
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Principles and trends in extreme fast charging lithium-ion batteries
Yu-Xing
Yao
a,
Lei
Xu
b,
Chong
Yan
*b and
Qiang
Zhang
 *a
*a
aTsinghua Center for Green Chemical Engineering Electrification, Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China
bAdvanced Research Institute of Multidisciplinary Science, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China
First published on 14th January 2025
Abstract
The booming electric vehicle (EV) industry is laying the cornerstone for decarbonized road transport, a sector responsible for one-sixth of global energy-related emissions. A critical barrier to the wider adoption of EVs is their ability to fast charge on a timescale comparable to refueling gasoline cars. In 2017, the US Department of Energy defined extreme fast charging (XFC), aiming to charge 80% battery capacity within 10 minutes or at 400 kW. The aim of this review is to discuss current trends and provide principles for fast charging battery research and development. We begin by comparing the charge time and power of the fastest-charging electric vehicle models on the recent markets to identify the technological gap. We then benchmark XFC battery performance in the literature based on three key parameters: charge rate, energy density, and cycle life under fast charging conditions, in an effort to standardize XFC battery data reporting. The crucial effects of electrode mass loading and cell format are highlighted. Next, a thorough analysis is conducted regarding limiting electrodes (cathode vs. anode) and their respective rate-limiting steps (mass transport vs. charge transfer) during XFC, which remains a long-standing controversy in the field and requires timely clarification. On this basis, a comprehensive perspective is presented on the most promising current strategies and future lines of research for enabling XFC-capable LIBs, focusing on electrode/electrolyte materials and battery state monitoring techniques. We anticipate that this review sharpens the focus of XFC research and serves as a guide for developing fast-charging energy storage systems including LIBs and beyond.
Broader contextElectric vehicles (EVs) powered by lithium-ion batteries (LIBs) have experienced surging market penetration. While the cost and range of current EVs are now at parity with internal combustion engine vehicles, the ability to charge as quickly as refueling is a critical challenge. Therefore, to increase the EV's ability to fast charge on a timescale comparable to refueling gasoline cars, in 2017, the US Department of Energy defined extreme fast charging (XFC), aiming to charge 80% battery capacity within 10 minutes or at 400 kW. Raising the charging speed of LIBs relies on materials chemistry innovations. XFC necessitates ultrafast Li ion transport within transition metal oxide cathodes, graphite anodes, liquid electrolytes, and across their interfaces in working LIBs. In this contribution, we begin by comparing the charge time and power of the fastest-charging electric vehicle models on the recent markets. A performance benchmark system to facilitate the evaluation or replication of XFC data is established across different cell chemistries and formats. An in-depth discussion on the kinetic contribution of ion transport processes under diverse testing conditions is presented. The rapid evolution of XFC technology is set to redefine multiple sectors beyond EVs, such as 3C portable electronics, electric Vertical Take-off and Landing (eVTOL), and grid-scale energy storage. |
1. Introduction
Electrifying the global transportation sector is crucial for unlocking significant emission reductions in the coming decades.1 Electric vehicles (EVs) powered by lithium-ion batteries (LIBs) have experienced surging market penetration in the past five years, comprising 18% of all cars sold in 2023, up from merely 2.6% in 2019. Projections indicate that EV sales must reach 65% by 2030 and 95% by 2035 to keep pace with the Net Zero Emissions by 2050 Scenario (NZE), setting ambitious targets for their competitiveness against internal combustion engine vehicles (ICEVs).2,3 While the cost and range of current EVs are now at parity with ICEVs, a critical challenge to mainstream adoption remains: the ability to charge as quickly as refueling, known as extreme fast charging (XFC).4,5 According to a 2017 report by the US Department of Energy, XFC aims to obtain 200 miles of driving range within 10 minutes or charge at 400 kW.6 At the battery level, this translates to charging a LIB to 80% state of charge (SOC) within 10 minutes, with charge rates of up to 4–6C (where xC means a full charge in 1/x hour). This definition is now widely accepted in the battery community.7–10To identify the technological gap between the current state and future goals of XFC, we compared the charging times from 10% to 80% capacity of 15 fastest-charging mass-produced EV models on the market (Fig. 1).11 These EV models are categorized based on their battery pack voltage. EVs with the traditional 400 V architecture can now achieve fast charging within 30 minutes. Increasing the pack voltage to 800 V reduces the current by half, thereby improving charging efficiency and enabling fast charging in under 20 minutes. The Hyundai IONIQ 6 is currently one of the fastest charging EVs with a charge time of merely 16 minutes. The Lotus Emeya boasts the highest charging power, with a peak power of up to 350 kW and an average power of 240 kW during charge. These data suggest that we are now on the verge of accomplishing the ultimate XFC goal.
 | ||
| Fig. 1 Charging times of 15 fastest-charging EV models available on the market. Data are from ref. 11. | ||
Further raising the charging speed of LIBs relies on materials chemistry innovations. For state-of-the-art LIB chemistry, XFC necessitates ultrafast Li ion transport within transition metal oxide cathodes, graphite anodes, liquid electrolytes, and across their interfaces. Insufficient ion transport kinetics can result in low active material utilization, localized Joule heating, and anode Li plating, all of which accelerate cell performance degradation and compromise safety.12,13 In recent years, new mechanistic understandings and engineering strategies to enable XFC have proliferated in academia, while the battery industry keeps refreshing new records for charging speed. At this pivotal juncture, it is necessary to address the most critical issues in the XFC battery field to bridge the gap between lab-scale innovations and industrial practicability and to refine the research focus in the near future.
This contribution summarizes principles and trends in designing XFC LIBs with a particular focus on the following issues: (1) certain battery performance metrics are often overlooked in laboratory research but are critical to practical applications.14 Herein, we establish a performance benchmark system to facilitate the evaluation or replication of XFC data across different cell chemistries and formats. (2) The rate-limiting factors during XFC have been a long-standing controversy, with most studies on the topic remaining inconclusive.15 We provide an in-depth discussion on the kinetic contribution of ion transport processes under diverse testing conditions. (3) Numerous strategies have been proposed in recent years to enhance the XFC performance of LIBs, while their efficacy and feasibility remain to be thoroughly examined. We offer a balanced perspective on these strategies and suggest the future lines of research.
2. Benchmarking the performance of fast charging batteries
XFC battery data comprise a series of interdependent test parameters that should be considered comprehensively to evaluate the electrochemical performance of XFC batteries. An allegedly superior battery performance reported in the scientific literature can be validated and reproduced only with a full transparency of these parameters.2.1. The trilemma of the charge rate, energy density, and cycle life
The best example to illustrate the trade-off nature of batteries is the trilemma of the charge rate, energy density, and cycle life.16 Efforts to increase each of these three parameters will inevitably sacrifice the other two, and the nature of battery design is to strike a balance between them. A high energy density necessitates high electrode mass loading or conversion-type materials such as silicon or metallic Li anodes; the former increases ion diffusion resistance at high charge rates and the latter shortens the cycle life.17,18 A high charge rate increases the Li plating risk and accelerates material degradation, also compromising the cycle life.7,19,20 Real vehicle XFC batteries must maintain both the energy density and cycle life under XFC conditions at an automotive-acceptable level.21 Lacking any of the three parameters when reporting XFC data is inadequate or even misleading. Fig. 2 displays the fast charging performance across different battery chemistries reported in all the scientific literature based on the three parameters.21–42 The dataset is compiled based on three stringent criteria: (1) the battery chemistry should be commercialized or awaiting imminent commercialization, leaving only Li-ion batteries, Na-ion batteries, Li metal batteries, and hybrid capacitors in the plot. (2) Only pouch and cylindrical cells with a total capacity of > 0.5 A h, an areal capacity of > 2.0 mA h cm−2, and a cycle number of > 100 are considered. (3) The cell energy density should be clearly reported or calculated based on the total mass of the test cell (including non-active materials, with no extrapolation to larger cell formats). | ||
| Fig. 2 Energy densities, charge rates and cycle numbers of fast charging batteries reported in the literature. Both the charge rate and energy density determine where the center of a bubble is located. The color and diameter of the bubbles represent the battery chemistry and cycle number, respectively. Data are from ref. 21–41. | ||
An automotive target zone highlighted by the orange shaded region in Fig. 2 is defined as a cell energy density of >250 W h kg−1 and a charge rate of >2C, with a cycle number preferably of >1000 under fast charging conditions. Li metal batteries featuring a metallic Li anode and a high-voltage cathode are the most sought-after candidates for achieving an ultra-high energy density of above 400 W h kg−1. Efforts need to be continuously made to improve the charge rate (<0.5C) and lifetime of Li metal batteries due to Li dendrite formation at large current densities.18 Na-ion batteries promise excellent fast charging capability and long cycle life but have an energy density ceiling of 180 W h kg−1 and therefore they could not outcompete LiFePO4 (LFP)-based LIBs in the EV market.43,44 Falling also in this category are supercapacitors or hybrid capacitors with ultrahigh power density but low energy density.45 Fabrication of Li-ion batteries with a graphite or silicon/graphite (Si/C) anode and an Ni-rich cathode is the most viable solution to XFC-capable EVs due to their all-round performance. The most impressive XFC performance reported to date is by Wang et al., who realized 12 (or 11)-minute fast charging of a 228 W h kg−1 LIB (3.4 mA h cm−2) to 75% (or 70%) SOC for more than 2000 (or 900) cycles.21 It should be noted that the authors adopted an asymmetric temperature modulation method and battery charging in this work was conducted at 60 °C with enhanced kinetics.
In XFC studies, one cannot overemphasize the importance of electrode mass loading, or areal capacity, because it is closely associated with the energy density and maximum charge rate of the LIB. Fig. 3a lists the energy densities and areal capacities of fast charging LIBs reported in the literature.21,31,32,34,36,38,39 The energy densities of 4 reference cell chemistries, including NCM811 | graphite, NCM811 | Li4Ti5O12 (LTO), NCM811 | Si, and NCM811 | Li, were estimated as a function of areal capacity based on a 50 A h pouch cell.21 For the NCM811 | graphite chemistry, its energy density increased from 234 W h kg−1 at 2.0 mA h cm−2 to 278 W h kg−1 at 4.0 mA h cm−2. Literature data points are located slightly below the NCM811 | graphite line due to its smaller cell capacity and lower cathode Ni content, but its energy density follows the same trend as the areal capacity increases. Numerical simulations by Gallagher et al. indicate that for an LiNi0.8Co0.15Al0.05O2 (NCA) | graphite cell with a fixed electrode mass loading (thickness), a rate threshold exists, above which the accessible areal capacity plunges (Fig. 3b).46 Such a rate threshold declines as the electrode thickens due to ion transport limitations.
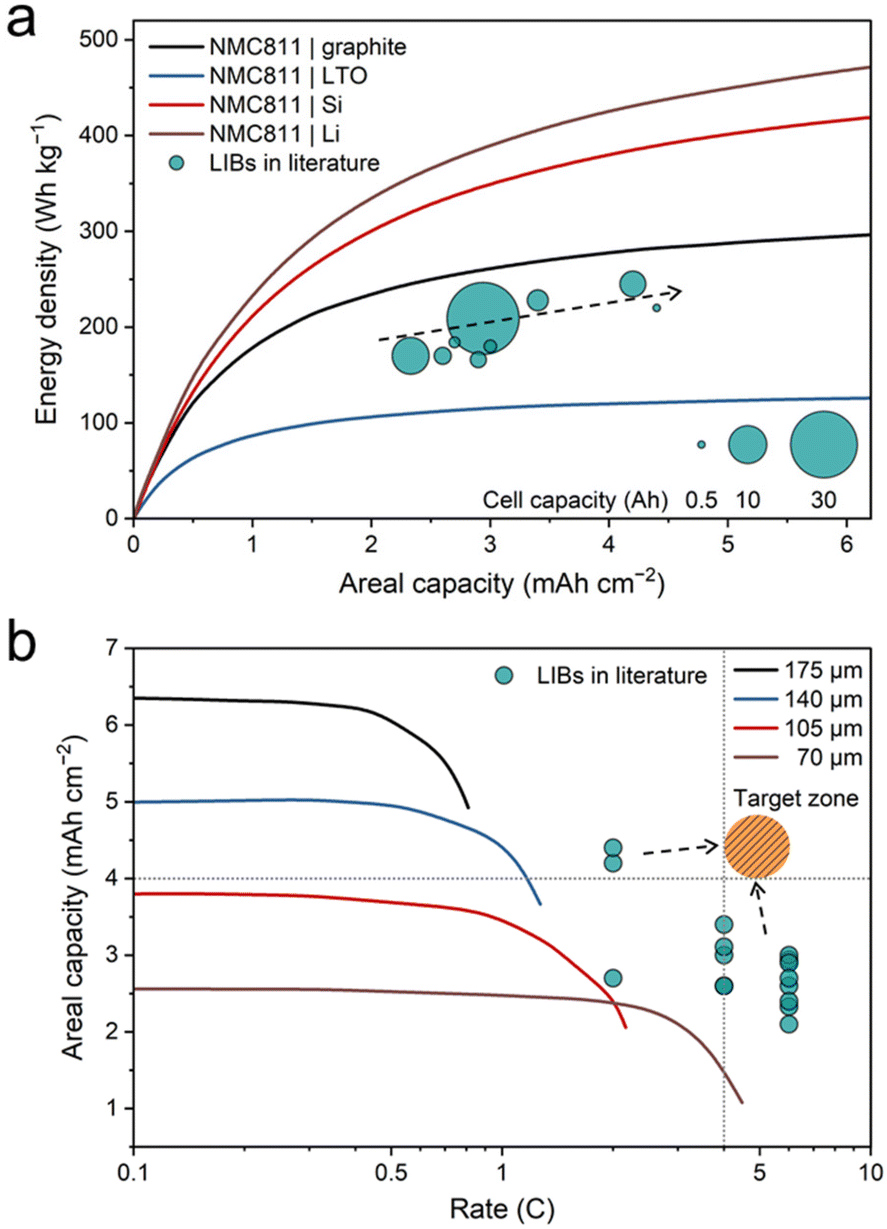 | ||
| Fig. 3 Dependence of the energy density and charge rate on the areal capacity of LIBs. (a) Energy densities and areal capacities of fast charging LIBs reported in the literature with reference to the theoretical values of 4 different cell chemistries. Reproduced from ref. 21, copyright 2022 NPG. The bubble size reflects the cell capacity. The energy densities of the solid lines were estimated in a 50 A h pouch cell. Data are from ref. 21, 31, 32, 34, 36, 38 and 39. (b) Areal capacities and charge rates of fast charging LIBs reported in the literature. Data are from ref. 21, 31, 32, 34, 36, 38, 39 and 47–53. Solid lines are simulated areal capacities during the rate capability test of an NCA | graphite cell with four different electrode mass loadings. Reproduced from ref. 46, copyright 2015 Electrochemical Society. | ||
In the past few years, the community has achieved significant results in the 2–3 mA h cm−2 range and promoted the fundamental understanding of XFC technology,21,31,32,34,36,38,39,47–53 bringing the research on XFC to a new chapter. LIBs designed for EV applications typically require an areal capacity of >3.0 mA h cm−2 for LFP or >4.0 mA h cm−2 for NCA and NCM, but charging such electrodes with a rate of >4C is beyond current capabilities. Recently, our group and Lee's group have attempted fast charging of electrodes of >4.0 mA h cm−2.36,38 However, in these two cases the charge rate is limited to 2C, falling short of the XFC requirement. Therefore, we call on the battery community to focus on fast charging of thick electrodes as highlighted by the orange shaded target zone in Fig. 3b, which constitutes the key technical bottleneck towards XFC. At the very least, we suggest that an A h-level pouch cell should be employed with a single-sided areal capacity higher than 3.0 mA h cm−2, with a charge rate not lower than 4C, while paying attention to the cycle life when conducting research on XFC.
2.2. Cell formats
A major barrier in benchmarking the XFC performance of LIBs across a large body of the literature is the variety of battery cell formats used for electrochemical measurements. Early studies often used Li | graphite half cells to assess the rate capability of graphite electrodes. However, the large polarization of the Li counter electrode at high discharge rates can drive the cell voltage down to the Li plating threshold of 0 V prematurely,54–56 leading researchers to grossly underestimate the capacity of graphite electrodes.57 In addition, the failure of half cells over long-term cycling is often induced by the failure of Li counter electrodes rather than working electrodes.58 One way to minimize the disruption of Li counter electrodes is to insert a reference electrode during testing. However, the limited durability of reference electrodes and their adverse impact on ion transport inside the cell induce additional problems for obtaining the desired electrochemical performance.59 For these reasons, three-electrode systems are more appropriate for electroanalytical measurements such as impedance and we highly recommend using full cells for evaluating XFC performance.Coin cells and pouch cells are the two most commonly used cell formats (Fig. 4a). The former is mA h-level, hand-made in a laboratory with minimum requirements of raw materials and equipment, while the latter is A h-level, often produced at the pilot scale and requires significant resources. For XFC studies, the difference between the pouch and coin cells lies not only in the scaled-up capacity, but also in their electrochemical performance. Son et al. demonstrated that with identical electrode materials and loadings, coin cells exhibit inferior rate capability compared with pouch cells due to their larger contact resistance between the spacer, spring, and casing (Fig. 4b).60 The larger voltage polarization in coin cells further triggered Li plating and aggravated capacity fade during cycling (Fig. 4c). Similar results were also obtained by Zhang et al., who noticed that a slight change in the coin cell configuration yields strikingly different fast charging performances.61,62 In addition, coin cells conceal a myriad of issues such as gas evolution, electrolyte dry-out, corrosion of non-active components, electrode swelling, and so on. Therefore, we strongly encourage researchers to adopt stacked pouch cells in XFC-related studies for industrially relevant results by seeking collaboration with cell manufacturers if possible. For new electrode material assessment or fundamental research purposes, readers could also refer to a series of manuals in fabricating highly reproducible Li-ion coin cells or single-layered pouch cells.63–66
 | ||
| Fig. 4 Differences in coin and pouch cells for assessing fast charging performance. (a) Structures of a coin cell and a pouch cell. (b) Rate and (c) cycling performances of ∼4.4 mA h cm−2 NCA | graphite coin and pouch cells. Reproduced from ref. 60, copyright 2023 Wiley-VCH. | ||
3. Identifying the rate-limiting factors of fast charging
The nature of XFC lies in achieving ultrafast ion transport in LIBs across multiple time and length scales, which requires a comprehensive understanding of the rate-limiting factors to design specific solutions effectively. However, opinions remain divided on this topic, as various researchers have identified different rate-limiting steps in their respective studies. Such controversy mainly arises from two reasons: (1) the rate-limiting step of a given system is contingent upon a combination of factors including cell chemistry, electrode mass loading, and charge rate. Discrepancies in test conditions have hindered researchers from reaching a unified conclusion. (2) It is experimentally challenging to decouple the kinetic contribution of various ion transport processes during fast charging. In this section, we attempt to settle this dispute by providing a detailed analysis on the limiting electrodes (cathode vs. anode) and their respective rate-limiting steps (mass transport vs. charge transfer).3.1. The limiting electrode
In most XFC studies, the graphite anode is identified as the limiting electrode because of its low working potential (65–200 mV vs. Li/Li+) and the resulting high propensity of Li plating at large charge rates.67 Li plating leads to active lithium loss and dendrite penetration, which constitute the main reason for capacity fade and safety issues under XFC. The layered cathode also undergoes gradual structural degradation during XFC, but its adverse impacts are far less severe compared with Li plating.68 However, the term “limiting” herein is used in the context of the failure mechanism rather than kinetics. In a three-electrode NCA | graphite coin cell, Zhang et al. discovered that the impedance of the cathode is significantly larger than the anode (Fig. 5a) and therefore the cell polarization is dominated by the cathode during XFC (Fig. 5b).69 This result can be partially attributed to the lower Li+ diffusivity in NCA (∼10−10 cm2 s−1) than in graphite (10−7–10−6 cm2 s−1), whereas the origin of the difference in charge transfer kinetics remains unclear.70 High cathode impedance leads to an early reach of the upper cut-off voltage, causing a transition from constant current (CC) charging to constant voltage (CV) charging, thereby reducing the charge speed. Therefore, Li+ extraction from the cathode is the kinetics-limiting process during XFC. | ||
| Fig. 5 Identifying the limiting electrode during XFC. (a) AC impedance of a three-electrode NCA | graphite Li-ion coin cell at 50% SOC. (b) Polarization potential of the NCA cathode and graphite anode during fast charging. Reproduced from ref. 69, copyright 2019 Wiley-VCH. Electrochemical performances and voltage profiles of NCA | graphite pouch cells at different charging rates containing (c) the EC/DMC LiPF6 electrolyte and (d) the EC/DMC LiTFSI electrolyte. LiTFSI reduces the impedance of the NCA cathode markedly and causes severe Li plating at the graphite anode under 4.0C charging. Reproduced from ref. 36, copyright 2023 Wiley-VCH. | ||
On the other hand, a high cathode-to-anode impedance ratio is the key to inhibiting Li plating during fast charging. Our group demonstrated this counterintuitive phenomenon by conducting fast charging tests of NCA | graphite pouch cells using two electrolytes: lithium hexafluorophosphate (LiPF6) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) dissolved in an ethylene carbonate (EC)/dimethyl carbonate (DMC) mixture.36 The standard LiPF6 cell has a high cathode-to-anode impedance ratio, whereas adding LiTFSI significantly reduces the cathode impedance by accelerating charge transfer kinetics.71 During fast charging, the LiPF6 cell exhibits large polarization and low charge acceptance in the CC phase. The early transition from CC to CV charging reduces the current and therefore inhibits Li plating, as indicated by the high coulombic efficiency (CE) during 4.0C charging (Fig. 5c). In sharp contrast, the LiTFSI cell retains high capacity during CC charging even at 4.0C due to the low cathode impedance. However, the graphite anode is unable to accept persistently a high Li+ input under 4.0C and the cell undergoes severe Li plating, as evidenced by the plunge in CE and capacity fade (Fig. 5d). In this particular case, the graphite anode becomes the limiting electrode and the kinetics of Li+ intercalation must be expedited to enable safe, fast charging without Li plating. In aged LIBs, the thickening of the SEI also reduces the cathode-to-anode impedance ratio and triggers Li plating.72 These examples shed fresh light on how the kinetic interplay between the cathode and anode dictates the fast charging performance of LIBs. A low overall cell impedance with a high cathode-to-anode impedance ratio is ideal for achieving Li-plating-free XFC. This can be achieved in many ways in practical battery engineering. For instance, combining a high cathode press density and a low anode press density can achieve such an impedance configuration by regulating the ion diffusion resistance in electrodes. More strategies of kinetics regulation are discussed in section 4.
3.2. The rate-limiting step
Identifying the rate-limiting step of a single electrode has been a persistent challenge due to the complex and intertwined kinetic processes involved during charge and discharge. These kinetic processes include electron conduction and Li+ transport, and the latter processes are further classified into 4 steps (Fig. 6a): (1) liquid-state Li+ transport in the bulk electrolyte; (2) charge transfer at the electrode/electrolyte interface, accompanied by Li+ (de)solvation; (3) Li+ transport through the SEI or cathode electrolyte interphase (CEI); and (4) solid-state Li+ diffusion in the active material.73 Steps (1) and (4) belong to mass transport, while steps (2) and (3) are often closely coupled and collectively referred to as charge transfer. A common approach for evaluating the kinetics of these processes is calculating their activation energy using temperature-dependent electrochemical impedance spectroscopy (EIS). Among the 4 steps mentioned above, charge transfer is widely believed to have the highest energy barrier (50–70 kJ mol−1).74 However, it is important to note that while a high energy barrier indicates high temperature sensitivity of a process, it does not necessarily equate to being the rate-limiting step. The most direct indicator is the overpotential contribution during cell operation, but it is experimentally challenging to separate the overpotential of individual processes. In light of this, Xiong et al. proposed an ion-intercalation reaction model that enables accurate decoupling of the complex kinetic processes in electrodes. A “phase diagram” of rate-limiting steps is obtained for a LiNi0.5Mn0.3Co0.2O2 (NMC532) electrode as a function of areal capacity and C-rate (Fig. 6b).75 Electrode kinetics is always controlled by solid-state Li+ diffusion at low rates (<1C); charge transfer becomes the rate-limiting step at medium rates (or at high rates with low areal capacities, as shown in Fig. 6c), and the charge transfer-controlled region rapidly contracts as the areal capacity increases; Li+ transport in liquid electrolytes is the dominant kinetic contributor at high rates, especially at high areal capacities. The XFC region (4–6C) highlighted in Fig. 6b suggests that Li+ transport in electrolytes is the rate-limiting step for an electrode areal capacity of >2.0 mA h cm−2. Fast charging LIBs reported in the literature are mostly located in the electrolyte Li+ transport region and near the borderline of the charge transfer region, highlighting the significance of expediting the kinetics of these two processes. A closer examination on the decomposed overpotential at high areal capacities (Fig. 6d and e) reveals a rather significant contribution of Li+ transport in separator pores, which is often overlooked, given the relatively low thickness of separators (∼25 μm) compared with electrodes (>100 μm).76 This is attributed to the sharp increase in Li+ flux at high rates, which causes intensified salt aggregation within the separator, hence impeding ion transport. It is worth noting that electron conduction accounts for only a small percentage of electrode polarization. Therefore, optimizing the electronic conductivity of LIB electrodes can reduce the cell internal resistance but will not bring substantial improvement to the XFC performance.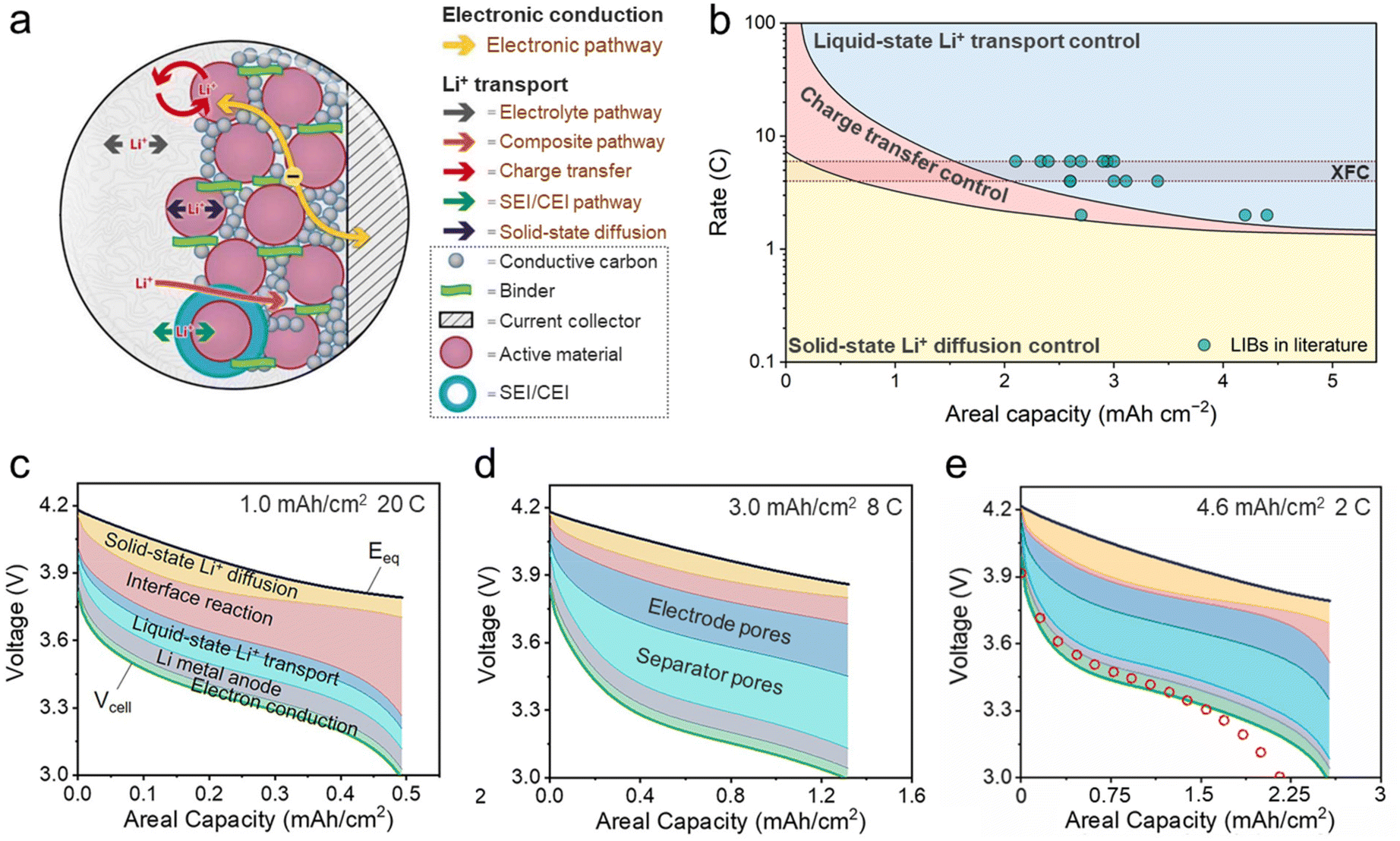 | ||
| Fig. 6 Identifying the rate-limiting step during XFC. (a) Kinetic processes of a LIB electrode. Reproduced from ref. 73, copyright 2016 Electrochemical Society. (b) The rate-limiting step of an NMC532 electrode as a function of areal capacity and C-rate by model prediction. This diagram also incorporates fast charging LIBs reported in the literature. Overpotential decomposition of an NMC532 | Li cell in the cases of (c) 1.0 mA h cm−2, 20C, (d) 3.0 mA h cm−2, 8C, and (e) 4.6 mA h cm−2, 2C. Each colored area between the equilibrium potential (Eeq) and the cell voltage (Vcell) represents the overpotential contribution of a certain kinetic process. Reproduced from ref. 75 and 76, copyright 2023 Elsevier. | ||
Although the above analysis is based on an NMC cathode, the rate-limiting step of a graphite anode generally follows the same pattern. Nevertheless, graphite exhibits several distinct features in terms of ion transport kinetics. First, because of the high structural anisotropy of layered graphite, Li+ entry is only allowed through the edge plane but kinetically prohibited through the basal plane, which significantly reduces the number of active intercalation sites.67 Second, the SEI on graphite is a non-negligible source of interfacial resistance.77–81 Li+ transport across the SEI can be the rate-limiting step, especially with excess film-forming electrolyte additives or upon cell aging.82 The CEI on a cathode, if it does exist, is often thinner and incomplete in coverage of the cathode surface and therefore less resistive than the SEI.83 Third, the charge transfer resistance of the graphite anode is ascribed to Li+ desolvation, an inverse process to Li+ solvation at the cathode during charge. Finally, the working potential of graphite is 65–200 mV vs. Li/Li+, which indicates that the overpotential of all the kinetic processes combined is confined within this narrow range; otherwise Li plating becomes thermodynamically favorable.4 For these reasons, most XFC strategies proposed in the literature are focused on the graphite anode.
4. Promising strategies to enable XFC
This section delves into recent advancements in XFC-enabling strategies, specifically examining (1) electrolyte design, (2) electrode materials and architecture engineering, and (3) Li plating detection and early safety warning. Rather than offering a comprehensive overview of all existing XFC strategies—a task already accomplished by several excellent review articles84–86—our goal is to highlight a select few with the greatest application potential. We particularly focus on their underlying mechanisms, design principles, and industrial practicability.4.1. Electrolytes
Electrolytes are probably the most important components during XFC because they directly determine the kinetics of 4 processes: (1) Li+ diffusion in electrode pores, (2) Li+ diffusion in separator pores, (3) Li+ desolvation, and (4) Li+ transport across the SEI/CEI. The basic requirements of XFC electrolytes include high ionic conductivity (σ) and Li+ transference number (t+), fast interfacial ion transport kinetics, and high interfacial stability.87 Meeting all these requirements simultaneously is extremely challenging due to their trade-off nature. Since liquid-state Li+ diffusion is the rate-limiting step for highly loaded electrodes during XFC, improving bulk electrolyte transport properties should be the top priority.88Fig. 7a shows σ and t+ targets for enabling XFC based on model prediction.89 Standard electrolytes based on EC and LiPF6 nowadays typically have an ionic conductivity of 8–12 mS cm−1 and a transference number below 0.4. However, cells with areal capacities of 3 and 4 mA h cm−2 require NextGen 1 (σ = 18 mS cm−1, t+ = 0.51) and NextGen2 (σ = 23 mS cm−1, t+ = 0.61) electrolytes, respectively, to ensure decent capacity retention during CC charging of up to 6C. Such highly conducting electrolytes can be achieved by employing low viscosity co-solvents or using lithium bis(fluorosulfonyl)imide (LiFSI) salt. For example, Dahn and co-workers showed that methyl acetate (MA) with 2 mol kg−1 LiPF6 has an ionic conductivity of 25 mS cm−1 at 20 °C and enables 4C fast charging of 29 mg cm−2 ultrathick NMC532 electrodes.90 Du et al. replaced LiPF6 with LiFSI in routine EC-based electrolytes, increasing the Li+ transference number from 0.384 to 0.495.91 However, most low viscosity co-solvents such as carboxylate esters and nitriles cannot form a stable SEI on graphite and have an innate tendency to co-intercalate, compromising the cell lifetime.87,92 LiFSI is highly corrosive against the Al current collector and stainless-steel cell casing even at a low concentration.50 In these circumstances, film-forming electrolyte additives are almost indispensable in stabilizing electrode/electrolyte interfaces. Considering the continuous consumption of electrolyte additives, interface instability and “rollover” failure may still occur when the battery approaches end-of-life, posing serious performance and safety concerns.93 | ||
| Fig. 7 Electrolyte design principles for XFC. (a) Electrolyte transport property requirements for achieving XFC at different areal capacities obtained by model predictions. Reproduced from ref. 89, copyright 2019 Electrochemical Society. (b) Solvation structures of routine electrolytes, (localized) high-concentration electrolytes, and weakly solvating electrolytes. Reproduced from ref. 94, copyright 2021 Wiley-VCH. (c) Solvation energies and solvation shell volumes of common solvents. (d) Schematics of the ligand channel-facilitated Li+ transport mechanism. Reproduced from ref. 97 and 78, copyright 2024 NPG. (e) Donor number versus the dielectric constant of solvents for constructing ideal weakly solvating electrolytes. Reproduced from ref. 99, copyright 2023 NPG. | ||
An important method to tailor stable interfaces for XFC is by exploiting anion-derived interfacial chemistry (Fig. 7b).94 In routine electrolytes, the primary solvation sheath of Li+ contains solvents with strong polarity such as EC. EC is not only responsible for dissociating Li salts and providing sufficient ionic conductivity but also for guaranteeing reversible Li+ (de)intercalation in graphite by forming a stable SEI. Since such an SEI carries the chemical signature of solvents in the primary solvation sheath, it is termed solvent-derived interfacial chemistry. Anion-derived interfacial chemistry refers to the SEI originating from anion decomposition, which is first discovered in highly concentrated electrolytes (HCEs) or localized highly concentrated electrolytes (LHCEs) with abundant contact ion pairs (CIPs) and aggregates (AGGs) in their solvation structures.95 Although superior XFC performances of HCEs and LHCEs are frequently reported in the literature, these results are often obtained at low areal capacities (<2 mA h cm−2) because HCEs and LHCEs have low ionic conductivities (<3 mS cm−1) due to the scarcity of charge carriers and prevailing neutral ion pairs. In addition, Li+ desolvation becomes more energy-consuming in HCEs and LHCEs due to the cleavage of ion pairs.96 The improved rate capabilities of HCEs and LHCEs reported in the literature may result from other factors such as a highly-conductive SEI or a high pre-exponential factor of charge transfer. The prohibitively high cost of concentrated salts in HCEs and fluorinated diluents in LHCEs also hinders their commercialization. An alternative path towards anion-derived interfacial chemistry is weakly solvating electrolytes (WSEs) that have attracted extensive research interest in recent years. Our group first proposed the concept of WSEs by formulating electrolytes with 1,4-dioxane, a practically non-polar solvent with a dielectric constant (ε = 2.2) even lower than that of benzene (ε = 2.3).94 The unique solvation structure of WSEs, i.e., loose solvent binding with Li+ and abundant ion pairs at low salt concentrations, affords rapid Li+ desolvation and an anion-derived inorganic-rich SEI with high Li+ conductivity. We also demonstrated excellent fast charging capability and suppressed Li plating in pouch and cylindrical LIBs with WSEs, showcasing their huge potential for next-generation XFC electrolytes.
The key to designing practical WSEs lies in resolving the dilemma between achieving high ionic conductivity and maintaining weak Li+–solvent binding. There are basically two principles in tackling this problem. (1) Building tiny solvation sheaths. Fan and co-workers screened a wide range of solvents in search of a promising combination of low solvation energy and small solvation shell volume (Fig. 7c).97 They found that dissolving 1.3 M LiFSI in fluoroacetonitrile (FAN) results in an exceptionally high ionic conductivity of 40.3 mS cm−1 at 25 °C and 11.9 mS cm−1 even at −70 °C. A ligand channel-facilitated mechanism is believed to underpin such anomalously high ion mobility (Fig. 7d). Solvents from the secondary solvation sheath are able to interact with Li+ due to the limited screening effect of small-sized, weakly solvating FAN molecules in the primary sheath, which creates ligand channels between neighboring Li+ solvates, thus enhancing transport.98 The FAN electrolyte also realizes fast desolvation and an anion-derived SEI, exhibiting excellent XFC performance. The only concern with small molecule electrolytes is their potentially poor high temperature storage and cycling performance, which need to be optimized through the use of functional additives or co-solvents. (2) Searching for high dielectric constant, low donor number (DN) solvents (Fig. 7e).99 A high ε weakens ion–ion interactions according to the Coulombic law, facilitating salt dissociation and improves bulk Li+ transport. A low DN weakens ion–solvent interactions and ensures rapid Li+ desolvation at the anode during charging. We anticipate more solvents to be discovered in the IV region in Fig. 7e for constructing WSEs with satisfactory XFC performance.
4.2. Electrode materials and architecture
Promising strategies on designing XFC electrodes can be generally classified into 3 types: (1) hybrid electrode materials. Hard carbon exhibits excellent fast charging capability compared with graphite, but suffers from low energy density due to the low initial CE and high average working potential.100 Chen et al. overcame this tradeoff by fabricating a hard carbon–graphite blend electrode (Fig. 8a), which achieves both high energy density and 6C fast charging of >1.0 A h pouch cells with an areal capacity of 3.0 mA h cm−2.39 (2) Surface modifications. Our group reported the microscopic mechanism of the improved rate capability of graphite with turbostratic carbon nanolayer coating (Fig. 8b).101 With the carbon coating, Li ions can diffuse across the basal plane before entering through the edge plane of graphite, substantially increasing the number of active sites for Li+ intercalation. This type of material has been widely applied in the battery industry. An artificial SEI with high Li+ conductivity also represents a promising route towards fast-charging anode materials.47,102 (3) Electrode architecture engineering. The ion transport ability of a porous structure can be quantified by the Macmullin number NM, which is defined as the ratio of the conductivity of an electrolyte (κ) and the effective conductivity (κeff) of the porous structure, or electrode tortuosity (τ) divided by porosity (ε):103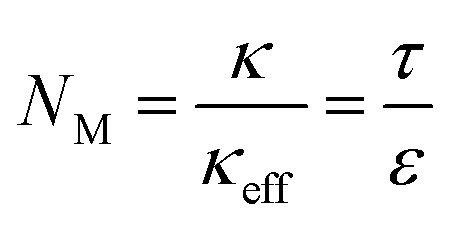 | (1) |
 | ||
| Fig. 8 Electrode engineering strategies for enabling XFC. (a) Hybrid anode consisting of graphite and hard carbon. Reproduced from ref. 39, copyright 2020 Wiley-VCH. (b) Nanolayer-coated graphite with enhanced interfacial ion transport kinetics. Reproduced from ref. 101, copyright 2020 Wiley-VCH. (c) Active material utilization versus the current density of a particle size–porosity dual-gradient structured graphite anode. Reproduced from ref. 106, copyright 2022 AAAS. | ||
The key to improving ion diffusivity is to reduce NM by either reducing tortuosity or increasing porosity. In practical cell design, this is usually achieved by simply reducing the press density of electrodes or applying high-porosity separators. From a structural engineering perspective, this can be achieved by a variety of approaches including laser patterning,31 magnetic alignment of electrode particles,104 pore structuring,105 and gradient electrode design.106 Among these approaches, gradient electrode design offers the best prospects in industrial applications. Lu et al. constructed a particle size–porosity dual-gradient structure in the graphite anode, which leads to a prominent increase in active material utilization at high charge rates (Fig. 8c).106 This work identifies higher porosity and smaller particle size at the top of the electrode than the bottom as the optimum structure for gradient electrodes, which can be realized by multi-layer coating during the electrode manufacturing process.107
4.3. Li plating detection and early safety warnings
Even with an optimum LIB design for XFC applications, localized Li plating may still occur during fast charging, giving rise to performance deterioration and thermal runaway.67 A non-destructive method with high temporal resolution for detecting Li plating is strongly needed for accurate battery state estimation and early safety warnings. Our group developed an operando quantified Li plating detection technique based on dynamic capacitance measurement (DCM) in working LIBs (Fig. 9).108 During charging, a single-frequency alternating current (AC) is superimposed on the direct current (DC) to calculate the value of CS, a capacitance reflecting the electrochemical active surface area (ECSA) of the electrodes. The characteristic frequency of AC perturbation is carefully chosen to mask the electrochemical response from the cathode and enables direct monitoring of the surface state of the graphite anode without the need for a reference electrode. When Li plating occurs, the dendritic morphology causes a spike in the ECSA of graphite, which is in turn reflected in the sharp increase of CS due to their approximate linear relationship. The DCM method can therefore capture the onset of Li plating without requiring extra equipment or time-consuming test procedures, offering great potential for incorporation into future battery management systems. | ||
| Fig. 9 The dynamic capacitance method for operando Li plating detection in working LIBs. Reproduced from ref. 108, copyright Wiley-VCH. | ||
5. Summary and perspectives
Fast charging is one of the few research areas in the battery field that attracts strong enthusiasm simultaneously from both academia and industry. Achieving the XFC target therefore hinges upon academic researchers publishing their mechanistic understandings and industry experts sharing their hands-on knowledge, thereby advancing XFC performance through concerted efforts. This review discusses principles and trends in designing next-generation fast charging LIBs from both academic and industrial viewpoints. We first explored the intricate interdependence among the energy density, charge rate, cycle life, electrode mass loading, and cell format to establish a performance benchmark for reporting XFC data. Following this, we elucidated the rate-limiting factors in XFC, offering a crucial guide for battery design. Lastly, we presented a balanced perspective on the promising strategies for enabling XFC, highlighting contributions from both our lab and peer researchers. We recommend further research be undertaken in the following areas:(1) Temperature adaptability of XFC batteries. In studies on XFC, batteries are typically tested at ambient or lower temperatures, while high temperature storage and cycling—common industry practices—are often overlooked in the scientific literature. For instance, electrolytes that enhance conductivity for XFC also demonstrate satisfactory performance at low temperatures.36,97,99 However, they frequently result in severe gas evolution or corrosion of cell casing at elevated temperatures (45–60 °C). While these issues may not be readily apparent in lab-made coin cells, they are critical to the functionality and safety of commercial cells. For example, gas evolution can lead to pouch cell swelling or activate current interruption devices (CIDs) or venting in cylindrical cells due to pressure buildup; the corrosion of cell casing can lead to metal deposition and internal micro-shorts.
(2) Understanding the nature of charge transfer. Researchers commonly correlate charge transfer with the desolvation of Li+ at the anode and use weakly solvating electrolytes to expedite this process, which contrasts with Li+ solvation at the cathode during charging. The charge transfer resistance at the cathode significantly contributes to the overall cell impedance, yet a universally accepted method to enhance its kinetics remains elusive. Wen et al. demonstrated that replacing LiPF6 with LiTFSI increases the cathode exchange current density by two orders of magnitude.71 However, their explanation focuses on the accelerated Li+–TFSI− dissociation during discharging rather than charging. The understanding of the interfacial ion transport mechanism, particularly the asymmetry between desolvation and solvation, is constrained by the lack of molecular-scale interface characterization tools or suitable interface structure models. Integrating interfacial molecular dynamics simulations with electrochemical analysis could provide further insights into the charge transfer mechanism and offer guidance for improving kinetics.109
(3) Developing Si-containing anodes. Blending Si-based materials (SiOx or Si–C composites) with graphite represents a major technical route to further increase the energy density of future Li-ion batteries due to the high gravimetric capacity of Si. This is also a facile route to improve the fast-charging capability because it reduces the mass loading and thickness of the anode, thus lowering the risk of Li plating. Future efforts should focus on addressing the intrinsic problems associated with high Si content (>5%) including cycling stability, volume expansion, calendar life, safety, and cost-effectiveness.
(4) Machine learning-assisted materials innovation. Despite more than 30 years since the invention of LIBs, commercial batteries still rely on a remarkably limited range of materials and molecules. For example, only a few hundreds of electrolyte molecules have been explored, which constitute merely a fraction of the vast array of small molecules, not to mention the almost infinite number of organic compounds. Compared with traditional time-consuming and costly experimental testing, machine learning (ML) enables high-throughput virtual screening of vast chemical space and prioritizes promising XFC electrolyte molecules based on predicted properties, significantly accelerating the discovery process.110 We anticipate that ML-generated fast charging electrolyte formulations will finally replace the long-standing EC-based systems that have stagnated for decades.
In the field of energy chemistry, advancements in fast charging can drive deeper research into the fundamental electrochemical processes, leading to a better understanding of ion transport, electrode reactions, and degradation mechanisms. These insights can also lend support to the R&D efforts of post-LIB battery systems. From an industrial standpoint, the rapid evolution of XFC technology is set to redefine multiple sectors beyond EVs, such as 3C portable electronics, electric Vertical Take-off and Landing (eVTOL), and grid-scale energy storage. We anticipate the next 3–5 years to be a golden age for the major expansion of XFC technologies across diverse applications, promoting global energy decarbonization into the fast lane.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Beijing Natural Science Foundation (L233004), the National Natural Science Foundation of China (22393903, 22393900, 22379014, and 22005172), the National Key Research and Development Program (2021YFB2500300), and the Tsinghua University Initiative Scientific Research Program. We thank Ye Xiao and Jia-Qi Huang for technical support and discussions.References
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef PubMed.
- Global EV Outlook 2024, https://www.iea.org/reports/global-ev-outlook-2024 (accessed 06–19, 2024).
- Y. Liu, H. Shi and Z.-S. Wu, Energy Environ. Sci., 2023, 16, 4834–4871 RSC.
- W. Cai, Y. X. Yao, G. L. Zhu, C. Yan, L. L. Jiang, C. He, J. Q. Huang and Q. Zhang, Chem. Soc. Rev., 2020, 49, 3806–3833 RSC.
- A. Meintz, J. C. Zhang, R. Vijayagopal, C. Kreutzer, S. Ahmed, I. Bloom, A. Burnham, R. B. Carlson, F. Dias, E. J. Dufek, J. Francfort, K. Hardy, A. N. Jansen, M. Keyser, A. Markel, C. Michelbacher, M. Mohanpurkar, A. Pesaran, D. Scoffield, M. Shirk, T. Stephens and T. Tanim, J. Power Sources, 2017, 367, 216–227 CrossRef CAS.
- S. Ahmed, I. Bloom, A. N. Jansen, T. Tanim, E. J. Dufek, A. Pesaran, A. Burnham, R. B. Carlson, F. Dias, K. Hardy, M. Keyser, C. Kreuzer, A. Markel, A. Meintz, C. Michelbacher, M. Mohanpurkar, P. A. Nelson, D. C. Robertson, D. Scoffield, M. Shirk, T. Stephens, R. Vijayagopal and J. Zhang, J. Power Sources, 2017, 367, 250–262 CrossRef CAS.
- M. Li, M. Feng, D. Luo and Z. Chen, Cell Rep. Phys. Sci., 2020, 1, 100212 CrossRef CAS.
- S. A. Khan, I. Hussain, A. K. Thakur, S. Yu, K. T. Lau, S. He, K. Dong, J. Chen, X. Li, M. Ahmad and J. Zhao, Energy Storage Mater., 2024, 65, 103144 CrossRef.
- W. Xie, X. Liu, R. He, Y. Li, X. Gao, X. Li, Z. Peng, S. Feng, X. Feng and S. Yang, J. Energy Storage, 2020, 32, 101837 CrossRef.
- E. J. Dufek, D. P. Abraham, I. Bloom, B.-R. Chen, P. R. Chinnam, A. M. Colclasure, K. L. Gering, M. Keyser, S. Kim, W. Mai, D. C. Robertson, M.-T. F. Rodrigues, K. Smith, T. R. Tanim, F. L. E. Usseglio-Viretta and P. J. Weddle, J. Power Sources, 2022, 526, 231129 CrossRef.
- Most fast charging electric vehicles, https://ev-database.org/compare/fast-charging-electric-vehicle-quickest, (accessed 06–19, 2024).
- A. Tomaszewska, Z. Y. Chu, X. N. Feng, S. O'Kane, X. H. Liu, J. Y. Chen, C. Z. Ji, E. Endler, R. H. Li, L. S. Liu, Y. L. Li, S. Q. Zheng, S. Vetterlein, M. Gao, J. Y. Du, M. Parkes, M. G. Ouyang, M. Marinescu, G. Offer and B. Wu, eTransportation, 2019, 1, 100011 CrossRef.
- D. Meng, Z. Xue, G. Chen, D. Zhou, Y.-S. He, Z.-F. Ma, Y. Liu and L. Li, Energy Environ. Sci., 2024, 17, 4658–4669 RSC.
- Y. Cao, M. Li, J. Lu, J. Liu and K. Amine, Nat. Nanotechnol., 2019, 14, 200–207 CrossRef.
- Y. Liu, Y. Zhu and Y. Cui, Nat. Energy, 2019, 4, 540–550 CrossRef.
- X. G. Yang and C. Y. Wang, J. Power Sources, 2018, 402, 489–498 CrossRef.
- K. Feng, M. Li, W. Liu, A. G. Kashkooli, X. Xiao, M. Cai and Z. Chen, Small, 2018, 14, 1702737 CrossRef.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef PubMed.
- F.-N. Jiang, S.-J. Yang, H. Liu, X.-B. Cheng, L. Liu, R. Xiang, Q. Zhang, S. Kaskel and J.-Q. Huang, SusMat, 2021, 1, 506–536 CrossRef.
- S. Liu, B. Gu, Z. Chen, R. Zhan, X. Wang, R. Feng and Y. Sun, J. Energy Chem., 2024, 91, 484–500 CrossRef.
- C. Y. Wang, T. Liu, X. G. Yang, S. Ge, N. V. Stanley, E. S. Rountree, Y. Leng and B. D. McCarthy, Nature, 2022, 611, 485–490 CrossRef PubMed.
- T. Broux, F. Fauth, N. Hall, Y. Chatillon, M. Bianchini, T. Bamine, J. B. Leriche, E. Suard, D. Carlier, Y. Reynier, L. Simonin, C. Masquelier and L. Croguennec, Small Methods, 2018, 3, 1800215 CrossRef.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- Y. Qiao, H. Yang, Z. Chang, H. Deng, X. Li and H. Zhou, Nat. Energy, 2021, 6, 653–662 CrossRef CAS.
- C. J. Niu, D. Y. Liu, J. A. Lochala, C. S. Anderson, X. Cao, M. E. Gross, W. Xu, J. G. Zhang, M. S. Whittingham, J. Xiao and J. Liu, Nat. Energy, 2021, 6, 723–732 CrossRef CAS.
- Y. Li, A. Vasileiadis, Q. Zhou, Y. Lu, Q. Meng, Y. Li, P. Ombrini, J. Zhao, Z. Chen, Y. Niu, X. Qi, F. Xie, R. van der Jagt, S. Ganapathy, M.-M. Titirici, H. Li, L. Chen, M. Wagemaker and Y.-S. Hu, Nat. Energy, 2024, 9, 134–142 CrossRef CAS.
- H. X. Li, Y. Gong, H. H. Zhou, J. Li, K. Yang, B. Y. Mao, J. C. Zhang, Y. Shi, J. H. Deng, M. X. Mao, Z. Y. Huang, S. Q. Jiao, Y. F. Kuang, Y. L. Zhao and S. L. Luo, Nat. Commun., 2023, 14, 6407 CrossRef CAS PubMed.
- Y. Feng, Y. Li, J. Lin, H. Wu, L. Zhu, X. Zhang, L. Zhang, C.-F. Sun, M. Wu and Y. Wang, Nat. Commun., 2023, 14, 3639 CrossRef CAS.
- J. Moon, H. C. Lee, H. Jung, S. Wakita, S. Cho, J. Yoon, J. Lee, A. Ueda, B. Choi, S. Lee, K. Ito, Y. Kubo, A. C. Lim, J. G. Seo, J. Yoo, S. Lee, Y. Ham, W. Baek, Y. G. Ryu and I. T. Han, Nat. Commun., 2021, 12, 2714 CrossRef CAS PubMed.
- M. He, A. E. L. Mejdoubi, D. Chartouni, M. Morcrette, P. Troendle and R. Castiglioni, J. Power Sources, 2023, 588, 233741 CrossRef CAS.
- K. H. Chen, M. J. Namkoong, V. Goel, C. L. Yang, S. Kazemiabnavi, S. M. Mortuza, E. Kazyak, J. Mazumder, K. Thornton, J. Sakamoto and N. P. Dasgupta, J. Power Sources, 2020, 471, 228475 CrossRef CAS.
- X.-G. Yang, T. Liu, Y. Gao, S. Ge, Y. Leng, D. Wang and C.-Y. Wang, Joule, 2019, 3, 3002–3019 CrossRef CAS.
- X. Rong, Y. Lu, X. Qi, Q. Zhou, W. Kong, K. Tang, L. Chen and Y. Hu, Energy Storage Sci. Technol., 2020, 9, 515–522 Search PubMed.
- Z. J. Du, Z. Z. Yang, R. M. Tao, V. Shipitsyn, X. Y. Wu, D. C. Robertson, K. M. Livingston, S. Hagler, J. Kwon, L. Ma, I. D. Bloom and B. J. Ingram, Batteries Supercaps, 2023, 6, e202300292 CrossRef CAS.
- Q. K. Zhang, S. Y. Sun, M. Y. Zhou, L. P. Hou, J. L. Liang, S. J. Yang, B. Q. Li, X. Q. Zhang and J. Q. Huang, Angew. Chem., Int. Ed., 2023, 62, e202306889 CrossRef.
- Y. X. Yao, X. Chen, N. Yao, J. H. Gao, G. Xu, J. F. Ding, C. L. Song, W. L. Cai, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202214828 CrossRef.
- Y. H. Zhang, P. Y. Zhao, Q. A. Nie, Y. Li, R. Guo, Y. F. Hong, J. K. Deng and J. X. Song, Adv. Mater., 2023, 35, 2211032 CrossRef.
- J. H. Choi, H. G. Lee, M. H. Lee, S. M. Lee, J. Kang, J. H. Suh, M. S. Park and J. W. Lee, Adv. Funct. Mater., 2024, 34, 2400414 CrossRef.
- K. H. Chen, V. Goel, M. J. Namkoong, M. Wied, S. Müller, V. Wood, J. Sakamoto, K. Thornton and N. P. Dasgupta, Adv. Energy Mater., 2020, 11, 2003336 CrossRef.
- L. Hu, X. Zhang, P. Zhao, H. Fan, Z. Zhang, J. Deng, G. Ungar and J. Song, Adv. Mater., 2021, 33, e2104416 CrossRef PubMed.
- Technology Benefits-Strong Performance, https://faradion.co.uk/technology-benefits/strong-performance/, (accessed 06–19, 2024).
- M. Bai, L. Yang, Q. Jia, X. Tang, Y. Liu, H. Wang, M. Zhang, R. Guo and Y. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 47490–47502 CrossRef PubMed.
- A. Rudola, R. Sayers, C. J. Wright and J. Barker, Nat. Energy, 2023, 8, 215–218 CrossRef.
- R. Usiskin, Y. Lu, J. Popovic, M. Law, P. Balaya, Y.-S. Hu and J. Maier, Nat. Rev. Mater., 2021, 6, 1020–1035 CrossRef.
- P. Simon and Y. Gogotsi, Nat. Mater., 2020, 19, 1151–1163 CrossRef PubMed.
- K. G. Gallagher, S. E. Trask, C. Bauer, T. Woehrle, S. F. Lux, M. Tschech, P. Lamp, B. J. Polzin, S. Ha, B. Long, Q. Wu, W. Lu, D. W. Dees and A. N. Jansen, J. Electrochem. Soc., 2015, 163, A138–A149 CrossRef.
- S. Tu, B. Zhang, Y. Zhang, Z. Chen, X. Wang, R. Zhan, Y. Ou, W. Wang, X. Liu, X. Duan, L. Wang and Y. Sun, Nat. Energy, 2023, 8, 1365–1374 CrossRef.
- H. Lee, H. An, H. Chang, M. Lee, S. Park, S. Lee, J. Kang, S. Byon, B. Koo, H. Lee, Y. M. Lee, J. Moon, S. Chae and H. Lee, Energy Storage Mater., 2023, 63, 102995 CrossRef.
- X. Wu, T. Liu, Y. Bai, X. Feng, M. M. Rahman, C.-J. Sun, F. Lin, K. Zhao and Z. Du, Electrochim. Acta, 2020, 353, 136453 CrossRef CAS.
- X. Y. Wu and Z. J. Du, Electrochem. Commun., 2021, 129, 107088 CrossRef CAS.
- Y. Hu, Z. Zhang and H. Wang, ChemistrySelect, 2022, 7, e202200470 Search PubMed.
- S. Yi, Z. L. Yan, X. D. Li, Z. Wang, P. P. Ning, J. W. Zhang, J. L. Huang, D. R. Yang and N. Du, Chem. Eng. J., 2023, 473, 145161 CrossRef CAS.
- H. P. Gao, Q. Z. Yan, J. Holoubek, Y. J. Yin, W. R. G. M. L. Bao, H. D. Liu, A. Baskin, M. Q. Li, G. R. Cai, W. K. Li, D. Tran, P. Liu, J. Luo, Y. S. Meng and Z. Chen, Adv. Energy Mater., 2023, 13, 2202906 CrossRef CAS.
- L. Xu, Y. Xiao, Y. Yang, R. Xu, Y.-X. Yao, X.-R. Chen, Z.-H. Li, C. Yan and J.-Q. Huang, Adv. Mater., 2023, 35, 2301881 CrossRef CAS.
- Y. Yang, L. Xu, S.-J. Yang, C. Yan and J.-Q. Huang, J. Energy Chem., 2022, 73, 394–399 CrossRef CAS.
- Y. Yang, X.-L. Zhong, L. Xu, Z.-L. Yang, C. Yan and J.-Q. Huang, J. Energy Chem., 2024, 97, 453–459 CrossRef CAS.
- S. S. Zhang, J. Electrochem. Soc., 2020, 167, 100510 CrossRef CAS.
- H. Kim, K. Lim, G. Yoon, J.-H. Park, K. Ku, H.-D. Lim, Y.-E. Sung and K. Kang, Adv. Energy Mater., 2017, 7, 1700418 CrossRef.
- Y. Xiao, L. Xu, C. X. Bi, S. Zhang, J. F. Ding, C. Yan and J. Q. Huang, Adv. Energy Mater., 2024, 14, 2304502 CrossRef.
- Y. Son, H. Y. Y. Cha, T. Y. Lee, Y. Kim, A. Boies, J. P. Cho and M. De Volder, Energy Environ. Mater., 2024, 7, e12615 CrossRef.
- S. S. Zhang, Energy Technol., 2022, 11, 2200370 CrossRef.
- Y. Zhang, S. Li, J. Shi, J. Lai, Z. Zhuang, J. Liu, W. Yang, L. Ma, Y.-P. Cai, J. Xu and Q. Zheng, J. Energy Chem., 2024, 94, 171–180 CrossRef.
- V. Murray, D. S. Hall and J. R. Dahn, J. Electrochem. Soc., 2019, 166, A329–A333 CrossRef.
- M. D. L. Garayt, M. B. Johnson, L. Laidlaw, M. A. Mcarthur, S. Trussler, J. E. Harlow, J. R. Dahn and C. Y. Yang, J. Electrochem. Soc., 2023, 170, 080516 CrossRef.
- J. T. Hu, B. B. Wu, S. J. Chae, J. Lochala, Y. J. Bi and J. Xiao, Joule, 2021, 5, 1011–1015 CrossRef.
- Y. Lu, X. Chen, X. Han, D. Guo, Y. Wang, X. Feng and M. Ouyang, J. Energy Chem., 2024, 99, 11–22 CrossRef.
- T. Waldmann, B. I. Hogg and M. Wohlfahrt-Mehrens, J. Power Sources, 2018, 384, 107–124 CrossRef.
- T. R. Tanim, Z. Yang, D. P. Finegan, P. R. Chinnam, Y. Lin, P. J. Weddle, I. Bloom, A. M. Colclasure, E. J. Dufek, J. Wen, Y. Tsai, M. C. Evans, K. Smith, J. M. Allen, C. C. Dickerson, A. H. Quinn, A. R. Dunlop, S. E. Trask and A. N. Jansen, Adv. Energy Mater., 2022, 10, 2103712 CrossRef.
- S. S. Zhang, InfoMat, 2019, 2, 942–949 CrossRef.
- Y. Bai, Q.-A. Huang, K. Wu and J. Zhang, J. Energy Chem., 2024, 92, 759–798 CrossRef.
- B. Wen, Z. Deng, P.-C. Tsai, Z. W. Lebens-Higgins, L. F. J. Piper, S. P. Ong and Y.-M. Chiang, Nat. Energy, 2020, 5, 578–586 CrossRef.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Joule, 2019, 3, 2322–2333 CrossRef.
- J. Kasnatscheew, U. Rodehorst, B. Streipert, S. Wiemers-Meyer, R. Jakelski, R. Wagner, I. C. Laskovic and M. Winter, J. Electrochem. Soc., 2016, 163, A2943–A2950 CrossRef.
- K. Xu, J. Electrochem. Soc., 2007, 154, A162–A167 CrossRef.
- R. Xiong, M. Zhou, L. Li, J. Xu, M. Li, B. yan, D. Li, Y. Zhang and H. Zhou, Energy Storage Mater., 2023, 54, 836–844 CrossRef.
- R. Xiong, Y. Yu, S. Chen, M. Li, L. Li, M. Zhou, W. Zhang, y. Bo, D. Li, H. Yang, Y. Zhang and H. Zhou, J. Power Sources, 2023, 553, 232296 CrossRef.
- Y. Yang, C. Yan and J. Huang, Acta Phys.-Chim. Sin., 2021, 37, 2010076 Search PubMed.
- S.-Y. Sun, X.-Q. Zhang, Y.-N. Wang, J.-L. Li, Z. Zheng and J.-Q. Huang, Mater. Today, 2024, 77, 39–65 CrossRef.
- L.-L. Jiang, C. Yan, Y.-X. Yao, W. Cai, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3402–3406 CrossRef PubMed.
- T. Li, X.-Q. Zhang, N. Yao, Y.-X. Yao, L.-P. Hou, X. Chen, M.-Y. Zhou, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22683–22687 CrossRef.
- Y.-X. Yao, J. Wan, N.-Y. Liang, C. Yan, R. Wen and Q. Zhang, J. Am. Chem. Soc., 2023, 145, 8001–8006 CrossRef PubMed.
- Y. X. Yao, N. Yao, X. R. Zhou, Z. H. Li, X. Y. Yue, C. Yan and Q. Zhang, Adv. Mater., 2022, 34, e2206448 CrossRef PubMed.
- M. Weiss, R. Ruess, J. Kasnatscheew, Y. Levartovsky, N. R. Levy, P. Minnmann, L. Stolz, T. Waldmann, M. Wohlfahrt-Mehrens, D. Aurbach, M. Winter, Y. Ein-Eli and J. Janek, Adv. Energy Mater., 2021, 11, 2101126 CrossRef.
- T. R. Tanim, P. J. Weddle, Z. Z. Yang, A. M. Colclasure, H. Charalambous, D. P. Finegan, Y. Y. Lu, M. Preefer, S. Kim, J. M. Allen, F. L. E. Usseglio-Viretta, P. R. Chinnam, I. Bloom, E. J. Dufek, K. Smith, G. Y. Chen, K. M. Wiaderek, J. N. Weker and Y. Ren, Adv. Energy Mater., 2022, 12, 2202795 CrossRef.
- S. Q. Li, K. Wang, G. F. Zhang, S. N. Li, Y. A. Xu, X. D. Zhang, X. Zhang, S. H. Zheng, X. Z. Sun and Y. W. Ma, Adv. Funct. Mater., 2022, 32, 2200796 CrossRef.
- S. Lei, Z. Q. Zeng, S. J. Cheng and J. Xie, Battery Energy, 2023, 2, 20230018 CrossRef.
- E. R. Logan and J. R. Dahn, Trends Chem., 2020, 2, 354–366 CrossRef.
- X. Chen and Q. Zhang, Acc. Chem. Res., 2020, 53, 1992–2002 CrossRef PubMed.
- A. M. Colclasure, A. R. Dunlop, S. E. Trask, B. J. Polzin, A. N. Jansen and K. Smith, J. Electrochem. Soc., 2019, 166, A1412–A1424 CrossRef.
- E. R. Logan, D. S. Hall, M. M. E. Cormier, T. Taskovic, M. Bauer, I. Hamam, H. Hebecker, L. Molino and J. R. Dahn, J. Phys. Chem. C, 2020, 124, 12269–12280 CrossRef.
- Z. Du, D. L. Wood and I. Belharouak, Electrochem. Commun., 2019, 103, 109–113 CrossRef.
- Y. Yu, H. Koh, Z. Zhang, Z. Yang, A. N. Alexandrova, M. Agarwal, E. A. Stach and J. Xie, Energy Environ. Sci., 2023, 16, 5904–5915 RSC.
- L. Ma, S. L. Glazier, R. Petibon, J. Xia, J. M. Peters, Q. Liu, J. Allen, R. N. C. Doig and J. R. Dahn, J. Electrochem. Soc., 2016, 164, A5008–A5018 CrossRef.
- Y. X. Yao, X. Chen, C. Yan, X. Q. Zhang, W. L. Cai, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4090–4097 CrossRef PubMed.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef.
- Y. Kondo, T. Abe and Y. Yamada, ACS Appl. Mater. Interfaces, 2022, 14, 22706–22718 CrossRef.
- D. Lu, R. H. Li, M. M. Rahman, P. Y. Yu, L. Lv, S. Yang, Y. Q. Huang, C. C. Sun, S. Q. Zhang, H. K. Zhang, J. B. Zhang, X. Z. Xiao, T. Deng, L. W. Fan, L. X. Chen, J. P. Wang, E. Y. Hu, C. S. Wang and X. L. Fan, Nature, 2024, 627, 101–107 CrossRef.
- C. Yan and J.-Q. Huang, Nature, 2024, 627, 42–43 CrossRef PubMed.
- J. J. Xu, J. X. Zhang, T. P. Pollard, Q. D. Li, S. Tan, S. Y. Hou, H. L. Wan, F. Chen, H. X. He, E. Y. Hu, K. Xu, X. Q. Yang, O. Borodin and C. S. Wang, Nature, 2023, 614, 694–700 CrossRef PubMed.
- L. J. Xie, C. Tang, Z. H. Bi, M. X. Song, Y. F. Fan, C. Yan, X. M. Li, F. Y. Su, Q. Zhang and C. M. Chen, Adv. Energy Mater., 2021, 11, 2101650 CrossRef.
- W. Cai, C. Yan, Y.-X. Yao, L. Xu, R. Xu, L.-L. Jiang, J.-Q. Huang and Q. Zhang, Small Struct., 2020, 1, 2000010 CrossRef.
- E. Kazyak, K. H. Chen, Y. X. Chen, T. H. Cho and N. P. Dasgupta, Adv. Energy Mater., 2022, 12, 2102618 CrossRef.
- J. Landesfeind, J. Hattendorff, A. Ehrl, W. A. Wall and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1373–A1387 CrossRef.
- L. S. Li, R. M. Erb, J. J. Wang, J. Wang and Y. M. Chiang, Adv. Energy Mater., 2019, 9, 1802472 CrossRef.
- R. Xiong, Y. Zhang, Y. Wang, L. Song, M. Li, H. Yang, Z. Huang, D. Li and H. Zhou, Small Methods, 2021, 5, 2100280 CrossRef PubMed.
- L. L. Lu, Y. Y. Lu, Z. X. Zhu, J. X. Shao, H. B. Yao, S. Wang, T. W. Zhang, Y. Ni, X. X. Wang and S. H. Yu, Sci. Adv., 2022, 8, eabm6624 CrossRef PubMed.
- J. Yang, Y. J. Li, A. Mijailovic, G. Y. Wang, J. Xiong, K. Mathew, W. Q. Lu, B. W. Sheldon and Q. L. Wu, J. Mater. Chem. A, 2022, 10, 12114–12124 RSC.
- L. Xu, Y. Xiao, Y. Yang, S. J. Yang, X. R. Chen, R. Xu, Y. X. Yao, W. L. Cai, C. Yan, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210365 CrossRef.
- N. Yao, X. Chen, Z. H. Fu and Q. Zhang, Chem. Rev., 2022, 122, 10970–11021 CrossRef.
- X. Chen, X. Liu, X. Shen and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24354–24366 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |