
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-functional Li+ diffusion network in high-nickel cathodes for solid-state Li metal batteries†
Meng
Ye
,
Zhian
Zhang
,
Jianhua
Chen
,
Qiuyue
Chen
,
Jiarui
Hu
,
Lang
Qiu
,
Fang
Wan
 * and
Xiaodong
Guo
*
* and
Xiaodong
Guo
*
School of Chemical Engineering, Sichuan University, Chengdu 610065, PR China. E-mail: wanfang2022@scu.edu.cn; xiaodong2009@scu.edu.cn
First published on 17th March 2025
Abstract
The lithiation/deintercalation of cathode materials leads to poor contact between the cathode particles in solid-state batteries. This process leads to fast capacity attenuation as there is no continuous ion transport medium to fill up the voids caused by the volume change of active materials. Herein, we designed a dehydrofluorination polyvinylidene fluoride (PVDF) coating layer for LiNi0.8Co0.1Mn0.1O2 (NCM) particles using residual Li2CO3 on the NCM surface to induce the dehydrofluorination reaction of PVDF. On the one hand, the in situ formed coating layer increased the contact area between the NCM particles and acted as a buffering barrier for the volume change of NCM, ensuring unobstructed Li+ transport during the lithiation/deintercalation process. On the other hand, the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C– and LiF generated via the dehydrofluorination reaction was beneficial for Li+ diffusion. As a result, a sturdy and fast Li+ transport network was constructed, and the electrochemical performance of the solid-state battery was greatly improved. Thus, this dual-functional Li+ transport network simultaneously alleviates the poor particle contact and limited Li+ transport in cathodes, offering a novel approach for achieving high-performance solid-state batteries.
C– and LiF generated via the dehydrofluorination reaction was beneficial for Li+ diffusion. As a result, a sturdy and fast Li+ transport network was constructed, and the electrochemical performance of the solid-state battery was greatly improved. Thus, this dual-functional Li+ transport network simultaneously alleviates the poor particle contact and limited Li+ transport in cathodes, offering a novel approach for achieving high-performance solid-state batteries.
Broader contextSolid-state batteries are the most promising energy storage devices for the future owing to their satisfactory security and energy density. However, there is no continuous medium for Li+ transport in their cathodes. Moreover, the volume change of cathode materials during the lithiation/deintercalation process leads to poor contact between particles, leading to further deterioration of the performance of solid-state batteries. This work constructs a dual-functional Li+ diffusion network by coating NCM811 with a PVDF layer. This Li+ diffusion network could promote Li+ transport efficiency by increasing the Li+ transport path and Li+ transport speeds. This strategy underscores the importance of constructing high-performance cathodes for solid-state batteries and provides a novel approach for achieving high-performance SSBs. |
1. Introduction
With the gradual increase in requirements for energy storage devices, solid-state batteries (SSBs) have drawn significant attention owing to their high safety and satisfactory energy density. An SSB uses a solid-state electrolyte (SSE) to replace the liquid electrolyte, which can not only solve the risk of electrolyte leakage but also provide the possibility for the stacking of cells, thereby achieving high energy density.1–3 Moreover, the high mechanical strength of SSE endows it with the ability to resist Li dendrites, making it compatible with Li metal anode with high theoretical energy density.4 For these reasons, SSBs are expected to substitute traditional secondary batteries for application in various scenarios in the future.5–7The interior of an SSB is entirely piled up by solids, which implies that there is no continuous medium to fill the voids between particles coming from the SSE–cathode interface, SSE–anode interface, SSE interior and cathode interior.8–11 As a consequence, ion transport paths are blocked by the large number of voids, and accordingly, rapid ion transport is limited.12 Resolving the issue of poor physical solid contact is the key to construct high-performance SSBs. The cathode electrolyte interface (CEI) layer13–17 and solid electrolyte interface (SEI) layer18–20 are usually built to increase the contact area at the interface to improve the contact between the SSE and electrode. SSE internal interface problems ordinarily exist in inorganic-based SSE. Hot pressing is an effective method to reduce the voids in SSE bulk.4,9,21 Beyond that, by combining organic and inorganic components, the voids between inorganic particles can be filled with continuous organic matrix, improving the internal interface of inorganic SSE.22–25 However, the situation is more complex for the cathode. On the one hand, the accumulation of particles results in voids in the bulk,26,27 hindering the ion diffusion. On the other hand, the volume of cathode particles undergoes significant changes during the lithiation and delithiation process.28,29 The volume effect leads to poor contact between particles,30–32 leading to further deterioration of the SSB performance. Inorganic SSEs with fast ion transport are often employed as additives to promote ion diffusion in the cathode.33,34 Another method to achieve unobstructed ion diffusion is by increasing the contact area of various components. Adding sintering additive can fill up the voids and increase the ion transport path.35,36 Additionally, organic SSEs can be used as surface contact modifiers because of their flexibility.37,38 However, the above strategies aim to address the issue of slow ion transport while ignoring the contact failure caused by volume changes. Even the soft polymer additives cannot adapt to the huge volume changes of cathode materials, especially the mainstream Ni-rich materials with high energy density. Therefore, constructing the sturdy and fast ion transport network with excellent volume adaptability for the cathode is of great significance.
In this study, polyvinylidene fluoride (PVDF) was selected as the coating layer for LiNi0.8Co0.1Mn0.1O2 (NCM). The residual Li2CO3 on the NCM surface induces partial dehydrofluorination of the PVDF chain to generate the coating layer for NCM in situ. The in situ generated layer increases the contact area between NCM particles to provide more Li+ transport pathways and firmly adheres to the NCM surface during the lithiation/delithiation process, ensuring satisfactory contact with the NCM when the volume changes. Favourable contact between particles ensures unobstructed Li+ transport at all times. More than that, the in situ dehydrofluorination reaction has generated the –CC– bond and LiF on the NCM surface. The –CC– bond has a more delocalized electron cloud, which adsorbs Li+ and facilitates its rapid transport. LiF is a good conductor for Li+, which is also beneficial for Li+ transport. Altogether, the PVDF coating layer has enhanced the performance of SSB from two aspects: the number of Li+ transport paths and Li+ diffusion rate. Analogously, the dehydrocyanation polyacrylonitrile (PAN) coating layer achieved the same accomplishments. This dual-function Li+ transport network addresses the challenges of poor particle contact and limited Li+ transport in cathodes and offers a novel approach for high-performance SSBs.
2. Results and discussion
A garnet-rich composite solid-state electrolyte (GRSE) with high hardness is favourable for resisting lithium dendrite, which is important for the anode stability of the SSB.39,40 Herein, a GRSE with 70 wt% Li6.4La3Zr1.4Ta0.6O12 (LLZTO) was employed. The surface of the GRSE is relatively uniform and flat, and the thickness is ∼52.5 μm (Fig. S1 and S2†). The GRSE has excellent mechanical properties. The maximum tensile strength of the film is 3.62 MPa and the elongation-at-break reaches 141.11% (Fig. S3a†). Moreover, the hardness of the GRSE is 5.94 MPa and the elastic modulus of the GRSE is 204.74 MPa (Fig. S3b†). The satisfactory mechanical strength provides the possibility of anode stability. Subsequently, the electrochemical properties were tested. The GRSE has good antioxidative stability, and the decomposition potential of the SE/GRSE/Li asymmetric battery is 4.62 V (Fig. S4†). The electrochemical stability window makes it applicable in the high voltage system. The Li+ migration number (tLi+) is 0.39 (Fig. S5†), and the ionic conductivity (δ) increases from 0.61 to 1.59 mS cm−1 with the temperature increasing from 30 to 80 °C (Fig. S6†). According to the δ result, the activation energy (Ea) is calculated as 0.18 eV (Fig. S7†), implying that Li+ can easily migrate in the GRSE. To confirm the stability of the GRSE to Li, the cycling stability of the Li–Li battery was tested. The battery can cycle stably for 1000 h (Fig. S8†). The overpotential initially increases, then decreases. After 100 h, it becomes stable. The unstable overpotential in the first 100 h is caused by FSI− decomposition into the SEI film. We disassembled the battery after 100 h cycling and investigated the interface. After cycling, the GRSE surface remains flat and the Li surface is covered by a uniform SEI layer (Fig. S9†). By analysing the component of the SEI layer, there is an obvious F 1s characteristic peak (Fig. S10†). It contains two substances: few –CF2CH2– and abundant LiF (Fig. S11†). –CF2CH2– comes from PVDF of the GRSE matrix, and LiF is caused by the decomposition of FSI−. LiF plays an important role in Li+ transport at the GRSE/Li interface.41,42 To sum up, GRSE has preeminent mechanical strength and acceptable electrochemical properties; thus, it has the potential to be used in high voltage SSBs.The GRSE was then matched to the NCM/Li battery. The discharge capacity is less than 100 mA h g−1 and decays rapidly at 0.5 C (Fig. 1a). After 15 cycles, the charge and discharge plateaus almost disappeared (Fig. S12†). Since the stability of the anode side has been demonstrated, the poor performance of the battery is caused by the cathode side. The morphology of the NCM cathode is displayed in Fig. 1b and S13a.† NCM and conductive carbon particles are bound together by PVDF binder. Between two NCM particles, PVDF acts as the Li+ transport medium, and the point–point contact mode between solids results in abundant voids. Unlike liquid electrolyte systems, SSEs cannot fill these voids. As a result, the Li+ migration pathway is blocked. During the lithiation/delithiation process, the volume of NCM constantly shrinks and expands, causing poorer contact between particles, and the cathode surface becomes rougher (Fig. S13b†). In addition, Li+ is extracted from the NCM crystal during the charge process, leading to the structural contraction. The volume of NCM decreases, causing contact failure between particles. As a result, the Li+ transport path decreases. During the subsequent discharge process, a portion of Li+ fails to insert into the NCM crystal structure. Therefore, the NCM battery displays low coulombic efficiency.
 | ||
| Fig. 1 (a) Cycling performance of the NCM/Li battery. (b) SEM image of the NCM cathode. (c) TEM image of NCM@PVDF cathode. (d) SEM image of NCM@PVDF cathode. 3D optical profile images of (e) NCM and (f) NCM@PVDF cathodes. (g) Pore area of NCM and NCM@PVDF particles. (h) Schematic of Li+ transport in NCM and NCM@PVDF cathodes. | ||
To address the issue of poor Li+ diffusion in the cathode, a soft PVDF coating layer was designed. A thin coating layer completely envelops the NCM particle (Fig. 1c). Compared to the NCM particle (Fig. S14†), the C element from PVDF and the S element from LiFSI uniformly distribute around NCM (Fig. S15†), indicating the uniformity of the PVDF coating layer. The coating layer makes the accumulation of cathode particles more compact; therefore, the surface of the NCM@PVDF cathode is smoother than the NCM cathode (Fig. 1d and S16†). This is because of the improved interface wettability between particles and PVDF binder. The voids in the cathode are decreased to a large extent, which provides more Li+ transport pathways. In addition, the height difference between the highest and lowest points of the NCM cathode is 21.58 μm, while that of the NCM@PVDF cathode is only 8.81 μm (Fig. 1e, f and S17†). The reduction in height difference reflects the compactness of particle accumulation, and the smoother surface is beneficial to the contact of the cathode and SSE. The automatic mercury porosimeter results reflect the contact of two particles more intuitively (Fig. 1g and Table S1†). The total pore area of the NCM particle is 0.404 m2 g−1, while that of the NCM@PVDF particle is only 0.145 m2 g−1. The smaller pore area reflects larger contact area between particles. However, the total pore volume of the NCM@PVDF particle is 0.2793 m3 g−1, which is slightly smaller than that of the NCM particle (0.2829 m3 g−1). This provides evidence that the coating layer is soft, which has a cushioning effect when the volume of NCM changes and avoids contact failure of particles (Fig. 1h). In short, the PVDF coating layer has improved the contact of cathode particles and ensured the effective contact of cathode/SSE, which is to the advantage of ion transport in both the cathode interior and cathode/SSE interface.
The generation mechanism of the PVDF coating layer was further investigated. The CC characteristic functional group was verified by Raman spectroscopy and Fourier transform infrared (FTIR) spectroscopy (Fig. 2a and b). The electron-rich CC bond is more favourable for Li+ transport compared to the C–C bond.43 Beside this, the original PVDF consists of an α-phase and β-phase, while only the β-phase PVDF was identified after coating (Fig. 2c). This indicates that the DMF solvent leads the phase transformation of PVDF.40 The configurations of the two kinds of PVDF are depicted in Fig. S18.† Compared to the α-phase, the molecular configuration of the β-phase PVDF is more ordered, and all the F atoms are on the same side of the polymer chain.44 This structure makes the Li+ transport path short and improves the transmission efficiency of Li+. X-ray photoelectron spectroscopy (XPS) results also prove the existence of a PVDF coating layer. The characteristic peaks of F 1s, N 1s, and S 2p are detected in NCM@PVDF (Fig. 2d). In addition, the –CF2CH2– characteristic peak can be found in NCM@PVDF, which is the important functional group of PVDF. Except for –CF2CH2–, there is a LiF characteristic peak in NCM@PVDF, which comes from the chemical reaction between Li2CO3 and HF (Fig. 2e). Li2CO3 is lithiophobic and related to low ionic conductivity, while LiF is lithiophilic and provides a fast channel for the transmission of Li+. Residual Li2CO3 on the NCM surface leads to partial dehydrofluorination of the PVDF chain (Fig. 2g), enhancing the interactions between PVDF and NCM. Apart from providing more Li+ transport channels, the PVDF coating layer improves the Li+ transport rate based on three aspects. Firstly, PVDF in situ dehydrofluorination on the NCM surface generates –CC– bonds, which is favourable for Li+ transport. Secondly, Li2CO3 with low ionic conductivity on the surface of NCM reacts with HF detached from PVDF chains to generate LiF with high ionic conductivity. Finally, the coating process has changed the configuration of PVDF from the α-phase to the β-phase, increasing Li+ transport efficiency by shortening the Li+ transport path.
 | ||
| Fig. 2 (a) Raman and (b, c) FTIR spectra of PVDF, NCM and NCM@PVDF. (d–f) XPS spectra of NCM and NCM@PVDF particles. (g) Schematic of PVDF coating layer promoting Li+ transport. | ||
Molecular dynamic simulation was conducted to theoretically validate and elucidate the experimental observations of enhanced Li+ transport facilitated by the dehydrofluorination PVDF coating layer. The simulation aims to investigate the differences in structure and electron distribution between PVDF and dehydrofluorination PVDF, as well as their effects on Li+ diffusion in the PVDF matrix and the NCM crystal. The structure of each component in the PVDF coating layer is exhibited in Fig. S19.†Fig. 3a and b display the simulated structure of the NCM cathode and the NCM@PVDF cathode, respectively. The electron cloud in dehydrofluorination PVDF is more delocalized because of –CC–; therefore LiFSI is more likely to be dissociated as Li+ and FSI−.45 In addition, Li+ in the NCM crystal structure can be captured by the electron cloud of –CC– due to electrostatic adsorption. As a result, there is more Li+ participating in transport, leading to more efficient Li+. The migration rate of Li+ in the two PVDF matrices are displayed in Fig. 3c. The Li+ migration rate of dehydrofluorination PVDF is 1.23 × 10−3 Å2 ps−1, which is six times more than that of PVDF (0.20 × 10−3 Å2 ps−1). The Li+ diffusion energy barriers in the NCM crystal of the two systems were also calculated. As shown in Fig. 3d and e, the PVDF repeating unit is at a distance from NCM, while the C atoms in dehydrofluorination PVDF bond with O atoms in the NCM crystal (Fig. 3g and h) because the electron distribution in –CC– is more delocalized. The optimized electron distribution plays a positive role in Li+ diffusion, with the Li+ diffusion energy barrier decreasing from 1.51 eV to 1.10 eV. In short, the dehydrofluorination PVDF coating layer promotes Li+ transport in the PVDF matrix and the NCM crystal.
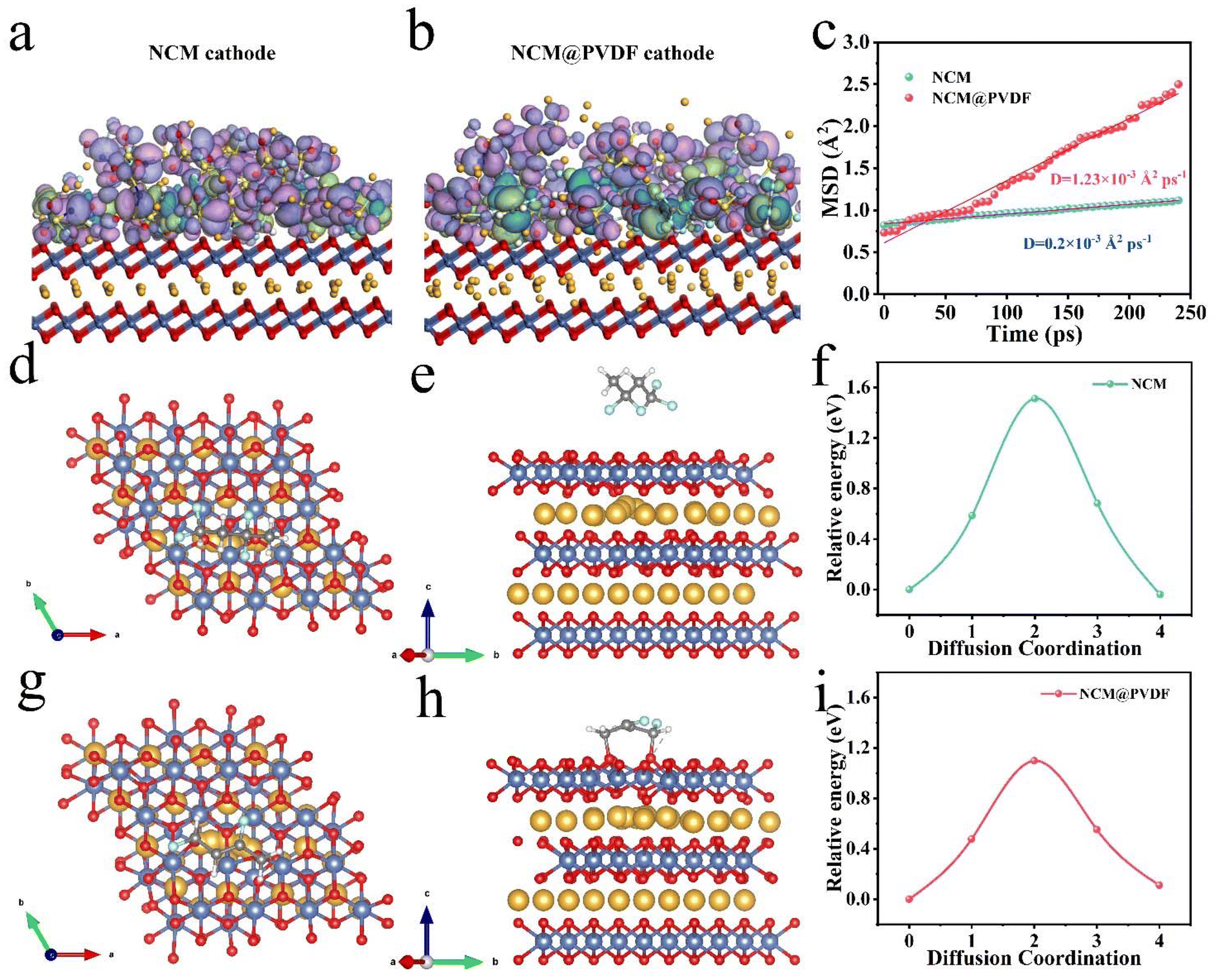 | ||
| Fig. 3 Simulation model of the (a) NCM surface and (b) NCM@PVDF surface. (c) Li+ migration rate in PVDF and dehydrofluorination PVDF matrix. The top and elevation view of the schematic of Li+ migration of (d, e) NCM and (g, h) NCM@PVDF. Diffusion energy barriers of Li+ in (f) NCM and (i) NCM@PVDF. | ||
SSBs were assembled with GRSE, and the electrochemical performances are illustrated in Fig. 4. The galvanostatic intermittent titration technique (GITT) plots show that the voltage polarization of NCM@PVDF is significantly weaker than that of NCM (Fig. 4a), indicating improved Li+ transport efficiency. Li+ experiences lower resistance when migrating in the NCM@PVDF cathode, leading to smaller polarization. The corresponding Li+ diffusion coefficient of the NCM@PVDF cathode is much higher than that of the NCM cathode (Fig. 4b). The transportation kinetics is enhanced and the Li+ diffusion coefficient of NCM@PVDF is much higher than that of NCM thanks to the efficient Li+ transport network. The Nyquist plots of the two batteries are shown in Fig. 4c and S20.† In general, the intercept of the plots with the Z' axis represents the internal resistance (Rs) of a battery, the first semicircle represents the interfacial impedance (Rse), and the second semicircle reflects the charge transfer impedance at the electrode surface (Rct). As the results show, no matter Rse or Rct of NCM@PVDF battery have significantly decreased, and Rs of NCM@PVDF battery is also smaller than that of NCM battery. The decreased impedance is the result of the improved contact in the inside of the cathode and the cathode/SSE interface. The smaller impedance is important for the battery to work at high current density. The impedance values of each part are listed in Table S2.† From the CV curves (Fig. 4d and S21†), it can be identified that the reversibility of oxidation peaks and reduction peaks of NCM@PVDF are excellent, and the NCM@PVDF battery reaches the stable state earlier than NCM battery. Taking the characteristic peaks of the H1-M phase transition as an example, the potential difference between the oxidation–reduction of NCM is 0.215 V, while that of NCM@PVDF is only 0.107 V. In addition, the phase transition processes of NCM occurs at 3.843 V, while that of NCM@PVDF occurs at 3.774 V, demonstrating that the deintercalation of Li+ in NCM@PVDF is easier. This provides evidence for the Li+ diffusion energy barriers results in Fig. 3. The cycling performance of the NCM@PVDF battery is superior to that of the NCM battery (Fig. 4e). The specific capacity of the NCM battery is low and approaches 0 mA h g−1 after 30 cycles and the voltage platform already disappears after 30 cycles (Fig. S22a†). While the NCM@PVDF battery displays slight capacity attenuation after 100 cycles, the voltage platform is maintained (Fig. S22b†). As mentioned above, the dehydrofluorination PVDF formats a rapid Li+ transport network and acts as a soft buffer layer for NCM. It avoids the interruption of Li+ transport paths caused by the volume change of NCM, ensuring the smoothness of Li+ transport. Compared to the original cathode, the morphology of the cycled NCM@PVDF cathode is well preserved (Fig. S23†). NCM@PVDF particles are clearly visible and their surface remains flat. This microstructure ensures the continuity of the Li+ transport network; therefore, the resistance of Li+ transport inside the cathode will not increase when the volume changes. As a result, the IR drops during cycling of the NCM battery significantly increases, while that of the NCM@PVDF battery almost remains at the same value (Fig. 4f and Table S3†). The rate capability has also been greatly improved (Fig. 4h). No matter how high the current density is, the specific capacity of the NCM@PVDF battery is much higher than that of the NCM battery. At the current density of 2 C, the specific capacity of the NCM battery is almost 0. From the corresponding charge–discharge profiles, the NCM battery clearly does not maintain the voltage platform at high current densities (Fig. 4h). However, the NCM@PVDF battery has fast charging and discharging owing to the efficient Li+ transport and the decreased resistance. The specific capacity reaches nearly 100 mA h g−1 at 2 C, and the voltage platform is obvious at each current density.
 | ||
| Fig. 4 (a) GITT plots and (b) Li+ diffusion coefficients of NCM and NCM@PVDF batteries. (c) Nyquist plots of NCM and NCM@PVDF batteries. (d) CV curves at a scan rate of 0.1 mV s−1. (e) Cycling performance at a current density of 1 C and (f) the corresponding IR drops. (g) Rate capability and (h, i) the corresponding GCD profiles. | ||
Similarly, PAN was also employed as the coating layer on NCM. The reaction mechanism was similar to that of PVDF (Fig. 5a). Li2CO3 can induce in situ dehydrocyanation of the PAN chain46,47 on the NCM surface, forming a soft coating layer for NCM particles to increase the contact area. The polymer layer provides buffering for NCM during the volume change process and the electron distribution of –CC– improves the Li+ transportation.46 There is a uniform coating layer on the surface of NCM particles (Fig. 5b). C, N, F, and S elements are contained in the coating layer (Fig. S24 and 25†). This indicates that PAN is successfully coated on the surface of NCM. The presence of the –CC– bond is proven by FTIR and Raman spectroscopy (Fig. 5c and d). In addition, the LiCN characteristic peak can be identified by further assaying the N 1s fine spectrum (Fig. 5e). This is due to the chemical reaction between HCN and Li2CO3. Compared with pristine NCM, there is no characteristic Li2CO3 peak in NCM@PAN (Fig. S26†), indicating that all the Li2CO3 which is adverse to Li+ transportation has been consumed by HCN. As a result, NCM@PAN displays an excellent cycling performance (Fig. 5f). The voltage platforms remain even after 100 cycles (Fig. 5g). Similar to the NCM@PVDF battery, the IR drop remains at almost the same value during cycling (Fig. 5h and Table S3†). The PAN coating layer facilitates unimpeded Li+ diffusion in the cathode; therefore, the battery exhibits a significantly improved rate capability (Fig. S27†). Thus, the dehydrocyanation PAN coating layer can also improve the contact between NCM particles and provide continuous Li+ transport in the cathode.
 | ||
| Fig. 5 (a) Schematic of the formation mechanism of the PAN coating layer. (b) TEM image of NCM@PAN. (c) FTIR and (d) Raman spectra of PAN, NCM, and NCM@PAN. (e) N 1s XPS spectrum of NCM@PAN. (f) Cycling performance and (g) the corresponding GCD profiles of NCM@PAN during different cycles. (h) IR drops at different cycle numbers. | ||
3. Conclusions
An in situ coating strategy is proposed to simultaneously address the issue of poor particle contact and limited Li+ transport in the cathode of the SSB. The PVDF was designed as the coating layer of NCM particles by the dehydrofluorination reaction of PVDF induced by the residual Li2CO3 on NCM surface. The in situ coating polymer layer increases the contact area between NCM particles, providing an abundant Li+ transmission path. In addition, the –CC– bond generated by the dehydrofluorination reaction of PVDF helps to adsorb Li+ and promotes its rapid transport. Besides, the product (HF) of the dehydrofluorination reaction consumes the residual Li2CO3 on the surface of NCM particles to generate LiF, which is also beneficial for Li+ transport. Profiting from the above advantages, a fast Li+ transport network is constructed in the cathode. As a result, the electrochemical performance of the SSB is greatly improved. Based on the above principle, PAN was employed as another coating layer through the dehydrocyanation reaction, and the electrochemical performance of NCM@PAN was comparable to that of NCM@PVDF. This in situ coating strategy provides a new approach for the construction of a high-performance SSB.
Author contributions
Meng Ye: conceptualization, visualization, writing-original draft; Zhian Zhang: data curation, investigation; Jianhua Chen: formal analysis, methodology; Qiuyue Chen: software; Jiarui Hu: data curation; Lang Qiu: formal analysis; Fang Wan: resources, validation, writing-review and editing; Xiaodong Guo: resources, project administration, writing-review and editing.Data availability
The data that support the findings of this work are available within the article and its corresponding ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by National Natural Science Foundation of China (22479099, 22425804), and Sichuan Science and Technology Program (2024NSFSC1161, 2024NSFTD0001).References
- S. C. Sand, J. L. M. Rupp and B. Yildiz, Chem. Soc. Rev., 2025, 54, 178–200 CAS
.
- J. Yu, Y. Wang, L. Shen, J. Liu, Z. Wang, S. Xu, H. M. Law and F. Ciucci, Adv. Mater., 2024, 2417796 Search PubMed
- J. Zhang, J. Fu, P. Lu, G. Hu, S. Xia, S. Zhang, Z. Wang, Z. Zhou, W. Yan, W. Xia, C. Wang and X. Sun, Adv. Mater., 2024, 2413499 Search PubMed
- S. Kalnaus, N. J. Dudney, A. S. Westover, E. Herbert and S. Hackney, Science, 2023, 381, eabg5998 CAS
- Y. S. Meng, V. Srinivasan and K. Xu, Science, 2022, 378, abq3750 Search PubMed
- X. Yang, K. R. Adair, X. Gao and X. Sun, Energy Environ. Sci., 2021, 14, 643–671 CAS
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 Search PubMed
- M. Botros and J. Janek, Science, 2022, 378, adf7524 CrossRef PubMed
- J. Sang, B. Tang, Y. Qiu, Y. Fang, K. Pan and Z. Zhou, Energy Environ. Mater., 2023, e12670 Search PubMed
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 CAS
- H. Xie, J. Feng and H. Zhao, Energy Storage Mater., 2023, 61, 102918 Search PubMed
- A. Banerjee, X. Wang, C. Fang, E. A. Wu and Y. S. Meng, Chem. Rev., 2020, 120, 6878–6933 CrossRef CAS PubMed
- O. Sheng, C. Jin, X. Ding, T. Liu, Y. Wan, Y. Liu, J. Nai, Y. Wang, C. Liu and X. Tao, Adv. Funct. Mater., 2021, 31, 2100891 CrossRef CAS
- M. K. Jangid, T. H. Cho, T. Ma, D. W. Liao, H. Kim, Y. Kim, M. Chi and N. P. Dasgupta, Nat. Commun., 2024, 15, 10233 CrossRef CAS PubMed
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2019, 5, 105–126 CrossRef
- W.-J. Kong, C.-Z. Zhao, L. Shen, S. Sun, X.-Y. Huang, P. Xu, Y. Lu, W.-Z. Huang, J.-L. Li, J.-Q. Huang and Q. Zhang, J. Am. Chem. Soc., 2024, 146, 28190–28200 Search PubMed
- J. Li, J. Yang, Z. Ji, M. Su, H. Li, Y. Wu, X. Su and Z. Zhang, Adv. Energy Mater., 2023, 13, 2301422 Search PubMed
- C. Wang, J. Wu, J. Hao, H. Shi, L. Yang, J. Wang, Z. Wang, X. Chen, J. Li, Y. Gao, X. Yan and Y. Gu, Adv. Funct. Mater., 2024, 2413176 Search PubMed
- T. Deng, L. Cao, X. He, A.-M. Li, D. Li, J. Xu, S. Liu, P. Bai, T. Jin, L. Ma, M. A. Schroeder, X. Fan and C. Wang, Chem, 2021, 7, 3052–3068 CAS
- J. Y. Liang, X. X. Zeng, X. D. Zhang, T. T. Zuo, M. Yan, Y. X. Yin, J. L. Shi, X. W. Wu, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 9165–9169 Search PubMed
- W. Li, J. A. Quirk, M. Li, W. Xia, L. M. Morgan, W. Yin, M. Zheng, L. C. Gallington, Y. Ren, N. Zhu, G. King, R. Feng, R. Li, J. A. Dawson, T. K. Sham and X. Sun, Adv. Mater., 2023, 36, 2302647 CrossRef PubMed
- L.-Z. Fan, H. He and C.-W. Nan, Nat. Rev. Mater., 2021, 6, 1003–1019 Search PubMed
- T. Jiang, P. He, G. Wang, Y. Shen, C. W. Nan and L. Z. Fan, Adv. Energy Mater., 2020, 10, 1903376 CAS
- Y. Xu, K. Wang, X. Zhang, Y. Ma, Q. Peng, Y. Gong, S. Yi, H. Guo, X. Zhang, X. Sun, H. Gao, S. Xin, Y. G. Guo and Y. Ma, Adv. Energy Mater., 2023, 13, 2204377 Search PubMed
- D. Cao, Q. Li, X. Sun, Y. Wang, X. Zhao, E. Cakmak, W. Liang, A. Anderson, S. Ozcan and H. Zhu, Adv. Mater., 2021, 33, 2105505 Search PubMed
- M. Balaish, J. C. Gonzalez-Rosillo, K. J. Kim, Y. Zhu, Z. D. Hood and J. L. M. Rupp, Nat. Energy, 2021, 6, 227–239 Search PubMed
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 Search PubMed
- H. Zhang, Z. Zeng, S. Cheng and J. Xie, eScience, 2024, 4, 100265 Search PubMed
- Z. Xue, F. Wu, M. Ge, X. Huang, Y. S. Chu, P. Pianetta and Y. Liu, eScience, 2024, 4, 100251 CrossRef
- Z. Gao, C. Zhao, K. Zhou, J. Wu, Y. Tian, X. Deng, L. Zhang, K. Lin, F. Kang, L. Peng, M. Wagemaker and B. Li, Nat. Commun., 2024, 15, 1503 CAS
- Z. Liu, Y. Zeng, J. Tan, H. Wang, Y. Zhu, X. Geng, P. Guttmann, X. Hou, Y. Yang, Y. Xu, P. Cloetens, D. Zhou, Y. Wei, J. Lu, J. Li, B. Liu, M. Winter, R. Kostecki, Y. Lin and X. He, Nat. Nanotechnol., 2024, 19, 1821–1830 CAS
- C.-J. Chen, W. K. Pang, T. Mori, V. K. Peterson, N. Sharma, P.-H. Lee, S.-H. Wu, C.-C. Wang, Y.-F. Song and R.-S. Liu, J. Am. Chem. Soc., 2016, 138, 8824–8833 CrossRef CAS PubMed
- Y. Liu, X. An, K. Yang, J. Ma, J. Mi, D. Zhang, X. Cheng, Y. Li, Y. Ma, M. Liu, F. Kang and Y.-B. He, Energy Environ. Sci., 2024, 17, 344–353 RSC
- C. Yang, Q. Wu, W. Xie, X. Zhang, A. Brozena, J. Zheng, M. N. Garaga, B. H. Ko, Y. Mao, S. He, Y. Gao, P. Wang, M. Tyagi, F. Jiao, R. Briber, P. Albertus, C. Wang, S. Greenbaum, Y. Y. Hu, A. Isogai, M. Winter, K. Xu, Y. Qi and L. Hu, Nature, 2021, 598, 590–596 CrossRef PubMed
- Y. Xiao, K. Turcheniuk, A. Narla, A. Y. Song, X. Ren, A. Magasinski, A. Jain, S. Huang, H. Lee and G. Yushin, Nat. Mater., 2021, 20, 984–990 CrossRef CAS PubMed
- H. Gao, L. Xue, S. Xin, K. Park and J. B. Goodenough, Angew. Chem., Int. Ed., 2017, 56, 5541–5545 CrossRef CAS PubMed
- Z. Cao, X. Yao, S. Park, K. Deng, C. Zhang, L. Chen and K. Fu, Sci. Adv., 2025, 11, eadr4292 CrossRef CAS PubMed
- W. Liu, C. Yi, L. Li, S. Liu, Q. Gui, D. Ba, Y. Li, D. Peng and J. Liu, Angew. Chem., Int. Ed., 2021, 60, 12931–12940 CrossRef CAS PubMed
- N. Meng, F. Lian, L. Wu, Y. Wang and J. Qiu, ACS Appl. Mater. Interfaces, 2024, 16, 41487–41494 CrossRef CAS PubMed
- H. Deng, F. He, T. Liu, M. Ye, F. Wan and X. Guo, Nanotechnology, 2024, 35, 195402 CrossRef CAS PubMed
- Z. Wu, Z. Jiang, S. Li, Z. Lu, M. Deng, C. Liu, C. Wei, Z. Peng, Z. Wang, L. Zhang and C. Yu, Nano Energy, 2025, 110840 CrossRef CAS
- L. Wang, J. Guo, Q. Qi, X. Li, Y. Ge, H. Li, Y. Chao, J. Du and X. Cui, Nano-Micro Lett., 2025, 17, 111 CrossRef CAS PubMed
- X. Zhang, T. Liu, S. Zhang, X. Huang, B. Xu, Y. Lin, B. Xu, L. Li, C.-W. Nan and Y. Shen, J. Am. Chem. Soc., 2017, 139, 13779–13785 CrossRef CAS PubMed
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS
- L. Zhao, X. Yu, J. Jiao, X. Song, X. Cheng, M. Liu, L.-L. Wang, J. Zheng, W. Lv, G. Zhong, Y.-B. He and F. Kang, Cell Rep. Phys. Sci., 2023, 4, 101382 CrossRef CAS
- W. P. Chen, H. Duan, J. L. Shi, Y. Qian, J. Wan, X. D. Zhang, H. Sheng, B. Guan, R. Wen, Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2021, 143, 5717–5726 CrossRef CAS PubMed
- Y. N. Yang, F. L. Jiang, Y. Q. Li, Z. X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24162–24170 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00031a |
| This journal is © The Royal Society of Chemistry 2025 |