
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stepwise volatilization induced by nature-sourced volatile solid additives improving the efficiency and stability of perovskite solar cells†
Jeewon
Park
 a,
Seoyoung
Kim
a,
Wonjun
Kim
a,
Zhe
Sun
a,
Byongkyu
Lee
a and
Changduk
Yang
*ab
a,
Seoyoung
Kim
a,
Wonjun
Kim
a,
Zhe
Sun
a,
Byongkyu
Lee
a and
Changduk
Yang
*ab
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulju-gun, Ulsan, 44919, South Korea. E-mail: yang@unist.ac.kr
bGraduate School of Carbon Neutrality, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulju-gun, Ulsan, 44919, South Korea
First published on 12th May 2025
Abstract
In contrast to the substantial efforts dedicated to additive engineering in the field of perovskite solar cells (PSCs), the exploration of volatile solid additives has received surprisingly little attention. This study introduces a novel approach for fabricating highly efficient and stable PSCs by employing “naturally sourced volatile solid additives” (CP and CQ), which exhibit distinct volatilization rates and varying degrees of Lewis base–acid interactions with perovskite. During the PSC fabrication process involving two-stage thermal annealing, CP completely volatilized after the initial annealing step, resulting in the formation of a densely packed PbI2 film. Conversely, owing to its robust Lewis base–acid interaction with perovskite through bidentate coordination, a certain amount of CQ tends to persist after the first annealing step. However, no residue was observed after the second annealing step (stepwise-volatilization mechanism). This process yields high-quality FA-based perovskite crystallinity characterized by large-grained perovskite and highly preferred crystallite orientation. Consequently, CQ-based PSCs achieve a promising power conversion efficiency (PCE) of 25.00% (independently certified at 24.89%) with exceptional MPPT stability (PCE retention of 90.1% after 1000 h, ISOS-L-1). This study underscores the viability of the naturally sourced volatile solid additives for ecofriendly manufacturing of PSCs.
Broader contextPerovskite solar cells (PSCs) are a promising candidate for next-generation photovoltaics owing to their excellent optoelectronic properties. Despite the current drastic advancements in PSCs, various defects can emerge owing to precursor compositions and processing conditions inherent to solution processing and rapid perovskite film growth. To address these sticking points, extensive research efforts have been devoted to additive engineering by controlling the film formation of perovskite layers. However, in the field of perovskite solar cells, the investigation of volatile solid additives has been rarely utilized compared to organic optoelectronics. Moreover, there are not many reports on nature-sourced additives. Herein, we report naturally derived volatile solid additives, namely camphor (CP) and camphorquinone (CQ), which improve the efficiency and stability of PSCs. The work introduces a “stepwise volatilization” to explain the working mechanism of CQ volatile additives on perovskite crystallization. As a result, the CQ-based PSC achieved an enhanced PCE of 25.00% compared to the 23.20% efficiency of the control device. This study provides a novel strategy using volatile natural additives to improve the photovoltaic performance and stability of PSCs. |
1. Introduction
Halide perovskites, characterized by the ABX3 crystal structure (where A = methylammonium (MA), formamidinium (FA), or cesium (Cs), B = lead or tin cation, X = a halogen anion), garnered significant attention due to their exceptional photoelectric properties.1–7 These properties include a high absorption coefficient (∼105), a small exciton binding energy of less than 100 meV, an extended diffusion length exceeding 1 μm, and a tunable bandgap spanning from 1.2 to over 3 eV.8–14 Remarkable progress has been achieved in the field of perovskite solar cells (PSCs) in only a few years. The power conversion efficiency (PCE) reached 26.08% through a range of strategies, including the development of novel functional materials, engineering of device interfaces, compositional tuning, and control of crystallization kinetics.15–19Despite the current rapid advancements and ongoing enhancements in PSCs, various defects can emerge owing to precursor compositions and processing conditions inherent to solution processing and rapid perovskite film growth. The introduction of additives (e.g., salts, molecules, polymers, and nanoparticles) has the potential to influence perovskite crystallization, film formation, bulk or surface defect passivation, and interface optimization in terms of structure and energetics.20–26 Consequently, extensive research efforts have been devoted to additive engineering to address these challenges by controlling the thermodynamics and growth kinetics of perovskite films.
Concurrently, in the organic electronics community, “volatile solid additives” have demonstrated their effectiveness in directly modulating active layer morphology, leading to improved characteristics in organic photovoltaics, including PCE, device stability, and reproducibility.27–35 While the use of volatile solid additives has become a widely adopted approach for achieving high-performance organic photovoltaics, it has surprisingly received limited attention in perovskite-based systems. Therefore, there is a pressing need for further research within the PSC community to explore volatile solid additives. This endeavor aims to develop advanced additive-related protocols and gain deeper insights into the underlying mechanisms governing the use of volatile solid additives.
In this study, we introduced naturally sourced volatile solid additives, namely camphor (CP) and camphorquinone (CQ), into the fabrication process of perovskite solar cells (PSCs). The PSCs were produced using a two-step sequential deposition technique that involved a two-stage thermal-annealing process.36–40 Both CP and CQ are polar Lewis-base molecules containing carbonyl groups, but they differ in the number of carbonyl groups (Fig. 1a for their structures). This distinction gives rise to varying rates of volatilization and different degrees of Lewis base–acid interaction with the perovskite material.
 | ||
| Fig. 1 Stepwise volatilization induced by volatile solid additives. (a) Chemical structures and electrostatic potential mappings of CP and CQ. (b) Schematic representation of the stepwise volatilization mechanism involving two-stage thermal annealing: an initial annealing at 70 °C for PbI2 precursor and a second annealing step at 150 °C for perovskite (PVK) crystallization. | ||
Following the initial annealing step at 70 °C, CP molecules were entirely volatilized from the PbI2 precursor, resulting in the formation of a tightly packed PbI2 film. In contrast, some CQ molecules remained within the PbI2 bulk film after the first annealing step. However, their complete volatilization occurred during the subsequent second annealing step at 150 °C, resulting in a stepwise-volatilization mechanism. This mechanism facilitated the development of high-quality FA-based perovskite crystallinity characterized by large-grained perovskite structures and a high degree of preferred crystallite orientation, enabling extremely high PCEs of 25.00% for a laboratory scale of 0.042 cm2 (independently certified at 24.89%). Moreover, the operational stability of CQ-treated PSCs was considerably enhanced, which retained over 90.1% of the initial PCE after 1000 h under maximum power point tracking conditions (ISOS-L-1).
2. Results and discussion
2.1. Volatilization and interaction in PbI2 precursor solution
In this research, a two-step sequential deposition process was employed to create the formamidinium-based (FA-based) perovskite film. The initial step involved the deposition of a PbI2 layer by spin-coating a PbI2 precursor solution (comprising a solvent mixture of dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) in a volume ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) onto the ITO/SnO2 substrate. This layer was then annealed at 70 °C for 1 min in a N2 atmosphere, constituting the first thermal-annealing step. Subsequently, an organic ammonium salt solution (containing formamidinium iodide (FAI), methylammonium iodide (MAI), and methylammonium chloride (MACl) in isopropyl alcohol) was spin-coated onto the PbI2 layer. This was followed by a 15-minute aging process at 150 °C under ambient atmospheric conditions (30–40% humidity), serving as the second thermal-annealing step. The solid additives were introduced into the PbI2 precursor solution.19,41
:1) onto the ITO/SnO2 substrate. This layer was then annealed at 70 °C for 1 min in a N2 atmosphere, constituting the first thermal-annealing step. Subsequently, an organic ammonium salt solution (containing formamidinium iodide (FAI), methylammonium iodide (MAI), and methylammonium chloride (MACl) in isopropyl alcohol) was spin-coated onto the PbI2 layer. This was followed by a 15-minute aging process at 150 °C under ambient atmospheric conditions (30–40% humidity), serving as the second thermal-annealing step. The solid additives were introduced into the PbI2 precursor solution.19,41
Fig. 1a provides an illustration of the molecular structures and electrostatic potential mapping of CP and CQ, along with their respective net dipole moments. These values were determined through density functional theory calculations using the B3LYP/6-31G* basis set.42 Notably, both CP and CQ exhibit strong negative electrostatic potential at the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) moiety. However, CQ possesses a larger net dipole moment (5.39 Debye) compared to CP (3.30 Debye), as shown in Table S1 (ESI†). This difference indicates that CQ, being a Lewis base, can effectively interact with uncoordinated Pb trap sites and FA+ ions present on the perovskite surface when introduced into the perovskite film.43,44
O) moiety. However, CQ possesses a larger net dipole moment (5.39 Debye) compared to CP (3.30 Debye), as shown in Table S1 (ESI†). This difference indicates that CQ, being a Lewis base, can effectively interact with uncoordinated Pb trap sites and FA+ ions present on the perovskite surface when introduced into the perovskite film.43,44
As shown in Fig. 2a for typical thermogravimetric analysis (TGA), relative to CQ, CP is a more volatile solid molecule with a clear weight loss starting at ∼40 °C. In the case of PbI2 samples mixed with these additives (in a 1:1 (wt/wt)), similar trends are observed. Their differential scanning calorimetry (DSC) measurements identify that the volatilization of CP and CQ rather than their decomposition also occurs during the thermal-annealing process (see Fig. S1, ESI†). Fig. S2 (ESI†) presents their isothermal TGA curves over time at 70 °C as the first annealing step of PSC fabrication, showing that the mass loss rate of CP is far higher than that of CQ. Besides, it is also found that CP has higher vapor pressure (500.64 Pa) determined by the Langmuir equation in comparison to CQ (68.20 Pa) (see Table S2 and Note S1 for details, ESI†). From the above results, we can conclude that CP has very high volatilization kinetics during the first thermal-annealing step.
 | ||
| Fig. 2 Volatility and interaction of CP and CQ additives. (a) TGA plots of CP and CQ (pristine and blended with PbI2). (b) The whole range and (c) magnified FTIR spectra of CP (RT), CQ (RT), PbI2-CP (70 °C), and PbI2-CQ (70 °C). (d) The whole range and (e) magnified FTIR spectra of CQ, PVK-CQ (150 °C), and PVK without CQ (150 °C). (f) 207Pb NMR spectra of PbI2, PbI2-CP, and PbI2-CQ, where the samples were calibrated by Pb(NO3)2, which represents −2959.7 ppm. (g) Schematic diagram of the PbI2 colloids (with or without additive). (h) DLS measurements of pristine PbI2, PbI2-CP, and PbI2-CQ. The inset shows the schematic diagram of the evolution of PbI2 flakelets. | ||
To further investigate the distinct volatilities of CP and CQ, Fourier transform infrared (FTIR) spectroscopy was utilized. Fig. 2b illustrates the stretching vibrations of the CO groups for CP and CQ at 1741 and 1750 cm−1, respectively. When both additives were heated at the same temperature of 70 °C, corresponding to the first thermal-annealing step, the CO peak in the CP-mixed PbI2 completely disappeared. Note that this volatilization of CP mainly occurred during the thermal annealing process rather than the spin-coating process (see Fig. S5, ESI†). In contrast, in the CQ-mixed PbI2, the CO peak still existed but shifted to a lower wavenumber of 1746 cm−1. This indicates the efficient formation of coordinating bonds between the CO group of CQ and Pb2+ through Lewis base–acid interactions, which reduces the electron density around the CO bond, thereby weakening its strength and resulting in a shift to a lower wavenumber.45,46 Additionally, it is noteworthy that the stretching vibrations of C–H peaks in 2950 cm−1 follow the same trend as that of the CO group (Fig. 2c). Interestingly, during the second thermal-annealing step at 150 °C, the CQ solid additive completely volatilized, as evidenced by the absence of the CO and C–H peaks (Fig. 2d and e). Note that the CN peak of perovskite film at 1712 cm−1 should be distinguished from the CO group of CQ.
To further confirm the complete evaporation of CP and CQ, 1H-nuclear magnetic resonance (NMR) analysis was performed on the ex situ taken films at each processing step, as shown in Fig. S6–S8 (ESI†). The experimental details are described in Note S2 (ESI†). The ex situ1H-NMR study clearly demonstrates that CP and CQ are completely volatilized during the first and second thermal annealing processes, respectively. Furthermore, the quantification of residual CP and CQ at each processing step was conducted using liquid chromatography-mass spectroscopy (LC-MS) (for details, see Fig. S9–S14 and Note S3, ESI†). As shown in Table S3 (ESI†), CP exhibits negligible residues, whereas most of CQ remains after the first annealing step. The remaining CQ is fully evaporated during the second annealing step. Collectively, these results identify CP as a volatile solid additive and CQ as a stepwise volatile solid additive, based on their distinct volatilization kinetics.
We conducted 207Pb NMR analysis in a deuterated DMF:DMSO solution (9:1, v/v) as shown in Fig. 2f. It is important to note that we characterized the PbI2 precursors and perovskites both without and with the optimal amounts of CP (30 mM) and CQ (3.0 mM) solid additives, respectively. These conditions were optimized for PSC fabrications, as detailed in the following photovoltaic section. The 207Pb resonance signal for pristine PbI2 was at 782.8 ppm, but it clearly shifted to a lower frequency in the additives-mixed PbI2 adducts. This shift was a result of the different Pb coordination environments induced by Lewis base–acid interactions.47 Note that the CQ-mixed PbI2 exhibited a more pronounced up-field shift, indicating a stronger Pb–CQ coordination environment.
Moreover, we performed 13C-NMR spectroscopy measurements to further the additive-PbI2 coordination interaction. Fig. S15 and S16 (ESI†) display the 13C-NMR resonance signals of the carbons in the CO bonds of CP and CQ molecules, respectively. In additive-mixed PbI2 solutions, the peaks clearly down-shifted with increasing additive concentrations. These results are due to the different chemical environments of the CO bond within the additive molecules after the Lewis base–acid interaction with the electron-deficient Pb2+ moiety. These observations align with the result of 207Pb NMR measurement showing the up-shifted resonance signals of the additive-Pb2+ adducts. Furthermore, in the results of diffusion ordered spectroscopy (DOSY) NMR measurements (Fig. S17–S20, ESI†), the diffusion coefficient (D) of CP and CQ decreased with increasing concentration of Pb2+ sites, where the D is related to the hydrodynamic radius of the nanostructure size in the PbI2-additive mixture (see Table S4 for details, ESI†). These results indicate the increased nanocrystal sizes of additive-mixed PbI2 colloidals, which are a result of the enhanced interaction between additives and Pb2+ sites. Collectively, we conclude that there are strong coordinative interactions between the CO bonds of the CP and CQ molecules and the Pb2+ site of the PbI2.
To assess the impact of CP and CQ on the size of PbI2 colloids, we conducted dynamic light scattering (DLS) measurements of the PbI2 precursor solutions (DMF:DMSO). Fig. 2g illustrates that the pristine PbI2 colloids consist of (001) and (101) facets. It is known that layered PbI2 structures can be exfoliated by solvent molecules through the (101) facet or vacancies on the (001) facet due to strong coordination between polar solvent molecules and Pb2+.48–55Fig. 2h presents histograms of the colloidal size distributions, showing that PbI2-pristine colloids exhibit flakelets along the (001) plane with a longitudinal dimension of ∼1 nm and no detectable lateral dimension. In contrast, the CP- and CQ-mixed PbI2 colloids display flakelets with similar longitudinal dimensions but significantly larger lateral dimensions of 726.7 nm and 2392.2 nm, respectively. We attribute the laterally enlarged PbI2 plates to additional coordination interactions between the CO group of the additives and Pb2+, which may hinder the intercalation of solvent molecules into the PbI2 (101) facets. The noticeable shift of the additive-mixed PbI2 in the ultraviolet-visible (UV-vis) spectra to a longer wavelength validates the interpretation from DLS data (Fig. S14, ESI†).56 In particular, the significant redshift in the CQ-mixed PbI2 can be partially attributed to the chromophore in the CQ molecule, which shows absorption in the 400 to 500 nm range in the pristine CQ (Fig. S21, ESI†).
2.2. Morphological characterization of the PbI2 layer
The surface morphologies of the PbI2 precursor films after the first thermal-annealing step were examined using scanning electron microscopy (SEM). As depicted in Fig. 3a, the PbI2-pristine film exhibits a compact texture with small grains. In contrast, the CP- and CQ-treated PbI2 films exhibited enlarged grain sizes with a greater number of pinholes (mean cluster sizes of 99.9 nm, 161.2 nm, and 180.5 nm for PbI2-pristine, CP-treated, and CQ-treated PbI2 films, respectively; Fig. 3b). This observation aligns with the changes in absorption and intensity shifts observed in their UV-vis spectra. Specifically, the CP- and CQ-treated PbI2 films exhibit higher absorptivity with red-shifting compared to the PbI2-pristine film (Fig. S22, ESI†). Additional evidence of relatively larger grains and more pinholes in CQ-treated PbI2 films is provided by their atomic force microscopy (AFM) images (Fig. 3a). The increased cluster sizes and porosities in the CP- and CQ-treated PbI2 films facilitate the formation of a mesoporous structure, enhancing the penetration of FAI into the PbI2 framework. This results in a high-quality perovskite film with fewer nucleation sites and larger perovskite grain sizes.26,50,57,58 To investigate the impact of CP and CQ solid additives on the crystallinity of PbI2 films, we conducted grazing incident wide angle X-ray scattering (GIWAXS). In this analysis, the incident angles were set to 0.13° and 1.0° to gather information on the near-surface region and the full depth of the films, respectively (Fig. 3c–e and Fig. S23, ESI†). At an incident angle of 0.13°, the PbI2-pristine film exhibits the PbI2 (001) peak at approximately q = 0.9 Å−1, consistent with previous reports.59–61 However, when treated with the solid additives, we observe not only additional shoulder peaks at q = ∼0.87 Å−1 and ∼0.96 Å−1 but also a shift in the position of the (001) peak. These results correlate with the broadening of cluster size distributions in CP- and CQ-mixed PbI2 (see Table S5 for a comparison of the standard deviation values, ESI†). Specifically, the (001) peak shifts to higher q vectors for CP-treated PbI2 and to lower q vectors for CQ-treated PbI2 films (Fig. S24 and S25, ESI†). These observations suggest that the CP and CQ treatments induce changes in the lattice d-spacing of the PbI2 surfaces. At a higher incident angle (1.0°), all the films exhibit no visible shoulder peaks, and the PbI2 (001) peak is almost identical but shifted to lower q vectors relative to what is observed at the low incident angle. Transmission electron microscopy (TEM) measurement was also applied to further elucidate the influence of the CP and CQ treatment on PbI2 lattice surfaces (Fig. S26, ESI†). The pristine PbI2 shows the lattice distance of 3.64 Å, which can match with the (110) plane of the hexagonal PbI2 phase. Note that the PbI2 (001) lattice does not appear since the ultrasonicated PbI2 powder dispersion method is involved to prepare appropriate TEM samples. Interestingly, the lattice distances of the CP- and CQ-treated PbI2 are changed to 3.46 Å and 3.97 Å, respectively. These results show good agreement with the opposite trend in the lattice d-spacing seen from the GIWAXS above. | ||
| Fig. 3 Effect of CP and CQ additives on PbI2 film morphology. (a) Top-view SEM images of PbI2-control, CP, and CQ with AFM images of the inset (4 μm × 4 μm). (b) Cluster size distribution of pristine PbI2, PbI2-CP, and PbI2-CQ. (c) Schematic illustration of the angle-dependent 2D-GIWAXS measurement. Line-cut profile of PbI2 (001) of the control, CP-, and CQ-PbI2 for (d) 0.13 and (e) 1.0° incident angles. (f) XPS spectra (Pb 4f) of PbI2 pristine, PbI2-CP, and PbI2-CQ. | ||
We conducted X-ray photoelectron spectroscopy (XPS) of the films to characterize the core-level binding energies (BEs) of the PbI2 film surfaces (Fig. 3f and Fig. S27, ESI†). The XPS pattern of Pb 4f for the PbI2-pristine film displays two dominant peaks at 137.88 and 142.68 eV, corresponding to the Pb 4f7/2 and Pb 4f5/2, respectively. Both peaks are shifted toward lower BEs for both CP- and CQ-treated PbI2 films, indicating an enriched electron environment of Pb in the additives-containing PbI2 lattices.62 A closer examination of the XPS data reveals a slightly larger shift for the CP-treated case, suggesting the formation of denser PbI2 film surfaces through the complete volatilization of CP after the first thermal-annealing step. Collectively, the data presented highlight the distinct roles played by CP and CQ solid additives during the processing of PbI2 films.
To elaborate, during the first thermal-annealing step: (i) CP entirely evaporates, resulting in the filling of voids with PbI2 and the consequent formation of a denser PbI2 film, particularly on the surfaces. (ii) In contrast, the complete volatilization of CQ molecules is constrained by their inherent Lewis base–acid interaction, as described above. Certain CQ molecules undergo partial volatilization, acting as a mediator for PbI2 surface packing similar to CP. However, a number of them remain within the PbI2 bulk film, serving as passivating agents for PbI2 defects owing to their inherently enhanced Lewis base–acid interaction, as discussed earlier. A schematic representation of the microstructure evolution during PbI2 film formation with CP and CQ is provided in Fig. S28 (ESI†).
2.3. Morphological characterizations of the perovskite film
To fabricate FA-based perovskite films, we initiated the reaction with FAI, followed by the second thermal-annealing process at 150 °C, as previously described. We conducted top-view and cross-sectional SEM analyses to assess the influence of CP and CQ on perovskite grain growth. As illustrated in Fig. 4a, the grain sizes were approximately ∼858 nm and ∼1183 nm in the CP- and CQ-treated perovskite films, respectively. This increase in grain size can be attributed to the reduced nucleation density resulting from the enlarged PbI2 cluster sizes. These grain sizes are significantly larger than those of the control perovskite film without any additives, which had an average grain size of approximately ∼608 nm (Fig. 4b and Table S6, ESI†). Relative to the control perovskite film, both CP- and CQ-treated perovskite films exhibit higher absorbance, partially reflecting the improved crystallinity driven by the larger grain sizes (Fig. S29, ESI†). | ||
| Fig. 4 Characterization of perovskite films. (a) Top-views and cross-sectional SEM images of the control, CP-, and CQ-treated perovskites (PVKs). (b) Grain size distribution of the control, CP-, and CQ-based PVKs. Polar intensity plots azimuthally along the ring at q = 1.00 Å−1 of the control, CP-, and CQ-PVKs for (c) 0.13° and (d) 1.0° incident angles. Schematic illustration for the proposed (110) facet stacking mode in (e) CP- and (f) CQ-treated PVK films. (g) TRPL plots of the control, CP-, and CQ-treated PVKs. | ||
To further investigate the crystal quality and orientation of the perovskite films, we analyzed depth GIWAXS at different incident angles (0.13° and 1.0°) and examined their polar intensity profiles of the integrated semicircle (110) peak at azimuthal angles ranging from 0° to 180° (Fig. S30, ESI†). The CP- and CQ-treated perovskite films exhibit increased intensities of the perovskite peaks compared to the control perovskite film. In Fig. 4c and d, intensity-corrected pole figures reveal two peaks at azimuthal angles near 35° & 145° and 90°, which correspond to the orientations of perovskite crystal stacking relative to the substrate. Across all the films, the intensity of the latter peak is higher than that of the former, regardless of the incident angles, indicating a greater prevalence of perovskite crystal stackings oriented perpendicular to the film plane. At a low incident angle (0.13°), the area ratios of the two peaks (the latter-to-the former) follow the sequence of the control (1.74) ≅ CP (1.84) < CQ (4.01)-treated perovskite films, suggesting a higher degree of preferred orientation in the CQ-treated perovskite film (Table S7, ESI†). This difference in preferred crystallographic orientations becomes more pronounced in the bulk films, as evidenced by the ratio values of the two peaks at a high incident angle (1.0°). These results demonstrate that CQ can effectively manipulate the crystallographic orientation, and the proposed stacking modes are schematically depicted in Fig. 4e and f. Time-resolved photoluminescence (TRPL) was further measured to estimate the carrier lifetimes of the control, CP-, and CQ-modified perovskite films using a picosecond laser with a wavelength of 450 nm (Fig. 4g). All three samples display mono-exponential decay related to trap-assisted recombination. The corresponding carrier lifetimes, obtained from TRPL fitting line, were 788 ns, 1126 ns, and 2130 ns for the control, CP-, and CQ-treated devices, respectively. These results indicated that the CQ additive improved the crystallinity and preferred orientation of the perovskite, prolonging charge carrier lifetime and reducing trap-assisted recombination.
We further investigated the effect of additive concentration on the morphological changes of PbI2 and perovskite films through SEM measurement (Fig. S31, ESI†). As the concentration of CP increases, both the number of PbI2 clusters and the perovskite grain size grow, achieving an optimal morphology at 30 mM. However, at a higher concentration (60 mM), excessively large and dense PbI2 clusters were formed, which are difficult to convert to a high-quality perovskite layer due to inhomogeneous and disturbed perovskite crystallization. This leads to the defects and non-uniform crystal growth of perovskite, ultimately degrading the device performance (see PSC data later). Similarly, in the case of CQ, concentrations up to 3 mM promoted enlarged and smoothed perovskite grains; however, at higher concentration of 6 mM, excessive amounts of CQ induced microporous surface structures in the perovskite layer. These voids significantly contributed to severe degradation in both device performance and operational stability. As shown in Fig. S32 (ESI†), the use of even higher CQ concentration (30 mM) leads to an increase in pores from the bottom to the top, which significantly reduced the operational stability (Fig. S33, ESI†).
2.4. In situ PL measurements of perovskite film formation
To gain a deep insight into the kinetics of volatile additives in FA-based perovskite crystallization, in situ PL measurements were performed during second thermal annealing at 150 °C using an excitation wavelength of 520 nm (Fig. 5a). After the FAI solution was spin-coated onto PbI2 films, the dark brown film was immediately transferred to a hot plate under controlled humidity (relative humidity of 40%). The FA-based perovskite crystallization starting with the annealing process was simultaneously monitored as a function of time in the PL response. Fig. 5b displayed the heat maps of the PL intensity for the control, CP-, and CQ-treated perovskite films (the detailed spectra are shown in Fig. S34, ESI†). All three samples exhibited instantaneous PL peaks in the initial stage of FA-based perovskite film formation (from 0 to 2500 ms). The initial evaporation of the remaining solvent in the precursor film leads to supersaturation of the solute and plenteous nuclei are formed at this stage.63–65 In the final stage, the PL spectra of the control perovskite film reached its maximum intensity of 1.081 × 104 for 13200 ms. Note that the PL intensity started to decrease after reaching the peak due to thermal quenching caused by continuous heating.66–68 Compared to the control perovskite film, both CP- and CQ-treated perovskite films exhibit an intermediate stage in the PL spectra between the initial stage and final stage (Fig. 5c). We speculate that this preliminary phase transition is attributed to the larger sized crystallites due to the CP and CQ additives, as confirmed by previous DLS measurements. These intermediate states are more pronounced in the CQ-based perovskite film (from 5000 to 10000 ms). In the final stage, the CQ-treated perovskite film shows the most delayed film formation time of 22890 ms with the highest maximum intensity of 1.770 × 104, indicating that the volatile CQ serves to retard the crystallization of the FA-based perovskite for the highly crystalline perovskite.62,69 As illustrated in Fig. 5d, upon transition from the intermediate to the final state, the volatile CQ molecules completely evaporate, filling the voids with perovskite, and finally resulting in the formation of a dense and vertically oriented perovskite film.
 | ||
| Fig. 5 In situ PL measurements during the FA-based PVK crystallization. (a) Schematic illustration of in situ PL monitoring during the FA-based PVK crystallization (150 °C). Heatmaps of in situ PL of the FA-based PVK film for (b) control, (c) PVK-CP, and (d) PVK-CQ. 3D colormap surfaces of in situ PL of the FA-based PVK film for (e) control, (f) PVK-CP, and (g) PVK-CQ. (h) Schematic summary showing the effects of CQ in crystallization kinetics. | ||
Additionally, the evolutions of film colors during the total process (from the initial PbI2 film to the final PVK film) were monitored over time to additionally exhibit the retardation of the CQ-assisted crystallization process (Fig. S35, ESI†). Compared to the initial control PbI2 film, the CQ-PbI2 film exhibited a more pronounced yellow and transition to a more orange color upon treatment with FAI solution. This more orange film was attributed to the much disordered PbI6 octahedra connectivity, which was a result of the interaction between CQ molecules and Pb2+ ions. During 5 min of FAI penetration, the CQ intermediate film darkened to a brown and transformed into a distinct black FA-based perovskite film after the second-stage annealing. This progression indicated that CQ treatment effectively retarded the crystallization process, resulting in a more compact and highly oriented perovskite structure.
2.5. Photovoltaic performances
We constructed planar n–i–p PSCs with the structure ITO or FTO/SnO2/FA0.92MA0.08PbI3 perovskite layer/OAI (2D treatment)/spiro-OMeTAD/Au (refer to Fig. 6a for the schematic). These PSCs were fabricated both with and without the inclusion of solid additives to investigate their impact on device performance. After screening PSC results at various additive concentrations (ranging from 15 to 60 mM), the optimal concentrations were determined to be 30 mM for CP and 3 mM for CQ, respectively (Fig. S36 and S37, ESI†). Detailed device fabrication procedures are outlined in the Experimental section. Note that even trace amounts of the solid additives in the PSCs, at different concentrations, induced significant variations in device performance, underscoring their pronounced influence on PSC characteristics. Fig. 6b illustrates the current density–voltage (J–V) curves for the optimized control, CP-treated, and CQ-treated PSCs (active area = 0.042 cm−2, under AM 1.5G 1-sun illumination), and the pertinent photovoltaic parameters are summarized in Table 1. For the optimized control device with a FA0.92MA0.08PbI3 perovskite, a PCE of 22.60% was achieved, with an open-circuit voltage (VOC) of 1.153 V, a short-circuit current (JSC) of 24.86 mA cm−2, and a fill-factor (FF) of 78.81%. Notably, significant performance enhancements were observed in the devices treated with optimal CP and CQ concentrations. In particular, the CQ-treated device achieved the enhanced PCE of 24.19%, accompanied by considerably increased VOC (1.187 V) and FF (81.57%) values, surpassing those of both the control and CP-treated devices. Importantly, all the devices exhibited negligible hysteresis (Fig. S38 and Table S8, ESI†), and the JSC values obtained from J–V scanning closely matched the values obtained by integrating the EQE spectra (24.32, 24.85, and 24.86 mA cm−2 for the control, CP-treated, and CQ-treated devices, respectively, Fig. 6d). For the control and CQ-based device, the stabilized power output and current density agree with those obtained from J–V measurements (Fig. S39 and S40, ESI†). To further validate these findings, Fig. S41 (ESI†) displays histograms of 24 individual devices for each case, confirming a narrower distribution of PSCs treated with additives compared to the control devices. We further optimized device fabrication to upgrade the champion device achieving higher PCE. The CQ-treated champion device achieved remarkable PCE of 25.00% with a VOC of 1.183 V, JSC of 25.95 mA cm−2, and FF of 81.45% (Fig. S42 and Table S9, ESI†), where the JSC value was well matched with the values obtained by integrating the EQE spectra (25.81 mA cm−2) (Fig. S43, ESI†). One of the best CQ-treated devices obtained a certified PCE of 24.89% from an independent institute, which is comparable to those obtained from the two-step-based state-of-the-art devices (Fig. S44 and Table S10, ESI†). Additionally, the CQ-treated large-area devices (active area: 1.0 cm2) were also fabricated to evaluate its scalability. As shown in Fig. 6c, the best-performing large-area device shows a high PCE of 22.50% with a VOC of 1.169 V, JSC of 25.20 mA cm−2, and FF or 76.39%. This indicates improvement in charge-carrier dynamics, including reduced charge recombination and enhanced charge transport properties in the CQ-treated device, which shows a similar trend to reported studies on the preferentially oriented perovskite-based solar cells (Table S11, ESI†). To exhibit universality of stepwise volatilization of the CQ additive in two-step-based fabrication methodology, we applied the CQ additive to the Cs-containing A-cation system (Cs0.05FA0.95PbI3) and mixed-halide perovskite system (Cs0.05FA0.95Pb(I0.95Br0.05)3).70,71 The corresponding J–V characteristics and photovoltaic parameters are summarized in Fig. S45 and Table S12 (ESI†), respectively. In common with all systems, the CQ additive effectively enhanced VOC and FF values. In particular, the CQ-treated Cs0.05FA0.95PbI3 system showed better performance than that of the FA0.92MA0.08PbI3-based previous system, which was attributed to the additional lattice stabilization through Cs-inclusion.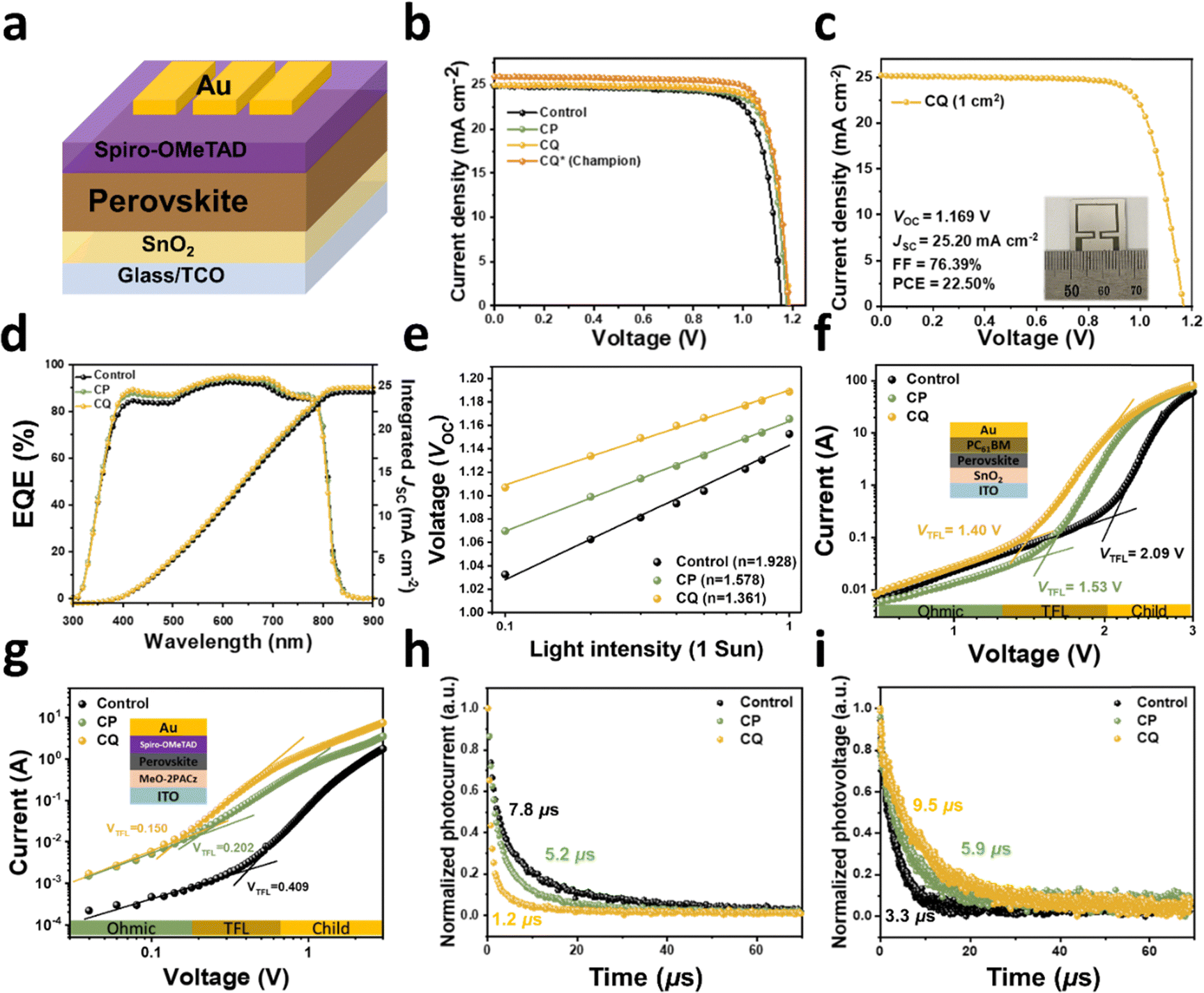 | ||
| Fig. 6 Photovoltaic performance. (a) N–i–p device architecture. (b) J–V curves of the control, CP- and CQ-treated devices (reverse scan). The champion device was fabricated with architecture of the FTO/chemical bath deposited SnO2 with the anti-reflecting film for enhanced current density. (c) J–V curve of the CQ-treated large-area device (area: 1 cm−2). (d) EQE spectra and integrated JSC plots. (e) The semi-logarithmic plots of light intensity versus VOC. SCLC measurements for the (f) electron-only devices (ITO/SnO2/perovskite/PC61BM/Au) and (g) hole-only devices (ITO/MeO-2PACz/perovskite/spiro-OMeTAD/Au). (h) TPC and (i) TPV measurements of the control, CP-, and CQ-based devices. | ||
| Device | V OC [V] | J SC [mA cm−2] | J SC cal [mA cm−2] | FF [%] | PCE [%] |
|---|---|---|---|---|---|
| a Calculated JSC by EQE measurements. b The average values and standard deviations obtained from 24 devices. c The champion device was fabricated with the architecture of FTO/chemical bath deposited SnO2/Cs0.05FA0.95PbI3/OAI/spiro-OMeTAD/Au (an anti-reflecting film was used to enhance the current density). | |||||
| Control | 1.153 (1.148 ± 0.017)b | 24.86 (24.49 ± 0.76)b | 24.32a | 78.81 (77.64 ± 2.46)b | 22.60 (21.84 ± 0.65)b |
| CP | 1.173 (1.163 ± 0.009)b | 24.96 (24.90 ± 0.56)b | 24.85a | 80.69 (80.00 ± 1.34)b | 23.63 (23.17 ± 0.48)b |
| CQ | 1.187 (1.170 ± 0.007)b | 24.99 (24.97 ± 0.35)b | 24.86a | 81.57 (80.29 ± 0.90)b | 24.19 (23.47 ± 0.57)b |
| Controlc (Champion) | 1.156 | 25.12 | 25.16 | 79.93 | 23.20 |
| CQc (Champion) | 1.183 | 25.95 | 25.81 | 81.45 | 25.00 |
| CQc (Certified) | 1.186 | 25.75 | — | 81.48 | 24.89 |
We also investigated the dependence of VOC and JSC on light intensity (PLight) to gain further insights into the recombination mechanisms within the devices, as depicted in Fig. 6e. The semi-logarithmic plot of PLightversus VOC can be analyzed using VOC ∝ (nkBT)/qln(PLight), where n, kB, T, and q represent the ideality factor, Boltzmann constant, temperature in Kelvin, and elementary charge, respectively. A lower slope approaching kBT/q indicates a reduced contribution from trap-assisted recombination.72,73 The calculated n values were found to be 1.69, 1.50, and 1.34 for the control, CP-, and CQ-treated devices, respectively. Additionally, the power-law equation JSC ∝ PLightα describes the PLightversus JSC relationship, where α denotes the extent of bimolecular recombination.74 The control device exhibits an α value of 0.989, whereas the CP- and CQ-treated devices have α values of 0.992 and 0.993, as evident from Fig. S46 (ESI†). Combined together, these results indicate that both trap-assisted and bimolecular recombinations are effectively mitigated by the presence of solid additives, especially in the case of CQ.
Furthermore, we conducted an assessment of how the incorporation of solid additives affects trap-state density (Nt) within the perovskite films. Dark J–V characteristics were collected for electron-only devices with the structure of ITO/SnO2/perovskite/[6,6]-phenyl-C61-butyric acid methyl ester (PC61BM)/Au and hole-only devices with the structure of ITO/MeO-2PACz/perovksite/spiro-OMeTAD/Au. As depicted in Fig. 6f and g, the dark J–V curves for all the perovskite devices exhibit a linear Ohmic response at low bias, followed by a trap-filled limit regime and a trap-free space-charge limited current (SCLC) regime. Nt and the trap-filled limit voltage (VTFL) were determined using the equation Nt = (2εε0VTFL)/(qL2), where ε and ε0 represent the relative dielectric constant of the perovskite (ε = 28.8) and the vacuum permittivity, respectively, and L denotes the thickness of the perovskite film (∼730 nm).75 For electron-only devices, the VTFL values for the control, CP-, and CQ-treated devices were 2.09, 1.53, and 1.40 V, corresponding to Nt values of 1.25 × 1016 cm−3, 0.92 × 1016 cm−3, and 0.84 × 1016 cm−3, respectively (Table S13, ESI†). In case of hole-only devices, the VTFL values for the control, CP-, and CQ-based devices were 0.41, 0.20, and 0.15 V, resulting in Nt values of 2.45 × 1015 cm−3, 1.20 × 1015 cm−3, and 0.90 × 1015 cm−3, respectively (Table S14, ESI†). These results confirm a reduction in trap density induced by the solid additives, consistent with the recombination studies mentioned earlier.
Transient photocurrent/photovoltage (TPC/TPV) decay curves were analyzed to investigate the behavior of charge carriers within the devices. TPC and TPV measurements were conducted under short-circuit and open-circuit conditions, respectively.76Fig. 6h reveals that photocurrent decay times, extracted from TPC, were 7.8 μs, 5.2 μs, and 1.2 μs for the control, CP-, and CQ-treated devices, respectively. The photovoltage decay time extracted from TPV (Fig. 6i) was 3.3 μs for the control device, but increased to 5.9 μs and 9.5 μs after treatment with CP and CQ, respectively. These results confirm the reduced recombination and improved charge extraction, especially in the case of CQ.
2.6. Stability evaluation
Finally, we compared the storage and thermal stabilities of the control, CP-, and CQ-treated devices. The storage stability test was conducted at 25 °C under 10% relative humidity without encapsulation, with the devices periodically exposed to simulated light for J–V testing. As depicted in Fig. 7a, the CP- and CQ-treated devices exhibited over 91.5% and 96.7% retention of their initial PCE values during 1608 hours, while the control device retained only 86.8%. Notably, the degradation in performance is primarily attributed to decreased FF, reflecting the decomposition of the perovskite crystals. We assessed the thermal stability of the devices at 85 °C in a nitrogen atmosphere. To exclude the influence of thermally unstable spiro-OMeTAD, the hole-transporting layer was replaced with PBDB-T-2F. Compared to a significant PCE degradation of 73.2% observed in the control device after continuous thermal stress for over 256 hours, the CP- and CQ-treated devices demonstrated excellent PCE retentions of 88.2% and 92.8% for over 312 hours, respectively, as depicted in Fig. 7b. Furthermore, we evaluated the operational stability of the encapsulated control and CQ-treated devices by maximum power point tracking (MPPT) under 1 sun illumination under ambient conditions (ISOS-L-1 protocol: temperature of 25 °C, ambient condition, Fig. 7c). The CQ-treated device maintained 90.1% of its initial PCE after 1000 h of MPPT, whereas the control device degraded to 80.8% of its original PCE after 500 h (Fig. S47 and Table S15, ESI†). | ||
| Fig. 7 Stability evaluation. (a) Storage stability of the control, CP-, and CQ-treated PSCs stored in a dry cabinet (∼25 °C, RH of ∼10%). (b) Thermal stability test of the control, CP-, and CQ-treated PSCs (∼85 °C, N2-filled glovebox). MPPT testing for the control and CQ-treated PSCs using (c) ISOS-L-1 and (d) ISOS-L-3, which was measured under full solar illumination (AM 1.5 G, 100 mW cm−2) without a UV filter. | ||
Additionally, the stability under combined heat and humidity stress was evaluated using the more rigorous ISOS-L-3 protocol, which involves MPPT at a temperature of 65 °C and a relative humidity of 50%. As shown in Fig. 5d, the CQ-treated device exhibits enhanced endurance against heat and humidity, maintaining 91.8% of its original PCE after 250 h, on the other hand, the control device degraded to 81.3% after 250 h. These improved stabilities can be attributed to the high-quality perovskite films with preferred orientation, as demonstrated previously, which provide a robust barrier against heat and light soaking conditions.
3. Conclusions
In summary, we introduced nature-sourced volatile solid additives, namely, CP and CQ, into PSCs to enhance their photovoltaic performance and stability. Through a comprehensive analysis using various characterization techniques, we observed distinct behaviors in the presence of these additives during the fabrication of PSCs via a two-stage thermal-annealing process. CP demonstrated complete volatilization during the first thermal-annealing step, resulting in the formation of a denser PbI2 film. In contrast, CQ exhibited a stepwise-volatilization mechanism due to its stronger Lewis base–acid interaction with perovskite, facilitated by its bidentate coordination capability. This unique behavior resulted in high-quality FA-based perovskite crystallinity characterized by large-grained perovskite structures and a highly preferred crystallite orientation. As a result, the treatment with these solid additives significantly improved the efficiency and stability of PSCs compared to the control devices, with CQ treatment being particularly notable. It achieved an impressive PCE of 25.00% with a certified PCE of 24.89% and demonstrated excellent PCE retentions of 90.1% after 1000 hours under MPPT conditions. This study reports a pioneering advancement in enhancing PSCs using nature-sourced solid additives, furthering the ecofriendly processability of PSCs. This approach shows promise for enhancing other perovskite-based optoelectronic devices as well.Author contributions
J. P. and C. Y. conceived the ideas and organized the work. J. P. conducted device fabrication and characterization and analyzed data. J. P. performed the morphology characterizations and analyzed the data. S. K. measured GIWAXS measurement and 207Pb NMR data. W. K. conducted TGA measurements. S. Z. performed AFM measurements. B. L. conducted zeta-sizer measurements. J. P. and C. Y. contributed to the preparation and revision of this manuscript.Data availability
The data that support the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (2021R1A2C3004202), and the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (20213091010010, Super Solar cells-development of double junction solar cells, breakthrough for the theoretical limit of silicon solar cell efficiency (>35%)).References
- H. Chen, A. Maxwell, C. Li, S. Teale, B. Chen, T. Zhu, E. Ugur, G. Harrison, L. Grater, J. Wang, Z. Wang, L. Zeng, S. M. Park, L. Chen, P. Serles, R. A. Awni, B. Subedi, X. Zheng, C. Xiao, N. J. Podraza, T. Filleter, C. Liu, Y. Yang, J. M. Luther, S. De Wolf, M. G. Kanatzidis, Y. Yan and E. H. Sargent, Nature, 2023, 613, 676–681 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- M. Jeong, I. W. Choi, E. M. Go, Y. Cho, M. Kim, B. Lee, S. Jeong, Y. Jo, H. W. Choi, J. Lee, J.-H. Bae, S. K. Kwak, D. S. Kim and C. Yang, Science, 2020, 369, 1615–1620 CrossRef CAS PubMed.
- M. Jeong, I. W. Choi, K. Yim, S. Jeong, M. Kim, S. J. Choi, Y. Cho, J.-H. An, H.-B. Kim, Y. Jo, S.-H. Kang, J.-H. Bae, C.-W. Lee, D. S. Kim and C. Yang, Nat. Photonics, 2022, 16, 119–125 CrossRef CAS.
- Q. Jiang, R. Tirawat, R. A. Kerner, E. A. Gaulding, Y. Xian, X. Wang, J. M. Newkirk, Y. Yan, J. J. Berry and K. Zhu, Nature, 2023, 623, 313–318 CrossRef CAS PubMed.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS PubMed.
- Y. Zhou, C. Fei, M. A. Uddin, L. Zhao, Z. Ni and J. Huang, Nature, 2023, 616, 712–718 CrossRef CAS PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef PubMed.
- J.-W. Lee, D.-J. Seol, A.-N. Cho and N.-G. Park, Adv. Mater., 2014, 26, 4991–4998 CrossRef CAS PubMed.
- C. Ma, M.-C. Kang, S.-H. Lee, S. J. Kwon, H.-W. Cha, C.-W. Yang and N.-G. Park, Joule, 2022, 6, 2626–2643 CrossRef CAS.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- C. K. MØLler, Nature, 1957, 180, 981–982 CrossRef.
- C. K. MØLler, Nature, 1958, 182, 1436 CrossRef.
- S. Pang, H. Hu, J. Zhang, S. Lv, Y. Yu, F. Wei, T. Qin, H. Xu, Z. Liu and G. Cui, Chem. Mater., 2014, 26, 1485–1491 CrossRef CAS.
- Y. Bai, Z. Huang, X. Zhang, J. Lu, X. Niu, Z. He, C. Zhu, M. Xiao, Q. Song, X. Wei, C. Wang, Z. Cui, J. Dou, Y. Chen, F. Pei, H. Zai, W. Wang, T. Song, P. An, J. Zhang, J. Dong, Y. Li, J. Shi, H. Jin, P. Chen, Y. Sun, Y. Li, H. Chen, Z. Wei, H. Zhou and Q. Chen, Science, 2022, 378, 747–754 CrossRef CAS PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS PubMed.
- J. Park, J. Kim, H.-S. Yun, M. J. Paik, E. Noh, H. J. Mun, M. G. Kim, T. J. Shin and S. I. Seok, Nature, 2023, 616, 724–730 CrossRef CAS PubMed.
- P. Shi, Y. Ding, B. Ding, Q. Xing, T. Kodalle, C. M. Sutter-Fella, I. Yavuz, C. Yao, W. Fan, J. Xu, Y. Tian, D. Gu, K. Zhao, S. Tan, X. Zhang, L. Yao, P. J. Dyson, J. L. Slack, D. Yang, J. Xue, M. K. Nazeeruddin, Y. Yang and R. Wang, Nature, 2023, 620, 323–327 CrossRef CAS PubMed.
- Y. Zhao, F. Ma, Z. Qu, S. Yu, T. Shen, H.-X. Deng, X. Chu, X. Peng, Y. Yuan, X. Zhang and J. You, Science, 2022, 377, 531–534 CrossRef CAS PubMed.
- X. Cao, L. Zhi, Y. Li, F. Fang, X. Cui, Y. Yao, L. Ci, K. Ding and J. Wei, J. Mater. Chem. C, 2017, 5, 7458–7464 RSC.
- C. Liu, X. Xu, Z. Zhang, S. Cao, J. Han, Y. Zheng, Z. Bi, Y. Zhu, S. Luo, G. Xu, S. Liu, K. Wang, Z. Ren, G. Li, Q. Deng and J. Liu, J. Solid State Chem., 2023, 326, 124195 CrossRef CAS.
- K. Liu, S. Rafique, S. F. Musolino, Z. Cai, F. Liu, X. Li, Y. Yuan, Q. Bao, Y. Yang, J. Chu, X. Peng, C. Nie, W. Yuan, S. Zhang, J. Wang, Y. Pan, H. Zhang, X. Cai, Z. Shi, C. Li, H. Wang, L. Deng, T. Hu, Y. Wang, Y. Wang, S. Chen, L. Shi, P. Ayala, J. E. Wulff, A. Yu and Y. Zhan, Joule, 2023, 7, 1033–1050 CrossRef CAS.
- R. Wang, A. Altujjar, N. Zibouche, X. Wang, B. F. Spencer, Z. Jia, A. G. Thomas, M. Z. Mokhtar, R. Cai, S. J. Haigh, J. M. Saunders, M. S. Islam and B. R. Saunders, Energy Environ. Sci., 2023, 16, 2646–2657 RSC.
- G. Zeng, G. Liu and X. Li, ACS Sustainable Chem. Eng., 2023, 11, 7664–7672 CrossRef CAS.
- P. Zhang, N. Gu, L. Song, X. Chen, P. Du, L. Zha, W.-H. Chen and J. Xiong, Nanoscale, 2022, 14, 5204–5213 RSC.
- T. Zhou, Z. Xu, R. Wang, X. Dong, Q. Fu and Y. Liu, Adv. Mater., 2022, 34, 2200705 CrossRef CAS PubMed.
- B. Fan, W. Zhong, W. Gao, H. Fu, F. R. Lin, R. W. Y. Wong, M. Liu, C. Zhu, C. Wang, H.-L. Yip, F. Liu and A. K. Y. Jen, Adv. Mater., 2023, 35, 2302861 CrossRef CAS PubMed.
- D. Hu, H. Tang, S. Karuthedath, Q. Chen, S. Chen, J. I. Khan, H. Liu, Q. Yang, J. Gorenflot, C. E. Petoukhoff, T. Duan, X. Lu, F. Laquai and S. Lu, Adv. Funct. Mater., 2023, 33, 2211873 CrossRef CAS.
- J. Oh, S. Jung, S.-H. Kang, G. Park, M. Jeong, S. Kim, S. Lee, W. Kim, B. Lee, S. M. Lee and C. Yang, J. Mater. Chem. A, 2022, 10, 20606–20615 RSC.
- J. Qin, Q. Yang, J. Oh, S. Chen, G. O. Odunmbaku, N. A. N. Ouedraogo, C. Yang, K. Sun and S. Lu, Adv. Sci., 2022, 9, 2105347 CrossRef CAS PubMed.
- L. Ye, Y. Cai, C. Li, L. Zhu, J. Xu, K. Weng, K. Zhang, M. Huang, M. Zeng, T. Li, E. Zhou, S. Tan, X. Hao, Y. Yi, F. Liu, Z. Wang, X. Zhan and Y. Sun, Energy Environ. Sci., 2020, 13, 5117–5125 RSC.
- R. Yu, H. Yao, L. Hong, Y. Qin, J. Zhu, Y. Cui, S. Li and J. Hou, Nat. Commun., 2018, 9, 4645 CrossRef PubMed.
- L. Zhong, S. Jeong, S. Lee, T. L. H. Mai, J. Park, J. Park, W. Kim and C. Yang, Chem. Commun., 2023, 59, 12108–12111 RSC.
- L. Zhong, S.-H. Kang, J. Oh, S. Jung, Y. Cho, G. Park, S. Lee, S.-J. Yoon, H. Park and C. Yang, Adv. Funct. Mater., 2022, 32, 2201080 CrossRef CAS.
- L. Zhong, Z. Sun, S. Lee, S. Jeong, S. Jung, Y. Cho, J. Park, J. Park, S.-J. Yoon and C. Yang, Adv. Funct. Mater., 2023, 2305450 CrossRef CAS.
- J. Burschka, N. Pellet, S.-J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- Y. Y. Kim, E. Y. Park, T.-Y. Yang, J. H. Noh, T. J. Shin, N. J. Jeon and J. Seo, J. Mater. Chem. A, 2018, 6, 12447–12454 RSC.
- L. Shen, P. Song, L. Zheng, L. Wang, X. Zhang, K. Liu, Y. Liang, W. Tian, Y. Luo, J. Qiu, C. Tian, L. Xie and Z. Wei, Adv. Mater., 2023, 35, 2301624 CrossRef CAS PubMed.
- S. Wang, H. Luo, Z. Gu, R. Zhao, L. Guo, N. Wang, Y. Lou, Q. Xu, S. Peng, Y. Zhang and Y. Song, Adv. Funct. Mater., 2023, 33, 2214834 CrossRef CAS.
- Z. Xiao, C. Bi, Y. Shao, Q. Dong, Q. Wang, Y. Yuan, C. Wang, Y. Gao and J. Huang, Energy Environ. Sci., 2014, 7, 2619–2623 RSC.
- Q. Jiang, Z. Chu, P. Wang, X. Yang, H. Liu, Y. Wang, Z. Yin, J. Wu, X. Zhang and J. You, Adv. Mater., 2017, 29, 1703852 CrossRef PubMed.
- S. M. Bouzzine, S. Bouzakraoui, M. Bouachrine and M. Hamidi, J. Mol. Struct.: THEOCHEM, 2005, 726, 271–276 CrossRef CAS.
- J. Guo, J. Sun, L. Hu, S. Fang, X. Ling, X. Zhang, Y. Wang, H. Huang, C. Han, C. Cazorla, Y. Yang, D. Chu, T. Wu, J. Yuan and W. Ma, Adv. Energy Mater., 2022, 12, 2200537 CrossRef CAS.
- R. Wang, J. Xue, L. Meng, J.-W. Lee, Z. Zhao, P. Sun, L. Cai, T. Huang, Z. Wang, Z.-K. Wang, Y. Duan, J. L. Yang, S. Tan, Y. Yuan, Y. Huang and Y. Yang, Joule, 2019, 3, 1464–1477 CrossRef CAS.
- Q. Cao, T. Wang, J. Yang, Y. Zhang, Y. Li, X. Pu, J. Zhao, H. Chen, X. Li, I. Tojiboyev, J. Chen, L. Etgar and X. Li, Adv. Funct. Mater., 2022, 32, 2201036 CrossRef CAS.
- Q. Yang, X. Liu, S. Yu, Z. Feng, L. Liang, W. Qin, Y. Wang, X. Hu, S. Chen, Z. Feng, G. Hou, K. Wu, X. Guo and C. Li, Energy Environ. Sci., 2021, 14, 6536–6545 RSC.
- J. Hu, R. A. Kerner, I. Pelczer, B. P. Rand and J. Schwartz, ACS Energy Lett., 2021, 6, 2262–2267 CrossRef CAS.
- P. Boonmongkolras, D. Kim, Esra M. Alhabshi, I. Gereige and B. Shin, RSC Adv., 2018, 8, 21551–21557 Search PubMed.
- T. Liu, Y. Jiang, M. Qin, J. Liu, L. Sun, F. Qin, L. Hu, S. Xiong, X. Jiang, F. Jiang, P. Peng, S. Jin, X. Lu and Y. Zhou, Nat. Commun., 2019, 10, 878 Search PubMed.
- C. Luo, G. Zheng, F. Gao, X. Wang, Y. Zhao, X. Gao and Q. Zhao, Joule, 2022, 6, 240–257 CrossRef CAS.
- M. E. O’Kane, J. A. Smith, R. C. Kilbride, E. L. K. Spooner, C. P. Duif, T. E. Catley, A. L. Washington, S. M. King, S. R. Parnell and A. J. Parnell, Chem. Mater., 2022, 34, 7232–7241 CrossRef PubMed.
- C. Ran, W. Gao, N. Li, Y. Xia, Q. Li, Z. Wu, H. Zhou, Y. Chen, M. Wang and W. Huang, ACS Energy Lett., 2019, 4, 358–367 Search PubMed.
- Y. Wu, G. Xu, J. Xi, Y. Shen, X. Wu, X. Tang, J. Ding, H. Yang, Q. Cheng, Z. Chen, Y. Li and Y. Li, Joule, 2023, 7, 398–415 Search PubMed.
- Y. Zhang, Y. Wang, X. Yang, L. Zhao, R. Su, J. Wu, D. Luo, S. Li, P. Chen, M. Yu, Q. Gong and R. Zhu, Adv. Mater., 2022, 34, 2107420 Search PubMed.
- H. Zhong, X. Liu, M. Liu, S. Yin, Z. Jia, G. Fu, S. Yang and W. Kong, Nano Energy, 2023, 105, 108014 CrossRef CAS.
- P. M. Moreno-Romero, A. N. Corpus-Mendoza, M. A. Millán-Franco, C. A. Rodríguez-Castañeda, D. M. Torres-Herrera, F. Liu and H. Hu, J. Mater. Sci.: Mater. Electron., 2019, 30, 17491–17503 CrossRef CAS.
- W. Shao, H. Wang, F. Ye, C. Wang, C. Wang, H. Cui, K. Dong, Y. Ge, T. Wang, W. Ke and G. Fang, Energy Environ. Sci., 2023, 16, 252–264 RSC.
- Y. Zhao, H. Tan, H. Yuan, Z. Yang, J. Z. Fan, J. Kim, O. Voznyy, X. Gong, L. N. Quan, C. S. Tan, J. Hofkens, D. Yu, Q. Zhao and E. H. Sargent, Nat. Commun., 2018, 9, 1607 CrossRef PubMed.
- T.-H. Kim, E. K. Jeon, Y. Ko, B. Y. Jang, B.-S. Kim and H.-K. Song, J. Mater. Chem. A, 2014, 2, 7600–7605 RSC.
- S. Song, S. J. Yang, W. Choi, H. Lee, W. Sung, C. Park and K. Cho, Adv. Energy Mater., 2020, 10, 2001759 CrossRef CAS.
- C. Zhu, X. Niu, Y. Fu, N. Li, C. Hu, Y. Chen, X. He, G. Na, P. Liu, H. Zai, Y. Ge, Y. Lu, X. Ke, Y. Bai, S. Yang, P. Chen, Y. Li, M. Sui, L. Zhang, H. Zhou and Q. Chen, Nat. Commun., 2019, 10, 815 CrossRef CAS PubMed.
- T. Yang, L. Gao, J. Lu, C. Ma, Y. Du, P. Wang, Z. Ding, S. Wang, P. Xu, D. Liu, H. Li, X. Chang, J. Fang, W. Tian, Y. Yang, S. Liu and K. Zhao, Nat. Commun., 2023, 14, 839 CrossRef CAS PubMed.
- L. Bi, Q. Fu, Z. Zeng, Y. Wang, F. R. Lin, Y. Cheng, H.-L. Yip, S. W. Tsang and A. K. Y. Jen, J. Am. Chem. Soc., 2023, 145, 5920–5929 CrossRef CAS PubMed.
- T. Huang, S. Tan, S. Nuryyeva, I. Yavuz, F. Babbe, Y. Zhao, M. Abdelsamie, M. H. Weber, R. Wang, K. N. Houk, C. M. Sutter-Fella and Y. Yang, Sci. Adv., 2021, 7, eabj1799 CrossRef CAS PubMed.
- K. Schötz and F. Panzer, J. Phys. Chem. A, 2021, 125, 2209–2225 Search PubMed.
- Y.-C. Lin, M. Bettinelli and M. Karlsson, Chem. Mater., 2019, 31, 3851–3862 CrossRef CAS.
- M. Liu, Q. Wan, H. Wang, F. Carulli, X. Sun, W. Zheng, L. Kong, Q. Zhang, C. Zhang, Q. Zhang, S. Brovelli and L. Li, Nat. Photonics, 2021, 15, 379–385 Search PubMed.
- D. Zhang, Y. Fu, H. Zhan, C. Zhao, X. Gao, C. Qin and L. Wang, Light: Sci. Appl., 2022, 11, 69 Search PubMed.
- F. Li, X. Deng, Z. Shi, S. Wu, Z. Zeng, D. Wang, Y. Li, F. Qi, Z. Zhang, Z. Yang, S.-H. Jang, F. R. Lin, S. W. Tsang, X.-K. Chen and A. K. Y. Jen, Nat. Photonics, 2023, 17, 478–484 Search PubMed.
- Y. Li, Y. Duan, J. Feng, Y. Sun, K. Wang, H. Li, H. Wang, Z. Zang, H. Zhou, D. Xu, M. Wu, Y. Li, Z. Xie, Z. Liu, J. Huang, Y. Yao, Q. Peng, Q. Fan, N. Yuan, J. Ding, S. Liu and Z. Liu, Angew. Chem., Int. Ed., 2024, 34, e202410378 Search PubMed.
- L. Yang, H. Zhou, Y. Duan, M. Wu, K. He, Y. Li, D. Xu, H. Zou, S. Yang, Z. Fang, S. Liu and Z. Liu, Adv. Mater., 2023, 35, 2211545 CrossRef CAS PubMed.
- D. Glowienka and Y. Galagan, Adv. Mater., 2022, 34, 2105920 CrossRef CAS PubMed.
- S. Ryu, N. Y. Ha, Y. H. Ahn, J.-Y. Park and S. Lee, Sci. Rep., 2021, 11, 16781 CrossRef CAS PubMed.
- S. Ryu, D. C. Nguyen, N. Y. Ha, H. J. Park, Y. H. Ahn, J.-Y. Park and S. Lee, Sci. Rep., 2019, 9, 19846 Search PubMed.
- M. Sajedi Alvar, P. W. M. Blom and G.-J. A. H. Wetzelaer, Nat. Commun., 2020, 11, 4023 Search PubMed.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2016, 2, 16177 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee03897e |
| This journal is © The Royal Society of Chemistry 2025 |