé's29 model results, and the work of Huang and Gustin.30 A weak correlation between HgII measured at SPL with [Hg]aq in a weekly precipitation sample is consistent with the source of HgII washout at this site being primarily entrainment of middle free tropospheric air into the storm system many days before reaching the site.51
| Linear correlation with log [Hg]aq | SPL (DCS) r2 | MD99 (DCS) r2 |
|---|---|---|
| Log sample volume | −0.02 | 0.30 |
| Hg0 | −0.03 | −0.01 |
| HgII | 0.09 | 0.00 |
| O3 | 0.24 | |
| Log PM10 scattering | 0.33 | |
| %RH | 0.39 | |
| Back trajectory height | 0.27 |
3.2 Beltsville, MD (MD99) Hg data
The dual-channel system HgII and Hg0 measurements at this site were aligned with hourly precipitation data to determine which measurements were grouped either wet or dry. Like the trend observed at SPL, HgII concentrations at MD99 were statistically significantly lower during wet periods with the difference between the medians of dry and wet being 7 pg m−3 (Table 1 and Fig. 3a). The proportion of HgII measurements classified as wet was 7.0%. Hg0 concentrations were significantly higher during wet periods with a significant difference between the medians of 0.049 ng m−3 (Table 1 and Fig. 3b). Seasonal effects for both HgII washout and Hg0 enhancement are evident in Fig. 3c and d. For HgII, washout occurred mainly from Feb to July, peaking in the months of April and June. For Hg0, the months of Jun.–Dec. showed the most enhancement.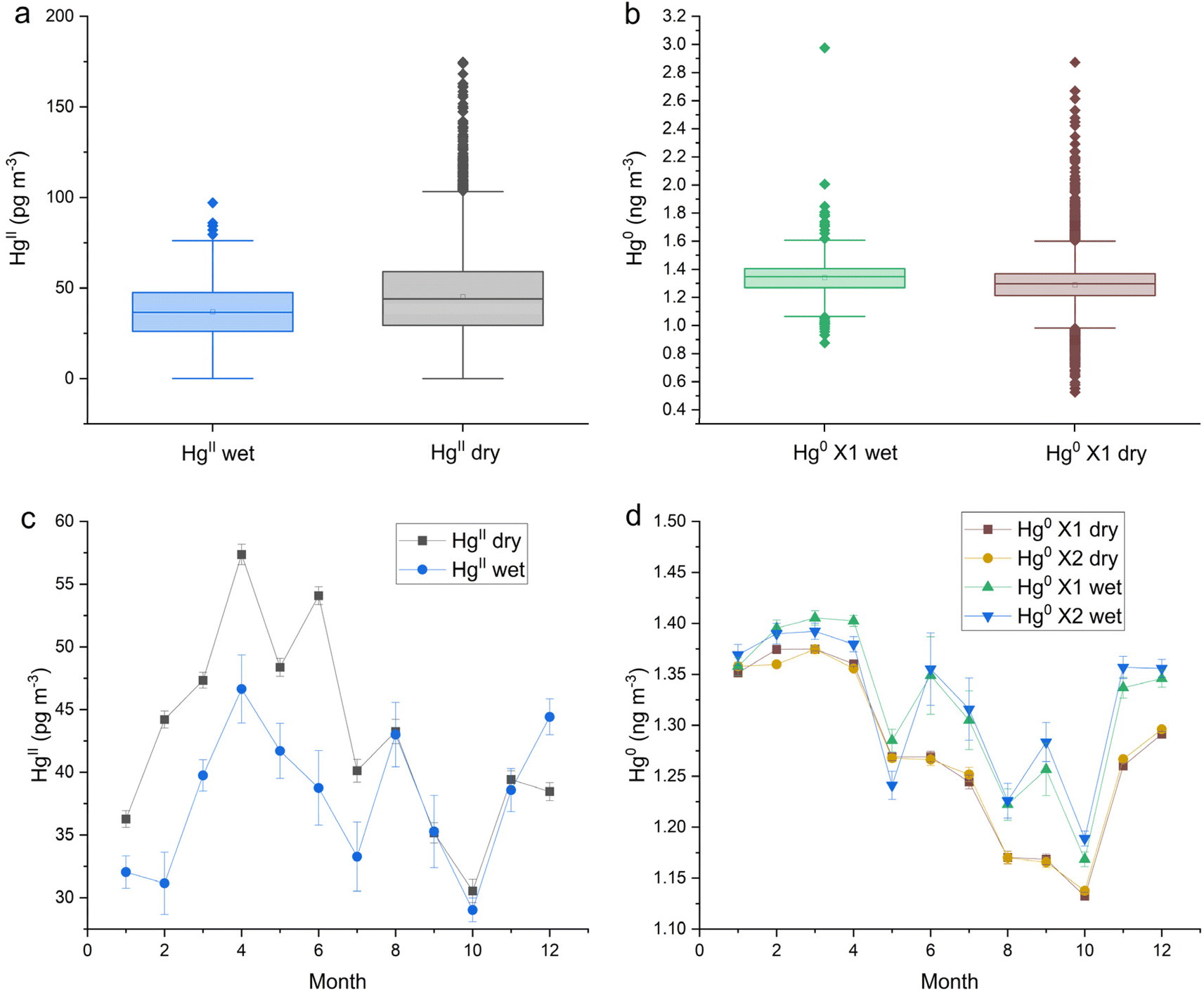 | ||
| Fig. 3 Beltsville, MD (MD99) box and whisker plots of (a) HgII and (b) Hg0 concentrations by precipitation state, and (c) HgII and (d) Hg0 monthly medians by precipitation state. These data were measured with a dual-channel measurement system. X1 and X2 refer to two Tekran 2537X instruments used. | ||
HgII and Hg0 concentrations at MD99 were compared with [Hg]aq concentrations in each weekly precipitation sample (Table 2). Log[Hg]aq was negatively correlated with log sample volume due to the dilution effect, with the relationship being significant. However, neither HgII nor Hg0 showed a significant correlation with log[Hg]aq at MD99. Principal component analysis for MD99 data (Appendix A, Fig. 7) showed that the first component is associated with elevated [Hg]aq and lower sample volume, i.e., the dilution effect.
3.3 Gaseous and aqueous Hg data from 16 co-located AMNet-MDN sites
Co-located AMNet-MDN sites were categorized based on patterns of mean Hg concentrations in seasonal and diel cycles (in Appendix A, see Table 4, Fig. 8, and 9), and each site was assigned one of four site types: Local Emissions, Suburban North, Suburban South, Rural North. At each site, GOM, PBM, and Hg0 concentrations were paired with the corresponding hour when precipitation was >0 to produce a data set that could be summarized based on precipitation state. Median HgII dry, median HgII wet, and the median dry-median wet difference for each AMNet site are shown in Fig. 4a–c, respectively, and for Hg0, these values are plotted in Fig. 4d. HgII washout ranged from 1–10 pg m−3 with an all-site mean of 4 pg m−3 (Table 3). Hg0 enhancement ranged from −0.037 to 0.120 ng m−3 with an all-site mean of 0.049 ng m−3, which was notably similar to that observed at the two DCS sites. We hypothesize that the pattern in Hg0 is related to emissions of Hg0 from surfaces when they become wet.58–61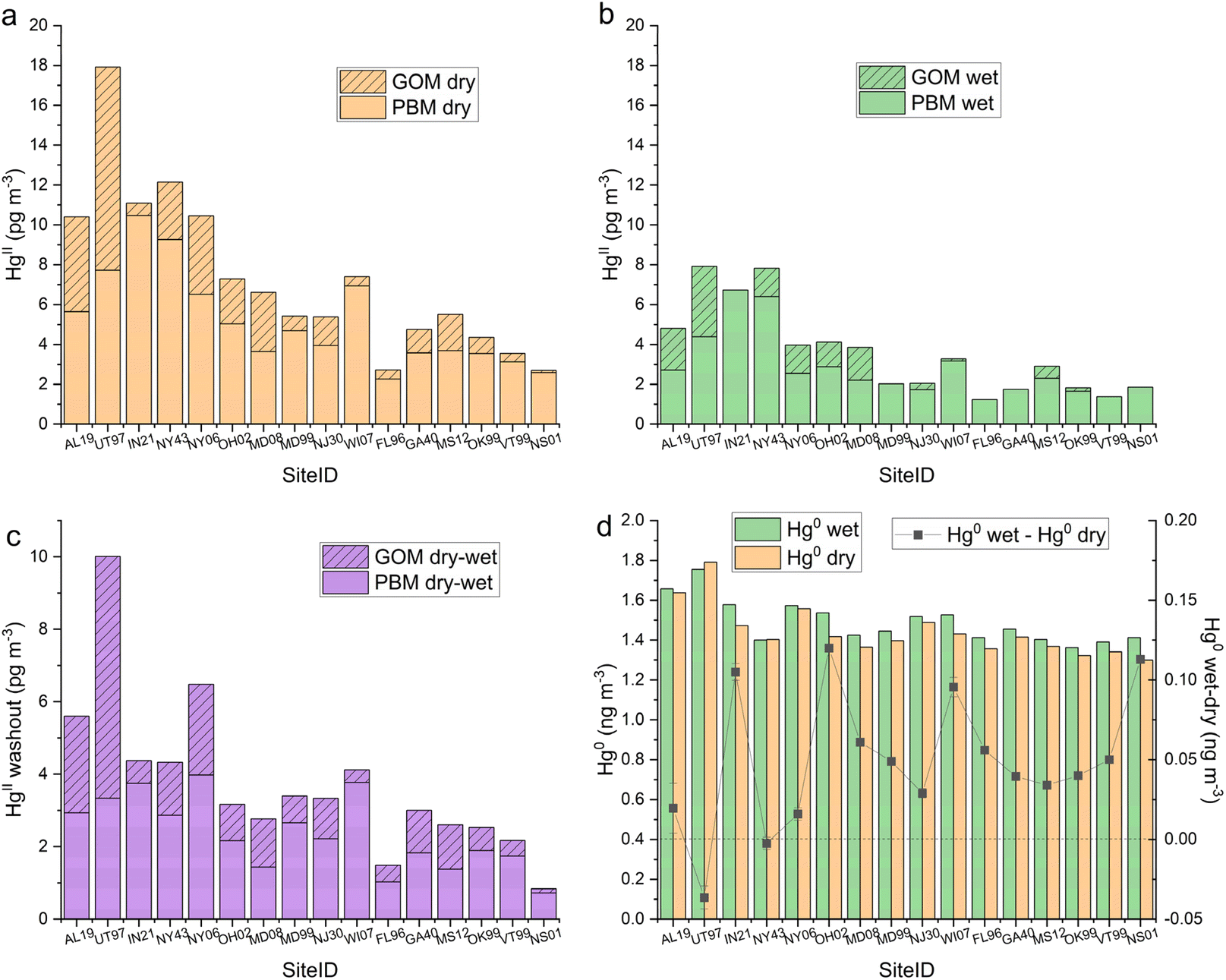 | ||
| Fig. 4 Comparison of median HgII concentrations, which is composed of GOM and PBM, at the 16 AMNet sites segregated by precipitation state: (a) dry, (b) wet, (c) the difference between median dry and median wet, and (d) median wet, median dry and wet-dry Hg0 concentrations. The total height of the bars in panels (a)–(c) represents GOM + PBM = HgII. The sites are ordered from left to right on the x-axes by site group (Local Emissions, Suburban North, Suburban South, and Rural North). | ||
| Site ID | Median HgII ambient HgII pg−3 | Median dry-median wet HgII “washout” | % washout GOM vs. PBM | Correction estimate | Median Hg0 ambient Hg0 ng m−3 | Median wet-dry Hg0 enhance Hg0 ng−3 | % increase Hg0 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PBM pg−3 | GOM pg−3 | HgII pg−3 | PBM | GOM | Ambient HgII/washout HgII | HgII corr fact | |||||
| AL19 | 10 | 3 | 3 | 6 | 55 | 59 | 1.8 | 2.8 | 1.810 | 0.020 | 1.1 |
| UT97 | 17 | 3 | 7 | 10 | 44 | 68 | 1.7 | 2.8 | 1.886 | −0.037 | −1.9 |
| IN21 | 11 | 4 | 1 | 4 | 37 | 114 | 2.5 | 2.0 | 1.508 | 0.105 | 7.0 |
| NY43 | 12 | 3 | 1 | 4 | 32 | 53 | 2.7 | 1.8 | 1.436 | −0.003 | −0.2 |
| NY06 | 10 | 4 | 3 | 6 | 64 | 68 | 1.5 | 3.2 | 1.684 | 0.016 | 1.0 |
| OH02 | 7 | 2 | 1 | 3 | 45 | 48 | 2.2 | 2.3 | 1.453 | 0.120 | 8.3 |
| Loc. Emis. | 11 | 3 | 2 | 6 | 46 | 68 | 2.1 | 2.5 | 1.629 | 0.037 | 2.5 |
| MD08 | 6 | 1 | 1 | 3 | 44 | 48 | 2.2 | 2.2 | 1.360 | 0.061 | 4.5 |
| MD99 | 5 | 3 | 1 | 3 | 59 | 119 | 1.5 | 3.2 | 1.400 | 0.049 | 3.5 |
| NJ30 | 5 | 2 | 1 | 3 | 60 | 84 | 1.5 | 3.2 | 1.490 | 0.029 | 1.9 |
| WI07 | 7 | 4 | 0 | 4 | 63 | 60 | 1.6 | 3.1 | 1.440 | 0.096 | 6.6 |
| Sub. North | 7 | 3 | 1 | 3 | 56 | 78 | 1.7 | 2.9 | 1.423 | 0.059 | 4.1 |
| FL96 | 3 | 1 | 0 | 1 | 45 | 73 | 2.0 | 2.5 | 1.370 | 0.056 | 4.1 |
| GA40 | 5 | 2 | 1 | 3 | 51 | 91 | 1.6 | 3.0 | 1.380 | 0.040 | 2.9 |
| MS12 | 4 | 1 | 1 | 3 | 51 | 98 | 1.5 | 3.2 | 1.360 | 0.034 | 2.5 |
| OK99 | 4 | 2 | 1 | 3 | 55 | 84 | 1.7 | 2.9 | 1.320 | 0.040 | 3.0 |
| Sub. South | 5 | 2 | 1 | 2 | 50 | 87 | 1.7 | 2.9 | 1.358 | 0.042 | 3.1 |
| VT99 | 4 | 2 | 0 | 2 | 55 | 117 | 1.6 | 3.0 | 1.350 | 0.050 | 3.7 |
| NS01 | 3 | 1 | 0 | 1 | 29 | 115 | 3.1 | 1.6 | 1.310 | 0.113 | 8.6 |
| Rur. North | 4 | 1 | 0 | 2 | 42 | 116 | 2.4 | 2.3 | 1.330 | 0.082 | 6.2 |
| All AMNet | 7 | 2 | 1 | 4 | 49 | 81 | 1.8 | 2.7 | 1.472 | 0.049 | 3.5 |
| MD99 | 43 | — | — | 7 | — | — | 5.8 | — | 0.049 | 3.9 | |
| SPL | 90 | — | — | 22 | — | — | 4.1 | — | 1.290 | 0.052 | 4.2 |
| DCS sites | 66 | 15 | 4.9 | 1.270 | 0.051 | 4.0 | |||||
The washout amount and the percent removal varied by Hg form with GOM percent removal being greater for 15 of 16 sites (all-site mean 81%) than PBM (Table 3). This is consistent with HgII being the more scavenge-able form. From the HgII washout pattern across sites shown in Fig. 4c, there is greater HgII washout for sites that are the Local Emissions category of site types and also have higher ambient HgII concentrations (Appendix A, Table 4). The mean HgII washout amount for Local Emission site group is 6 pg m−3, compared to 2 pg m−3 for the two Rural North sites.
