
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA pulsed laser photolysis – pulsed laser induced fluorescence study of the kinetics and mechanism of the reaction of HgBr with NO2 and O2†
Dieter
Bauer
a,
Deanna
Donohoue
ab and
Anthony
Hynes
 *a
*a
aDepartment of Atmospheric Sciences, Rosenstiel School of Marine, Atmospheric and Earth Science, 4600 Rickenbacker Causeway, Miami, FL 33149, USA. E-mail: ahynes@miami.edu
bDepartment of Chemistry, Lawrence University, 711 East Boldt Way, Appleton, WI 54911, USA
First published on 8th April 2025
Abstract
The kinetics of the reactions of mercurous bromide (HgBr) with NO2 and O2 have been studied using the pulsed laser photolysis – pulsed laser induced fluorescence technique in nitrogen, air and helium at room temperature and as a function of pressure. For reaction with NO2, temporal profiles showed good pseudo-first order behavior and we see a three-body recombination and obtain rate coefficients of ∼1–7 × 10−11 cm3 per molecules per s over the pressure range 50–700 Torr in nitrogen. As expected, He is a less efficient 3rd body and rates are somewhat slower. We monitored the presence of a reduction channel regenerating Hg(0) and saw no evidence for it occurring. We obtained temporal profiles of HgBr at pressures of up to 500 Torr of O2 demonstrating that laser induced fluorescence has adequate sensitivity as a concentration diagnostic in laboratory studies. The temporal profiles showed no evidence for any reaction between HgBr and O2 at room temperature.
Environmental significanceMercury is a potent neurotoxin and global pollutant. Concerns about its impact on the environment have led to 128 countries signing the Minamata Convention on Mercury, a legally binding international treaty, with the goal of reducing anthropogenic emissions of Hg. In the atmosphere mercury exists primary as gas phase elemental mercury, Hg(0). To be efficiently deposited and incorporated into terrestrial ecosystems Hg(0) must undergo atmospheric oxidation to stable mercuric compounds and multiple models have been developed to predict mercury cycling. However, the elementary reactions and the rate coefficients that are used in these models are based on computational chemistry and there is very little experimental data to support the calculated rates and reaction mechanisms. In this work we present results on a laboratory study of the kinetics and mechanism of the reaction of HgBr with NO2, one potential route to a stable mercuric reaction product. |
Introduction
A detailed understanding of the biogeochemical cycling of mercury and the routes to the production of organomercury compounds in terrestrial ecosystems is a critical issue from a human health perspective. Direct exposure to mercury is primarily through the ingestion of methylmercury from fish consumption.1 Wet or dry deposition of oxidized mercury is an important step in a complex process that involves both chemistry and microbiology and eventually produces alkyl mercuric compounds. Consequently, an understanding of the overall budget and mechanisms of chemical transformation of mercury in both its elemental and combined forms is critically important. Hg(0) is almost insoluble in water, has a low deposition rate, and estimates of its atmospheric lifetime are variable ranging from a few months to more than one year.2,3 The typical background concentrations of Hg(0) in unpolluted environments range from 1.2–2 ng m−3 in the northern hemisphere and 1–1.5 ng m−3 in the southern hemisphere where 1 ng m−3 is ∼3 × 106 atoms per cm3 or ∼120 ppq (parts per quadrillion). Concentrations in both hemispheres have decreased over the past two decades.4 Observations of Hg(0) concentrations in polar regions have demonstrated that Hg(0) undergoes rapid chemical reaction in the polar spring, and it is now generally accepted that the initial step in the Hg(0) oxidation process, in the polar atmosphere and globally, is the three body recombination of Hg(0) with Br atoms to give mercurous bromide, HgBr.5However, HgBr is unstable and will dissociate to reactants unless it undergoes further reaction to form a stable mercuric compound. Over the past decade multiple models of the atmospheric oxidation process have been developed to explain the global distribution of Hg(0) concentrations, and deposition measurements of oxidized mercury. Nevertheless, the experimental database on the rate coefficients and mechanisms of potentially important atmospheric reactions is very limited or nonexistent in which case models rely on ab initio calculations. Ab initio calculations are challenging because of the necessity to account for the relativistic effects associated with the heavy mercury atom. Dibble and coworkers have reported calculations and experimental measurements concluding that HgBr can react rapidly with both NO2 and HO2.6,7 Based on this work, Horowitz et al.8 concluded that the Hg(0) + Br recombination followed by reaction of HgBr with NO2 and HO2 is the primary oxidation mechanism for Hg(0) on a global scale.
| HgBr + NO2 → product | (R1) |
| HgBr + NO2 → BrHgNO2 | (R1a) |
| HgBr + NO2 → Hg + BrNO2 | (R1b) |
A more recent work suggests reaction with O3 may be more important.9 In this work we report measurements of the rate coefficients and mechanism of the reaction of HgBr with NO2 and O2 and compare this with recently published measurements from Dibble and coworkers.6,10 Although we see no evidence for reaction with O2 at room temperature we demonstrate that laser induced fluorescence has adequate sensitivity to monitor HgBr in laboratory studies at high O2 concentrations.
Experimental section
The experimental approach utilized the pulsed laser photolysis – pulsed laser induced fluorescence technique and is similar to multiple other studies published from this laboratory.11 The 4th harmonic of a Nd-YAG laser is used to photolyze a suitable precursor generating a low concentration of the free radical of interest, which is then monitored using laser induced fluorescence with a frequency doubled Nd-YAG pumped dye laser. The lasers operate at a repetition frequency of 10 Hz and the time delay between the lasers is varied to obtain a temporal profile of the free radical. Most of our previous work has focused on kinetic studies of the OH radical, but the basic approach in this work was very similar. A schematic of the experimental setup is shown in Fig. 1. The free radical of interest, HgBr, was produced by photolysis of HgBr2 using the fourth harmonic of a Nd-YAG laser at 266 nm.| HgBr2 + hν266 nm → HgBr + Br | (R2) |
 | ||
| Fig. 1 Schematic of the experimental setup. | ||
The HgBr radical was monitored using an approach described by Donohoue,12 exciting the D2П3/2–X2Σ (2–0) transition at ∼256 nm. The D2П3/2 (2) vibrational level undergoes collision induced transfer to the B2Σ state and the fluorescence from the B2Σ–X2Σ transition in the visible wavelength region is observed at 485–505 nm.13 The fluorescence is primarily characterized by transitions from a highly excited B2Σ state to the ground state of the X2Σ level. The maximum fluorescence was observed at ∼502 nm, mainly the (22,0) transition. However, the observed peak contains fluorescence from all transitions, which have a Δv = 22 such as (22,0), (23,1), and (24,2). Resolved fluorescence measurements12 indicate that the intensity of the B–X band increases by a factor of three relative to the D–X band as pressure is increased from 80 to 400 tour in N2 but we are not aware of any quantitative measurements of the rate of collision induced crossing between the D and B states. The B2Σ–X2Σ fluorescence was monitored with a photomultiplier tube (PMT) using a filter pack consisting of a 500 ± 8 nm interference filter and a long pass filter. Hg(0) was detected by exciting the 63P1–61S0 transition at 253.7 nm and by monitoring the resonance fluorescence with a PMT equipped with a 254 nm interference filter. The photomultiplier outputs were amplified, and fed to a 200 MHz digital oscilloscope to obtain the integrated voltage for, typically, 64 laser shots. The temporal profile of the HgBr was obtained by varying the delay between the photolysis and probe lasers using a digital delay generator. The whole experimental sequence was controlled by a computer using a program written in python which set and controlled the delay sequence and read the integrated PMT voltage at each delay setting. The experimental procedure involved scanning from short to long delay times, and then returning to a normalization delay. The sequence was repeated twice with different delay times, again returning to the normalization delay after each delay scan. The probe laser was then blocked and scattered light from the photolysis laser was monitored as a function of delay time. The sets of delay scans were then normalized to give a complete concentration time profile and, finally, the photolysis scatter was subtracted from the normalized concentration time profile. The photolysis scatter was less than 5% of the maximum signal an was only measurable for 20 μs. Experiments were performed in a Pyrex reaction vessel at the temperature in the laboratory which was 22 °C. The reaction vessel was 8 cm in length and 2.5 cm in diameter. Four side arms were attached to the center. The photolysis and probe laser beams counter propagated through two of the side arms and fluorescence was detected perpendicular to the laser beams through a third side arm. The experiments were carried out under slow flow conditions with flows along the long axis of the cell ranging from 1 to 5 standard liters per minute (SLPM). This corresponded to linear flow rates between 15 and 70 cm s−1. Gas flow rates were measured using calibrated mass flow controllers and mass flow meters. Pressures were measured using capacitance manometers. HgBr2 was introduced into the reactant mixture by flowing the premixed gases through a Pyrex tube containing a porcelain boat filled with solid HgBr2 powder. This tube was located immediately before the reaction vessel and was not heated. We estimated that we obtained an HgBr2 concentration of ∼4 × 1011 molecules per cm3 in the gas mixture which was determined by pyrolysis and measurement of [Hg(0)] as discussed below. This is about an order of magnitude lower than calculated equilibrium vapor pressures (4.3 × 1012, 303 K (ref. 6)) and (2 × 1014, 338 K (ref. 9)).
The gas mixture then passed through an absorption cell, cold trap and pump. The different length and design of the absorption cells is discussed below.
In situ measurements of stable reactants
In this experimental approach the accuracy of the rate coefficient is dependent on the accuracy with which the concentration of the reactants in pseudo first order excess can be determined. In our experience the most accurate determination is obtained by monitoring the excess reactant in situ using absorption spectroscopy. Clearly this requires a suitable absorption feature with an adequate absorption cross section. In prior work on the kinetics of the reaction of OH with NO2, we monitored the NO2 concentration using absorption cells located before and after the reaction vessel, using absorption of the 365 nm line of a Hg lamp and absorption of the output of a Deuterium lamp between 200 and 380 nm using a monochromator and an optical multichannel analyzer.11 In our prior work the absorption cell lengths were limited to 100 cm and we obtained excellent agreement between concentrations using the atomic line, broadband absorption and flow calculations. To extend the range of concentrations that can be monitored in situ we used 2 different absorption cells with lengths of 0.83 and 2.5 m. This allowed us to measure very low concentrations of NO2, and it also allows us to measure low concentrations of Hg(0) and provide an absolute calibration of our Hg(0) LIF signal. NO2 was purified by freeze–pump cycles at 77 K and mixtures containing approximately 3% NO2 in nitrogen or helium were prepared manometrically in 20 L Pyrex bulbs. An accurate bulb concentration was then measured using the absorption of the 365 nm mercury line which has a well established11,14 cross-section of (5.75 ± 0.17) × 10−19 cm2. We wanted to have the option to use a 405 nm diode laser for absorption measurements, so a second absorption cell was placed in series with the 365 nm cell and the absorption cross-section at 405 nm was determined relative to the 365 nm absorption cross-section. We obtained an absorption cross section of 4.8 × 10−19 cm2. Both the 365 nm mercury line and the 405 nm diode laser line were then used for in situ measurements of the NO2 concentration during experiments. Initial experiments at lower NO2 were performed using the 405 nm diode laser which was multi passed through an 83 cm absorption cell with typically six passes giving an absorption path length of 5 m. A beam splitter directed a small fraction of the 405 nm beam onto a photomultiplier tube to monitor the variability in diode laser output. After passage through the cell the transmitted beam was also monitored using a second photomultiplier tube. Output currents were measured with a picoameter and an oscilloscope and fed to a computer with measurements being recorded at 1 Hz. A similar approach was used for the 365 nm measurements.Results and discussion
HgBr2, HgBr, and Hg(0) background concentration measurements
We have measured the concentration of the HgBr2 precursor using pyrolytic conversion of HgBr2 to Hg(0). In this approach, developed by Landis et al.,15 the gas mixture is flown over a KCl coated annular denuder which is heated to 500 °C in a tube furnace. We used an annular denuder which is identical to that described by Landis et al.Fig. 2 shows a measurement of the Hg(0) concentration profile in the 2.5 m absorption cell as a function of time using absorption of the 253.7 nm mercury line. After an initial flow of pure N2 through the absorption cell to establish an I0, the HgBr2 source and absorption cell are connected in line at 900 s. We see an increase in Hg(0) which corresponds to a background level of ∼3 × 1010 atoms per cm3 and this value was used to calibrate the mercury concentrations in Fig. 6 and 7. We speculate that this background Hg(0) comes from the dissociation of HgBr2. At approximately 1100 s the denuder is heated to 500 °C and we see a spike in Hg(0) as the accumulated HgBr2 decomposes eventually giving a close to steady state concentration of ∼4 × 1011 atoms per cm3 which corresponds to the HgBr2 concentration at a flow of 5 SLPM. The typical 266 nm photolysis energy in the reaction volume is 15–20 mJ with a diameter of 0.5 cm. There is considerable uncertainty in the absorption cross section of HgBr2 at 266 nm based on the literature measurements which were made at elevated temperatures.16 We assume an absorption cross section of 1 × 10−18 cm2 which gives a photodissociation efficiency of ∼10% and an initial HgBr concentration of 4 × 1010 molecules per cm3 assuming a quantum yield of 1.| HgBr + NO2 → products | (R1) |
 | ||
| Fig. 2 Hg(0) temporal profile during pyrolysis of HgBr2. | ||
Experiments were carried out under pseudo first-order conditions. Typical NO2 concentrations ranged between 1 × 1014 to 5 × 1015 molecules per cm3, with an estimated HgBr concentration of ∼4 × 1010 molecules per cm3. There it's a significant uncertainty in this number but it's clear that the HgBr concentration is several orders of magnitude lower than the NO2 concentration.
For reaction (R1) the HgBr temporal profiles were analyzed assuming simple pseudo-first order behavior to extract a pseudo-first order rate,
[HgBr]t = [HgBr]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−k′t) exp(−k′t) | (1) |
 | ||
| Fig. 3 (a) and (b) Temporal profiles of HgBr at several NO2 concentrations at a total pressure of 400 Torr of N2. | ||
 | ||
| Fig. 4 Pseudo-first order decay rates as a function of NO2 concentration for all 400 Torr data. The plot shows rates at NO2 concentrations calculated using both flow (flowconc) and in situ absorption (absconc) measurements. | ||
The pressure dependence of reaction (R1) was studied in nitrogen, helium and one measurement was made in air. Fig. 5 shows the pressure dependence of the rate coefficients obtained using NO2 concentrations calculated from both absorption measurements and flow calculations. The rate coefficients are listed in Table S2 in the ESI.† There is typically good agreement between the flow and absorption measurements, but the rates obtained using flows are systematically a few percent higher than those obtained from absorption. It is important to note that both sets of measurements ultimately depend on the use of an absorption measurement to calculate NO2 concentration (i.e. in the initial 3% mixture in the 20 L Pyrex bulb and then directly in situ). We typically see larger zero offsets in the regression lines that used concentrations based on flows which is suggestive of a small systematic error in the flow calibrations at low flows. Consequently, we regard the rate coefficients based on concentrations determined by the in situ absorption measurements as being more reliable. There is a significant pressure dependence of the rate coefficient in both molecular nitrogen and helium. The single measurement in air is essentially identical to the measurement in nitrogen. As expected nitrogen shows a greater third body efficiency than helium although the scatter in the He data is larger than in the N2 data, and, based on the single measurement in air, it appears that there is not a significant difference in the third body efficiency between oxygen and nitrogen.
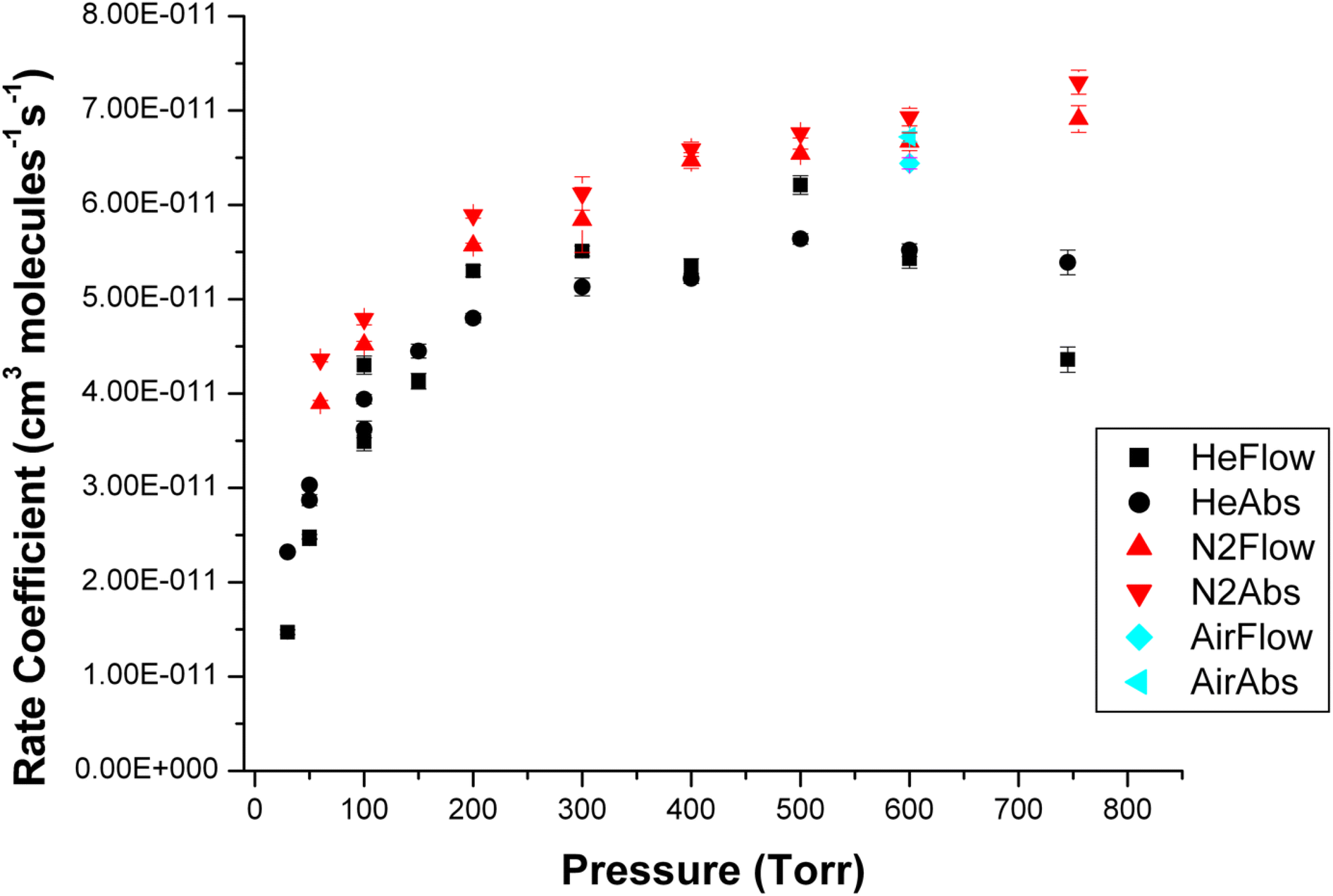 | ||
| Fig. 5 Pressure dependence of the rate coefficients for reaction (R1) in N2, air and He. Rates are shown for NO2 concentrations calculated using both flow (i.e. HeFlow) and in situ absorption (i.e. HeAbs) measurements. | ||
 | ||
| Fig. 6 Hg(0) concentration as a function of laser shots before and after the introduction of the photolysis laser. | ||
To look for the presence of a reduction channel we need to monitor Hg(0) formation after photolysis of an HgBr2/NO2 mixture. Fig. 7 shows the temporal profile of Hg(0) measured by LIF at 253.7 nm in 100 Torr of He in the presence of NO2 at a concentration of 3 × 1015 molecules per cm3. The profile is obtained by taking a pre-photolysis point and then several post photolysis points at different delays and then repeating the sequence. At this pressure we obtain a rate coefficient for reaction (R1) of ∼4 × 10−11 cm3 per molecules per s. Hence the reaction proceeds with a pseudo-first order rate of 120000 s−1 and a 1/e time of ∼8 μs. The reaction has essentially proceeded to completion in 40 μs. If we use the background concentration of Hg(0) of ∼7 × 1010 atoms per cm3, we can calibrate the concentration axis of Fig. 7 and estimate the importance of the reduction channel (R1b) by monitoring Hg(0) production. The pre-photolysis signal is (6.83 ± 0.21) × 1010 atoms per cm3. The average of the post photolysis signal is (7.05 ± 0.16) × 1010 atoms per cm3. Statistically we see no increase in Hg(0) as a function of delay time after photolysis suggesting that channel (R1b) is not significant.
 | ||
| Fig. 7 Hg(0) as a function of delay time with an NO2 concentration of 3 × 1015 molecules per cm3 at a total pressure of 100 Torr in He. | ||
| HgBr + O2 ⇌ HgBrOO | (R3) |
To estimate the significance of any reaction between HgBr and O2, we measured HgBr temporal profiles at several O2 partial pressures in N2/O2 mixtures at a total pressure of 500 Torr. Fig. 8 show a series of temporal profiles of the HgBr LIF signal with the signal normalized so that the initial HgBr LIF signal is 100 arbitrary units in each decay. Fig. 8 shows the temporal profiles on logarithmic scale. The decays do not follow simple pseudo-first order kinetics. We see an initial rise in the [HgBr] followed by decays at a rate of ∼50 s−1 which show no dependence on O2 concentration. The decays are dominated by diffusion of the HgBr from the reaction volume and reaction (R3) is too slow to be of any significance at room temperature. Lower temperature studies are required to determine the possible significance of the reaction at upper tropospheric temperatures. The initial rise in the HgBr concentration may be an indication of radiative cascading of vibrationally excited HgBr formed in the photolysis of HgBr2. Fig. 9 shows a laser excitation spectrum of the HgBr product formed in the 213 nm photolysis of HgBr2.12 The HgBr product was monitored via the D2П3/2–X2Σ transition with fluorescence detection using a PMT with a 262 nm filter. The spectrum shows production of vibrationally excited HgBr with population of the v = 1–4 levels in the X2Σ ground electronic state. Both Wu et al.6 and Gómez Martín et al.9 published laser excitation spectra of HgBr with photolysis of HgBr2 at 266 nm and HgBr detection using the B2Σ–X2Σ transition. These excitation spectra also show transitions from vibrationally excited HgBr. None of these spectra were obtained under conditions that would likely show the nascent vibrational excitation of the HgBr product. This type of vibrational excitation can be problematic in kinetic studies if cascading of high vibrational levels to the ground vibrational state occurs on a time scale that is close to the removal of the ground state by reaction. Because of the very large rate coefficient for reaction (R1) we do not believe this is a potential artifact in these studies.
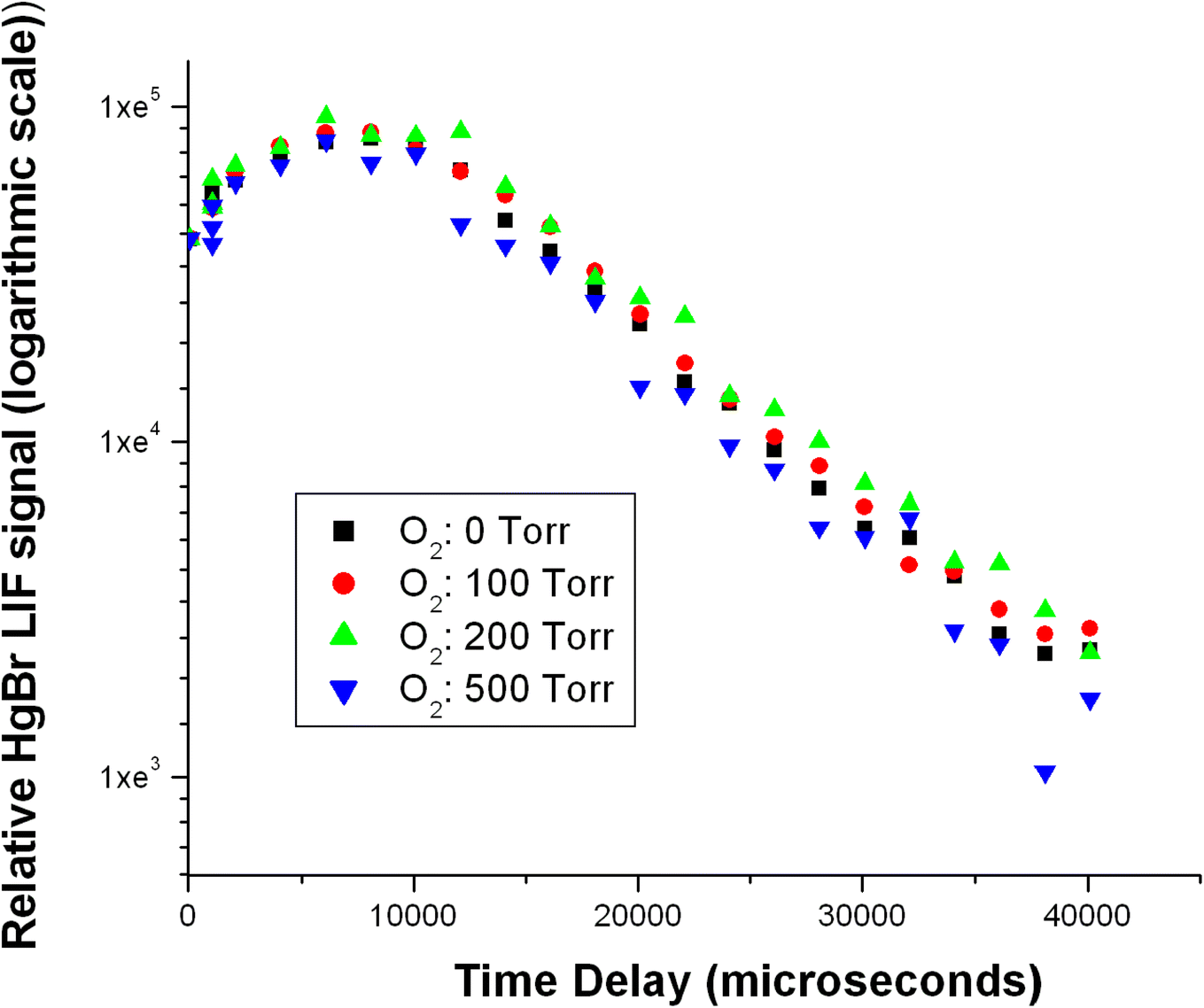 | ||
| Fig. 8 Temporal profiles of HgBr in N2/O2 mixtures at a total pressure of 500 Torr at several partial pressures of O2. | ||
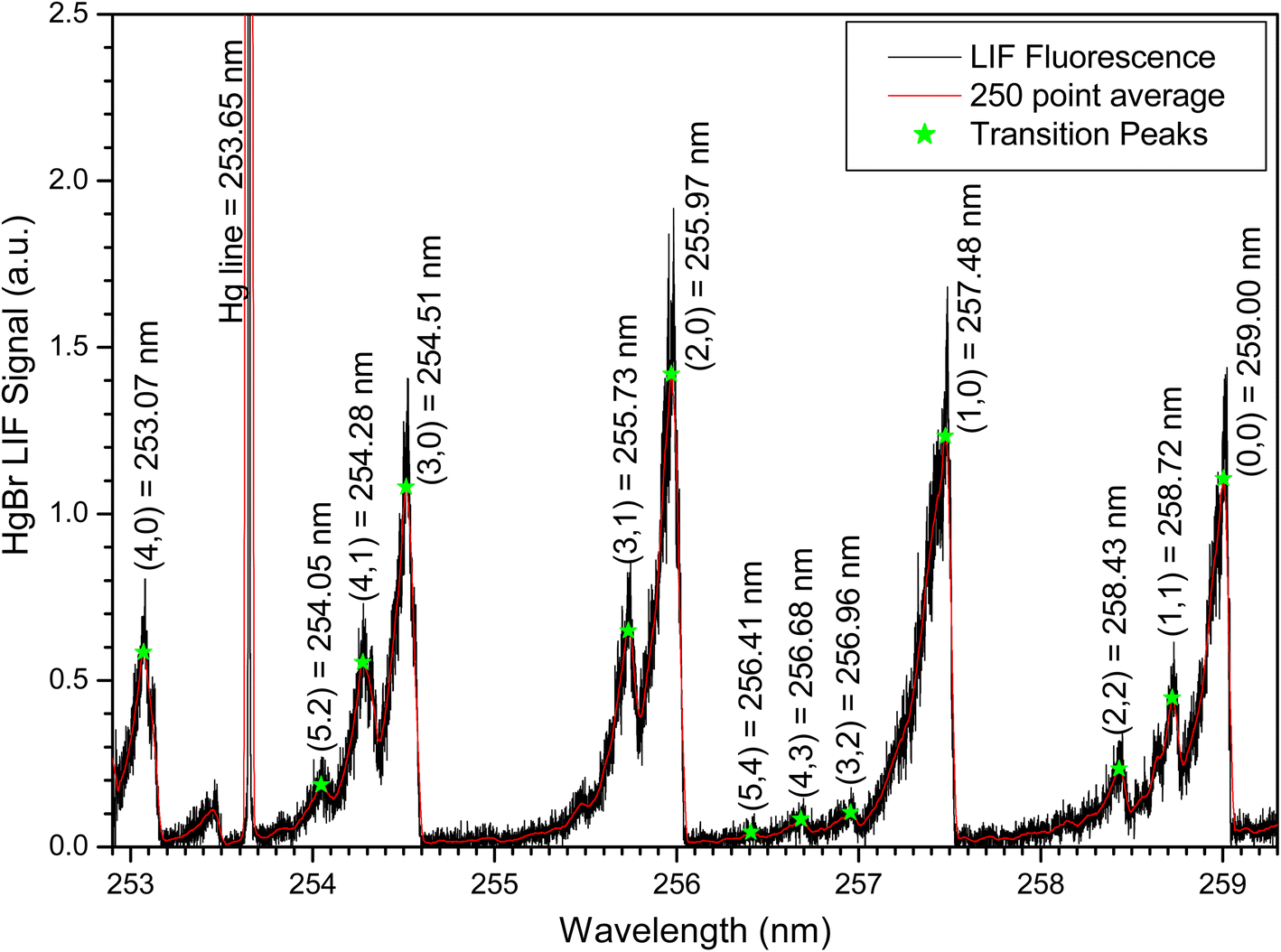 | ||
| Fig. 9 Laser excitation spectrum of the HgBr product formed in the 213 nm photolysis of HgBr2.12 | ||
Comparison with previous work
In this work we use a similar approach to that recently published by Wu et al.6 although there are some differences that we believe are significant. A comparison of our results in N2 with those of Wu et al. are shown in Fig. 10. The slight difference in temperature, 313 K for Wu et al. versus 295 K in this work, cannot account for the difference which is greater than a factor of 2 at 500 Torr. A significant difference between the two studies is the relationship between the propagating laser beams and the direction of flow of the reactant gas. In our work the photolysis and probe lasers counter propagate in a direction that is perpendicular to the direction of gas flow. This means that the gas in the reaction volume is photolyzed only once and replaced by a fresh gas mixture for every laser shot. In contrast, with Wu et al., the photolysis laser is propagating along the direction of flow. | ||
| Fig. 10 Comparison of rate coefficients for reaction (R1) based on experimental measurements from this work and that of Wu et al.6 and calculations from Jiao and Dibble.7 | ||
Wu et al. state some concerns with their flow configuration noting that “The accumulation of photolytically produced species from multiple laser shots could represent a more serious concern, as the residence time (1.6–1.8 s) is much longer than the laser pulse interval (0.1 s).”
They also note that their pseudo first order plots have large Y intercepts suggesting that a significant loss of BrHg occurs due to side-reactions with other species. They conclude that “Apparently, there are sources of variation or deterministic errors that are not captured by the error bars. The apparent negative curvature of some of the data in these plots further suggests that side reactions are causing significant loss of BrHg.”
In our work the values of k′ at [NO2] = 0, shown in Table S2,† are reasonable, particularly for the data in N2 with NO2 measured by absorption. We should also note that Wu et al. measured a smaller range of k′, measuring pseudo-first rates of up to k′ = 30000 s−1. Our results are in better agreement with the calculations of Jiao and Dibble but lie below their high pressure values. Additional work at lower temperatures should shed further light on this.
In their attempt to study reaction (R3) Wu et al.10 found that the D2П3/2 state is quenched rapidly and state that: “To study the reaction of BrHg + O2 at high [O2], a different approach, such as cavity ringdown spectroscopy, is needed to quantify and track absolute [BrHg(![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) )] without the issue of fluorescence quenching.” Our work demonstrates that with a well-designed detection system LIF can detect HgBr in 500 Torr of O2 with good sensitivity.
)] without the issue of fluorescence quenching.” Our work demonstrates that with a well-designed detection system LIF can detect HgBr in 500 Torr of O2 with good sensitivity.
Possible systematic errors
NO2 has an absorption cross section of 2 × 10−20 cm2 at 266 nm which will result in photolysis of ∼0.4% of the NO2 at the photolysis power used in our experiments. At the highest concentration to NO2 used in our experiments, ∼5 × 1015 molecules per cm3 we generate ∼2 × 1013 molecules per cm3 of NO and O products. Even if these products reacted with HgBr at a gas kinetic rate they would produce pseudo first order loss rates of ∼1000 s−1, a negligible value compared with the experimental loss rate of ∼500000 s−1.
Systematic errors can arise in this type of study if the excess reactant contains a significant amount of impurity that reacts rapidly with HgBr. Another potential systematic error can arise if the photolysis of the HgBr2 precursor produces a reactant that reacts rapidly with HgBr and, as mentioned above, relaxation of vibrationally excited HgBr can be problematic.
NO2 was purified by freeze–pump cycles at 77 K prior to making reactant mixtures but we were not able to analyze our NO2 sample for impurities. We do not believe in impurities in our NO2 sample can account for the difference in the measurements reported in this study and that of Wu et al.6 In particular if we had significant impurities we would not expect the pressure dependence of the reaction to show good three body recombination behavior. This would require the impurity reactant to show an identical pressure dependence which is unlikely.
Any systematic errors associated with the products of HgBr2 photolysis or background levels of Hg(0) would not scale with NO2 concentration but would rather produce a rapid decay of HgBr in the absence of NO2 and give a large intercept on k′ versus NO2 pseudo first order plots. We see no evidence for any systematic errors resulting from this. Similarly Fig. 8 shows some evidence for vibrational relaxation but this is much too small and much too slow to have any impact on the kinetic studies in the presence of NO2.
Atmospheric implications
Horowitz et al.8 presented a model for Hg(0) redox chemistry that included rates for HgBr with NO2 and HO2 calculated by Jiao and Dibble.7 The model found that atomic bromine (Br) of marine organobromine origin is the main atmospheric Hg(0) oxidant and that second-stage HgBr oxidation is mainly by the NO2 and HO2 radicals. The resulting chemical lifetime of tropospheric Hg(0) against oxidation was 2.7 months, shorter than in previous models. The breakdown of HgBr oxidation in the model is 62% by NO2, 30% by HO2 and with less than 10% oxidized via reaction with ClO, BrO and OH.Subsequently reaction (R4) was proposed by Saiz-Lopez et al.20 as an extremely fast reaction essentially proceeding at the hard sphere collision rate.
| HgBr + O3 → HgBrO + O2 | (R4) |
Since ozone concentrations are much higher than other potential reactants such as NO2, reaction (R4) would become the dominant sink for HgBr under most tropospheric conditions. Following the Saiz-Lopez et al. hypothesis, Shah et al.21 prosed that steric constraints would lower the rate coefficient of reaction (R4), but they still predicted a fast rate coefficient of 3 × 10−11 cm3 per molecule per s. They incorporated (R4) with this rate coefficient into the GEOS-Chem global atmospheric chemistry model and found that, using this fast rate, ozone is the dominant sink for HgBr. Gomez Martin et al. recently published an experimental study of reaction (R4) and obtained a rate coefficient of (7.5 ± 0.6) × 10−11 cm3 per molecule per s. The potential problem in the study of (R4) lies in the fact that the photolysis wavelength that is used to photolyze the HgBr precursor, HgBr2, overlaps the peak of the O3 absorption spectrum and a significant fraction of the O3 is photolyzed producing a mixture of O1D and O3P. The O1D is rapidly quenched to O3P producing an extremely high concentration of O3P. In the work of Gómez Martin et al.9 they calculated that ∼50% of the O3 present was photolyzed producing equal concentrations of O3 and O3P. This study was conducted at low pressures of N2 with secondary reactions of O atoms dominating the experimental profiles of HgBr. A complex numerical analysis of the experimental temporal profiles was required to extract rate coefficients. We measured the rate coefficient for (R4) in N2/O2 mixtures at pressures between 455–750 Torr with O2 pressures that varied between 270 and 430 Torr. Under these conditions O atoms are rapidly converted to O3 and do react with HgBr. Hence we obtain good pseudo first order decays of HgBr and obtain a preliminary rate coefficient of (6.6 ± 0.7) × 10−11 cm3 per molecule per s in excellent agreement with the value reported by Gómez Martin et al.9 These two experimental studies clearly confirm reaction (R4) is fast and based on the model study of Shah et al.21 confirms that reaction of HgBr with O3 is the dominant sink of HgBr in the atmosphere.
Conclusions
We have measured the pressure dependent rate coefficient for the reaction of HgBr with NO2, reaction (R1), in N2 and He bath gases with a single measurement in air. Our results in N2 are between a factor of 2 and 3 larger than those reported in recent work by Wu et al.6 and 10 to 15% smaller than the values from the theoretical calculations by Jiao and Dibble.7We monitored Hg(0) production and found no evidence for a reduction channel (R1b). We measured temporal profiles of HgBr in O2 and found that there is no reaction. Our work demonstrates that with a well-designed detection system LIF can detect HgBr in 500 Torr of O2 with good sensitivity.
Although we measure fast rate coefficients for reaction (R1) recent work on the reaction of O3 with HgBr suggests this will be the dominant oxidation process for HgBr in the atmosphere.
Data availability
The most critical data, the pressure and bath gas dependence of the rate coefficients for reaction (R1) are listed in the ESI,† will be uploaded to an accessible database upon the manuscript's publication.Author contributions
Conceptualization: AJH, DB; formal analysis: AJH, DB; funding acquisition: AJH; detection design: DD; writing: AJH, DB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported the National Science Foundation under award number 2003976. We thank Elliot Atlas for helpful discussions and logistical support and Nicholas Forcone for support in the development of the data acquisition software.References
- United Nations Environment Program, Global Mercury Assessment 2018, United Nations, 2019, https://wedocs.unep.org/20.500.11822/27579, accessed April 2025 Search PubMed.
- C. J. Lin and S. O. Pehkonen, The chemistry of atmospheric mercury: a review, Atmos. Environ., 1999, 33, 2067 CrossRef CAS.
- W. H. Schroeder and J. Munthe, Atmospheric mercury—an overview, Atmos. Environ., 1998, 32, 809–822 CrossRef CAS.
- F. Slemr, E. G. Brunke, R. Ebinghaus and J. Kuss, Worldwide trend of atmospheric mercury since 1995, Atmos. Chem. Phys., 2011, 11, 4779–4787 CrossRef CAS.
- D. L. Donohoue, D. Bauer, B. Cossairt and A. J. Hynes, Temperature and pressure dependent rate coefficients for the reaction of Hg with Br and the reaction of Br with Br: a pulsed laser photolysis pulsed laser induced fluorescence study, J. Phys. Chem. A, 2006, 110, 6623–6632 CrossRef CAS PubMed.
- R. Wu, C. Wang and T. S. Dibble, First experimental kinetic study of the atmospherically important reaction of BrHg + NO2, Chem. Phys. Lett., 2020, 759, 137928 CrossRef CAS.
- Y. Jiao and T. S. Dibble, First kinetic study of the atmospherically important reactions BrHg + NO2 and BrHg + HOO, Phys. Chem. Chem. Phys., 2017, 19, 1826–1838 RSC.
- H. M. Horowitz, D. J. Jacob, Y. Zhang, T. S. Dibble, F. Slemr, H. M. Amos, J. A. Schmidt, E. S. Corbitt, E. A. Marais and E. M. Sunderland, A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget, Atmos. Chem. Phys., 2017, 17, 6353–6371 CrossRef CAS.
- J. C. Gómez Martín, T. R. Lewis, K. M. Douglas, M. A. Blitz, A. Saiz-Lopez and J. M. C. Plane, The reaction between HgBr and O3: kinetic study and atmospheric implications, Phys. Chem. Chem. Phys., 2022, 24, 12419–12432 RSC.
- R. Wu, P. J. Castro, K. C. Gaito, T. S. Dibble and C. Wang, Combined experimental and computational kinetics studies for the atmospherically important BrHg radical reacting with NO and O2, J. Phys. Chem. A, 2022, 126, 3914–3925 CrossRef CAS PubMed.
- L. D'Ottone, P. Campuzano-Jost, D. Bauer and A. J. Hynes, A pulsed laser photolysis-pulsed laser induced fluorescence study of the kinetics of the gas-phase reaction of OH with NO2, J. Phys. Chem. A, 2001, 105, 10538–10543 CrossRef.
- D. L. Donohoue, PhD thesis, University of Miami, 2008.
- J. H. Parks, Laser action on the B2Σ+1/2 → X2Σ+1/2 band of HgBr at 5018 Å, Appl. Phys. Lett., 1977, 31, 297 CrossRef CAS.
- P. H. Wine, N. M. Kreutter and A. R. Ravishankara, Flash photolysis-resonance fluorescence kinetics study of the reaction OH + NO2 + M → HNO3 + M, J. Phys. Chem., 1979, 83, 3191–3195 CrossRef CAS.
- M. S. Landis, R. K. Stevens, F. Schaedlich and E. M. Prestbo, Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air, Environ. Sci. Technol., 2002, 36, 3000–3009 CrossRef CAS PubMed.
- J. Maya, Ultraviolet absorption cross sections of HgI2, HgBr2, and tin(II) halide vapors, J. Chem. Phys., 1977, 67, 4976–4980 CrossRef CAS.
- M. B. Williams, P. Campuzano-Jost, A. J. Pounds and A. J. Hynes, Experimental and theoretical studies of the reaction of the OH radical with alkyl sulfides: 2 kinetics and mechanism of the OH initiated oxidation of methylethyl and diethyl sulfides; observations of a two channel oxidation mechanism, Phys. Chem. Chem. Phys., 2007, 9, 4370–4382 RSC.
- M. B. Williams, P. Campuzano-Jost, B. M. Cossairt, A. J. Hynes and A. J. Pounds, Experimental and theoretical studies of the reaction of the OH radical with alkyl sulfides: 1 direct observations of the formation of the OHDMS adduct – pressure dependence of the forward rate of addition and development of a predictive expression at low temperature, J. Phys. Chem. A, 2007, 111, 89–104 CrossRef CAS PubMed.
- D. Amedro, A. J. C. Bunkan, M. Berasategui and J. N. Crowley, Kinetics of the OH + NO2 reaction: rate coefficients (217–333 K, 16–1200 mbar) and fall-off parameters for N2 and O2 bath gases, Atmos. Chem. Phys., 2019, 19, 10643–10657 CrossRef CAS.
- A. Saiz-Lopez, O. Travnikov, J. E. Sonke, C. P. Thackray, D. J. Jacob, J. Carmona-García, A. Frances-Monerris, D. Roca-Sanjuan, A. U. Acuna, J. Z. Davalos, C. A. Cuevas, M. Jiskra, F. Wang, J. Bieser, J. M. C. Plane and J. S. Francisco, Photochemistry of oxidized Hg(I) and Hg(II) species suggests missing mercury oxidation in the troposphere, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 30949–30956 CrossRef CAS PubMed.
- V. Shah, D. J. Jacob, C. P. Thackray, X. Wang, E. M. Sunderland, T. S. Dibble, A. Saiz-Lopez, I. Cernusak, V. Kello, P. J. Castro, R. Wu and C. Wang, Improved mechanistic model of the atmospheric redox chemistry of mercury, Environ. Sci. Technol., 2021, 55, 14445–14456 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ea00148f |
| This journal is © The Royal Society of Chemistry 2025 |