
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of particle size on the slurry-based processability and conductivity of t-Li7SiPS8†
Duc Hien Nguyen ab,
Lars Grunenberga,
Igor Moudrakovskia,
Kathrin Küstera and
Bettina V. Lotsch*abc
ab,
Lars Grunenberga,
Igor Moudrakovskia,
Kathrin Küstera and
Bettina V. Lotsch*abc
aMax Planck Institute for Solid State Research, Heisenbergstraße 1, 70569 Stuttgart, Germany
bLMU Munich, Butenandstraße 5-13, 81377 Munich, Germany
cExcellence Cluster e-conversion, Lichtenbergstraße 4a, 85748 Garching, Germany. E-mail: b.lotsch@fkf.mpg.de
First published on 6th May 2025
Abstract
All-solid-state batteries (ASSBs) promise higher energy and power densities and improved safety over lithium-ion batteries (LIBs) by using non-flammable solid electrolytes (SEs), with thiophosphate-based SEs having the highest ionic conductivities. In this study, we present the slurry-based processing of tetragonal Li7SiPS8 (t-Li7SiPS8) into freestanding SE-sheets using six different slurry formulations based on anisole, p-xylene and toluene as solvents and polyisobutene (PIB) and two hydrogenated nitrile butadiene rubbers (HNBR-17 and HNBR-34) as binders, respectively. We systematically investigate their chemical compatibility and morphology and show the effect of different particle size distributions on the slurry-based processability. Depending on the particle size, significant differences are observed in sheet homogeneity and relative density. Specifically, higher ionic conductivities are observed for sheets based on larger particles, likely stemming from smaller inter-particle grain boundary effects as demonstrated by electrochemical impedance spectroscopy (EIS). We confirm the positive correlation between Li diffusivity and particle size through pulsed field gradient nuclear magnetic resonance (PFG NMR) experiments, with larger particle sizes resulting in higher diffusivities. Our study suggests a beneficial effect of larger particles for SEs in terms of transport properties and can be considered as a strategy to maximize the performance of future ASSBs.
Broader contextThis study investigates the slurry-based processability of the Earth-abundant superionic conductor tetragonal Li7SiPS8 using different slurry formulations, and highlights the significant effect of particle size on the transport properties of the obtained Li7SiPS8-sheets. |
1. Introduction
To date, lithium-ion batteries (LIBs) are considered the dominant technology for electrical energy storage, powering most electronic devices while demonstrating high reliability and cyclability.1 However, LIBs are approaching their capacity limits, and with increasing safety and environmental concerns, other energy storage systems are being considered for future battery generations.2 One promising contender is the all-solid-state battery (ASSB), in which a nonflammable solid electrolyte (SE) replaces the flammable organic electrolyte found in conventional LIBs. This can significantly improve battery safety, yield higher energy densities by roughly 70%, and facilitate faster charging compared to conventional LIBs.3,4 These properties are particularly relevant in applications such as electric vehicles and mobile devices, where fast charging times and high energy densities are required. Among the different SE types, sulfide-based and particularly thiophosphate-based SEs have gained much attention since the discovery of Li10GeP2S12 (LGPS) with an ionic conductivity of up to 12 mS cm−1, exceeding those of polymer-based, oxide-based and liquid electrolytes.5 Besides their high conductivity, these materials possess favorable mechanical properties such as lattice softness and ductility.6–8 This allows for better contact with the electrodes, effectively lowering interfacial resistance, and better adaptability to volume changes during battery cycling.9,10 Most ASSB cells reported are usually based on newly developed pelletized SEs with much attention being catered to the analysis of interfacial properties or cycling behavior and degradation phenomena.11–13 The thicknesses of the pelletized SEs need to be ideally less than 100 μm to be competitive with current LIBs.14 For the large-scale production of ASSB cells, however, pelletized SEs are unsuitable due to the challenges of their implementation into continuous production lines.15 Thus, ASSB cells are often limited to small-scale laboratory cells. In contrast to this, thin sheet-like SE-based layers can be produced from slurry-based processing, which is more attractive for industrial scalability.16 In combination with a densification step via calendaring, a continuous roll-to-roll process can be established, which is promising in reducing costs and thus attractive for industrial applications. General challenges in slurry-based processing involve, for example, achieving a uniform dispersion of SE particles to prevent agglomeration, obtaining dense sheets with uniform thicknesses, or finding the right blend of solvent and binder with good viscosity suited for processing, and most crucially, preventing the decomposition of the SE. In recent years, more studies on appropriate slurry formulations for the fabrication of SE-based separator sheets have been conducted.17–23 Sulfide-based SE layers based on crystalline LGPS and glass-ceramic LPS were first reported by Nam et al. in 2015. These layers were processed with polymer scaffolds (non-woven porous polymeric materials), which enabled flexibility and exhibited a thickness of around 70 μm.20 Attempts to reduce the cost of LGPS by substituting Ge with Si, making it more viable for large-scale production, were reported with Li9.54Si1.74P1.44S11.7Cl0.3 exhibiting a remarkable room temperature ionic conductivity of 25 mS cm−1.24,25In a recent study,26 tetragonal Li7SiPS8 (t-Li7SiPS8), which is isostructural to LGPS and first reported by Harm et al.,27 was subjected to a variety of solvents, ranging from protic polar to aprotic non-polar. The authors concluded that aprotic solvents with donor numbers smaller than 15 kcal mol−1 are suitable for wet-processing of t-Li7SiPS8, as only an amorphous side phase was observed to react with the solvents to form polysulfide species. Based on these results, we present the slurry-based processing of t-Li7SiPS8 to fabricate freestanding SE-sheets using anisole, p-xylene and toluene as solvents and polyisobutene (PIB) and two hydrogenated nitrile butadiene rubbers (HNBR-17 and HNBR-34) as binders, respectively. With a total of six different slurry formulations, we systematically investigate the morphology of the processed SE-sheets and demonstrate that only negligible chemical decomposition of t-Li7SiPS8 occurs, highlighting the compatibility between our SE, solvent and binder. Moreover, we show the impact of different particle size distributions of t-Li7SiPS8 on the slurry-based processability and the resulting SE-sheet morphology while also examining their influence on the ionic conductivities via electrochemical impedance spectroscopy (EIS). Finally, we show a positive correlation between diffusivity and particle size through pulsed field gradient nuclear magnetic resonance (PFG NMR) experiments and demonstrate that larger particle sizes result in higher diffusivities and, by extension, higher ionic conductivities.
2. Results and discussion
2.1. Effect of the binder and solvent on t-Li7SiPS8 sheets
Fig. 1 depicts the sheets fabricated from the fraction with a particle size distribution of <50 μm and processed with different binders and solvents. In our experiments, the solubility of the binders in the solvents was identified as a critical prerequisite for successful sheet fabrication. With combinations of HNBR-34/p-xylene, HNBR-34/toluene and PIB/anisole, reasonable processing was not possible due to the insolubility of the binders in the respective solvents, thus resulting in overall six slurry formulations only (see the ESI†). Processing with HNBR-17 results in homogeneous sheets with well-defined edges regardless of the solvent used (Fig. 1a–c). The opposite is observed when HNBR-34 is used as the binder with anisole (Fig. 1d). While still homogeneously dense, the resulting sheet is less defined with jagged edges. It should be noted that the sheet initially shows well-defined edges directly after doctor blading but forms jagged edges and shrinks as it dries. Apparently, the high polarity of the solvent and binder is unsuitable for ensuring good adhesion to the substrate foil. Processing with PIB (Fig. 1e and f) also results in homogeneous sheets with well-defined edges similar to the case with HNBR-17. From these observations, it appears that the type of binder has a significant influence on the resulting sheet quality, with less polar binders (here: HNBR-17 and PIB) being more favorable. Among the investigated solvents, toluene is preferred over anisole and p-xylene due to its higher vapor pressure, which leads to easier evaporation under ambient conditions. | ||
Fig. 1 Abbreviations used in the figure: ani for anisole, px for p-xylene, and tol for toluene. Top-view photographs of t-Li7SiPS8 sheets processed with: (a) HNBR-17 in anisole, (b) HNBR-17 in p-xylene, (c) HNBR-17 in toluene, (d) HNBR-34 in anisole, (e) PIB in p-xylene and (f) PIB in toluene. The depicted sheets were obtained from the fraction having a particle size distribution of <50 μm and a SE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :binder ratio of 90:10 wt%. :binder ratio of 90:10 wt%. | ||
Scanning electron microscopy (SEM) was employed to investigate further the identified morphological differences in the different fabricated sheets (Fig. S2†). When t-Li7SiPS8 is processed with HNBR-17 in anisole (Fig. S2a†), a rough surface with voids is observed at low magnification (top row). The rough surface coupled with voids likely leads to lower ionic conductivities due to the lack of intimate particle contact, effectively decreasing the number of pathways for Li diffusion. The surface particles appear to be interconnected and individual grains are visually indistinguishable from each other. Interestingly, at higher magnification (middle row), a translucent binder layer, connecting individual particles, is visible. Processing with HNBR-17 in p-xylene (Fig. S2b†) and toluene (Fig. S2c†) results in similarly rough surfaces due to the random but homogeneous distribution of agglomerated particles. Unlike before, no flattened morphology or translucent binder layer can be observed. The processing of t-Li7SiPS8 with HNBR-34 in anisole (Fig. S2d†) appears to be denser compared to its HNBR-17 counterpart. Finally, processing with PIB in p-xylene (Fig. S2e†) and toluene (Fig. S2f†) yields less rough-appearing sheets compared to those obtained with the HNBR-17 equivalent.
Next, energy dispersive X-ray spectroscopy (EDX) mappings of carbon in the sheets (Fig. S2,† bottom row) were recorded. As the binder is the sole carbon source, the mapping qualitatively reflects the binder distribution within the sheets. It can be observed that the binders are homogeneously distributed across all sheets. While this suggests the effectiveness of the slurry mixing procedure, the homogeneous binder distribution may also lower the ionic conductivity if all SE particles are encapsulated by the binder, which acts as a resistive interface. The mappings of the remaining elements are summarized in Fig. S3† and, similar to the carbon mapping, show a homogeneous distribution.
2.2. Effect of particle size fractions on t-Li7SiPS8 sheets
To discuss the effect of the different particle size fractions on the t-Li7SiPS8 sheets, some key terms related to crystallites (also primary particles), aggregates (also secondary particles), agglomerates, and their relationship need to be defined for a better understanding, as shown exemplarily in Fig. 2. While aggregates consist of firmly fused primary particles that are randomly oriented in spatial directions (indicated by the gray lines as thought Miller planes), agglomerates are loosely bound assemblies of either primary particles and/or aggregates. Here, we differentiate between intra-particle grain boundary (blue line, within an aggregate/secondary particle between primary particles) and inter-particle grain boundary (between aggregates).28 For the purpose of this study, “particles” will refer to secondary particles unless otherwise specified.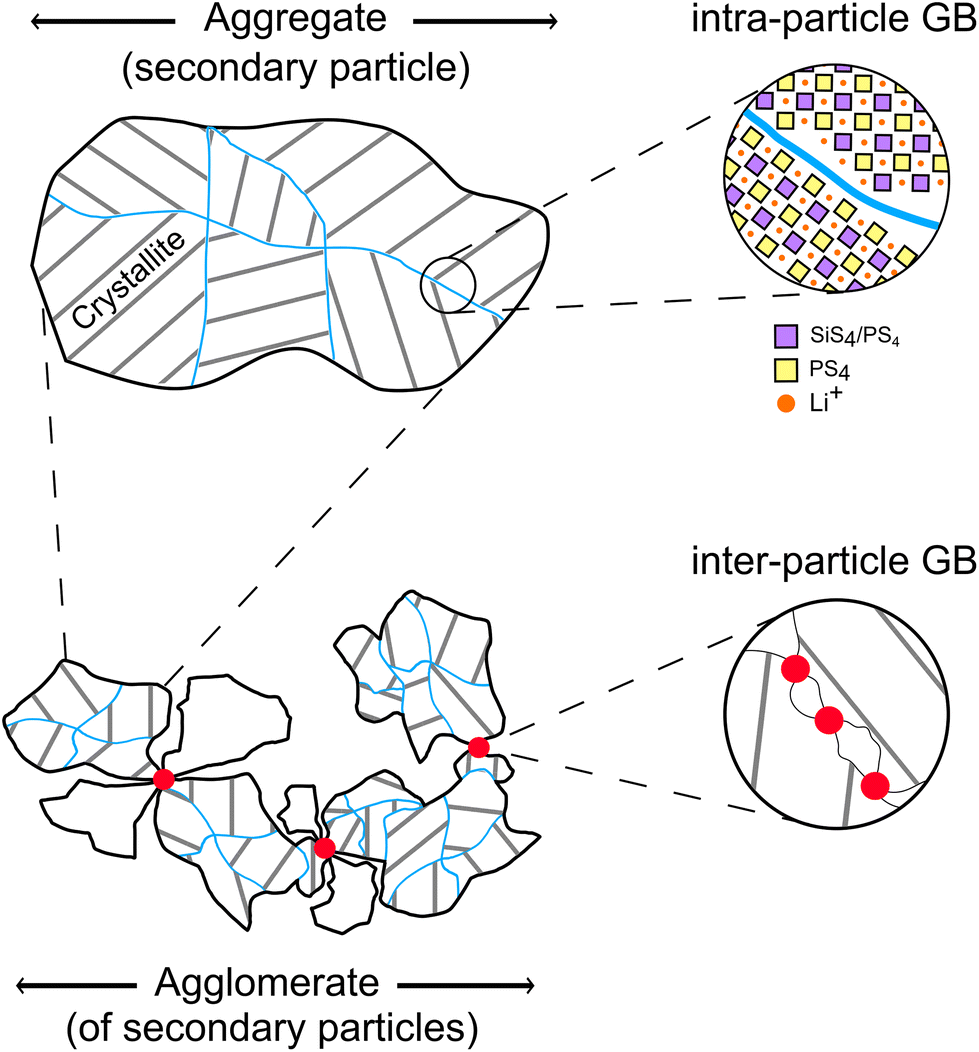 | ||
| Fig. 2 Top: Illustration of an aggregate (secondary particle) consisting of various primary particles (crystallite) with different orientations (gray lines as thought Miller planes) and their intra-particle grain boundaries (blue lines). Bottom: Illustration of an agglomerate, consisting of several aggregates. The red dots depict inter-particle grain boundaries. It should be noted that the abbreviation GB stands for grain boundary. | ||
Initially, the quality of the sheets varied largely in terms of packing density and homogeneity. SEM images (Fig. S4†) revealed that the reason for this was the broad and random particle size distribution of t-Li7SiPS8 obtained from manual grinding of the product after solid-state synthesis. To investigate the effect of particle size distribution on the sheet quality, the material was sieved and divided into three different fractions with particle size distributions of <50 μm, 50–100 μm and >100 μm. The effect of the particle size distribution on the obtained sheets is exemplarily given for the couple HNBR-17 and p-xylene. Sheets composed of >100 μm particles do not have well-defined edges (Fig. 3a). Instead, the sheet is inhomogeneous, coarse, and dense only at its top. Its density gradually decreases along the sheet, especially at the sides. Apparently, the time between the application of the slurry to the applicator and the doctor blade is sufficient for the larger, heavier particles to sediment, leading to an inhomogeneous particle distribution within the slurry. In contrast, the quality of the sheet improves as the particle size distribution decreases (Fig. 3b and c). This observation is particularly apparent for the sheet comprised of <50 μm particles (Fig. 3c), where no faded areas related to a loss in particle density can be observed. SEM images corroborate the findings from the optical inspection. Even at moderate magnifications, it is obvious that the sheets consisting of particles >100 μm have a rough and inhomogeneous morphology. This is also reflected in the mechanical stability during sample handling, as these sheets tear apart easily. On the other hand, the most convenient sample handling was observed for sheets made of particles <50 μm. This can be ascribed to their uniform and dense packing with overall homogeneously distributed particles. At higher magnifications, it can be seen that all sheets, regardless of their sieving fraction, have similar morphologies and that the particles are composed of aggregates of different sizes.
 | ||
| Fig. 3 Comparison of t-Li7SiPS8 sheets (with HNBR-17 in p-xylene) containing different fractions of particles (top row): (a) >100 μm, (b) 50–100 μm and (c) <50 μm and the corresponding SEM images highlighting the deviating morphologies at moderate magnifications. At higher magnifications, a similar morphology with similar-sized agglomerates is observed. | ||
2.3. Chemical stability of t-Li7SiPS8 sheets
Another important factor in slurry-based processing is the chemical inertness of t-Li7SiPS8 to the binder and solvent. Therefore, slurries containing <50 μm-sized t-Li7SiPS8 particles were stirred for an extended period (3 days) at room temperature and then processed the same way as described before. From the X-ray powder diffraction (XRPD) pattern, it can be seen that the crystal structure of the processed t-Li7SiPS8 (Fig. 4a) is still preserved even after 3 days of stirring, as the reflections are sharp and no new reflections or lack thereof are observed. However, when compared to the diffraction pattern of pristine t-Li7SiPS8 (Fig. 4b), an increased diffuse background intensity is observed and can be attributed to the amorphous binder and/or partial amorphization of t-Li7SiPS8. The results indicate that the long-range order of the crystal structure of t-Li7SiPS8 is not noticeably affected by the processing procedure. | ||
| Fig. 4 (a) XRPD pattern of an exemplary sheet, processed from t-Li7SiPS8 stirred for an extended period of 3 days with HNBR-17 in p-xylene. It shows an amorphous background between 0 and 35 stemming from the binder. (b) XRPD pattern of pristine t-Li7SiPS8 powder. (c) 31P NMR spectra of t-Li7SiPS8 processed sheets with HNBR-17 in toluene (orange), anisole (red), p-xylene (blue) and pristine t-Li7SiPS8 powder (black). The gray peaks are attributed to PS43− units and the pink peaks are assigned to amorphous Li3PS4-type ortho-thiophosphate and polymeric PS3− side phases. XPS signals in the S 2p and P 2p regions of the t-Li7SiPS8 sheets processed with: HNBR-17 (d, h) in p-xylene, (e, i) in anisole, (f, j) in toluene and (g, k) pristine t-Li7SiPS8 powder. The orange peaks are attributed to PS43− and SiS43− units while the blue peaks are assigned to Li2SiS3 and P-[S]n-P-type anions. | ||
To complement the diffraction data, 7Li, 29Si (Fig. S5a and b,† respectively) and 31P solid-state NMR (ss-NMR) was performed to investigate changes in the local structure of the compounds. In the 31P NMR spectrum (Fig. 4c), the signal at 94 ppm corresponds to isolated PS43− units at the 4d site (occupied by Si and P), while the signal at 73 ppm is attributed to the 2b phosphorus site (solely occupied by P). The additional signals at 86 ppm and 84 ppm are attributed to the amorphous Li3PS4-type ortho-thiophosphate and the polymeric PS3− side phases, respectively.26 After slurry processing, neither shifts nor additional peaks are observed, thus further confirming the stability of t-Li7SiPS8 to the processing conditions. Interestingly, the amorphous phase appears to be slightly affected by slurry processing as the phase fraction decreases from 16 at% in the pristine sample to 12 at% after toluene (Fig. 4c, orange) and anisole (Fig. 4c, red) exposure. In a previous study, reduced amorphous phase content was observed when t-Li7SiPS8 was treated with solvents with donor numbers (DN) ≥15 kcal mol−1.26 Water impurities can increase the DN value of the solvents used in this study. However, to approximate industrial applications, the solvents were not further dried or purified. The structural studies revealed that neither the local environment nor the bulk structure of t-Li7SiPS8 appears to be affected by the slurry-based processing.
To obtain further insights into the surface of the SE-sheets, X-ray photoelectron spectroscopy (XPS) in the S 2p and P 2p region was conducted on the sheets processed with HNBR-17 in every solvent combination (Fig. 4d–f and h–k) and compared to the pristine sample (Fig. 4g and k). The orange marked peaks at 161.3 eV in the S 2p spectrum and at 131.8 eV in the P 2p spectrum are assigned to PS43− and SiS44−, respectively. The blue marked peaks between 162.5–163.5 eV (S 2p region) and 132–132.6 eV (P 2p) are attributed to Li2SiS3 and P-[S]n-P-type anions.13,29–31 Interestingly, no Li2SiS3 or P-[S]n-P-type compounds are found on the pristine sample in the S 2p spectrum, but the latter is found in the P 2p spectrum. These compounds are likely unavoidable side products that form during synthesis, as demonstrated in a previous study.13 Aside from these peaks, no additional features are observed that could potentially hint at other (decomposition) products. Based on XRPD, ss-NMR, and XPS analysis, we conclude that t-Li7SiPS8 is largely stable under our processing conditions and maintains its structural and compositional integrity.
2.4. Conductivity measurements on t-Li7SiPS8 sheets
Although the morphology and chemical stability of the t-Li7SiPS8 sheets certainly play an important role, the ionic conductivity is the decisive factor in evaluating their performance. It has already been shown that the ionic conductivity suffers when polymeric binders are incorporated. This effect is even more severe when the binder encapsulates the SE particles rather than being present as granular domains between the particles.32–38 However, the conductivity of several SEs can also be increased with denser packing of particles as demonstrated in previous studies.22,23,39 As the cell pressure affects the packing density, all EIS measurements for lithium ion conductivity were performed with an operating cell pressure of 4 MPa at room temperature. The operation cell pressure denotes the pressure applied during measurement. The Nyquist plots of exemplary sheets (HNBR-17 in toluene) consisting of different particle size fractions are compared with that of pristine t-Li7SiPS8 powder and summarized in Fig. S6.† For better comparison, the plots were normalized to the cell constant. As expected, due to the absence of a binder, pristine t-Li7SiPS8 shows the lowest resistance, while for the sheets it increases with decreasing particle size. In other words, t-Li7SiPS8 sheets composed of smaller-sized particles suffer from higher resistance, which we assume to be related to an increased inter-particular grain boundary resistance.Fig. 5a summarizes the ionic conductivities of the pristine t-Li7SiPS8 powder (black) and the sheets prepared from different particle size fractions using different solvents and binders. The highest conductivity is observed for the pristine t-Li7SiPS8 powder (3 mS cm−1). In contrast, the conductivities of the sheets are consistently one order of magnitude lower. As implied previously by the Nyquist plot, the conductivities are consistently higher for sheets incorporating larger-sized particles than smaller-sized ones, regardless of the binder or solvent used. Processing with either HNBR-17 (Fig. 5a, red) or PIB (Fig. 5a, green) results in similar conductivities. In both instances, toluene seems to have a slight advantage over the other solvents, reaching up to 0.3 mS cm−1. Out of all solvent/binder combinations, sheets processed with HNBR-34 in anisole (Fig. 5a, blue) result in the lowest conductivities. Using a more polar binder and solvent in the slurry formulation results in a lower ionic conductivity of the sheet. The conductivity of a sheet obtained by PIB in toluene and unsieved t-Li7SiPS8 particles (Fig. 5b, purple) is also included. While the conductivity is higher than those obtained with sheets composed of <50 μm particles, it is also accompanied by a considerably larger error bar compared to the other sheets. This can be explained by the inhomogeneous particle size distribution after manual grinding, leading to morphologically inhomogeneous SE-sheets. Upon comparing SEM images of the uncompressed (before EIS measurement) and compressed (after EIS measurement) sheets (Fig. S7a–c),† distinct differences in morphology are evident. Notably, for sheets composed of particles >100 μm (Fig. S7a†), the initial rough morphology with discernible individual aggregates appears significantly flattened after compression. A similar trend of surface flattening is observed in sheets composed of smaller particles. At higher magnifications, we can see several strings, presumably stemming from the binder, which were not visible in the uncompressed state. Interestingly, the sheet with particles <50 μm (Fig. S7c†) exhibits a greater number of voids compared to the sheet with particles in the 50–100 μm range (Fig. S7b†) after compression. The lowest number of voids is observed in the sheet containing particles >100 μm. This observation is supported by the calculation of relative densities (Fig. S7d†) of the different sheets. Sheets composed of >100 μm particles generally exhibit higher final relative densities compared to those with particles <50 μm. In the former case, this can be attributed to particle fragmentation into smaller units that fill the voids, thus improving the microstructure. However, for sheets with smaller particle sizes, the explanation is less straightforward. One potential reason may be the absence of significant fragmentation, where instead, the binder may stretch and separate individual aggregates. Although the relative density of a sample is not a strong descriptor for its impedance,39 the EIS measurements showed a clear discrepancy based on particle size.
 | ||
| Fig. 5 (a) Conductivities of pristine t-Li7SiPS8 pellets (black, no solvent, and no binder) and t-Li7SiPS8/binder sheets of different slurry formulations (red: HNBR-17, blue: HNBR-34, green: PIB in their respective solvents) and different particle size fractions. The conductivity of an exemplary sheet (PIB in toluene) consisting of unsieved t-Li7SiPS8 particles (purple) is also shown for comparison. Error bars reflect the deviations among three individual samples. (b) Tracer diffusion coefficient as a function of diffusion time as obtained by 7Li PFG NMR measurements of t-Li7SiPS8 sheets (with HNBR-17 in toluene) comprised of different particle sizes. | ||
Therefore, we also employed PFG NMR spectroscopy on sheets composed of different particle size fractions. With this method it is possible to determine the 7Li tracer diffusion coefficients, DtrNMR, for lithium diffusion as a function of observation (diffusion) times, ΔNMR (Fig. 5c). For an unrestricted diffusion process, DtrNMR would remain constant for all ΔNMR. However, we observe a steady decrease in the experimental diffusion coefficients with increasing diffusion times, irrespective of the particle size fractions in the sheets. This suggests that diffusion barriers restrict the diffusion process. Using the formula
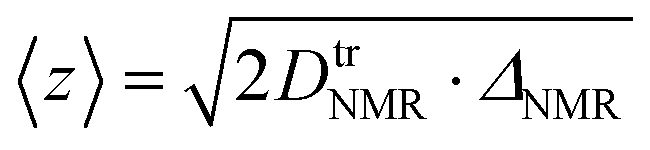 | (1) |
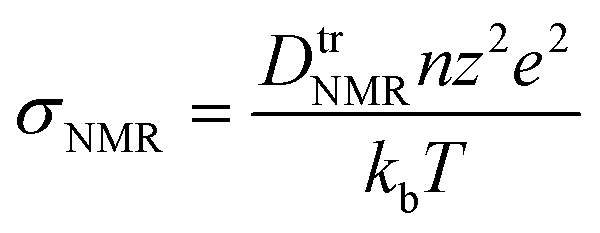 | (2) |
μm (DtrNMR = 2.1 × 10−11 m2 s−1) is approximately 1.4 times higher than that for the sheet with <50 μm particles (DtrNMR = 1.5 × 10−11 m2 s−1). These values are one order of magnitude higher than those of previously reported benchmark LGPS (2.2 × 10−12 m2 s−1)25,43 or a hybrid solid electrolyte of t-Li7SiPS8/Li6PS5Br (5.5 × 10−12 m2 s−1)44 but similar to Li5.5PS4.5Cl (1.01 × 10−11 m2 s−1).45 The fact that differences in diffusivity are observed even at these short times indicates that intrinsic (within primary particles) bulk diffusivities, which should be similar across all particle sizes, are not being measured. At longer observation times (150 ms), sheets with smaller particles continue to exhibit lower diffusivities, suggesting that 7Li diffusion is more strongly impeded by inter-particle grain boundaries in these samples. This behavior is consistent with EIS measurements, which further support the conclusion that larger secondary particles, with fewer inter-particle grain boundaries, allow for easier Li diffusion. In contrast, the increased inter-particle grain boundaries in sheets with smaller particles likely create more resistance to ion transport. In summary, the observed diffusivities of our slurry-processed material exceed reported benchmarks by almost an order of magnitude. This demonstrates that slurry processing is an effective method for SE materials, enabling the combination of high Li diffusivity in SE-sheets, a prerequisite for the performance of all-solid-state batteries, with industry-ready continuous processing options.
3. Conclusions
In this study, six different slurry formulations based on three different binder materials, HNBR-17, HNBR-34, and PIB, in three different solvents, anisole, p-xylene, and toluene, were evaluated for their compatibility to form solid electrolyte sheets with t-Li7SiPS8. We found that processing with less polar binders such as HNBR-17 and PIB was more favorable in obtaining homogeneous sheets than with the polar binder HNBR-34.The chemical stability of t-Li7SiPS8 towards the processing parameters was demonstrated by XRPD, MAS-NMR, and XPS, respectively. In addition, XPS revealed that the surface of t-Li7SiPS8 sheets remains mainly unaffected and no evidence of accumulation of decomposition products at the surface was found.
Fabrication of sheets from the as-prepared t-Li7SiPS8 powder after solid-state synthesis resulted in irregular sheets with poor reproducibility, demonstrating the necessity of sieving. We used three different sieved size fractions (<50 μm, 50–100 μm and >100 μm) and found that the quality and the homogeneity of the processed sheets are heavily dependent on the sieved particle size fractions. Specifically, sheets containing particles of a smaller particle size distribution showed higher stability in handling and overall better homogeneity due to a denser packing, whereas the opposite was observed for sheets made of larger-sized particles.
In contrast, EIS measurements revealed that higher conductivity values are obtained with sheets made of larger particle sizes, while the conductivity was found to decrease with decreasing particle size. SEM images and relative density calculations after EIS measurements further confirm that sheets composed of >100 μm particles exhibit higher relative densities, likely due to the fragmentation of aggregates that fill voids. In contrast, sheets with smaller particles (<50 μm) show increased voids, potentially caused by binder stretching, leading to lower relative densities. 7Li PFG NMR spectroscopy results support these findings, demonstrating higher diffusivities in sheets made of larger particles. This is attributed to fewer inter-particle grain boundaries, which we assume create more resistances for ion transport. In contrast, smaller particles exhibit lower diffusivity due to a higher number of inter-particle grain boundaries and increased binder interaction, both of which limit Li diffusion. To conclude, the slurry processing of t-Li7SiPS8 to fabricate SE-sheets was investigated. While t-Li7SiPS8 is cheaper than LGPS, it is also susceptible to hydrolysis, hence, slurry processing needs to proceed under an inert atmosphere. Furthermore, we observe a correlation between diffusivity and particle size and attribute the higher conductivities for larger particle-sized sheets to lower inter-particle grain boundaries and less contact with the binder and to an increased relative density. Although sheets with smaller particles offer advantages in terms of processing and uniformity compared to sheets consisting of larger particles, the enhanced ionic diffusivity of the latter highlights the importance of optimizing particle size and the microstructure for improved Li transport.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge financial support from the Max Planck Society, the German Federal Ministry of Research and Education (BMBF), projects 03XP0177B (MPI-FestBatt I) and 03XP0430B (MPI-FestBatt II), the Center for Nanoscience (CeNS) and the Deutsche Forschungsgemeinschaft via the Cluster of Excellence e-conversion (EXC2089). Our gratitude goes to V. Duppel for SEM-EDX imaging and Sebastian Bette for XRPD data interpretation. We thank Christian Schneider and Maximilian Plaß for their support on EIS data interpretation and for the scientific discussions.References
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- W. Zhang, D. A. Weber, H. Weigand, T. Arlt, I. Manke, D. Schröder, R. Koerver, T. Leichtweiss, P. Hartmann and W. G. Zeier, et al., ACS Appl. Mater. Interfaces, 2017, 9, 17835–17845 CrossRef CAS PubMed.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama and K. Kawamoto, et al., Nat. Mater., 2011, 10, 682–686 CrossRef CAS PubMed.
- A. Sakuda, A. Hayashi and M. Tatsumisago, Sci. Rep., 2013, 3, 2261 CrossRef PubMed.
- J. Lau, R. H. DeBlock, D. M. Butts, D. S. Ashby, C. S. Choi and B. S. Dunn, Adv. Energy Mater., 2018, 8, 1800933 CrossRef.
- M. Dixit, N. Muralidharan, A. Parejiya, C. Jafta, Z. Du, S. M. Neumayer, R. Essehli, R. Amin, M. Balasubramanian and I. Belharouak, ACS Appl. Mater. Interfaces, 2022, 14, 44292–44302 CrossRef CAS PubMed.
- F. Han, T. Gao, Y. Zhu, K. J. Gaskell and C. Wang, Adv. Mater., 2015, 27, 3473–3483 CrossRef CAS PubMed.
- N. C. Rosero-Navarro, T. Kinoshita, A. Miura, M. Higuchi and K. Tadanaga, Ionics, 2017, 23, 1619–1624 CrossRef CAS.
- S. Wenzel, T. Leichtweiss, D. Krüger, J. Sann and J. Janek, Solid State Ionics, 2015, 278, 98–105 CrossRef CAS.
- L. M. Riegger, R. Schlem, J. Sann, W. G. Zeier and J. Janek, Angew. Chem., Int. Ed., 2021, 60, 6718–6723 CrossRef CAS PubMed.
- L. M. Riegger, S.-K. Otto, M. Sadowski, S. Jovanovic, O. Kötz, S. Harm, L. G. Balzat, S. Merz, S. Burkhardt and F. H. Richter, et al., Chem. Mater., 2022, 34, 3659–3669 CrossRef CAS.
- K. Kerman, A. Luntz, V. Viswanathan, Y.-M. Chiang and Z. Chen, J. Electrochem. Soc., 2017, 164, A1731 CrossRef CAS.
- Y. S. Jung, D. Y. Oh, Y. J. Nam and K. H. Park, Isr. J. Chem., 2015, 55, 472–485 CrossRef CAS.
- L. Zhou, K.-H. Park, X. Sun, F. Lalère, T. Adermann, P. Hartmann and L. F. Nazar, ACS Energy Lett., 2018, 4, 265–270 CrossRef.
- T. Liu, L. Zhang, J. Li, Y. Li, K. Lai, S. Zhang, G. Zhao, D. Liu, Z. Xi and C. Liu, et al., J. Electroanal. Chem., 2023, 928, 117032 CrossRef CAS.
- H. Liu, P. He, G. Wang, Y. Liang, C. Wang and L.-Z. Fan, Chem. Eng. J., 2022, 430, 132991 CrossRef CAS.
- D. H. Kim, Y.-H. Lee, Y. B. Song, H. Kwak, S.-Y. Lee and Y. S. Jung, ACS Energy Lett., 2020, 5, 718–727 CrossRef CAS.
- Y. J. Nam, S.-J. Cho, D. Y. Oh, J.-M. Lim, S. Y. Kim, J. H. Song, Y.-G. Lee, S.-Y. Lee and Y. S. Jung, Nano Lett., 2015, 15, 3317–3323 CrossRef CAS PubMed.
- N. Riphaus, P. Strobl, B. Stiaszny, T. Zinkevich, M. Yavuz, J. Schnell, S. Indris, H. A. Gasteiger and S. J. Sedlmaier, J. Electrochem. Soc., 2018, 165, A3993–A3999 CrossRef CAS.
- A. Tron, R. Hamid, N. Zhang, A. Paolella, P. Wulfert-Holzmann, V. Kolotygin, P. López-Aranguren and A. Beutl, J. Energy Storage, 2023, 66, 107480 CrossRef.
- C. Sedlmeier, T. Kutsch, R. Schuster, L. Hartmann, R. Bublitz, M. Tominac, M. Bohn and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 070508 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- A. Kuhn, O. Gerbig, C. Zhu, F. Falkenberg, J. Maier and B. V. Lotsch, Phys. Chem. Chem. Phys., 2014, 16, 14669–14674 RSC.
- A.-K. Hatz, R. Calaminus, J. Feijoo, F. Treber, J. Blahusch, T. Lenz, M. Reichel, K. Karaghiosoff, N. M. Vargas-Barbosa and B. V. Lotsch, ACS Appl. Energy Mater., 2021, 4, 9932–9943 CrossRef CAS.
- S. Harm, A.-K. Hatz, I. Moudrakovski, R. Eger, A. Kuhn, C. Hoch and B. V. Lotsch, Chem. Mater., 2019, 31, 1280–1288 CrossRef CAS.
- D. Walter, Nanomaterials, 2013, 9–24 CAS.
- F. Walther, S. Randau, Y. Schneider, J. Sann, M. Rohnke, F. H. Richter, W. G. Zeier and J. Janek, Chem. Mater., 2020, 32, 6123–6136 CrossRef CAS.
- F. Walther, R. Koerver, T. Fuchs, S. Ohno, J. Sann, M. Rohnke, W. G. Zeier and J. Janek, Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- F. J. Simon, M. Hanauer, F. H. Richter and J. Janek, ACS Appl. Mater. Interfaces, 2020, 12, 11713–11723 CrossRef CAS PubMed.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto and S. Ryu, et al., Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- K. T. Kim, D. Y. Oh, S. Jun, Y. B. Song, T. Y. Kwon, Y. Han and Y. S. Jung, Adv. Energy Mater., 2021, 11, 2003766 CrossRef CAS.
- A. Sakuda, K. Kuratani, M. Yamamoto, M. Takahashi, T. Takeuchi and H. Kobayashi, J. Electrochem. Soc., 2017, 164, A2474 CrossRef CAS.
- K. Lee, S. Kim, J. Park, S. H. Park, A. Coskun, D. S. Jung, W. Cho and J. W. Choi, J. Electrochem. Soc., 2017, 164, A2075 CrossRef CAS.
- T. Ates, M. Keller, J. Kulisch, T. Adermann and S. Passerini, Energy Storage Mater., 2019, 17, 204–210 CrossRef.
- D. Y. Oh, D. H. Kim, S. H. Jung, J.-G. Han, N.-S. Choi and Y. S. Jung, J. Mater. Chem. A, 2017, 5, 20771–20779 RSC.
- Y. J. Nam, D. Y. Oh, S. H. Jung and Y. S. Jung, J. Power Sources, 2018, 375, 93–101 CrossRef CAS.
- C. Schneider, C. P. Schmidt, A. Neumann, M. Clausnitzer, M. Sadowski, S. Harm, C. Meier, T. Danner, K. Albe and A. Latz, et al., Adv. Energy Mater., 2023, 13, 2203873 CrossRef CAS.
- O. Kwon, M. Hirayama, K. Suzuki, Y. Kato, T. Saito, M. Yonemura, T. Kamiyama and R. Kanno, J. Mater. Chem. A, 2015, 3, 438–446 RSC.
- X. Liang, L. Wang, Y. Jiang, J. Wang, H. Luo, C. Liu and J. Feng, Chem. Mater., 2015, 27, 5503–5510 CrossRef CAS.
- D. A. Weber, A. Senyshyn, K. S. Weldert, S. Wenzel, W. Zhang, R. Kaiser, S. Berendts, J. Janek and W. G. Zeier, Chem. Mater., 2016, 28, 5905–5915 CrossRef CAS.
- A. Kuhn, V. Duppel and B. V. Lotsch, Energy Environ. Sci., 2013, 6, 3548–3552 RSC.
- R. Calaminus, S. Harm, D. H. Fabini, L. G. Balzat, A.-K. Hatz, V. Duppel, I. Moudrakovski and B. V. Lotsch, Chem. Mater., 2022, 34, 7666–7677 CrossRef CAS.
- P. Adeli, J. D. Bazak, K. H. Park, I. Kochetkov, A. Huq, G. R. Goward and L. F. Nazar, Angew. Chem., Int. Ed., 2019, 58, 8681–8686 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00005j |
| This journal is © The Royal Society of Chemistry 2025 |