Relationships between AMNet atmospheric Hg concentration and [Hg]aq in a weekly precipitation sample are presented in Fig. 5. Each HgIIvs. [Hg]aq data pair represents the weekly averaged GOM + PBM (both wet and dry) and the weekly Hg concentration in the wet deposition sample. In Fig. 5a the mean of weekly average HgII concentration is weakly linearly correlated to median [Hg]aq across all sites (r2 = 0.2, p = 0.05). AL19 and UT97 have the highest HgII and [Hg]aq concentrations, whereas NS01 and VT99 have the lowest. IN21 and NY43 are positively shifted above the trend, perhaps due to the larger proportion of PBM as HgII that was observed at these two sites, since PBM is less easily scavenged by precipitation compared to GOM.41 In contrast the Suburban South group had low HgII concentrations and elevated [Hg]aq consistent with convective storms that scavenge high altitude RM in the SE U.S. region29,31 and possibly greater levels of instrument bias due to the humidity.62,63 The linear trend shown in Fig. 5b for all weekly average HgIIvs. [Hg]aq data for the six Local Emissions sites grouped together is significant (r2 = 0.04, p < 0.05), albeit weak, but one must remember that a stronger correlation would probably be seen if it were not for the negative bias in the HgII measurements. Uncorrected AMNet HgII measurements are likely underestimating the contribution from local emissions to wet deposition of Hg.
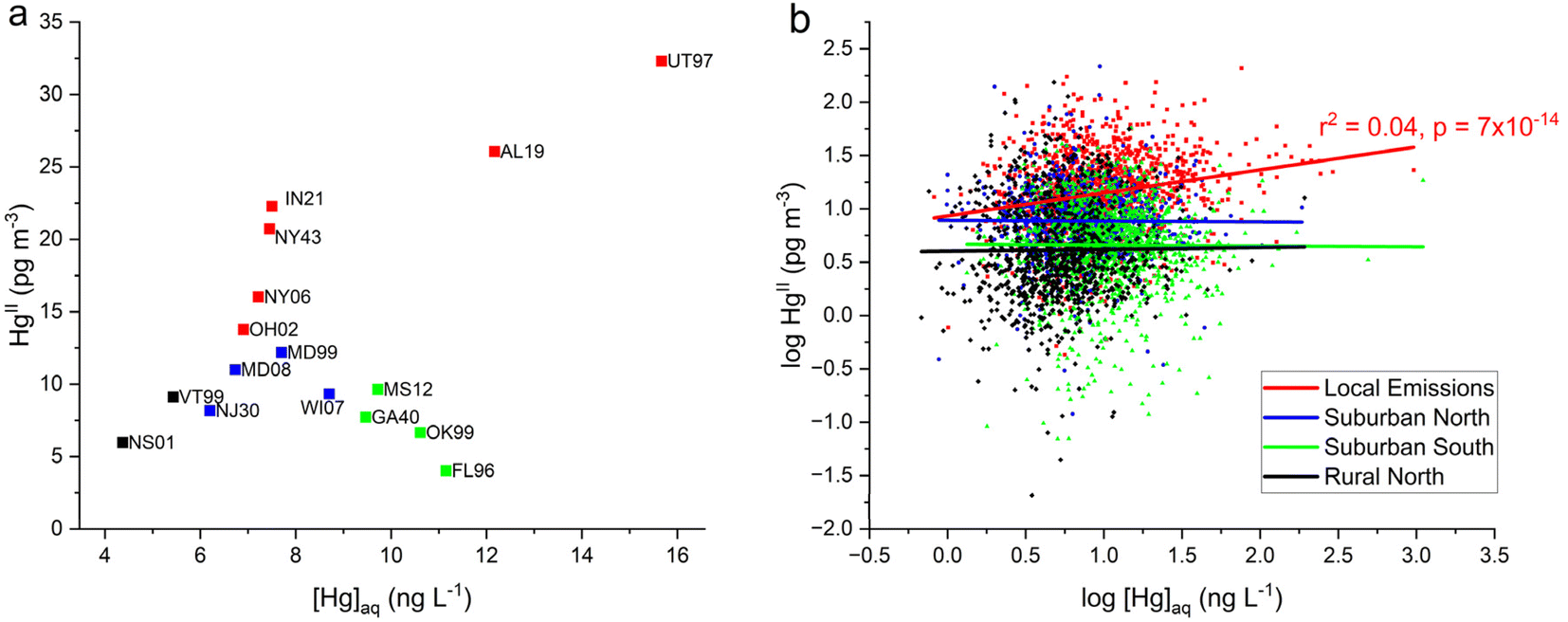 | ||
| Fig. 5 Comparison of AMNet and MDN data for the 16 co-located sites grouped into 4 site types. (a) Mean of HgII weekly averages (not segregated by precipitation state) vs. median [Hg]aq by site. (b) Log of mean of weekly HgII (GOM + PBM) concentrations vs. log of weekly [Hg]aq at. Linear regression statistics in (b) are shown for the data from Local Emissions sites; the other fits shown were not significantly different from zero. | ||
3.4 Ambient HgIIvs. washout HgII
Here we investigate whether HgII measurements made at two sites with the DCS method could be used to correct the older measurements made with KCl-denuders across the network. To address this, the relationship was explored between the median ambient HgII concentration and the median dry–median wet HgII (washout HgII) concentrations at each site. We tested whether at any given site using the same measurement method, there would be a constant fraction of ambient HgII scavenged during precipitation, when averaging over at least an annual cycle. Washout HgII concentrations for each site were calculated using eqn (1) and these values are plotted on the x-axis with median ambient HgII concentration for each site on the y-axis in Fig. 6a for the two DCS sites and in Fig. 6b for the 16 AMNet sites (note the different scales in the x- and y-axes). Linear fits for each group of sites were forced through zero and the resulting slopes are 4.8 ± 0.9 and 1.8 ± 0.1 for the DCS and AMNet sites, respectively. Standard deviations of the ambient HgII and the washout HgII concentrations are included in Fig. 6a to indicate the uncertainty in the slope that is derived from two DCS data points. Despite the low number of DCS sites, the pattern is clear that the slope from these sites is a factor 2.7 higher than for the AMNet sites.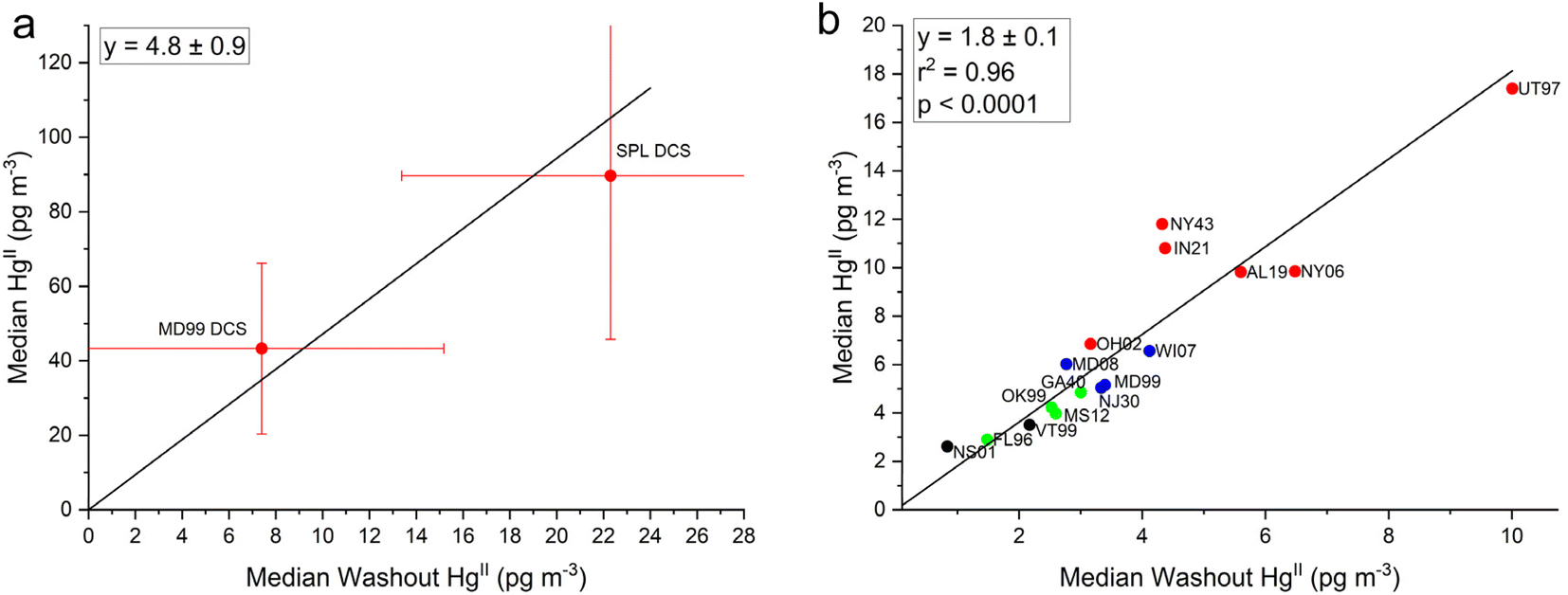 | ||
| Fig. 6 Median ambient HgII concentrations plotted against median dry-wet HgII concentrations for (a) the two dual-channel system (DCS) sites and (b) the 16 AMNet sites color coded by site type (see Fig. 5). The slopes are forced through zero and the error bars in (a) represent the standard deviation of all measurements in the y-direction and the standard deviation propagated from error in the dry and wet measurements in the x-direction. | ||
Based on the observations, it is expected then, that if all the instruments were behaving similarly the ratio of long term median ambient HgII to washout HgII concentration should be the same for all the instruments. Using this assumption allows for a potential correction factor which was derived for each AMNet site by using eqn (3) and dividing the HgII ambient/HgII washout ratio for the reference sites (DCS) by the same ratio for the AMNet site. This produces site-specific correction factors between 2–3 for each of the AMNet sites (Table 3). Prior work to address the low bias in AMNet GOM measurements using a scavenging ratio approach showed that the mean low bias across nine AMNet sites is factor of 2.3,42 a similar result obtained with the method shown here, suggesting a possible consensus in the directions and magnitude of the correction needed. While the results of this and the Cheng and Zhang42 studies produced similar results, the benefit of the washout approach described here is that it does not rely upon the assumption that there is a strong empirical relationship between atmospheric oxidized mercury and mercury wet deposition, as was previously assumed.42
The washout approach described here assumes a mass balance of HgII around the sampling site and it must be noted that other processes may be occurring that give rise to uncertainties in this method. These include in situ Hg redox reactions and transport of air enriched or depleted in HgII, especially when these processes might be different under wet and dry conditions. Future measurements and modeling should focus on the response of HgII and Hg0 concentrations around the sampling site during dry and wet periods to understand how certain this mass balance approach could be.
4 Conclusions
This study analyzed the behavior of atmospheric Hg as measured with two methods in relation to the effects of precipitation. Two sites made long-term measurements with a dual-channel system for quantifying atmospheric HgII (SPL and MD99), and these data were used as a basis for comparison of HgII washout, defined as median [HgII]dry–median [HgII]wet, with older GOM, PBM, and Hg0 data from 16 co-located sites in the AMNet and MDN networks (also with long-term data) as part of the National Atmospheric Deposition Program. A reduction in HgII concentrations during times of precipitation compared to dry conditions (i.e. HgII washout) was observed at all sites regardless of the measurement method, indicating this is a robust feature of the atmosphere due to the effects of precipitation. Similarly, Hg0 concentrations were higher during times of precipitation at nearly all sites (15 of 17) and likely is related to emissions of Hg0 from surfaces when they become wet. Measurements with the dual-channel system showed that on average, ambient HgII concentrations were related to the magnitude of HgII washout at a given site, with the ratios for SPL and MD99 (the dual-channel system sites) being 4.1 and 5.8, respectively. These relatively similar ratios between DCS sites were observed even though SPL had ambient HgII concentrations that were 2.1 times higher than MD99. When the same median ambient HgII/median washout HgII ratio was calculated for the 16 AMNet sites, it was found that the average ratio for the DCS sites was 2.7 times larger than the average for the AMNet sites. If it is assumed that the ratios from the same type of measurement method should be roughly similar, then it is possible to derive a correction factor using DCS data from a limited number of sites to apply to the AMNet sites. Site-specific correction factors were calculated between 2–3, which compared similarly with factors derived using a scavenging ratio approach42 giving another potential solution to improving the accuracy of semi quantitative older data.Correcting older HgII data is particularly important because a relationship was found between HgII in the air and [Hg]aq in wet deposition, and thus the proportion of local emissions scavenged from the atmosphere and wet deposited would be underestimated if the currently available measurements were used to calculate a flux. Furthermore, uncorrected observations are often used to validate chemical models; therefore, to improve source attribution studies and global model validity, it is critical to improve the accuracy of the available data even if the correction method itself is an estimate. Future work should make at least one-year worth of measurements using the DCS at multiple AMNet sites to calculate HgII washout and compare with ambient HgII concentrations in order verify if the correction factors suggested here still agree.
Data availability
The raw data from Storm Peak Laboratory are publicly available from at https://doi.org/10.5281/zenodo.10699270. The data from Beltsville, MD are also publicly available and can be obtained by sending an email to E-mail: Winston.luke@noaa.gov; . The AMNet and MDN data are publicly available through the NADP website https://nadp.slh.wisc.edu/.Author contributions
Peter Weiss-Penzias – conceptualization, investigation, data compilation and analysis, supervision, and lead writing of original draft and the final manuscript. Seth Lyman – collection of measurement data, design, construction, and testing of dual channel instrument, contributor to writing of the original draft and the final manuscript. Tyler Elgiar – collection of measurement data, construction, and testing of dual channel instrument. Lynne Gratz – collection of measurement data, contributor to writing of the original draft and the final manuscript. Winston Luke – collection of measurement data, construction, and testing of dual channel instrument. Gabriel Quevedo – data compilation and analysis. Nicole Choma – data compilation. Mae S. Gustin – conceptualization, resources, supervision, contributor to writing of original draft and the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Appendices
Appendix A: seasonal and diel cycles of AMNet-MDN Hg data
A list of the 16 co-located sites considered in this study, their measurement periods, and summary statistics for Hg are shown in Appendix Table 4. Medians were calculated for [Hg]aq to minimize the effect of single high sample since N = 4 or fewer for each month. The mean monthly concentrations of PBM, GOM, and Hg0 from AMNet sites, and the median monthly concentrations of [Hg]aq from the co-located MDN sites are shown in Fig. 7. Fig. 8 shows the mean hourly concentrations of PBM, GOM, and Hg0 from AMNet sites. Among the 16 sites, PBM concentrations were >75th percentile compared to all sites data at NY06, NY43, IN21, and UT97, GOM concentrations were >75th percentile at AL19, MD08, NY43, and UT97, and Hg0 concentrations were >75th percentile at AL19, NJ30, NY06, and UT97. Most sites showed a springtime maximum for GOM, a wintertime maximum for PBM, an early springtime maximum for Hg0, and a summertime maximum for [Hg]aq (Fig. 7). Across all sites, the magnitude of the seasonal cycle for Hg0 was in the 0.2–0.4 ng m−3 range, and GOM and PBM varied by 2–60 pg m−3, although these data are biased low, and the magnitude of the bias is dependent on which HgII forms are present and the ambient atmospheric chemistry. Trends such as these could also be due to trends in underlying non-Hg chemistry that impacts KCl-denuder performance, rather than due to trends in GOM itself. Concentrations of Hg in wet deposition displayed a warm-season maximum at all sites32 with UT97 displaying the highest summertime concentrations, probably due to its arid climate and low sample volume.23| Site ID | Site group | Date range (m/d/y) | Weekly total precip. median mm | Hg conc. median ng L−1 | Hg conc. std dev. ng L−1 | PBM median pg m−3 | PBM std dev. pg m−3 | GOM median pg m−3 | GOM std dev. pg m−3 | HgII median pg m−3 | HgII std dev. pg m−3 | Hg0 median ng m−3 | Hg0 std dev. ng m−3 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AL19 | LE | 4/11/12–12/16/15 | 75.3 | 12.2 | 19.1 | 5 | 34 | 5 | 90 | 10 | 125 | 1.810 | 2.310 |
| UT97 | LE | 12/2/08–9/27/16 | 30.7 | 15.7 | 66.1 | 8 | 45 | 10 | 28 | 17 | 73 | 1.886 | 0.860 |
| IN21 | LE | 5/3/16–12/31/19 | 240.1 | 7.5 | 10.2 | 10 | 39 | 1 | 4 | 11 | 44 | 1.508 | 0.350 |
| NY43 | LE | 9/30/08–2/2/16 | 144.2 | 7.5 | 7.1 | 9 | 19 | 3 | 11 | 12 | 31 | 1.436 | 0.380 |
| NY06 | LE | 9/2/08–2/2/16 | 195.4 | 7.2 | 6.8 | 6 | 18 | 4 | 13 | 10 | 32 | 1.684 | 0.410 |
| OH02 | LE | 1/16/09–2/26/20 | 173.6 | 6.9 | 7.1 | 5 | 16 | 2 | 12 | 7 | 27 | 1.453 | 0.230 |
| MD08 | SN | 1/6/09–1/2/19 | 160 | 6.7 | 6.8 | 3 | 5 | 3 | 13 | 6 | 17 | 1.360 | 0.210 |
| MD99 | SN | 1/6/09–7/15/14 | 159.2 | 7.7 | 11.1 | 5 | 72 | 1 | 46 | 5 | 118 | 1.400 | 0.260 |
| NJ30 | SN | 10/18/16–8/18/20 | 213.9 | 6.2 | 9.5 | 4 | 7 | 1 | 5 | 5 | 12 | 1.490 | 0.280 |
| WI07 | SN | 7/25/14–12/22/15 | 95.7 | 8.7 | 6.8 | 6 | 10 | 1 | 4 | 7 | 13 | 1.440 | 0.240 |
| FL96 | SS | 12/28/10–12/29/15 | 106.2 | 11.2 | 11.2 | 2 | 4 | 1 | 4 | 3 | 8 | 1.370 | 0.170 |
| GA40 | SS | 1/2/09–12/29/15 | 72.2 | 9.5 | 46.9 | 4 | 9 | 1 | 7 | 5 | 16 | 1.380 | 0.260 |
| MS12 | SS | 3/9/10–3/30/20 | 189.6 | 9.7 | 10.2 | 3 | 12 | 1 | 8 | 4 | 20 | 1.360 | 0.180 |
| OK99 | SS | 1/20/09–5/26/15 | 136.5 | 10.6 | 24.2 | 3 | 4 | 1 | 4 | 4 | 8 | 1.320 | 0.210 |
| VT99 | RN | 1/3/09–12/29/15 | 143.5 | 5.4 | 6.5 | 3 | 16 | 0 | 4 | 4 | 20 | 1.350 | 0.220 |
| NS01 | RN | 1/27/09–11/20/18 | 238.7 | 4.4 | 9.2 | 3 | 9 | 0 | 2 | 3 | 11 | 1.310 | 0.230 |
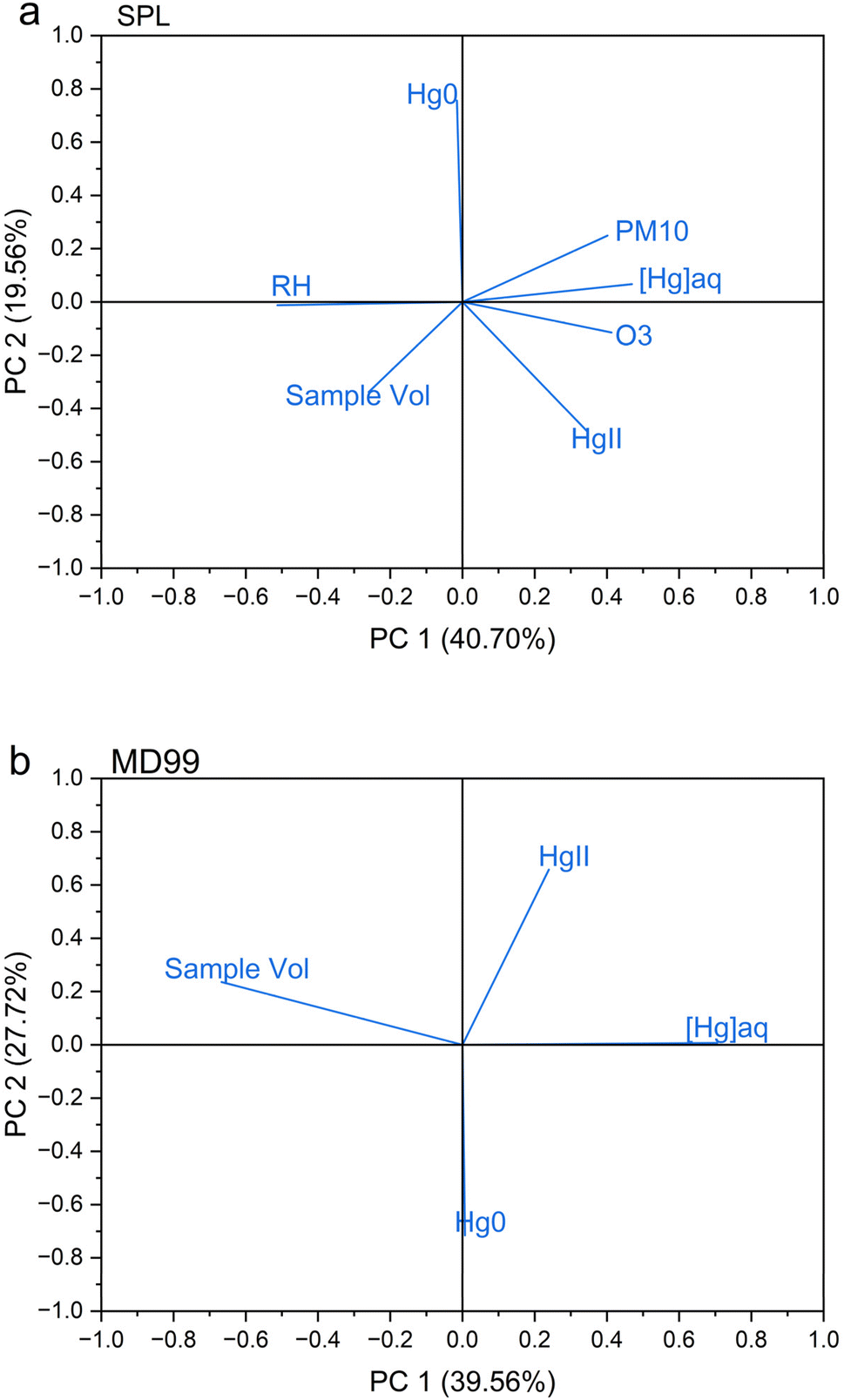 | ||
| Fig. 7 Principal component analysis of hourly data collected at the two DCS measurement sites (a) SPL, and (b) MD99, averaged over the hours of measurable precipitation during a weekly MDN sample. | ||
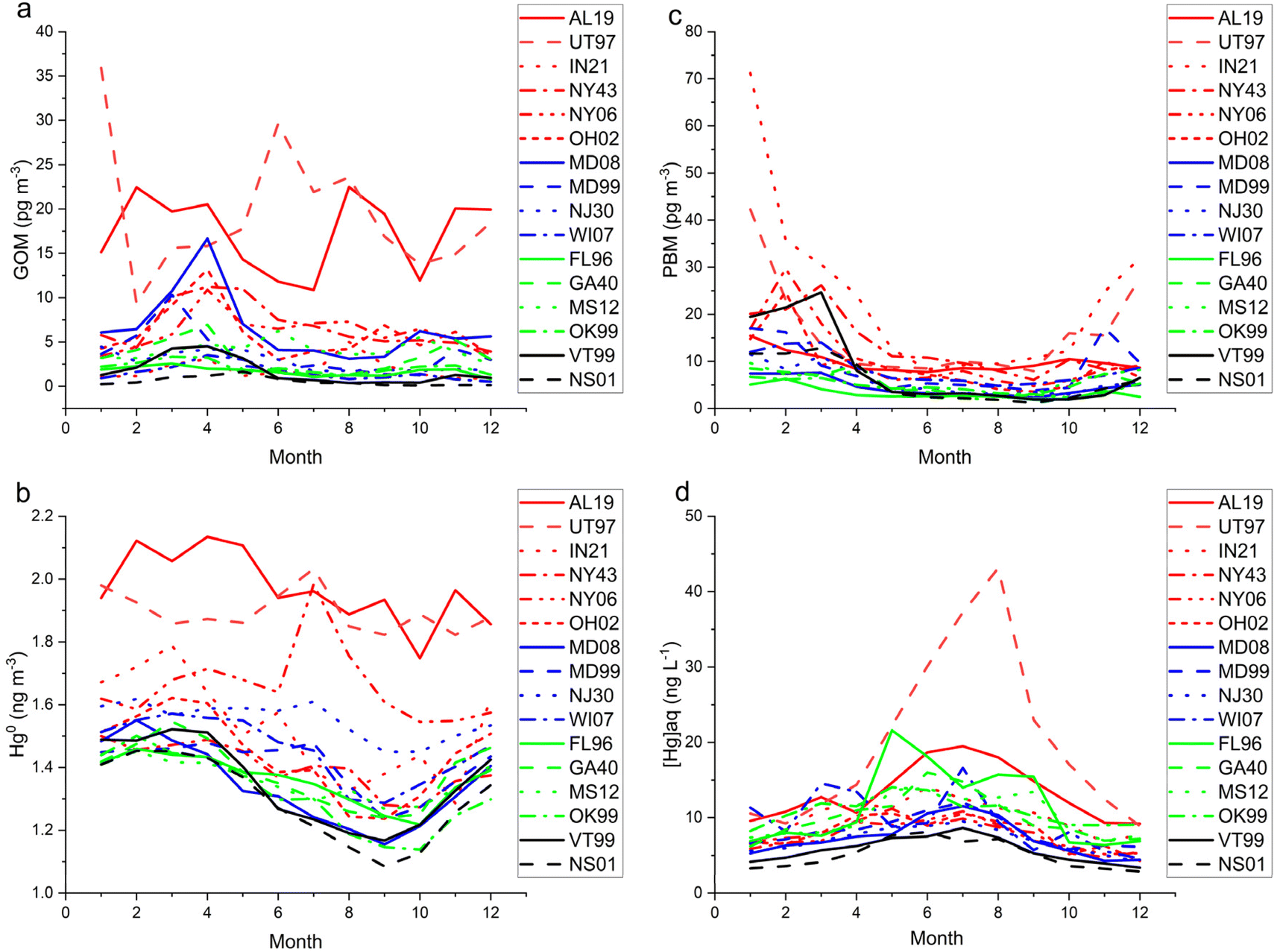 | ||
| Fig. 8 Mean monthly (a) GOM, (b) PBM, (c) GEM, and (d) median monthly [Hg]aq for the 16 AMNet and MDN sites color-grouped by site type (red = Local Emissions, blue = Suburban North, green = Suburban South, and black = Rural North). | ||
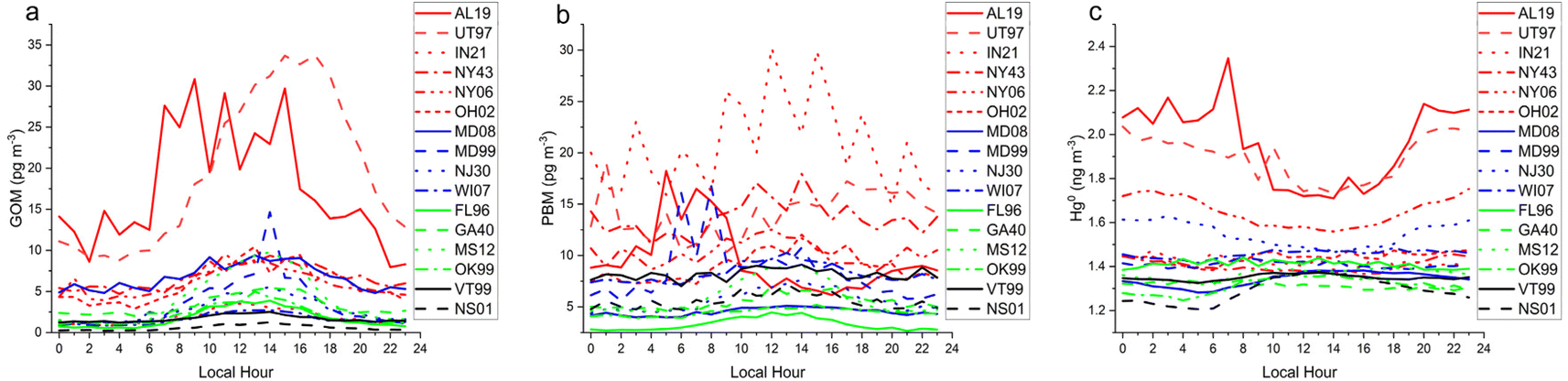 | ||
| Fig. 9 Mean hourly averages of (a) GOM, (b) PBM, and (c) GEM for the 16 AMNet sites color-grouped by site type, see caption for Fig. 1. | ||
The diel cycles of GOM demonstrated that it is a daytime form at least in the boundary layer, reaching a maximum at nearly every site in the afternoon (Fig. 8). AL19 is known to be impacted by local sources of GOM64 and thus displayed an earlier-shifted maximum compared to the other sites, reflecting the daily breakup of the nocturnal boundary layer.65 The diel pattern at UT97 is of similar magnitude as AL19, consistent with industrial sources in its vicinity,66 but is shifted later and smoother, indicating photochemistry or deep convective mixing is more important at this site. The PBM diel pattern is noisier than that for GOM, but generally shows a daytime increase in concentrations, for example at OH02 the amplitude of the cycle is 5 pg m−3 with a peak at 14:00 local time. Nocturnal boundary layer trapping of pollutants is evident in the diel pattern of PBM at AL19 with a peak in concentrations at 04:00 local time. IN21 displayed the highest PBM concentrations of any site; the small number of samples and the 3 hours measurement cycle produced the sawtooth pattern in Fig. 8. Hg0 diel patterns show a daytime minimum for the more polluted sites which could reflect a combination of plant uptake and the mixing of cleaner air from aloft throughout the day, and a daytime maxima or no diel pattern for the more background sites. Looking across all sites, the magnitude of the diel cycle for Hg0 is in the 0.1–0.2 ng m−3 range, and GOM and PBM vary by 2–20 pg m−3 although, as stated before, these data are biased low.
Acknowledgements
Robert Larson and Mark Kuether at Wisconsin State Hygiene Laboratory, and Greg Weatherbee at the US Geological Survey helped with attaining NADP data. Gannet Hallar, Ian McCubbin, Maria Garcia, and Dan Gilchrist of the University of Utah and SPL facilitated access to the laboratory and collected ancillary measurements. Sarah N. Dunham-Cheatham assisted with data compilation. Funding for the project came from the National Science Foundation Division of Atmospheric & Geospace Sciences, grant numbers 2043042, 1951513, 1951514, 1951515, and 1951632.References
- T. W. Clarkson, Environmental contaminants in the food chain, Am. J. Clin. Nutr., 1995, 61, 682S–686S CrossRef CAS PubMed.
- D. Mergler, H. A. Anderson and L. H. Chan, et al., Methylmercury exposure and health effects in humans: a worldwide concern, Ambio, 2007, 36, 3–11 CrossRef CAS PubMed.
- C. A. Eagles-Smith, J. J. Willacker and S. J. Nelson, et al., A National-Scale Assessment of Mercury Bioaccumulation in United States National Parks Using Dragonfly Larvae As Biosentinels through a Citizen-Science Framework, Environ. Sci. Technol., 2020, 54, 8779–8790 CrossRef CAS PubMed.
- P. A. Ariya, M. Amyot and A. Dastoor, et al., Mercury physicochemical and biogeochemical transformation in the atmosphere and at atmospheric interfaces: a review and future direction, Chem. Rev., 2015, 115, 3760–3802 CrossRef CAS.
- A. Saiz-Lopez, S. P. Sitkiewicz and D. Roca-Sanjuán, et al., Photoreduction of gaseous oxidized mercury changes global atmospheric mercury speciation, transport and deposition, Nat. Commun., 2018, 9, 4796 CrossRef.
- X. Yang, M. Jiskra and J. E. Sonke, Experimental rainwater divalent mercury speciation and photoreduction rates in the presence of halides and organic carbon, Sci. Total Environ., 2019, 697, 133821 CrossRef CAS.
- V. Shah, D. J. Jacob and C. P. Thackray, et al., Improved Mechanistic Model of the Atmospheric Redox Chemistry of Mercury, Environ. Sci. Technol., 2021, 55, 14445–14456 CrossRef CAS.
- A. Saiz-Lopez, O. Travnikov and J. E. Sonke, et al., Photochemistry of oxidized Hg(I) and Hg(II) species suggests missing mercury oxidation in the troposphere, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30949–30956 CrossRef CAS.
- S. N. Lyman, T. Elgiar, M. S. Gustin, S. M. Dunham-Cheatham, L. M. David and L. Zhang, Evidence against Rapid Mercury Oxidation in Photochemical Smog, Environ. Sci. Technol., 2022, 56, 11225–11235 CrossRef CAS PubMed.
- S. N. Lyman, I. Cheng, L. E. Gratz, P. Weiss-Penzias and L. Zhang, An updated review of atmospheric mercury, Sci. Total Environ., 2020, 707, 135575 CrossRef CAS.
- J. Zhou and D. Obrist, Global Mercury Assimilation by Vegetation, Environ. Sci. Technol., 2021, 55, 14245–14257 CrossRef CAS.
- L. Zhang, P. Blanchard, D. A. Gay, E. Prestbo, M. Risch, D. Johnson, J. Narayan, R. Zsolway, T. Holsen and E. Miller, Estimation of speciated and total mercury dry deposition at monitoring locations in eastern and central North America, Atmos. Chem. Phys., 2012, 12, 4327–4340 CrossRef CAS.
- D. O'Connor, D. Hou and Y. S. Ok, et al., Mercury speciation, transformation, and transportation in soils, atmospheric flux, and implications for risk management: A critical review, Environ. Int., 2019, 126, 747–761 CrossRef PubMed.
- J. Munthe, R. A. Bodaly and B. A. Branfireun, et al., Recovery of mercury-contaminated fisheries, Ambio, 2007, 36, 33–44 CrossRef CAS.
- C. A. Eagles-Smith, J. T. Ackerman and J. J. Willacker, et al., Spatial and temporal patterns of mercury concentrations in freshwater fish across the Western United States and Canada, Sci. Total Environ., 2016, 568, 1171–1184 CrossRef CAS.
- R. Ferrara, B. Maserti, A. Petrosino and R. Bargagli, Mercury levels in rain and air and the subsequent washout mechanism in a central Italian region, Atmos. Environ., 1986, 1967, 125–128 CrossRef.
- G. E. Glass, J. A. Sorensen, K. W. Schmidt, G. R. Rapp, D. Yap and D. Fraser, Mercury deposition and sources for the upper Great Lakes region, Water, Air, Soil Pollut., 1991, 56, 235–249 CrossRef CAS.
- J. Huang, M. B. Miller, P. Weiss-Penzias and M. S. Gustin, Comparison of gaseous oxidized Hg measured by KCl-coated denuders, and nylon and cation exchange membranes, Environ. Sci. Technol., 2013, 47, 7307–7316 CrossRef CAS.
- C. D. McClure, D. A. Jaffe and E. S. Edgerton, Evaluation of the KCl denuder method for gaseous oxidized mercury using HgBr2 at an in-service AMNet site, Environ. Sci. Technol., 2014, 48, 11437–11444 CrossRef CAS PubMed.
- M. S. Landis, A. F. Vette and G. J. Keeler, Atmospheric mercury in the Lake Michigan basin: influence of the Chicago/Gary urban area, Environ. Sci. Technol., 2002, 36, 4508–4517 CrossRef CAS.
- E. M. White, M. S. Landis, G. J. Keeler and J. A. Barres, Investigation of mercury wet deposition physicochemistry in the Ohio River Valley through automated sequential sampling, Sci. Total Environ., 2013, 448, 107–119 CrossRef CAS.
- L. E. Gratz, G. J. Keeler and E. K. Miller, Long-term relationships between mercury wet deposition and meteorology, Atmos. Environ., 2009, 43, 6218–6229 CrossRef CAS.
- P. S. Weiss-Penzias, D. A. Gay, M. E. Brigham, M. T. Parsons, M. S. Gustin and A. Ter Schure, Trends in mercury wet deposition and mercury air concentrations across the U.S. and Canada, Sci. Total Environ., 2016, 568, 546–556 CrossRef CAS.
- Y. M. Wang, D. Y. Wang, B. Meng, Y. L. Peng, L. Zhao and J. S. Zhu, Spatial and temporal distributions of total and methyl mercury in precipitation in core urban areas, Chongqing, China, Atmos. Chem. Phys., 2012, 12, 9417–9426 CrossRef CAS.
- Z. Ye, H. Mao and C. T. Driscoll, Primary effects of changes in meteorology vs. anthropogenic emissions on mercury wet deposition: a modeling study, Atmos. Environ., 2019, 198, 215–225 CrossRef CAS.
- C. Pearson, D. Howard, C. Moore and D. Obrist, Mercury and trace metal wet deposition across five stations in Alaska: controlling factors, spatial patterns, and source regions, Atmos. Chem. Phys., 2019, 19, 6913–6929 CrossRef CAS.
- X. Nie, C. Wu, H. Zhang, Y. Li, T. Li and Y. Wang, Atmospheric wet deposition of mercury in urban Jinan, eastern China: Speciation, scavenging process and potential sources, Ecotoxicol. Environ. Saf., 2023, 251, 114529 CrossRef CAS.
- H. Zhou, C. Zhou, P. K. Hopke and T. M. Holsen, Mercury wet deposition and speciated mercury air concentrations at rural and urban sites across New York state: Temporal patterns, sources and scavenging coefficients, Sci. Total Environ., 2018, 637–638, 943–953 CrossRef CAS PubMed.
- V. Shah and L. Jaeglé, Subtropical subsidence and surface deposition of oxidized mercury produced in the free troposphere, Atmos. Chem. Phys., 2017, 17, 8999–9017 CrossRef CAS.
- J. Huang and M. S. Gustin, Evidence for a free troposphere source of mercury in wet deposition in the Western United States, Environ. Sci. Technol., 2012, 46, 6621–6629 CrossRef CAS.
- A. S. Kaulfus, U. Nair, C. D. Holmes and W. M. Landing, Mercury Wet Scavenging and deposition differences by precipitation type, Environ. Sci. Technol., 2017, 51, 2628–2634 CrossRef CAS.
- E. M. Prestbo and D. A. Gay, Wet deposition of mercury in the US and Canada, 1996–2005: Results and analysis of the NADP mercury deposition network (MDN), Atmos. Environ., 2009, 43, 4223–4233 CrossRef CAS.
- D. A. Jaffe, S. Lyman, H. Amos, M. S. Gustin, J. Huang, N. E. Selin, L. Levin, A. Ter Schure, R. P. Mason, R. Talbot and A. Rutter, et al., Progress on understanding atmospheric mercury hampered by uncertain measurements, Environ. Sci. Technol., 2014, 48, 7204–7206 CrossRef CAS PubMed.
- S. N. Lyman, D. A. Jaffe and M. S. Gustin, Release of mercury halides from KCl denuders in the presence of ozone, Atmos. Chem. Phys., 2010, 10, 8197–8204 CrossRef CAS.
- N. Allen, J. Gacnik, S. M. Dunham-Cheatham and M. S. Gustin, Tests of alternate membranes and alternate configurations for the Reactive Mercury Active System, Atmos. Environ., 2024, 318, 120240 CrossRef CAS.
- A. Luippold, M. S. Gustin, S. M. Dunham-Cheatham and L. Zhang, Improvement of quantification and identification of atmospheric reactive mercury, Atmos. Environ., 2020, 224, 117307 CrossRef CAS.
- S. N. Lyman, L. E. Gratz, S. M. Dunham-Cheatham, M. S. Gustin and A. Luippold, Improvements to the Accuracy of Atmospheric Oxidized Mercury Measurements, Environ. Sci. Technol., 2020, 54, 13379–13388 CrossRef CAS.
- X. Bu, H. Zhang, G. Lv, H. Lin, L. Chen and X. Yin, et al., Comparison of reactive gaseous mercury collection by different sampling methods in a laboratory test and field monitoring, Environ. Sci. Technol. Lett., 2018, 5, 600–607 CrossRef CAS.
- Y. Tang, S. Wang and G. Li, et al., Elevated Gaseous Oxidized Mercury Revealed by a Newly Developed Speciated Atmospheric Mercury Monitoring System, Environ. Sci. Technol., 2022, 56, 7707–7715 CrossRef CAS PubMed.
- S. Chen, X. Qiu, L. Zhang, F. Yang and P. Blanchard, Method development estimating ambient oxidized mercury concentration from monitored mercury wet deposition, Atmos. Chem. Phys., 2013, 13, 11287–11293 CrossRef CAS.
- I. Cheng, L. Zhang and H. Mao, Relative contributions of gaseous oxidized mercury and fine and coarse particle-bound mercury to mercury wet deposition at nine monitoring sites in North America, J. Geophys. Res. Atmos., 2015, 120, 8549–8562 CrossRef CAS.
- I. Cheng and L. Zhang, Uncertainty Assessment of Gaseous Oxidized Mercury Measurements Collected by Atmospheric Mercury Network, Environ. Sci. Technol., 2017, 51, 855–862 CrossRef CAS PubMed.
- M. S. Gustin, S. M. Dunham-Cheatham, J. Huang, S. Lindberg and S. N. Lyman, Development of an understanding of reactive mercury in ambient air: A review, Atmosphere, 2021, 12, 73 CrossRef CAS.
- M. S. Gustin, S. M. Dunham-Cheatham and N. Allen, et al., Observations of the chemistry and concentrations of reactive Hg at locations with different ambient air chemistry, Sci. Total Environ., 2023, 904, 166184 CrossRef CAS.
- M. M. Lynam, J. T. Dvonch, J. A. Barres, M. S. Landis and A. S. Kamal, Investigating the impact of local urban sources on total atmospheric mercury wet deposition in Cleveland, Ohio, USA, Atmos. Environ., 2016, 127, 262–271 CrossRef CAS.
- M. Muntean, G. Janssens-Maenhout, S. Song, A. Giang, N. E. Selin and H. Zhong, et. al., Evaluating EDGARv4. tox2 speciated mercury emissions ex-post scenarios and their impacts on modelled global and regional wet deposition patterns, Atmos. Environ., 2018, 184, 56–68 CrossRef CAS.
- X. Faïn, D. Obrist, A. G. Hallar, I. Mccubbin and T. Rahn, High levels of reactive gaseous mercury observed at a high elevation research laboratory in the Rocky Mountains, Atmos. Chem. Phys., 2009, 9, 8049–8060 CrossRef.
- D. Obrist, A. G. Hallar, I. McCubbin, B. B. Stephens and T. Rahn, Atmospheric mercury concentrations at Storm Peak Laboratory in the Rocky Mountains: Evidence for long-range transport from Asia, boundary layer contributions, and plant mercury uptake, Atmos. Environ., 2008, 42, 7579–7589 CrossRef CAS.
- T. R. Elgiar, S. N. Lyman, T. D. Andron, L. Gratz, A. G. Hallar, M. Horvat, S. Vijayakumaran, T. O'Neil, R. Volkamer and I. Živković, Traceable Calibration of Atmospheric Oxidized Mercury Measurements, Environ. Sci. Technol., 2024, 58, 10706–10716 CrossRef CAS PubMed.
- S. M. Dunham-Cheatham, S. Lyman and M. S. Gustin, Comparison and calibration of methods for ambient reactive mercury quantification, Sci. Total Environ., 2023, 856, 159219 CrossRef CAS.
- E. J. Derry, T. R. Elgiar, T. Y. Wilmot, N. W. Hoch, N. S. Hirshorn, P. Weiss-Penzias, C. F. Lee, J. C. Lin, A. G. Hallar, R. Volkamer and S. N. Lyman, Elevated oxidized mercury in the free troposphere: Analytical advances and application at a remote continental mountaintop site, Atmos. Chem. Phys., 2024, 24, 9615–9643 CrossRef CAS.
- L. J. Ambrose, Improved methods for signal processing in measurements of mercury by Tekran® 2537A and 2537B instruments, Atmos. Meas. Tech., 2017, 10, 5063–5073 CrossRef.
- M. B. Miller, M. S. Gustin and G. C. Edwards, Reactive mercury flux measurements using cation exchange membranes, Atmos. Meas. Tech. Discuss., 2018, 2018, 1–28 Search PubMed.
- X. Ren, W. T. Luke, P. Kelley, M. D. Cohen, M. L. Olson, J. Walker, R. Cole, M. Archer, R. Artz and A. A. Stein, Long-Term Observations of Atmospheric Speciated Mercury at a Coastal Site in the Northern Gulf of Mexico during 2007–2018, 2020, Atmosphere, 11, 268 CrossRef CAS.
- S. M. Dunham-Cheatham, S. Lyman and M. S. Gustin, Evaluation of sorption surface materials for reactive mercury compounds, Atmos. Environ., 2020, 242, 117836 CrossRef CAS.
- N. Allen, J. Gačnik, S. M. Dunham-Cheatham and M. S. Gustin, Interaction of reactive mercury with surfaces and implications for atmospheric mercury speciation measurements, Atmos. Environ., 2024, 318, 120240 CrossRef CAS.
- X. Lan, R. Talbot, M. Castro, K. Perry and W. Luke, Seasonal and diurnal variations of atmospheric mercury across the US determined from AMNet monitoring data, Atmos. Chem. Phys., 2012, 12, 10569–10582 CrossRef CAS.
- X. Song and B. Van Heyst, Volatilization of mercury from soils in response to simulated precipitation, Atmos. Environ., 2005, 39, 7494–7505 CrossRef CAS.
- M. S. Gustin, S. E. Lindberg and P. J. Weisberg, An update on the natural sources and sinks of atmospheric mercury, Appl. Geochem., 2008, 23, 482–493 CrossRef CAS.
- C. Briggs and M. S. Gustin, Building upon the conceptual model for soil mercury flux: Evidence of a link between moisture evaporation and Hg evasion, Water, Air, Soil Pollut., 2013, 224, 1744 CrossRef.
- M. S. Gustin, J. A. Ericksen, D. E. Schorran, D. W. Johnson, S. E. Lindberg and J. S. Coleman, Application of controlled mesocosm for understanding mercury plant-soil-air exchange, Environ. Sci. Technol., 2004, 38, 6044–6050 CrossRef CAS PubMed.
- J. Huang, M. B. Miller, P. Weiss-Penzias and M. S. Gustin, Comparison of gaseous oxidized Hg measured by KCl-coated denuders, and nylon and cation exchange membranes, Environ. Sci. Technol., 2013, 47, 7307–7316 CrossRef CAS.
- S. Lyman, C. Jones and T. O'Neil, et al., Automated Calibration of Atmospheric Oxidized Mercury Measurements, Environ. Sci. Technol., 2016, 50, 12921–12927 CrossRef CAS.
- U. S. Nair, Y. Wu, J. Walters, J. Jansen and E. S. Edgerton, Diurnal and seasonal variation of mercury species at coastal-suburban, urban, and rural sites in the southeastern United States, Atmos. Environ., 2012, 47, 499–508 CrossRef CAS.
- H. Mao and R. Talbot, Speciated mercury at marine, coastal, and inland sites in New England–Part 1: Temporal variability, Atmos. Chem. Phys., 2012, 12, 5099–5112 CrossRef CAS.
- C. Peterson and M. S. Gustin, Mercury in the air, water and biota at the Great Salt Lake (Utah, USA), Sci. Total Environ., 2008, 405, 255–268 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |