
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interphase design from ionic liquid cation mixtures and multi-mode surface analysis for safe and stable Na metal batteries†
Lixu
Huang
 a,
Mahin
Maleki
a,
Manuel
Salado
abe,
José M.
Porro
be,
Viktor
Petrenko
be,
Anton
Le Brun
f,
Kilian Shani
Fraysse
a,
Hua
Li
c,
Xuedong
Zhang
d,
Rob
Atkin
c,
Jianyu
Huang
d,
Fangfang
Chen
a,
Faezeh
Makhlooghiazad
a,
Patrick C.
Howlett
a and
Maria
Forsyth
*a
a,
Mahin
Maleki
a,
Manuel
Salado
abe,
José M.
Porro
be,
Viktor
Petrenko
be,
Anton
Le Brun
f,
Kilian Shani
Fraysse
a,
Hua
Li
c,
Xuedong
Zhang
d,
Rob
Atkin
c,
Jianyu
Huang
d,
Fangfang
Chen
a,
Faezeh
Makhlooghiazad
a,
Patrick C.
Howlett
a and
Maria
Forsyth
*a
aInstitute for Frontier Materials (IFM), Deakin University, Burwood, Victoria 3125, Australia. E-mail: maria.forsyth@deakin.edu.au
bBCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain
cSchool of Molecular Sciences, University of Western Australia, Crawley, Western Australia, Australia
dClean Nano Energy Center, State Key Laboratory of Metastable Materials Science and Technology, Yanshan University, Qinhuangdao 066004, P. R. China
eIKERBASQUE, Basque Foundation for Science, 48009 Bilbao, Spain
fAustralian Centre for Neutron Scattering, Australian Nuclear Science and Technology Organisation (ANSTO), Lucas Heights, NSW 2234, Australia
First published on 14th May 2025
Abstract
Enhancing the cycling performance of sodium metal batteries requires deliberate electrolyte design to optimize interfacial structuring and sodium deposition. This study explores novel ionic liquid (IL) electrolyte mixtures containing 20 mol% phosphonium cations ((tributylmethyl phosphonium) P1444+ and (trimethyl isobutyl phosphonium) P111i4+) as co-solvent additives in a base (N-propyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide) C3mpyrFSI electrolyte. The effects of these mixtures on electrochemical performance, physicochemical properties, interfacial nano-structuring, and sodium deposition morphology are investigated using a multifaceted approach, including cell cycling, cyclic voltammetry (CV), differential scanning calorimetry (DSC), ionic conductivity measurements, and in situ techniques such as neutron reflectometry (NR), atomic force microscopy (AFM), and optical microscopy (OM). The addition of the larger P1444+ cation, while decreasing ionic conductivity, surprisingly exhibits reduced polarisation overpotential and significantly extended lifespan and improved solid electrolyte interphase (SEI) formation kinetics. Our results, supported by NR, AFM, and in situ optical studies, reveal that incorporating P1444+ as a co-solvent disrupts interfacial nano-structuring. Under applied negative potentials, this disruption increases the presence of Na+ cations at the interface and their coordinated FSI− anions, leading to enhanced SEI formation kinetics as evidenced by CV results. This facilitates improved cycling stability and more uniform sodium deposition morphology. This work highlights the potential of mixed-cation ILs in achieving long-term performance and stability in sodium metal batteries. By shedding light on the poorly understood mechanisms underlying SEI-related performance improvements, it provides new strategies for optimizing interfacial structuring and electrolyte design.
Broader contextThe pursuit of high-performance sodium metal batteries requires the development of advanced electrolyte formulations capable of addressing critical challenges related to interfacial stability, cycling efficiency, and sodium deposition morphology. While lithium-ion batteries currently dominate the market, the abundance and cost-effectiveness of sodium make it a promising alternative for large-scale energy storage applications. This study introduces a novel approach through the use of mixed-cation ionic liquid (IL) in highly concentrated NaFSI electrolytes to control the interfacial chemistry which in turn controls the solid electrolyte interphase (SEI) and electrode cycling. By integrating cutting-edge in situ characterization techniques such as neutron reflectometry (NR), optical microscopy (OM), and atomic force microscopy (AFM), this work reveals how these mixed-cation electrolytes modulate interfacial nano-structuring, enhance SEI formation kinetics, and promote more uniform sodium deposition. The findings demonstrate that the incorporation of larger phosphonium cations reduces polarization overvoltage and significantly improves cycling stability. These insights provide a deeper understanding of the SEI formation process and interfacial phenomena, offering practical strategies for the design of next-generation electrolytes with optimized performance. By addressing key challenges in sodium battery technology, this research contributes to the broader goal of sustainable and scalable energy storage solutions, ultimately supporting the transition to more affordable and environmentally friendly energy systems. |
Introduction
In recent years, batteries have been actively developed as a critical technology to address the intensifying greenhouse effects and climate change. Increasing attention has been directed towards sodium batteries, recognized for their low cost and inherent stability.1 Sodium (Na) metal anodes are widely regarded as highly promising candidates for next-generation, high-energy sodium-based batteries due to their exceptional theoretical specific capacity (1166 mA h g−1) and low standard electrode potential.2 In addition to the electrode, the selection of the electrolyte is crucial for the development of the sodium batteries.Ionic liquids (ILs), composed of large organic cations and charge-delocalized anions, offer unique properties such as negligible vapor pressure, low flammability, and high thermal stability, making them a promising alternative to conventional electrolytes.3 Additionally, the modular design of ILs enables the tuning of their physicochemical and electrochemical properties by altering cation–anion combinations or mixing different ILs.
Among ILs, systems based on imidazolium and pyrrolidinium cations are the most extensively studied.4 Modifications to cation structures, such as the introduction of asymmetric alkyl chains, allow for adjustments in melting point, viscosity, and ionic conductivity. Non-aromatic pyrrolidinium cations, like C3mpyr+, exhibit higher reductive stability than imidazolium-based counterparts, making them particularly suitable for battery applications.5 Phosphonium-based ILs, such as P111i4FSI, offer additional advantages over nitrogen-based ILs, including higher ionic conductivity and enhanced electrochemical stability.6 Previous studies revealed that mixtures of phosphonium ILs with high NaFSI concentrations achieve superior performance in sodium batteries, with stable Na stripping/plating and SEI properties. Comparative studies also showed that phosphonium ILs provide better cycling stability and capacity than pyrrolidinium ILs, particularly when water is added to improve SEI characteristics.7
It is recognised that the SEI is a controlling factor in the electrochemical performance of Li and Na batteries. Moreover, the structuring of the electrolyte at the electrode interface (electrical double layer, EDL) has also been shown to be important in determining the electrochemical behaviour.8 Several methods have been used to study electrolyte structuring at interfaces including atomic force microscopy (AFM),9,10 Raman spectroscopy,11,12 molecular dynamics (MD) simulations,9,13,14 neutron reflectometry (NR)15 and differential capacitance (DC).16 In combination, these techniques provide a powerful approach to studying the interface and correlating to cell performance.
In addition to using chemical modification of the ionic species to control IL properties, mixtures of cations and/or anions can also be used as cost-effective methods to design new electrolyte systems. For example, early work by Lopes et al.17 and Annat et al.18 studied mixtures of ILs where the anion was kept constant while the cations varied in chemistry and composition. These studies showed that such mixing is a viable method to control physicochemical properties such as conductivity and ion diffusion in IL mixtures. In the area of electrolytes for lithium batteries, mixtures of anions have been shown to have a major impact on both physical properties, including conductivity and Li diffusion, and electrochemistry.19–22 In the case of Na batteries, it was demonstrated that combining different fluorosulfonamide anions (FSI, TFSI and FTFSI) with dicyanamide led to significant differences in sodium metal cycling. These differences were attributed to differences in ion speciation within these mixtures in addition to changes in the interfacial structuring which impacted SEI formation.
Additives, including FEC,23,24 DME25etc.26,27 (or co-solvents) have also been shown to influence physicochemical and electrochemical behaviour of IL electrolytes for both lithium and sodium batteries. However, little work has been reported on the use of different cations as co-solvents in ionic liquid-based electrolytes.
The IL electrolyte, super concentrated C3mpyrFSI with 50 mol% NaFSI, has been well-studied and shown to support Na metal and Na/NFP battery cycling at current densities up to 1 mA cm−2 for 1 mA h cm−2. However, this was a significantly lower performance than the equivalent P111i4FSI super concentrated electrolyte (42 mol% NaFSI due to lower solubility).7 The reason for this difference was not clear in our previous work, however, there was a hint that the SEI layer composition was modified in the presence of the P111i4+ cation.6 Prior work also indicated that the near-electrode structuring of the IL electrolyte also affected the SEI and subsequent electrochemical cycling.28,29 Begić et al. demonstrated that the chemical structure of the IL cation in DCA-based ILs also affected the electrolyte structuring and the subsequent Zn electrochemistry.30 It, therefore, seems logical that mixing of cations could modify the near-electrode ion structuring and the subsequent electrochemistry.
This paper will demonstrate the potential for adding IL co-solvent to control bulk physical properties, interfacial structuring and electrochemical behaviour for sodium battery electrochemistry. To investigate the potential for making a significant improvement to Na battery cycling, we consider the influence of several IL cations (see Fig. S1†) to the base C3mpyrFSI electrolyte at a fixed concentration of 20 mol% in ILs (based our previous work with P111i4+ cation, which showed a slight enhancement).6,31 We compare the previously studied small phosphonium with a larger IL cation, P1444+ as well as the influence of adding an ether oxygen to the pyrrolidinium, which has been suggested to improve Li cycling.32,33 A further reason for using 20 mol% co-solvent/additive is that for this larger phosphonium cation, the NaFSI solubility significantly decreases due to the increase in hydrophobicity. We will discuss a multifaceted study of these novel IL systems including NR, AFM and in situ optical cell to compare the influence of adding different IL additives/co-solvent to the base C3mpyrFSI electrolyte keeping the NaFSI content constant at 42 mol%. A significant enhancement in cell cycling stability was observed with the addition of a larger tributyl, methyl phosphonium P1444+ cation to the base IL which correlated with changes in deposition morphology and interfacial structuring near the electrode surface.
Results and discussion
Fig. 1 presents the cycling profiles of all electrolyte mixtures where P111i4+, P1444+ and C2O1mpyr+ IL cations are incorporated into the base C3mpyrFSI IL electrolyte. The four samples were evaluated in sodium symmetric cells under constant current conditions of 1 mA cm−2/1 mA h cm−2 over extended cycles at 50 °C to compare their electrochemical performance. The electrochemical performance of the mixtures demonstrated significant variations in polarisation overpotential and cycling lifespan. Pure C3mpyrFSI exhibited short-circuiting after 96 cycles, while the addition of C2o1mpyrFSI led to higher overpotentials and rapid shorting. Incorporating P111i4FSI improved the electrochemical performance by doubling the cycling lifespan. Surprisingly, the inclusion of the larger cation, P1444+ led to a remarkable enhancement, extending cycling stability, by more than fivefold achieving at least 552 cycles without any evidence of shorting. | ||
| Fig. 1 Cycling profile for Na/Na coin cells of the mixtures. | ||
The initial cycling profiles further highlighted these trends: the C3mpyrFSI/P1444FSI mixture exhibited the lowest overpotential, followed by pure C3mpyrFSI and the C3mpyrFSI/P111i4FSI mixture, which showed similar overpotentials, while the C3mpyrFSI/C2o1mpyrFSI mixture showed the highest overpotential. These differences remained consistent beyond 10 cycles. The cycling profiles were characterized by pronounced voltage spikes in C3mpyrFSI/C2o1mpyrFSI mixture at the beginning and end of each cycle, leading to a curved profile. The observed “peaking” behaviour during initial cell polarization can be attributed to spatially varying surface kinetics at the electrode/electrolyte interphase, where mass-transport limitations are minimal.34,35 Notably, this “peaking” behaviour was less pronounced in the C3mpyrFSI/P1444FSI curve. In contrast, the C3mpyrFSI/C201mpyrFSI sample exhibited two distinct plateaus during Na stripping, indicating a two-step dissolution process. As reported in previous studies,36 such behaviour occurs when Na initially dissolves from surface dendrites, which present lower interfacial impedance. After these dendrites are depleted, stripping proceeds from the bulk, where the higher interfacial resistance leads to a second plateau at elevated potential. This transition in dissolution pathway gives rise to the characteristic “double step” feature observed in the polarization curve.
To further investigate the influence of IL co-solvent on electrochemical behaviour, cyclic voltammetry measurements were performed in a two electrode Cu/Na cell. The CV results, shown in Fig. 2, indicate that the onset potentials for sodium deposition in C3mpyrFSI/P1444FSI, C3mpyrFSI/P111i4FSI, and C3mpyrFSI electrolytes are −0.16 V, −0.21 V, and −0.25 V, respectively. These results suggest that sodium deposition occurs at a less negative potential in the C3mpyrFSI/P1444FSI electrolyte, reflecting enhanced electrochemical performance. Additionally, focussing on the peaks prior to Na deposition (Fig. 2b), the CV curves reveal that the cathodic peaks of the C3mpyrFSI/P1444FSI mixture both appear at different potentials compared to the other two samples and are larger. As discussed by Begić et al.37 for Zn electrochemistry, the onset potential likely reflects a different electrode/electrolyte interface and/or may be attributed to differences in SEI composition. Previous experimental and computational studies suggest that in ionic liquid systems, the anion-originated reactions are responsible for the formation of the SEI layer which itself controls metal (Li/Na) deposition.14 To further elucidate the electrochemical reactions occurring in the SEI formation region of the electrochemical window, three-electrode CVs were conducted on a gold electrode, as depicted in Fig. 3.
 | ||
| Fig. 2 (a) Cyclic voltammetry curves for the mixtures with 42 mol% NaFSI system at 1st cycle. (b) The enlarged reduction process. | ||
 | ||
| Fig. 3 Three-electrode cyclic voltammetry curves for mixed ILs. (a) C3mpyrFSI/P111i4FSI mixture, (b) C3mpyrFSI/P1444FSI mixture, (c) enlarged view of the first and second cycles prior to the onset of Na deposition. | ||
As shown in Fig. 3a and b, the electrolyte containing P1444FSI (b) demonstrates greater stability and reversibility over multiple cycles compared to the electrolyte with the P111i4FSI cosolvent (a), which exhibits reduced activity after the first cycle. During the first cycle, the reduction current density in the P111i4FSI-containing electrolyte reaches approximately −8 mA cm−2, which is higher than the current density observed in the P1444FSI-containing electrolyte (∼−6 mA cm−2). However, in subsequent cycles (cycles 2–6), the reduction and oxidation current densities exhibit a greater increase and more stable behaviour in the P1444FSI-containing electrolyte, likely due to the formation of a more stable interphase.
A closer examination of the anion-derived reaction region during the first and second cycles in both systems (Fig. 3c) reveals multiple irreversible reactions occurring in the first cycle. These reactions can be attributed to electrolyte degradation and interphase formation.
Previous work has identified these reduction peaks as being associated with the electrolyte decomposition, in particular, the anion (FSI or TFSI).38 However, identifying the origin of these peaks is challenging due to the complexity of the anion coordination environment in the mixed cation system, which is affected by multiple factors, such as salt concentration, the IL cation chemistry and the electrode voltage. The different cation–anion interaction and anion coordination structures can influence the HOMO–LUMO energy levels.14,39
Interestingly, the peaks observed in the first cycle differ between the two systems in terms of current densities and potential ranges. The P1444FSI-containing electrolyte exhibits overall higher current densities in this system, suggesting more favourable kinetics for this electrochemical process which we attribute to the SEI formation step. Considering the lower overpotential and improved cycling in the Na/Na symmetric cell data shown in Fig. 1, we suggest that a less resistive and more homogeneous SEI is able to form in this mixed cation system.
To determine whether the bulk physicochemical properties also have a positive effect on the low overpotential and long lifespan of the C3mpyrFSI/P1444FSI mixture, we compared the glass transition temperature, Tg, and ionic conductivity for the base C3mpyrFSI/42 mol% NaFSI with the mixtures containing either P111i4+ or P1444+ cations (Fig. 4).
 | ||
| Fig. 4 (a) Glass transition temperature (b) temperature-dependent ionic conductivity. | ||
After mixing with 42 mol% NaFSI, only the glass transition temperatures (Tg) were detected, as shown in Fig. S5† and Fig. 4a. The Tg of C3mpyrFSI was the highest, but the differences among the three samples were less than 2 °C, suggesting a relatively minor impact from the mixtures. Incorporating P111i4FSI into the C3mpyrFSI/42 mol% NaFSI system resulted in a slight increase in ionic conductivity, as shown in Fig. 4b.
Addition of P1444+ reduced the ionic conductivity as would be expected due to its larger molecular structure. The ionic conductivity of the C3mpyrFSI/P1444FSI mixture was the lowest of the three systems, reflecting higher resistance within the bulk electrolyte. Moreover, to have a deeper understanding of the electrolyte resistance in the coin cell setup compared to the flooded electrolyte cell can also be considered, EIS measurements of the cells before cycling and after the first cycle shown in Fig. S2 and Table S1† presents EIS fitting results after first cycle.
The C3mpyrFSI/P1444FSI mixture shows the lowest overall resistance among the samples, which corresponds to its minimal overpotential observed in the cycling profile. Although the ionic conductivity of the C3mpyrFSI/P1444FSI mixture was the lowest, reflecting higher resistance within the bulk electrolyte, its fitted bulk resistance in Table S1† was lower. This finding suggests that the addition of P1444FSI reduces resistance associated with ion transport through the separator and across the sodium metal electrode interface.
As shown in Fig. S2,† the wettability of C3mpyrFSI/P1444FSI with the Solupor separator was significantly better than that of C3mpyrFSI/P111i4FSI and C3mpyrFSI. When 60 μL of electrolyte was dropped onto the separator, the C3mpyrFSI/P1444FSI mixture penetrated the separator within 5 minutes, spreading into a flat droplet. In contrast, C3mpyrFSI/P111i4FSI and C3mpyrFSI formed rounded droplets and did not readily penetrate the separator. This improved wettability of the C3mpyrFSI/P1444FSI mixture effectively enhanced electrolyte permeation into the Solupor separator, correlating with the observed reduction in resistance for ion transport through the separator.40
Thus far we have observed that the addition of the large P1444+ IL cation has a significant positive influence on the Na metal electrochemistry despite its lower overall ionic conductivity. The CV data shows dramatic differences between the two phosphonium mixtures with a large irreversible cathodic reaction at approximately −1.65 V vs. Ag/AgCl present in the P1444+ system not evident for the P111i4+. This cathodic peak originates from the chemical species at the electrode interface and leads to the formation of a favourable SEI. In the following section we probe the interfacial structuring in various mixtures on model electrodes (Au) and correlate this with the observed electrochemical behaviour.
Interphase ion layering
In situ NR experiments were conducted under different voltage applications (0 V and −0.7 V) to investigate the interface between the solid electrode and the described electrolytes. The measured neutron reflectivity signal, R, versus scattering wave vector (q) curves for various electrolyte-electrode interfaces under different voltage applications are presented in Fig. 5, along with the corresponding fitted results and Table 1 presents the scattering length density (SLD) value (estimated for each component in studied materials (see Table S2†)) for each system at open circuit potential (OCP) and −0.7 V. Notably, the SLD of the C3mpyrFSI/P1444FSI mixture is negative at both potentials whereas for base C3mpyrFSI system and the C3mpyrFSI/P111i4FSI mixture the values are positive with the latter being significantly lower than that of the base sample. The thickness values extracted from the raw data and presented in Table 1, indicate that, at OCP the values are similar within the uncertainty range, however, upon applying a negative potential the C3mpyrFSI/P1444FSI mixture doesn't appear to change significantly whereas the thickness of the other two systems increases. These differences in behaviour have important implications for the chemistry at the interface observed when scanning to negative potentials in the CV data described earlier.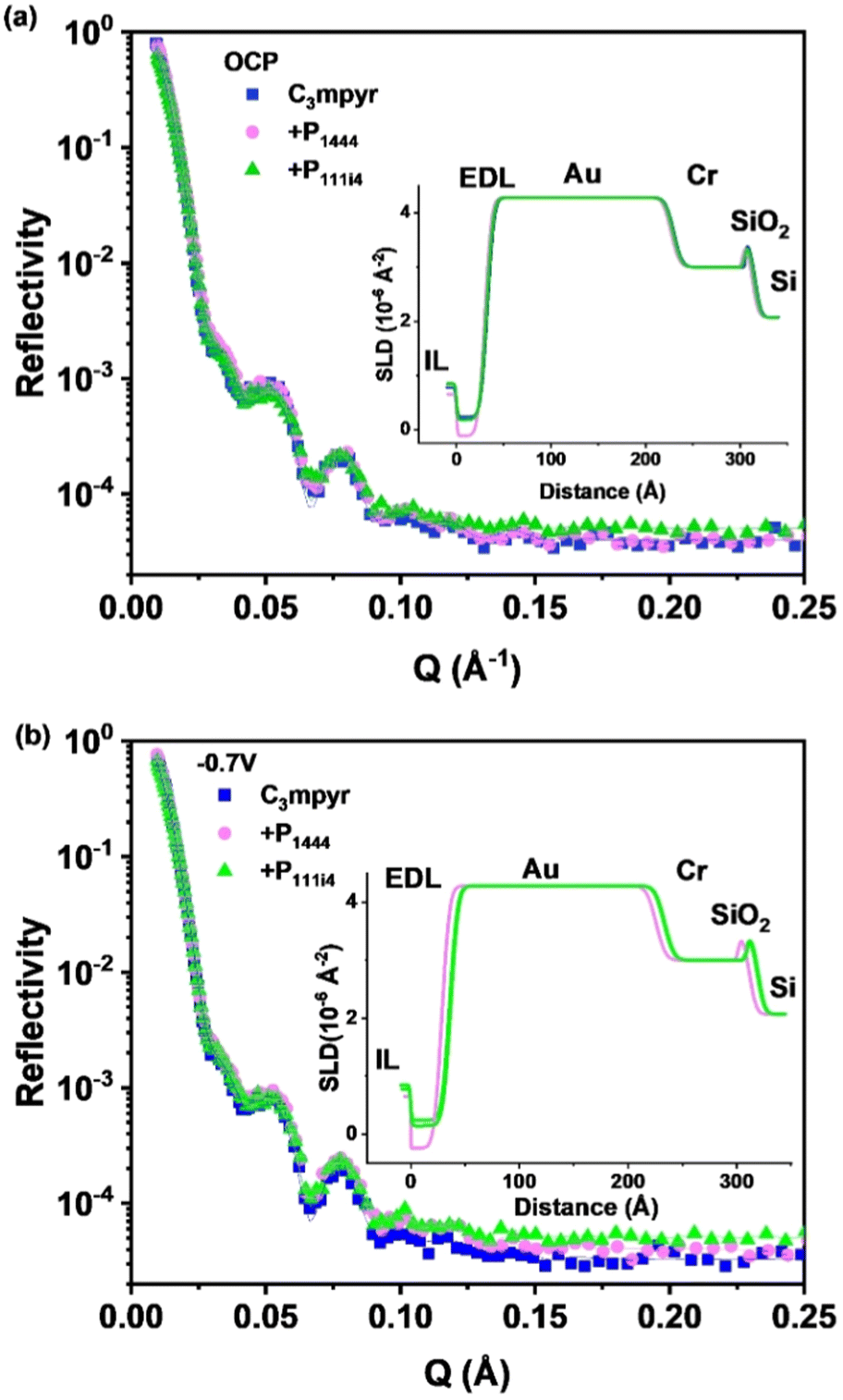 | ||
| Fig. 5 Neutron reflectometry curves for electrolytes/electrode interfaces for (a) 0 V; (b) −0.7 V vs. OCP. Solid lines are fitting curves with four-slab model with varied parameters only for EDL. Insets are SLD profiles. | ||
| Potential (vs. OCP) | Samples (with 42 mol% NaFSI) | SLD (10−6 Å−2) | Thickness (Å) |
|---|---|---|---|
| 0 V | 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 C3mpyrFSI:P1444FSI :20 C3mpyrFSI:P1444FSI |
−0.12 ± 0.09 | 30 ± 2 |
| 80:20 C3mpyrFSI:P111i4FSI |
0.18 ± 0.07 | 32 ± 2 | |
| C3mpyrFSI | 0.23 ± 0.06 | 33 ± 2 | |
| −0.7 V | 80:20 C3mpyrFSI:P1444FSI |
−0.24 ± 0.09 | 29 ± 2 |
| 80:20 C3mpyrFSI:P111i4FSI |
0.15 ± 0.05 | 36 ± 2 | |
| C3mpyrFSI | 0.24 ± 0.05 | 36 ± 2 |
For the C3mpyrFSI/P1444FSI mixture, the SLD decreases under an applied negative potential. Considering that the SLD of the cations is negative and that of the anions is positive (as shown in Table S1†), this indicates that more cations are attracted to the electrode. However, the thickness remains nearly unchanged, which could be attributed to a larger number of Na+ ions being attracted compared to P1444+, as Na+ is significantly smaller than the other cations. For the C3mpyrFSI/P111i4FSI mixture, there is almost no change (3% decrease) in SLD between OCP and −0.7 V, while EDL thickness increases. In contrast, for pure C3mpyrFSI, a 10% increase in both SLD and EDL thickness is observed under a negative potential, likely due to the attraction of cations to the electrode. At the same potential, the SLD and EDL thickness of the C3mpyrFSI/P1444FSI mixture are much smaller than those of the other samples. This observation suggests that more Na+ ions and fewer P1444+ and C3mpyr+ ions are attracted to the electrode, which may contribute to the reduced charge transfer resistance. Furthermore, the increase in Na+ most likely also increases the fraction of coordinated FSI− anions (in NaxFSIy clusters) at the interface. These coordinated FSI− species have been shown to more readily undergo reduction to form a favorable SEI.14
AFM measurements were performed to examine the interfacial nanostructure adjacent to an Au(111) electrode at OCP and OCP −0.7 V, focusing on the effect of adding different phosphonium-based cations to the base electrolyte of C3mpyrFSI + 42 mol% NaFSI (Fig. 6). Previous studies by Rakov et al.41 and Fraysse et al.16 have investigated the interfacial structure of the C3mpyrFSI system at various salt concentrations and the P111i4FSI + 42 mol% NaFSI electrolyte. Their findings reveal that increasing the salt concentration or using a phosphonium-based cation results in a less structured or unstructured EDL. Additionally, they report that the P111i4FSI + 42 mol% NaFSI electrolyte shows a thinner innermost layer at OCP +0.5 V compared to the C3mpyrFSI system which exhibits an innermost layer with a thickness of 0.55 nm, comparable to the diameter of the FSI anion (0.53 nm), suggesting the presence of small P111i4+ cations along with the FSI− anion in the innermost layer in P111i4FSI + 42 mol% NaFSI system.16
 | ||
| Fig. 6 Interfacial layering structure of mixed ILs through AFM force–distance two-dimensional histogram with probability colour bar on the right-hand. (a and b) at OCP; (c and d) at OCP-0.7V for C3mpyrFSI/P111i4FSI mixture and C3mpyrFSI/P1444FSI mixture. | ||
In our study, as shown in Fig. 6, the EDL overall does not show significant ordering in either system. At OCP, the C3mpyrFSI/P1444FSI mixture with 42mol% NaFSI maintains a similar interfacial layering structure to C3mpyrFSI + 50mol% NaFSI at OCP as reported by Rakov et al.,41 exhibiting three distinct layers with similar thicknesses. However, the push-through force of the innermost layer decreases significantly. In contrast, the C3mpyrFSI/P111i4FSI mixture with 42mol% NaFSI demonstrates a considerably reduced interfacial nanostructure, featuring only a single, weaker layer. This layer, with a thickness of ∼0.7 nm, is thicker than those observed in both pure C3mpyrFSI and the C3mpyrFSI/P1444FSI mixture (∼0.5 nm), aligning with the neutron reflectometry (NR) data presented in Table 1. The enhanced interfacial structure observed in the C3mpyrFSI/P1444FSI mixture compared to the C3mpyrFSI/P111i4FSI mixture suggests minimal participation of the larger P1444 cation in the innermost layer, thereby preserving the interfacial structure integrity.
Applying a negative potential (OCP −0.7 V) induces distinct changes in the interfacial structuring, particularly evident in both the apparent separation distance and rupture forces of the innermost ionic layers, Fig. 6c and d. Although both systems show an increase in the innermost layer thickness, they demonstrate opposing trends in the forces required to rupture these layers – decreasing for C3mpyrFSI/P1444FSI but increasing for C3mpyrFSI/P111i4FSI. These contrasting behaviours suggest different interfacial nanostructures. The observed changes for the C3mpyrFSI/P1444FSI system align well with our NR measurements, which reveal a decrease in SLD values under negative polarization. This decrease in SLD can be attributed to enhanced Na+ participation at the interface, leading to disruption of the original IL nanostructure. The larger P1444+ cation appears to facilitate this process by allowing more efficient Na+ access to the electrode surface, as evidenced by both AFM and NR data. However, for the C3mpyrFSI/P111i4FSI system at the negative potential of OCP −0.7 V, the smaller P111i4+ cation, compared to P1444+, enables more efficient packing with FSI− anions and better coordination with Na+ ions at the interface. The compact size of P111i4+ allows it to effectively compete with Na+ ions for interfacial positions while maintaining structural integrity, resulting in a more ordered interfacial layer that requires greater force to disrupt. In contrast, the larger P1444+ cation disrupts this ordered structure, leading to decreased rupture forces and facilitating Na+ access to the electrode surface.
Apart from force–distance curves, surface imaging of the gold electrode upon applying a negative potential of OCP −2.3 V was captured, as shown in Fig. 7a–c for the C3mpyrFSI/P1444FSI mixture, the C3mpyrFSI/P111i4FSI mixture, and for P111i4FSI + 42 mol% NaFSI, respectively. As mentioned earlier, previous studies indicate that neat P111i4FSI + 42 mol% NaFSI demonstrates significantly better performance than neat C3mpyrFSI + 50 mol% NaFSI.42
 | ||
| Fig. 7 Surface morphology of the formed solid interphase using ILs through AFM surface imaging at OCP −2.3 V for (a) C3mpyrFSI/P1444FSI mixture, (b) C3mpyrFSI/P111i4FSI mixture and (c) P111i4FSI + 42 mol% NaFSI. | ||
In the mixed systems, as shown in Fig. 7a and b, the C3mpyrFSI/P1444FSI electrolyte forms a morphology more similar to P111i4FSI + 42 mol% NaFSI, Fig. 7c, at the same potential (OCP −2.3 V), compared to the C3mpyrFSI/P111i4FSI electrolyte.
The similarities between the interface surface morphologies of these phosphonium systems suggests that these globular morphologies may be correlated with better cycling performance, through their contribution to the SEI formation process and/or in enabling better Na+ ion flux to the electrode surface.
Surface morphology
Changes in the electrode surface were recorded in the attached videos, and the corresponding cycling profiles of the open cell were analysed, as shown in Fig. 8. These observations provide insight into the impact of different electrolyte compositions on the morphology and behaviour of the sodium electrode during cycling. | ||
| Fig. 8 In situ optical micrographs obtained for flooded optical cell cycled at 0.5 mA cm−2. (a) Cycling profile and morphology are shown at pristine; (b) after 10 min (Na deposition); (c) after first half-cycle; (d) after first cycle. | ||
The flooded optical cell was cycled at two conditions: 0.5 mA cm−2/1 mA h cm−2 for the first cycle and 1 mA cm−2/1 mA h cm−2 for the second. Initially, the C3mpyrFSI/P1444FSI mixture exhibited the lowest polarisation overpotential, while the C3mpyrFSI/P111i4FSI mixture and the base C3mpyrFSI system showed similar overpotentials. This trend in overpotential remained consistent throughout cycling, aligning with the cycling profile in Fig. 1 using the sodium symmetric coin cell setup. Additionally, the “peaking” behaviour was less significant for the C3mpyrFSI/P1444FSI mixture in the flooded optical cell. When −1 mA cm−2 current density was applied, the polarisation of the C3mpyrFSI/P111i4FSI mixture and the base C3mpyrFSI system cells increased rapidly. In contrast, the C3mpyrFSI/P1444FSI mixture maintained stable and low overpotentials, demonstrating enhanced stability and performance under higher current densities.
The addition of C3mpyrFSI/P111i4FSI and C3mpyrFSI resulted in the formation of numerous bubbles on the pristine electrode surface. These bubbles are either reaction of residual water in the electrolyte, or entrapped gases from the electrolyte drops which escape with time. Upon applying a deposition potential, these microbubbles coalesce and eventually disappear. In contrast, the addition of C3mpyrFSI/P1444FSI introduced fewer bubbles, as evident in the videos. This difference could be attributed to less water absorption due to more hydrophobic character of P1444 IL cation and/or better immediate spreading of the electrolyte on the metal surface, resulting from improved contact between C3mpyrFSI/P1444FSI and the sodium electrode; improved contact is thought to promote more homogeneous sodium deposition.43 Supporting this observation, Fig. 8c, d, and the SEM images in Fig. S3† illustrate the uniform deposition and less aggregated pattern achieved with C3mpyrFSI/P1444FSI which is consistent with the AFM images (Fig. 7) showing more evenly distributed interphase morphology using this electrolyte.
Fig. 8b demonstrates that after applying a current density of 0.5 mA cm−2 for 10 minutes, the sodium metal particles formed in the presence of C3mpyrFSI/P1444FSI are smaller compared to those formed with other electrolytes. Over time, as shown in Fig. 8c and d, these deposits grow progressively smoother, leading to a thinner and less porous sodium deposition layer.35,44,45 This improved morphology is consistent with the electrochemical behaviour in Fig. 1 and the CV data in Fig. 2 and 3 described above, even though in this case a flooded optical cell was used, without any pressure controlling the surface morphology. We can surmise that the smoother, more uniform particle deposition obtained when using C3mpyrFSI/P1444FSI relates to surface modification in the presence of the P1444+ cation and the preferential availability of Na+ ions as suggested by the NR and AFM results.
Discussion and conclusions
In recent years, studies employing ionic liquid (IL) electrolytes have demonstrated that despite having similar bulk compositions and properties, ILs can exhibit significantly different cycling performances in batteries. These disparities are often attributed to differences in the SEI they form. However, the underlying mechanisms governing these differences at the nanoscale level on electrode surfaces remain poorly understood.It has also been observed that super-concentrated electrolytes can disrupt the electrode interface by increasing the aggregation of NaxFSIy clusters at the interface. Since increasing the salt concentration requires ILs with high salt solubility, this study explores novel mixtures of ILs with a fixed salt concentration. By incorporating different cations as co-solvents in an IL, we demonstrate that this approach disrupts the interfacial structuring of the electrolytes at the electrode interface without significantly altering bulk physicochemical properties, such as ionic conductivity. Notably, incorporating the larger phosphonium cation, P1444+, results in significant improvements in cycling performance, as confirmed by our coin cell and optical cell cycling results.
Cyclic voltammetry (CV) measurements reveal that the addition of the P1444+ cation to the base electrolyte, C3mpyrFSI with 42 mol% NaFSI, promotes more favourable kinetics for irreversible reactions involved in SEI formation. NR and AFM studies further confirm that the introduction of different cations induces a disruption in the innermost interfacial nanostructuring of the electrolyte, akin to the effects observed with increased salt concentration in the literature.14 These findings suggest that the improved performance of the P1444FSI/C3mpyrFSI mixture arises from increased number of Na+ cation at the interface and their coordinated FSI−, effectively excluding IL cations from this region.
Surface imaging of the SEI formed on a model gold electrode using AFM, as well as Na deposition morphology studies in the flooded Na|Na optical cells, reveal that the addition of P1444+ cations leads to a more homogeneous and less aggregated SEI and Na deposition morphology.
Overall, this study employs a multifaceted, multiscale approach to highlight the potential of using ILs as co-solvents to modify the interfacial nanostructuring of electrolytes. This strategy improves SEI formation and cycling performance without the need to increase salt concentration, providing a promising pathway for advancing next-generation energy storage technologies by improving the performance and properties like density and reducing the cost.
Methods
Sample preparation
P111i4FSI and C3mpyrFSI were supplied by the Boron Molecular company with 99.9% purity. P1444FSI was purchased from IoLiTec with 99% purity, sodium bis(fluorosulfonyl)imide (NaFSI, 99.9%) and N-methyl-N-methoxymethylpyrrolidinium bis(fluorosulfonyl)imide (C2o1mpyrFSI) were purchased from Solvionic. The structures of the cations and anion are presented in Fig. S1.† To minimize water contamination, the three ILs were dried under vacuum at 50 °C for 48 hours, reducing water content to less than 50 ppm, as confirmed by Karl Fischer titration (831 Karl Fischer Coulometer with Hydranal® Coulomat AG as the titrant). C3mpyrFSI was mixed with P111i4FSI, P1444FSI, or C2o1mpyrFSI in an 80:20 molar ratio, with a fixed NaFSI concentration of 42 mol%. Pure C3mpyrFSI was also mixed with 42 mol% NaFSI for comparison purposes. All sample preparations were carried out inside an argon-filled glove box. The prepared electrolytes were then stored in sealed vials under argon inside the glove box.
Electrochemical experiments
Na/Na symmetric coin cells (components CR2032) were purchased from Hohsen Corporation, Japan. The electrodes were two 8 mm diameter sodium metal electrodes (Sigma-Aldrich, purity 99.9%). A 16 mm diameter polyethylene separator (gratis, Lydall, 7P03A, 50 mm thickness, 85% 0.3 mm porosity) soaked with the electrolyte solution was used. A 0.5 mm spacer and 1.4 mm spring were used to ensure contact. After assembly, the cells were rested for 24 hours at 50 °C before cycling at 1 mA cm−2/1 mA h cm−2 for long cycles at 50 °C. Electrochemical impedance spectroscopy (EIS) was employed to monitor changes on the sodium electrode surface, with measurements performed using a VMP3 multichannel potentiostat (Bio-Logic) over a frequency range of 50 mHz–1 MHz and a 10 mV voltage amplitude. Data analysis was conducted using EC-Lab software (Z Fit version 10.44).Cu|Na coin cells were prepared for conducting cyclic voltammetry (CV) experiments. The working electrode was copper (12.7 mm diameter), and the counter and pseudo-reference electrode was sodium metal (8 mm diameter). The copper electrode was punched and washed with 1M HCl solution for 2 minutes followed by washing with deionized (DI) water for 2 minutes and finally with acetone for 2 minutes. The electrodes were then vacuum-dried for 24 hours at 100 °C and transferred into the glovebox. The rest of the assembly followed the same procedure as the Na/Na symmetric cells. All the measurements were carried out at 50 °C and at 20 mV s−1 scan rate. The onset potential was determined by intersection of the tangent to the exponentially increasing current part of the current curve with the linear extrapolation of the baseline current.
To eliminate any contributions from chemically formed SEI in Cu|Na coin cells during CV measurements, and also to have a reference for more accurate comparison of potentials, an alternative set of CV measurements was performed using a three-electrode cell configuration on gold surface. This setup consisted of a platinum (Pt) coil-shaped electrode as the counter electrode (CE), refillable silver triflate (AgOTf) reference electrodes (RE) filled with the respective ionic liquid (IL) electrolytes containing ∼5 mM AgOTf, and gold (Au) electrodes (3 mm diameter) as the working electrode (WE). Prior to the measurements, the electrodes underwent mechanical polishing using a micro-disc cloth covered with alumina (Al2O3) slurry (0.3 μm), followed by rinsing with deionized water and ethanol. Subsequently, the electrodes were sonicated in ethanol for 5 minutes, dried with argon (Ar) flow, and placed in an oven at 60 °C for 20 minutes. Additionally, the Pt electrodes were annealed using a butane flame. All measurements were conducted under a dry argon atmosphere inside a glovebox (H2O ∼ 0 ppm, O2 < 1 ppm) at 50 °C, with a scan rate of 20 mV s−1.
Physicochemical properties
Surface morphology
 | ||
| Fig. 9 In situ electrochemical/video optical microscopy setup. | ||
Interphase ion layering
sinθ, the component of the momentum transfer vector along the normal to the interface plane where λ is neutron wavelength and θ is incident angle of neutron beam to the planar surface. The specular reflectivity of neutrons was measured at the neutron reflectometer Spatz47 which operates in time-of-flight regime (using a 2.5 to 19 Å range of wavelengths) at the 20 MW OPAL reactor (ANSTO, Australia) viewing the cold-neutron source. The sample was placed on a custom electrochemical cell48 with potentials applied through a BioLogic VSP-300 potentiostat (BioLogic, Grenoble, France). A neutron beam passes through a single-crystal silicon substrate (thickness of 7 mm) with low resistivity (<1 Ω.cm) to reflect from the interface between a liquid sample and an electrode film (thickness below 300 Å) deposited on the substrate prior to the experiment. The electrode was a double layer of metals e-beam evaporation of 8 ± 0.8 nm Cr and 20 ± 2 nm Au on a single-crystal silicon block with dimensions of diameter 50.8 mm (2-inch) and roughness <5 Å for the working surface (plane orientation 〈111〉). A silicon single crystal with inlet and outlet tubes was used as a backing wafer to contain the bulk liquid and the electrode and backing wafer were separated using a PTFE gasket. The electrochemical cell was filled by the different IL mixture samples inside an argon filled glove box under moisture and oxygen-controlled conditions (H2O level: <1 ppm and O2 level: <10 ppm). To achieve the largest possible dynamic range of qz, two neutron incident angles (0.85° and 3.5° using a 20 mm footprint) for the same sample were used and the data from the two angles was combined to generate the final data to cover the entire range of qz (0.008–0.24 Å−1). Reflectivity curves were measured with a ΔQ/Q resolution ∼5%. The NR data were fitted by a number of structural models with variation of SLD, thickness and roughness for each layer/layers at the interface using the RefnX package49 to minimize the difference between the experimental and theoretical curves. A sketch of the NR experiment and interface structure are presented in Fig. 10.
 | ||
| Fig. 10 Sketch of the NR experiment with approximate sizes of the layers. | ||
Taking into account materials composition, gravimetric density and volume of the different ion molecules, neutron scattering length density (SLD) was estimated for each component in studied materials (see Table S2†).
The initial structure of the Si substrates with electrode layers was determined by fitting the NR measurements at the air/solid interface. The thicknesses of the Au and Cr layers showed good agreement with the manufacturer's specifications. Additionally, a thin SiO2 layer between the Si block and the Cr layer was identified and incorporated into the model. These structural parameters for the Si block, Au, Cr, and SiO2 layers were subsequently fixed during the fitting of the solid/liquid interface data. A new additional layer was introduced at the IL/solid interface and SLD, thickness and roughness of this layer were varied during the fitting process.
The working electrode consisted of Au(111) surfaces, comprising a 300 nm thick atomically smooth gold film on mica, obtained from Georg Albert PVD – Beschichtungen. Two 0.25 mm platinum wires (Alfa, 99.99%) were used as the reference and counter electrodes. AFM probes of type HQ:NSC36/Cr-Au BS, featuring gold-coated soft tapping mode cantilevers with a spring constant of approximately 0.8 N m−1, were employed for the measurements.
Before the measurements, the gold surfaces and AFM probes were cleaned by sequential rinsing with deionized water and ethanol, followed by drying under nitrogen (N2) flow. Both were subsequently treated in a UV-O3 plasma cleaner for 20 minutes.
Measurements were performed at open circuit potential and under applied negative potentials to the working electrode. These potentials were precisely controlled using a Pine WaveNano potentiostat, operated via AfterMath software in a three-electrode configuration.
To reduce variability and ensure consistency in the AFM measurements, the same type of soft cantilever was used throughout, and the approach speed was kept low and constant across all samples. These settings were chosen based on literature and optimized to detect subtle ionic layering near the electrode surface. For neutron reflectometry, measurements were performed using the same cell setup and electrode structure, and data from two incident angles were combined to extend the q-range. All fittings were carried out using a consistent model across samples. While surface-sensitive techniques like AFM and NR can be affected by sample preparation or measurement conditions, the procedures used here were carefully controlled to ensure comparability and reliability of the results.
Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript. L. H. – experimental design, methodology, investigation, data analysis, writing original draft. M. M. – conducted AFM experiments (under the guidance of K. S. F., R. A. and H. L.), contributed to experimental design, methodology, investigation, data analysis, and writing the original draft for CV (three-electrode setup) and AFM. M. S., J. M. P. and V. P. – experimental design, methodology, investigation, data analysis, writing original draft for NR. J. H. and X. Z. – methodology guidance for in situ OM. F. C. and F. M. – supervising, scientific discussion and understanding concept. P. H. – conceptualization, supervising, scientific discussion and understanding concept. M. F. – conceptualization, supervising, scientific discussion and understanding concept.Data availability
Neutron reflectometry: https://doi.org/10.5281/zenodo.14713708.Videos for in situ optical microscopy: https://doi.org/10.26187/deakin.28282061.
The remaining data supporting this article are included as part of the ESI.†
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Australian Research Council (ARC) for funding under Discovery Project DP210101172, Future Energy Storage Technologies (StorEnergy) IC180100049, and Deakin University Battery Research and Innovation Hub facilities. M. S. acknowledges the funding from the European Union H2020 Program under the Marie Curie global fellowship (ROCHE, 101026163). J. M. P. acknowledges the Ramón y Cajal grant RYC2023-044841-I funded by MICIU/AEI/10.13039/501100011033 and by the FSE+. Neutron beamtime was awarded by ANSTO under proposal 17200. Spatz operations are supported by the National Collaborative Research Infrastructure Strategy (NCRIS), an Australian Government initiative. J. H. and X. Z. ‘s work was supported by the National Natural Science Foundation (NSFC) of China (U20A20336, and 21935009), and the Hebei Natural Science Foundation (B2024203054). The authors extend their thanks to Luke O'Dell, Agilio Padua, and Michel Armand for their supervision, scientific discussions, and conceptual insights.References
- K. Matsumoto, J. Hwang, S. Kaushik, C.-Y. Chen and R. Hagiwara, Advances in sodium secondary batteries utilizing ionic liquid electrolytes, Energy Environ. Sci., 2019, 12(11), 3247–3287 RSC.
- B. Sun, P. Xiong, U. Maitra, D. Langsdorf, K. Yan, C. Wang, J. Janek, D. Schroder and G. Wang, Design Strategies to Enable the Efficient Use of Sodium Metal Anodes in High-Energy Batteries, Adv. Mater., 2020, 32(18), e1903891 CrossRef PubMed.
- K. Dong, X. Liu, H. Dong, X. Zhang and S. Zhang, Multiscale Studies on Ionic Liquids, Chem. Rev., 2017, 117(10), 6636–6695 CrossRef CAS PubMed.
- K. Karuppasamy, J. Theerthagiri, D. Vikraman, C.-J. Yim, S. Hussain, R. Sharma, T. Maiyalagan, J. Qin and H.-S. Kim, Ionic Liquid-Based Electrolytes for Energy Storage Devices: A Brief Review on Their Limits and Applications, Polymers, 2020, 12(4), 918 CrossRef CAS PubMed.
- Z. Xue, L. Qin, J. Jiang, T. Mu and G. Gao, Thermal, electrochemical and radiolytic stabilities of ionic liquids, Phys. Chem. Chem. Phys., 2018, 20(13), 8382–8402 RSC.
- S. A. Ferdousi, L. A. O'Dell, J. Sun, Y. Hora, M. Forsyth and P. C. Howlett, High-Performance Cycling of Na Metal Anodes in Phosphonium and Pyrrolidinium Fluoro(sulfonyl)imide Based Ionic Liquid Electrolytes, ACS Appl. Mater. Interfaces, 2022, 14(13), 15784–15798 CrossRef CAS PubMed.
- M. Hilder, P. C. Howlett, D. Saurel, E. Gonzalo, A. Basile, M. Armand, T. Rojo, M. Kar, D. R. MacFarlane and M. Forsyth, The effect of cation chemistry on physicochemical behaviour of superconcentrated NaFSI based ionic liquid electrolytes and the implications for Na battery performance, Electrochim. Acta, 2018, 268, 94–100 CrossRef CAS.
- C. M. Schott, P. M. Schneider, K.-T. Song, H. Yu, R. Götz, F. Haimerl, E. Gubanova, J. Zhou, T. O. Schmidt, Q. Zhang, V. Alexandrov and A. S. Bandarenka, How to Assess and Predict Electrical Double Layer Properties. Implications for Electrocatalysis, Chem. Rev., 2024, 124(22), 12391–12462 CrossRef CAS PubMed.
- X. Mao, P. Brown, C. Cervinka, G. Hazell, H. Li, Y. Ren, D. Chen, R. Atkin, J. Eastoe, I. Grillo, A. A. H. Padua, M. F. Costa Gomes and T. A. Hatton, Self-assembled nanostructures in ionic liquids facilitate charge storage at electrified interfaces, Nat. Mater., 2019, 18(12), 1350–1357 CrossRef CAS PubMed.
- J. M. Black, M. Zhu, P. Zhang, R. R. Unocic, D. Guo, M. B. Okatan, S. Dai, P. T. Cummings, S. V. Kalinin, G. Feng and N. Balke, Fundamental aspects of electric double layer force-distance measurements at liquid-solid interfaces using atomic force microscopy, Sci. Rep., 2016, 6, 32389 CrossRef CAS PubMed.
- G. Yang, I. N. Ivanov, R. E. Ruther, R. L. Sacci, V. Subjakova, D. T. Hallinan and J. Nanda, Electrolyte Solvation Structure at Solid-Liquid Interface Probed by Nanogap Surface-Enhanced Raman Spectroscopy, ACS Nano, 2018, 12(10), 10159–10170 CrossRef CAS PubMed.
- Y. Gu, E.-M. You, J.-D. Lin, J.-H. Wang, S.-H. Luo, R.-Y. Zhou, C.-J. Zhang, J.-L. Yao, H.-Y. Li, G. Li, W.-W. Wang, Y. Qiao, J.-W. Yan, D.-Y. Wu, G.-K. Liu, L. Zhang, J.-F. Li, R. Xu, Z.-Q. Tian, Y. Cui and B.-W. Mao, Resolving nanostructure and chemistry of solid-electrolyte interphase on lithium anodes by depth-sensitive plasmon-enhanced Raman spectroscopy, Nat. Commun., 2023, 14(1), 3536 CrossRef CAS PubMed.
- N. Kumar and J. M. Seminario, Lithium-Ion Model Behavior in an Ethylene Carbonate Electrolyte Using Molecular Dynamics, J. Phys. Chem. C, 2016, 120(30), 16322–16332 CrossRef CAS.
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Engineering high-energy-density sodium battery anodes for improved cycling with superconcentrated ionic-liquid electrolytes, Nat. Mater., 2020, 19(10), 1096–1101 CrossRef CAS PubMed.
- H. Wang, D. Ning, L. Wang, H. Li, Q. Li, M. Ge, J. Zou, S. Chen, H. Shao and Y. Lai, In Operando Neutron Scattering Multiple–Scale Studies of Lithium–Ion Batteries, Small, 2022, 18(19), 2107491 CrossRef CAS PubMed.
- K. S. Fraysse, L. Huang, H. Li, R. Atkin, A. Padua, M. Armand, P. C. Howlett and M. Forsyth, On the Parasitic Surface Adsorption of Pyrrolidinium and Phosphonium-based Ionic Liquids Preventing Accurate Differential Capacitance Measurements, J. Electrochem. Soc., 2024, 171(10), 106506 CrossRef CAS.
- J. N. Canongia Lopes, T. C. Cordeiro, J. M. S. S. Esperança, H. J. R. Guedes, S. Huq, L. P. N. Rebelo and K. R. Seddon, Deviations from Ideality in Mixtures of Two Ionic Liquids Containing a Common Ion, J. Phys. Chem. B, 2005, 109(8), 3519–3525 CrossRef PubMed.
- G. Annat, M. Forsyth and D. R. MacFarlane, Ionic liquid mixtures-variations in physical properties and their origins in molecular structure, J. Phys. Chem. B, 2012, 116(28), 8251–8258 CrossRef CAS PubMed.
- H. Wu, K. Han, W. Hu, W. Feng, M. Armand, Z. Zhou and H. Zhang, Ionic liquids with sulfinyl-functionalized imide anion and their lithium electrolytes: (I) Physical and electrochemical properties, J. Power Sources Adv., 2024, 28, 100154 CrossRef CAS.
- V. Chaudoy, J. Jacquemin, F. Tran-Van, M. Deschamps and F. Ghamouss, Effect of mixed anions on the transport properties and performance of an ionic liquid-based electrolyte for lithium-ion batteries, Pure Appl. Chem., 2019, 91(8), 1361–1381 CrossRef.
- V. Lesch, S. Jeremias, A. Moretti, S. Passerini, A. Heuer and O. Borodin, A combined theoretical and experimental study of the influence of different anion ratios on lithium ion dynamics in ionic liquids, J. Phys. Chem. B, 2014, 118(26), 7367–7375 CrossRef CAS PubMed.
- M. Martinez-Ibañez, N. Boaretto, L. Meabe, X. Wang, H. Zhu, A. Santiago, O. Zugazua, M. Forsyth, M. Armand and H. Zhang, Revealing the Anion Chemistry Effect on Transport Properties of Ternary Gel Polymer Electrolytes, Chem. Mater., 2022, 34(16), 7493–7502 CrossRef.
- N. N. Intan and J. Pfaendtner, Effect of Fluoroethylene Carbonate Additives on the Initial Formation of the Solid Electrolyte Interphase on an Oxygen-Functionalized Graphitic Anode in Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2021, 13(7), 8169–8180 CrossRef CAS PubMed.
- T. Gao, J. Bian, F. Huang, S. Ling, Z. Li, H. Yuan, H. Lin, L. Kong, B. Deng, Y. Zhao and Z. Lu, Enhanced cycling, safety and high-temperature performance of hybrid Li ion/Li metal batteries via fluoroethylene carbonate additive, Mater. Chem. Phys., 2024, 314, 128868 CrossRef CAS.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett, Y. S. Meng and M. Forsyth, Interphase control for high performance lithium metal batteries using ether aided ionic liquid electrolyte, Energy Environ. Sci., 2022, 15(5), 1907–1919 RSC.
- H. Ye, Y.-X. Yin, S.-F. Zhang, Y. Shi, L. Liu, X.-X. Zeng, R. Wen, Y.-G. Guo and L.-J. Wan, Synergism of Al-containing solid electrolyte interphase layer and Al-based colloidal particles for stable lithium anode, Nano Energy, 2017, 36, 411–417 CrossRef CAS.
- S. Park, S. Y. Jeong, T. K. Lee, M. W. Park, H. Y. Lim, J. Sung, J. Cho, S. K. Kwak, S. Y. Hong and N. S. Choi, Replacing conventional battery electrolyte additives with dioxolone derivatives for high-energy-density lithium-ion batteries, Nat. Commun., 2021, 12(1), 838 CrossRef CAS PubMed.
- D. A. Rakov, J. Sun, P. V. Cherepanov, K. Arano, P. C. Howlett, A. N. Simonov, F. Chen and M. Forsyth, The impact of electrode conductivity on electrolyte interfacial structuring and its implications on the Na0/+ electrochemical performance, Energy Environ. Sci., 2023, 16(9), 3919–3931 RSC.
- T. Pathirana, D. A. Rakov, F. Chen, M. Forsyth, R. Kerr and P. C. Howlett, Improving Cycle Life through Fast Formation Using a Superconcentrated Phosphonium Based Ionic Liquid Electrolyte for Anode-Free and Lithium Metal Batteries, ACS Appl. Energy Mater., 2021, 4(7), 6399–6407 CrossRef CAS.
- S. Begić, F. Chen, E. Jónsson and M. Forsyth, Water as a catalyst for ion transport across the electrical double layer in ionic liquids, Phys. Rev. Mater., 2020, 4(4), 045801 CrossRef.
- L. Huang, F. Makhlooghiazad, L. A. O'Dell, P. C. Howlett and M. Forsyth, Investigating the role of mixed-cation ionic liquid electrolytes in sodium battery efficiency and stability, Mater. Adv., 2024, 5(17), 6899–6909 RSC.
- A. Warrington, L. A. O'Dell, O. E. Hutt, M. Forsyth and J. M. Pringle, Structure and interactions of novel ether-functionalised morpholinium and piperidinium ionic liquids with lithium salts, Energy Adv., 2023, 2(4), 530–546 RSC.
- A. Warrington, M. Hasanpoor, A. Balkis, P. C. Howlett, O. E. Hutt, M. Forsyth and J. M. Pringle, Investigation of properties and performance of three novel highly concentrated ether-functionalised ionic liquid electrolytes for lithium metal batteries, Energy Storage Mater., 2023, 63, 102984 CrossRef.
- K.-H. Chen, K. N. Wood, E. Kazyak, W. S. LePage, A. L. Davis, A. J. Sanchez and N. P. Dasgupta, Dead lithium: mass transport effects on voltage, capacity, and failure of lithium metal anodes, J. Mater. Chem. A, 2017, 5(23), 11671–11681 RSC.
- K. N. Wood, E. Kazyak, A. F. Chadwick, K. H. Chen, J. G. Zhang, K. Thornton and N. P. Dasgupta, Dendrites and Pits: Untangling the Complex Behavior of Lithium Metal Anodes through Operando Video Microscopy, ACS Cent. Sci., 2016, 2(11), 790–801 CrossRef CAS PubMed.
- A. Aleshin, S. Bravo, K. Redquest and K. N. Wood, Rapid Oxidation and Reduction of Lithium for Improved Cycling Performance and Increased Homogeneity, ACS Appl. Mater. Interfaces, 2021, 13(2), 2654–2661 CrossRef CAS PubMed.
- S. Begić, H. Li, R. Atkin, A. F. Hollenkamp and P. C. Howlett, A comparative AFM study of the interfacial nanostructure in imidazolium or pyrrolidinium ionic liquid electrolytes for zinc electrochemical systems, Phys. Chem. Chem. Phys., 2016, 18(42), 29337–29347 RSC.
- P. C. Howlett, E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Electrochemistry at Negative Potentials in Bis(trifluoromethanesulfonyl)amide Ionic Liquids, Z. fur Phys. Chem., 2006, 220(10), 1483–1498 CrossRef CAS.
- D. A. Rakov, J. Sun, S. A. Ferdousi, P. C. Howlett, A. N. Simonov, F. Chen and M. Forsyth, Polar Organic Cations at Electrified Metal with Superconcentrated Ionic Liquid Electrolyte and Implications for Sodium Metal Batteries, ACS Mater. Lett., 2022, 4(10), 1984–1990 CrossRef CAS.
- A. Davoodabadi, C. Jin, D. L. Wood III, T. J. Singler and J. Li, On electrolyte wetting through lithium-ion battery separators, Extreme Mech. Lett., 2020, 40, 100960 CrossRef.
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Engineering high-energy-density sodium battery anodes for improved cycling with superconcentrated ionic-liquid electrolytes, Nat. Mater., 2020, 19(10), 1096–1101 CrossRef CAS PubMed.
- S. A. Ferdousi, L. A. O'Dell, J. Sun, Y. Hora, M. Forsyth and P. C. Howlett, High-performance cycling of na metal anodes in phosphonium and pyrrolidinium fluoro (sulfonyl) imide based ionic liquid electrolytes, ACS Appl. Mater. Interfaces, 2022, 14(13), 15784–15798 CrossRef CAS PubMed.
- J. Kim, G. R. Lee, R. B. K. Chung, P. J. Kim and J. Choi, Homogeneous Li deposition guided by ultra-thin lithiophilic layer for highly stable anode-free batteries, Energy Storage Mater., 2023, 61, 102899 CrossRef.
- Q. Wang, C. Zhao, S. Wang, J. Wang, M. Liu, S. Ganapathy, X. Bai, B. Li and M. Wagemaker, Clarifying the Relationship between the Lithium Deposition Coverage and Microstructure in Lithium Metal Batteries, J. Am. Chem. Soc., 2022, 144(48), 21961–21971 CrossRef CAS PubMed.
- Y. Ji, H. Sun, Z. Li, L. Ma, W. Zhang, Y. Liu, L. Pan, W. Mai and J. Li, Salt engineering toward stable cation migration of Na metal anodes, J. Mater. Chem. A, 2022, 10(48), 25539–25545 RSC.
- E. M. Barsoukov and J. R. MacDonald, Impedance Spectroscopy Theory Experiment And Applications, 2004 Search PubMed.
- A. P. Le Brun, T. Y. Huang, S. Pullen, A. R. J. Nelson, J. Spedding and S. A. Holt, Spatz: the time-of-flight neutron reflectometer with vertical sample geometry at the OPAL research reactor, J. Appl. Crystallogr., 2023, 56(Pt 1), 18–25 CrossRef CAS PubMed.
- Y. Lauw, T. Rodopoulos, M. Gross, A. Nelson, R. Gardner and M. D. Horne, Electrochemical cell for neutron reflectometry studies of the structure of ionic liquids at electrified interface, Rev. Sci. Instrum., 2010, 81(7), 074101 CrossRef CAS PubMed.
- A. R. J. Nelson and S. W. Prescott, refnx: neutron and X-ray reflectometry analysis in Python, J. Appl. Crystallogr., 2019, 52(Pt 1), 193–200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5eb00052a |
| This journal is © The Royal Society of Chemistry 2025 |