
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-fused core-linked star-shaped oligomer acceptors for stable binary organic solar cells with over 19% efficiency†
Cheng
Sun‡
a,
Jianxiao
Wang‡
a,
Fuzhen
Bi
 abc,
Huanxiang
Jiang
d,
Chunming
Yang
e,
Yonghai
Li
*abc,
Junhao
Chu
abc and
Xichang
Bao
*abc
abc,
Huanxiang
Jiang
d,
Chunming
Yang
e,
Yonghai
Li
*abc,
Junhao
Chu
abc and
Xichang
Bao
*abc
aKey Laboratory of Photoelectric Conversion and Utilization of Solar Energy, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China. E-mail: liyh@qibebt.ac.cn; baoxc@qibebt.ac.cn
bLaboratory of Solar Energy, Shandong Energy Institute, Qingdao 266101, China
cQingdao New Energy Shandong Laboratory, Qingdao 266101, China
dCollege of Textiles and Clothing, State Key Laboratory of Bio-fibers and Eco-textiles, Qingdao University, Qingdao 266071, China
eShanghai Synchrotron Radiation Facility Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai 201204, China
First published on 22nd November 2024
Abstract
Star-shaped oligomer acceptors are promising candidates for high-performance and robust organic solar cells (OSCs). However, the limited diversity of this community of acceptors leaves a significant knowledge gap regarding their structure–performance relationship. Herein, we designed two new star-shaped oligomer acceptors, namely 3BY and 3QY by introducing non-fused central units to bridge the Y-acceptor arms. The structural variability of the non-fused cores provides an available platform to finely regulate the aggregation properties of oligomers. In particular, the triazine center of 3QY allows multisite intramolecular non-covalent interactions, which can not only improve the molecular self-assembly, but also refine the pre-aggregation of the polymer donor and film-forming kinetics of the heterojunction blend. Finally, assembled PM6:3QY solar cells realized a very impressive efficiency of up to 19.27%, far outperforming that of PM6:3BY (17.75%) and ranking the highest efficiencies among the reported OSCs based on oligomer acceptors. Meanwhile, the considerable molecular sizes of the star-shaped molecules retard molecular diffusion, affording notable device stability with a large T80% over 3000 h for PM6:3QY device under thermal stress. This study establishes a reliable structure-performance relationship and demonstrates the great potential of non-fused core-bridged star-shaped oligomers for the fabrication of high-efficiency OSCs with long-term stability.
Broader contextThe past several years have witnessed the exciting development of organic solar cells (OSCs) due to the emergence of innovative small molecular acceptors. However, the inherent large diffusion coefficients of small molecules usually deteriorate the heterojunction morphology, resulting in unsatisfactory device stability and uncertain application prospects. Therefore, retarding molecular diffusion by extension of the molecular size is considered a reliable solution for obtaining robust heterojunction textures and solar cells. Star-shaped oligomer acceptors with three-dimensional extended molecular configurations have emerged as a good candidate for high-performance and stable OSCs. However, their limited diversity creates a significant gap toward understanding the structure–performance relationship, and thus hinders the advances of this community material. In this study, we report two new star-shaped oligomer acceptors, namely 3BY and 3QY, featuring non-fused cores to link the surrounding arms. The non-fused parts endow improved flexibility and adaptability to alleviate their constraint on the oligomer configurations and aggregations. Of them, 3QY with multisite intramolecular non-covalent interactions displays refined film-forming kinetics and heterojunction textures, realizing an outstanding efficiency up to 19.27% (the highest value among oligomer acceptors) and a notable T80% over 3000 h under continuous thermal annealing. This study presents a promising approach for high-performance and stable OSCs by engineering the star-shaped oligomers with versatile non-fused cores. |
Introduction
Organic solar cells (OSCs) with conspicuous merits of light-weight, translucence, and flexibility have promising applications in wearable electronic devices, indoor Internet of Things, and building integrated photovoltaics.1–3 The ongoing innovation of acceptors has flourished the development of OSCs. In particular, OSCs based on polymeric donors and small molecular acceptors have achieved over 19% power conversion efficiencies (PCEs)4–6 and even reached 20% very recently based on self-assembled monolayer hole-transporting materials.7–9 However, bulk-heterojunction (BHJ) solar cells containing abundant small molecular acceptors often suffer from insufficient device stability, ascribed to the degeneration of the morphology caused by the rapid diffusion of small molecules under various stresses.10–13 Therefore, there remains an urgent demand for innovative acceptors to modulate film-forming kinetics and heterojunction textures for more satisfactory PCEs and long-term stabilities.14–18Oligomer acceptors are a class of promising candidates to break through the current challenges faced by organic photovoltaics.19–22 Compared to small molecular acceptors, oligomers are typically equipped with relatively larger molecular sizes and molecular weights (MWs), which dictate greater glass-transition temperatures (Tg) and thus lower diffusion coefficients for robust morphologies.23–25 Currently, most oligomer acceptors, including dimers and trimers, feature linear molecular configurations, which endow them with polymer-like characteristics and intense molecular aggregations.26,27 Nevertheless, OSCs with over 18% efficiency and improved stabilities have been reported. For example, Sun recently designed a linear heterotrimer acceptor, namely TQT, by integrating two different Y-monomers with benzothiadiazole and quinoline core moieties,28 and the assembled PM6:TQT-based device realized a decent PCE of 18.52% with exceptional device stability. It should be noted that linear oligomer acceptors with uniform molecular ordering usually exhibit strong self-aggregation properties, making it greatly difficult to optimize the film-forming kinetics of BHJ systems for achieving the optimum exciton/charge states.29–35 Under these circumstances, star-shaped oligomers are receiving increasing attention as an alternative to manage molecular aggregation.
Typical star-shaped oligomer acceptors are equipped with at least three spatially arranged acceptor arms, which can provide multiple-dimension channels through orbital hybridizations for exciton dissociation and charge transport. Meanwhile, their enlarged MW and molecular size are conducive to elevating Tg, which can help enhance device stability.36–39 Fully conjugated multiple-arm frameworks have been realized by fusing two or more Y-acceptors at the benzothiadiazole (BT) moiety through a dehydration reaction between diamine intermediates and ketones. For example, Li and coworkers reported the fully fused star-shaped oligomer DP-BTP,26 which showed a greater Tg than that of a small molecule acceptor and thus enhanced device stability. However, the binary device exhibited a moderate PCE of 15.08% due to its excessive self-aggregation behavior. Another molecular design strategy is to connect the endcaps of Y-acceptors with multisite central moieties through high-yield transition metal-catalyzed coupling reactions. Recently, Kim and Wei separately reported one three-arm oligomer by employing the identical electron-donating fused benzotrithiophene (TBDT) as the central part.38,39 As expected, the resultant OSC demonstrated improved long-term stability due to its greater Tg parameter. More impressively, the oligomer G-Trimer device realized an efficiency of 18.39%, which was further improved to 19.01% by employing a 2PACZ hole-transport layer. These results suggest the great promise of star-shaped acceptors obtained by rational molecular design for high-performance and robust solar cells. However, one possible demerit of such a kind of oligomer is that the constraint of the central parts may disturb configuration relaxation, which may deteriorate the film-forming procedures and molecular orientations.40–43 Unfortunately, star-shaped oligomers have been under researched and under developed so far, meaning there is limited molecular diversity among them and insufficient information about their structure–efficiency relationship. In particular, the core moieties are expected to generate multifaceted impacts on the molecular configurations, self-aggregations, and intermolecular interactions, which could collectively control the heterojunction microstructures and exciton/charge kinetics of solar cells. Therefore, an in-depth study of the mechanisms of star-shaped acceptors remains highly required.
Herein, we designed and report two new star-shaped acceptors, namely 3BY and 3QY, featuring non-fused central regions (see the chemical structures in Fig. 1(a)). The design concepts are briefly discussed as follows. (i) Non-fused cores with more flexibility and adaptability were employed to alleviate the constraint effect on the entire molecular configurations and aggregation properties. (ii) Phenyl- and triazine-based central parts were constructed separately, to investigate for one thing, the opposite electronic effect, and for another, the configuration modulation of the non-covalent intramolecular interactions. (iii) Triazine units with multiple nitrogen atoms are expected to enhance intermolecular interactions. Encouragingly, it was found that the release of molecular tension between the central parts and Y-arms optimized the self-aggregation characteristics and molecular ordering. Meanwhile, the moderately limited configuration flexibility of 3QY enabled appropriate interactions with the donor PM6 skeleton, allowing the preferred pre-aggregation and solution-to-film transformation kinetics for admirable phase separation. Besides, the larger contribution of delocalized singlet exciton species to charge transfer in the PM6:3QY system further improved the exciton/charge behaviors. Eventually, an outstanding PCE of up to 19.27% was achieved from the assembled PM6:3QY binary device, superior to that of the PM6:3BY device (17.75%) and ranking the highest among OSCs based on oligomer acceptors reported to date. The improved Tg of 3QY compared to that of 3BY enabled enhanced device stability, with a greater evaluated T80% of over 3000 h for the PM6:3QY device compared to that of PM6:3BY (720 h) under continuous thermal stress. These discoveries suggest the significant effects of manipulations of the molecular center on the photoelectronic performance of star-shaped acceptors and highlight the great potential of non-fused cores for efficient and robust organic solar cells.
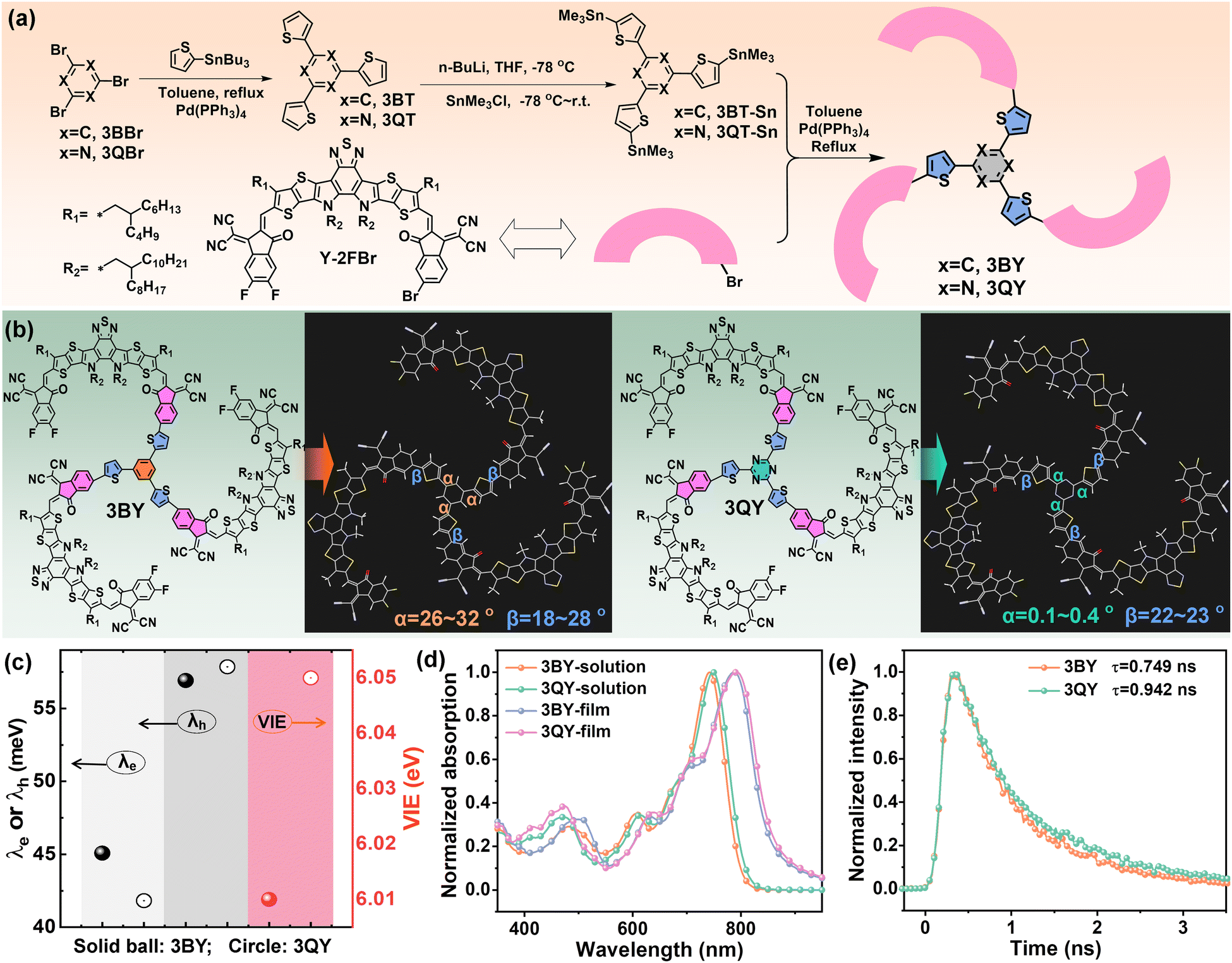 | ||
| Fig. 1 (a) Synthetic routes of the two star-shaped oligomers, 3BY and 3QY. (b) Chemical structures and optimized molecular configurations. (c) Calculated internal reorganization energies (λe, λh) and VIE. (d) Absorption spectra of the oligomers in dilute chloroform solutions and as thin films. (e) TRPL spectra of the neat film. | ||
Results and discussion
The synthetic approaches for the two oligomers are given in Fig. 1(a), with the synthesis details and characterizations provided in the ESI.† By connecting the core 3BT or 3QT with an asymmetrical acceptor Y-2FBr through a Stille coupling, the two oligomers 3BY and 3QY were obtained with notable yields of over 70% for the coupling procedure. At first, we investigated the core-mediated molecular configurations based on density functional theory (DFT). Fig. 1(b) gives the optimized molecular configurations of the two oligomers, which showed comparable fan-shaped architectures with the three Y-blades relatively evenly surrounding the central region, affording quasi three-dimensional (3D) molecular structures (Fig. S1, ESI†). We then focused on the core segments 3BT and 3QT to disclose the distinctions. As depicted in Fig. 1(b), the two molecules had similar dihedral angles between the thiophene linkers and Y-arms (labeled as β), but were dramatically different in terms of the dihedral angles between the benzene or triazine center and the adjacent thiophenes (labeled as α). The 3BT part showed a largely twisted molecular structure with significant α values of 26°32°. On the contrary, the 3QT segment had excellent molecular planarity with negligible α values smaller than 0.4°, which were attributed to the diminished steric hindrance and further enhanced non-covalent N⋯H interactions induced by the triazine center.44,45 In addition, it seems that the electronic property of the core segments had limited impacts on the molecular frontier orbitals of the oligomers (Fig. S2, ESI†). This conclusion was further confirmed by comparing the highest occupied molecular orbitals (HOMO) and lowest unoccupied molecular orbitals (LUMO) of the two acceptors obtained from DFT calculations and cyclic voltammetry (CV) measurements (Fig. S3, ESI†). We also calculated the internal reorganization energy (λint), which is an important metric of energetic cost for structural relaxation after ionization (Table 1 and Fig. 1(c)). Interestingly, 3QY exhibited a smaller electron internal reorganization energy (λe) of 41.82 meV than that of 3BY with a λe of 45.08 meV. According to Marcus–Levich–Jortner formalism, which describes the relationship between the electron-transfer rate and internal reorganization energy,46–48 3QY should favor a faster electron-transfer rate and higher intermolecular electron mobility. Likewise, the equally large hole internal reorganization energies (λh) (up to 57 meV) suggest the inferior hole-transport capacities of the two acceptors. Besides, 3QY was equipped with a slightly greater vertical ionization energy (VIE) of 6.05 eV than that of 3BY (6.01 eV), which is in favor of harvesting electrons from donors more efficiently.| MW (g mol−1) | λ max (nm) |
E
optg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (eV)
(eV) |
HOMO/LUMO (eV)CV | λ e (meV) | λ h (meV) | VIE (eV) | ||
|---|---|---|---|---|---|---|---|---|
| Solution | Film | |||||||
| a Calculated from the onset wavelength of the film absorption spectra. | ||||||||
| 3BY | 5660 | 742 | 787 | 1.43 | −5.65/−3.80 | 45.08 | 56.92 | 6.01 |
| 3QY | 5663 | 748 | 792 | 1.42 | −5.67/−3.83 | 41.82 | 57.86 | 6.05 |
The basic photophysical properties were investigated by ultraviolet-visible near-infrared absorption spectroscopy (UV-vis), steady-state photoluminescence (PL), and time-resolved photoluminescence (TRPL) spectroscopy. Albeit with very similar absorption profiles, 3QY demonstrates mildly red-shifted absorption profiles in both dilute solution and thin film (Fig. 1(d)), revealing the enhanced intra- and inter-molecular interactions by the triazine segment. Based on the onset wavelengths of film absorption, the optical bandgaps (Eoptg) of 3BY and 3QY were determined to be 1.43 and 1.42 eV, respectively. The PL spectra of the neat films (Fig. S4, ESI†) suggested the remarkably stronger emission from 3QY, which can help suppress the undesirable non-radiative energy according to previous literature.49 Besides, the TRPL spectrum of 3QY suggested a slightly delayed decay process, yielding a longer lifetime (τ) of 0.942 ns than that of 3BY (Fig. 1(e)). The prolonged fluorescence lifetime of 3QY could be expected to suppress geminate recombination and was partially correlated with its less intrachain disorder.
In the following, we focus on the aggregation behaviors of the two star-shaped acceptors in dilute solutions, thin films, and film-forming processes. Variable temperature absorptions were initially measured to evaluate the temperature-dependent-aggregation (TDA) properties. As given in Fig. 2(a) and (b), both the acceptors exhibited comparable TDA properties in chlorobenzene solutions. Upon heating, the 0–0 absorption peaks were gradually blue-shifted with a generally reduced intensity, suggesting de-aggregated intermolecular relaxation. The film absorption spectra were further combined to determine the TDA properties. Apparently, the final 0–0 peaks (110 °C) lay in between the 0–0 and 0–1 peaks of the film absorptions. This confirmed the notable pre-aggregations of the two oligomers, which could be collectively attributed to their partial 3D configurations, considerable MW over 5600 Da (Table 1), and resultant polymer-like behaviors with moderate molecular entanglements. In addition, 3BY had a routine TDA propensity with a gradually reduced absorbance, which however, decreased first and then increased for 3QY (Fig. 2(c)). This counterintuitive aggregation property may suggest that the 3QY molecule could find adjacent molecules to partly rearrange in hot solutions where intense molecular movements can occur. The unique phenomenon of 3QY could be correlated with its distinctive triazine center, featuring strong electronegativity and a N-heteroatom effect.
 | ||
| Fig. 2 (a) and (b) Temperature-dependent absorptions of 3BY and 3QY in dilute chlorobenzene (CB) solutions, and (c) corresponding evolutions of the maximum absorptions. (d) and (e) 2D patterns of the GIWAXS measurements. (f) Overlapping peak fitting and resolving of the (010) diffractions in the OOP direction. (g) and (h) 2D in situ absorption patterns and (i) corresponding time-dependent maximum absorption shifts of 3BY and 3QY casted from chloroform solutions (10 mg mL−1). | ||
Next, grazing incidence wide-angle X-ray scattering (GIWAXS) measurements were conducted to study the crystallinity and molecular orientations of the neat films.50 As shown in Fig. 2(d) and (e), and Fig. S5 (ESI†), both the oligomers delivered typical face-on orientations with dominating π–π diffractions (010) favoring intermolecular charge hopping. For more specific information, the (010) peak was resolved into two separate diffraction components, labeled as peak 1 and peak 2, respectively (Fig. 2(f)). The parameters of the π–π stacking spacing (dπ–π) and crystal coherence lengths (CCLs) for each peak are summarized in Table S1 (ESI†). Judging from the limited molecular spacings of peak 1 (d = ∼3.60 Å) and peak 2 (d = ∼3.80 Å), both the diffraction components should arise from π–π stacking but with different aggregation structures. In particular, peak 2 delivered much higher crystallinity with a doubled CCL of ∼2.20 nm compared to peak 1 (1.03 nm). Therefore, it is reasonable to define peak 1 and peak 2 as sub-crystalline and crystalline diffractions, respectively. As suggested in Fig. 2(f) and Table S1 (ESI†), both the crystalline and sub-crystalline components were slightly enhanced from 3BY to 3QY. From 3BY to 3QY, the crystalline profile was enhanced with a reduced dπ–π from 3.83 to 3.80 Å and increased CCL from 2.19 nm to 2.26 nm. The improved molecular ordering of 3QY should arise from its outstanding planar configuration of the core region assisted by intramolecular conformational locks. Synergistically contributed by the preferred molecular orientation and lower internal reorganization energy (Table 1), a greater electron mobility of the 3QY neat film of 0.83 × 10−4 cm2 V−1 s−1 was achieved compared to that of 3BY (0.59 × 10−4 cm2 V−1 s−1) based on the space-charge-limited current (SCLC) model (Fig. S6 and Table S2, ESI†).
To figure out the film-formation characteristics of the two star-shaped oligomers, in situ absorption measurements during spin-coating from chloroform solutions were performed.51,52Fig. 2(g) and (h) present the 2D-absorption contour plots. Generally, the film-formation procedure can be divided into three individual steps, including solution-state, solution-to-film transformation, and post-film stage.53 In the transformation stage, where molecules reach a critical concentration and start to aggregate into orderly aggregations brick by brick, one can extract the time and magnitude of absorption shifts for monitoring the film-forming kinetics.5 The peak shifts for the maximum absorptions of 3BY and 3QY are shown in Fig. 2(i) with the corresponding absorption spectra provided in Fig. S7 (ESI†). During the transformation stage, both the oligomers displayed similar red-shifts by ∼34 nm, but, however, experienced rather different transformation durations; where by 3BY endured a slow and gradual transition with a relevant time of 162 ms, while 3QY went through a quick and straight transition with the duration dramatically shortened to 85 ms. The stepwise transformation of 3BY may correspond to the emergence of different aggregation structures, which would result in more irregular and nonuniform aggregates. Contrarily, 3QY experienced a more homogeneous nucleation pathway, generating ordered assemblies quickly and completely, as partly supported by the above results based on GIWAXS (Fig. 2(f)). The accelerated film-formation of 3QY should be attributed to the stronger intermolecular interactions modulated by the unique molecular center.
Conventional BHJ solar cells with an architecture of glass/ITO/PEDOT:PSS/active layer/PDINN/Ag were fabricated to explore the photovoltaic performances of the two star-shaped acceptors. Here, polymer PM6 was employed as a donor to match the acceptors. Details of the device fabrication and optimization are provided in the ESI,† with relevant data collected in Table S3 (ESI†). The optimal current density–voltage (J–V) plots and photovoltaic parameters are shown in Fig. 3(a) and Table 2, respectively. The PM6:3BY device demonstrated an optimized PCE of 17.75%, with a large open-circuit voltage (VOC) of 0.969 V, short-circuit current density (JSC) of 23.92 mA cm−2, and fill factor (FF) of 76.59%. More impressively, the PM6:3QY device realized a champion PCE of up to 19.27%, with VOC of 0.951 V, improved JSC of 26.36 mA cm−2, and FF of 76.86%. Notably, the efficiency of the PM6:3QY device was certified by the National Center of Inspection on Solar Photovoltaic Products Quality (CPVT). The certified PCE of the PM6:3QY solar cell reached 18.80%, with a VOC of 0.955 V, JSC of 26.02 mA cm−2, and FF of 75.62% (Table 2 and Fig. S8, ESI†). Fig. 3(b) displays some reported high-performance OSCs (PCE ≥ 17%) based on oligomer acceptors in the previous literature compared with this study, with the relevant photovoltaic parameters summarized in Table S4 (ESI†). It can be observed that most of the enumerated PCEs were below the line of 19%, and the efficiency of 3QY undoubtedly ranked as the highest among all the reported oligomer acceptors so far, including linear and star-shaped oligomers. Fig. 3(c) depicts the EQE profiles of PM6:3BY and PM6:3QY solar cells, which suggests an improvement of PM6:3QY in nearly the full wavelength range from 300–900 nm and hence its more efficient exciton utilization efficiency. Besides, the EQE profiles were globally consistent with the absorption spectra of the corresponding active layers (Fig. S9, ESI†). Based on the EQE spectra, the JEQESC of PM6:3BY and PM6:3QY devices were found to be 22.86 and 25.10 mA cm−2, respectively, with small deviations of less than 5% compared to the recorded JSC. In previous work, Li and colleagues reported two typical star-shaped acceptors named G-Trimer-C6C8 and G-Trimer-C8C10, respectively.39 The G-Trimer acceptors, with chemical structures shown in Fig. S10 (ESI†), featured a completely rigid π-conjugated electron-rich core benzotrithiophene unit. As reported, the PM6:G-Trimer series solar cells with a universal PEDOT:PSS hole-transport layer outputted high PCEs over 18%. However, the corresponding VOCs were only 0.899 and 0.911 V, both of which were significantly smaller than those of PM6:3BY (0.969 V) and PM6:3QY (0.951 V). These results highlight the amazing regulation of the non-fused cores of star-shaped oligomers on energetics modification for elevated photovoltages and photovoltaic efficiencies. As is well-acknowledged, the VOCs of solar cells are controlled by energy losses (Eloss) during the period of photoelectronic conversion.54 Based on the onset wavelength of the EQE spectra, very small Eloss values of 0.465 and 0.481 eV were determined for the PM6:3BY and PM6:3QY devices. The photocurrent density (Jph) vs. effective voltage (Veff) was plotted to probe the charge generation and collection properties of solar cells. As suggested in Fig. 3(d), both PM6:3BY and PM6:3QY devices exhibited a very high exciton dissociation probability (ηdiss) of over 99%. However, the latter one had a greater charge collection probability (ηcoll) of 90.30% than that of PM6:3BY (88.59%), partly supporting the larger JSC of the PM6:3QY device. In the following, we report our investigations of the stability of solar cells based on 3BY and 3QY. The two optimal unencapsulated devices were put on a hot plate set at 80 °C in a glove box. As depicted in Fig. 3(e), after continuous annealing for 1315 h, the PM6:3QY device could maintain a larger PCE preservation of 84% compared to that of PM6:3BY (77%). The T80% of the PM6:3QY device could be extrapolated to be 3056 h, suggesting satisfactory long-term device stability under continuous thermal stress. As shown in Fig. S11 (ESI†) and Fig. 3(f), the improved stability of the PM6:3QY device could be attributed to the more robust Tg of 3QY (160 °C) than that of 3BY (132 °C). In spite of the similar molecular weights of 3BY and 3QY, the greater Tg of 3QY could be further correlated with its stronger crystallinity and the intermolecular interactions of the acceptor with a more coplanar molecular configuration arising from the distinctive triazine center.
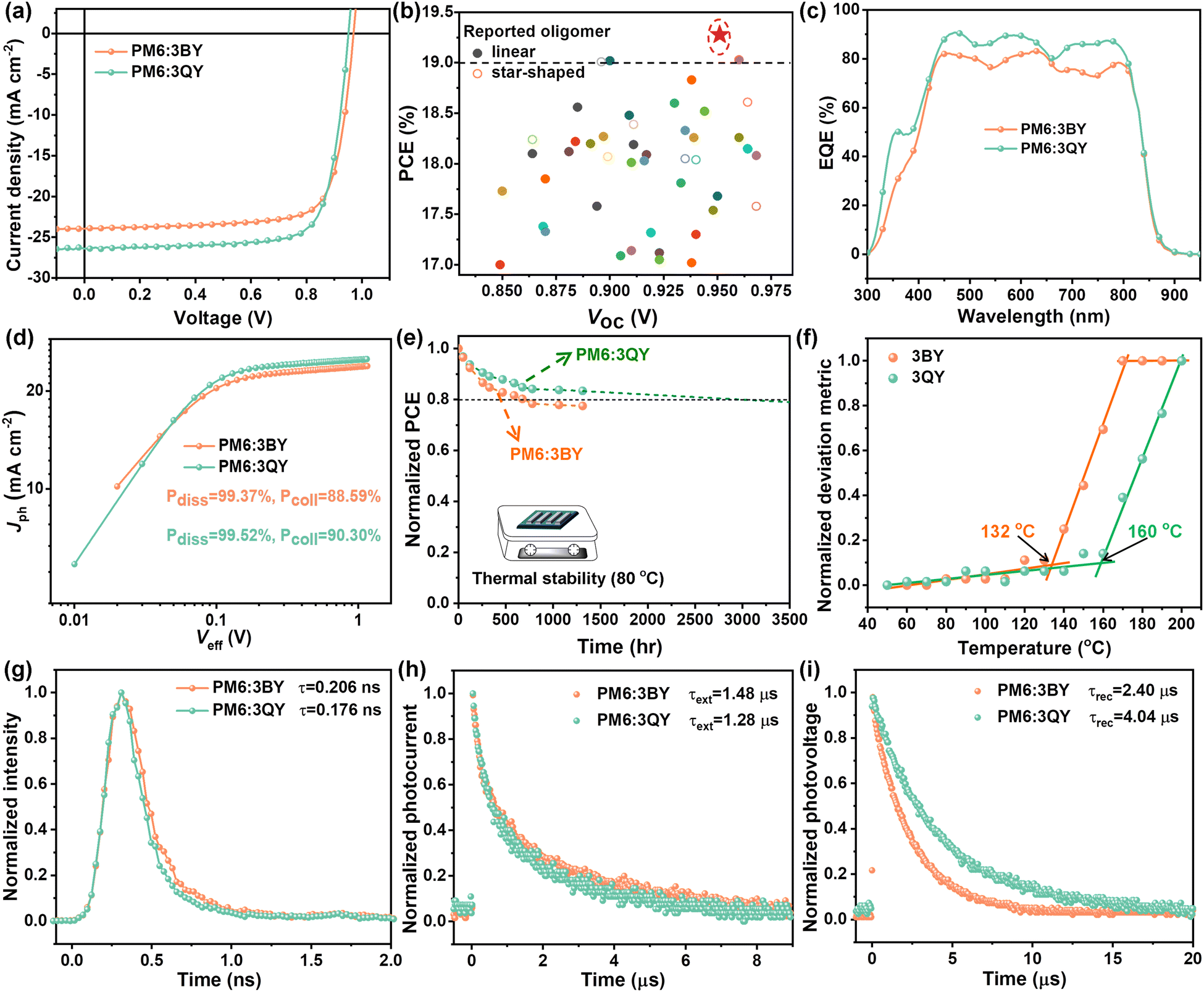 | ||
| Fig. 3 (a) Optimal J–V plots of PM6:3BY and PM6:3QY solar cells. (b) Comparison of the typical OSCs (PCE ≥ 17%) based on oligomer acceptors in the literature and 3QY. (c) Corresponding EQE profiles. (d) Jphvs. Veff plots. (e) Thermal stabilities under continuous heating at 80 °C. (f) Evolutions of the deviation metric as a function of annealing temperature for 3BY and 3QY films. (g) TRPL decay plots of PM6:3BY and PM6:3QY blend films. (h) and (i) Normalized transient photocurrent and photovoltage decay plots. | ||
| Active layer | V OC (V) | J SC (JEQESC) (mA cm−2) | FF (%) | PCEa (%) | E loss (eV) |
|---|---|---|---|---|---|
| a Average parameters with standard deviations obtained from 10 devices. b Calculated based on the onset wavelength of the EQE spectra. c Certificated by the National Center of Inspection on Solar Photovoltaic Products Quality (CPVT). | |||||
| PM6:3BY | 0.969 | 23.92 (22.86) | 76.59 | 17.75 (17.57 ± 0.18) | 0.465 |
| PM6:3QY | 0.951 | 26.36 (25.10) | 76.86 | 19.27 (19.06 ± 0.20) | 0.481 |
| PM6:3QYc | 0.955 | 26.02 | 75.62 | 18.80 | |
We next investigated the exciton dissociation, charge extraction, and recombination kinetics. As indicated in Fig. 3(g), the TRPL spectrum of the PM6:3QY blend showed a faster decay, resulting in a smaller fluorescence lifetime of 0.176 ns, compared to that of the PM6:3BY blend (0.206 ns). By integrating the PL spectra of neat acceptors and donor:acceptor blends, we could find that PM6:3QY had a higher quench efficiency of 97% than that of PM6:3BY (92%) (Fig. S4, ESI†). These results suggest the more efficient charge transfer at the interfaces between PM6 and 3QY, which should partly arise from the greater VIE of 3QY than that of 3BY (Table 1). In the following, we explore the charge-recombination mechanism of the two solar cells by plotting JSC–Plight and VOC–Plight curves through measuring J–V under gradient light intensities (Plight). As Fig. S12(a) (ESI†) shows, the two devices exhibited negligible bimolecular recombination based on the large pre-exponential factor α approaching 1, according to the equation JSC ∝ Pαlight.55,56 However, considering the smaller slope (s = kT/q) value of the PM6:3QY device obtained from the equation VOC ∝ nkT/qlnPlight, where k, T, and q represent the Boltzmann constant, Kelvin temperature, and elementary charge, respectively, the PM6:3QY device should suffer relatively less trap-assisted recombination compared to that of PM6:3BY (Fig. S12(b), ESI†).57,58 Transient photocurrent (TPC) and transient photovoltage (TPV) measurements were next carried out to investigate the charge extraction and recombination dynamics.59 As shown in Fig. 3(h), the slightly smaller charge-extraction times (τext) of the PM6:3QY (1.28 μs) device than that of PM6:3BY (1.48 μs) suggested its faster charge-extraction process. As acquired from the TPV decay plots (Fig. 3(i)), the PM6:3QY solar cell delivered a notably delayed decay, resulting in a prolonged charge-carrier lifetime or recombination time of free charges (τrec) of 4.04 μs compared to that of PM6:3BY (2.40 μs). A larger charge-carrier lifetime is crucial to reduce the charge-recombination probability, and well supports the excellent photovoltaic performance of PM6:3QY.
Femtosecond transient absorption (fs-TA) spectra were further investigated to study the charge-transfer kinetics.60 The 2D maps of the TA spectra of the two blend films are given in Fig. 4(a) and (d), with the corresponding spectra for the decay times displayed in Fig. S13 and S14 (ESI†). Comparable spectra could be observed for the two blend films with notable acceptor ground-state-bleaching (GSB) signals at 700–850 nm. The GSB absorption of PM6 emerged at 640 nm and increased in the first 10 ps. As the blend films were excited exclusively for the acceptors, the GSB appearance of PM6 confirmed successful hole transfer (HT) from the acceptors to PM6. By fitting the HT kinetics with a biexponential function, we could obtain the timescale for exciton dissociation and diffusion. As Fig. 4(b) depicts, the smaller component (τ1) and the larger component (τ2) correspond to the time required for exciton dissociation at the donor/acceptor interfaces, and the time for exciton diffusion toward the interfaces prior to dissociation, respectively. Both the two blends had comparable τ1 of ∼0.3 ps and thus equally ultrafast exciton dissociation; however, PM6:3QY exhibited a greater τ2 than that of PM6:3BY, which could be attributed to the improved crystallinity of the former blend. The positive absorption centered at 913 nm belonged to local exciton (LE) of the acceptors, for which 3QY had a slightly higher lifetime than that of 3BY. By integrating the LE of the acceptors and GSB of PM6, near-simultaneous transformation could be observed. Besides, another notable positive absorption at greater than 1200 nm caught our attention, which was attributed to the excitation species of the delocalized singlet exciton (DSE). The DSE species is a critical intermediate state from the LE to charge separated states, which facilitates the generation of free charge carriers even under a small energetic offset.61 As shown in Fig. 4(c), the decay of the LE and rise of DSE occurred at a timescale of ∼0.4 ps in the two blend films, suggesting ultrafast conversion from the short-lived LE state to long-lived DSE intermediate. For comparison, we measured the TA spectra of neat acceptors of 3BY and 3QY under an identical pump wavelength (Fig. S15–S18, ESI†). Notably, more conspicuous DSE signals were detected in the neat acceptors. To describe the evolutions of this significant intermediate species, we compared the decay kinetics of DSE sites in the neat acceptors and blend films (Fig. 4(e)). The high DSE concentrations in the neat acceptors were sharply quenched in the blend systems, accompanied with notable curtate excited state lifetimes. Specifically, both the neat acceptors had fairly long-lived DSE states with over 1000 ps lifetimes. In comparison, the lifetimes decreased to 730 ps and 625 ps in the PM6:3BY and PM6:3QY blends, respectively. The synchronous quenching of both the intensity and exciton lifetime in the blend films suggested the participation of DSE intermediates in the HT pathways, which was more effective in the PM6:3QY system. This contribution could be one important factor to the greater JSC of photovoltaic devices.61 In addition, we also plotted the decay kinetics of the excited-state absorption (ESA) located at ∼550 nm (Fig. 4(f)), which revealed a similar evolution tendency with the DSE absorption and thus an efficient HT contribution in solar cells.
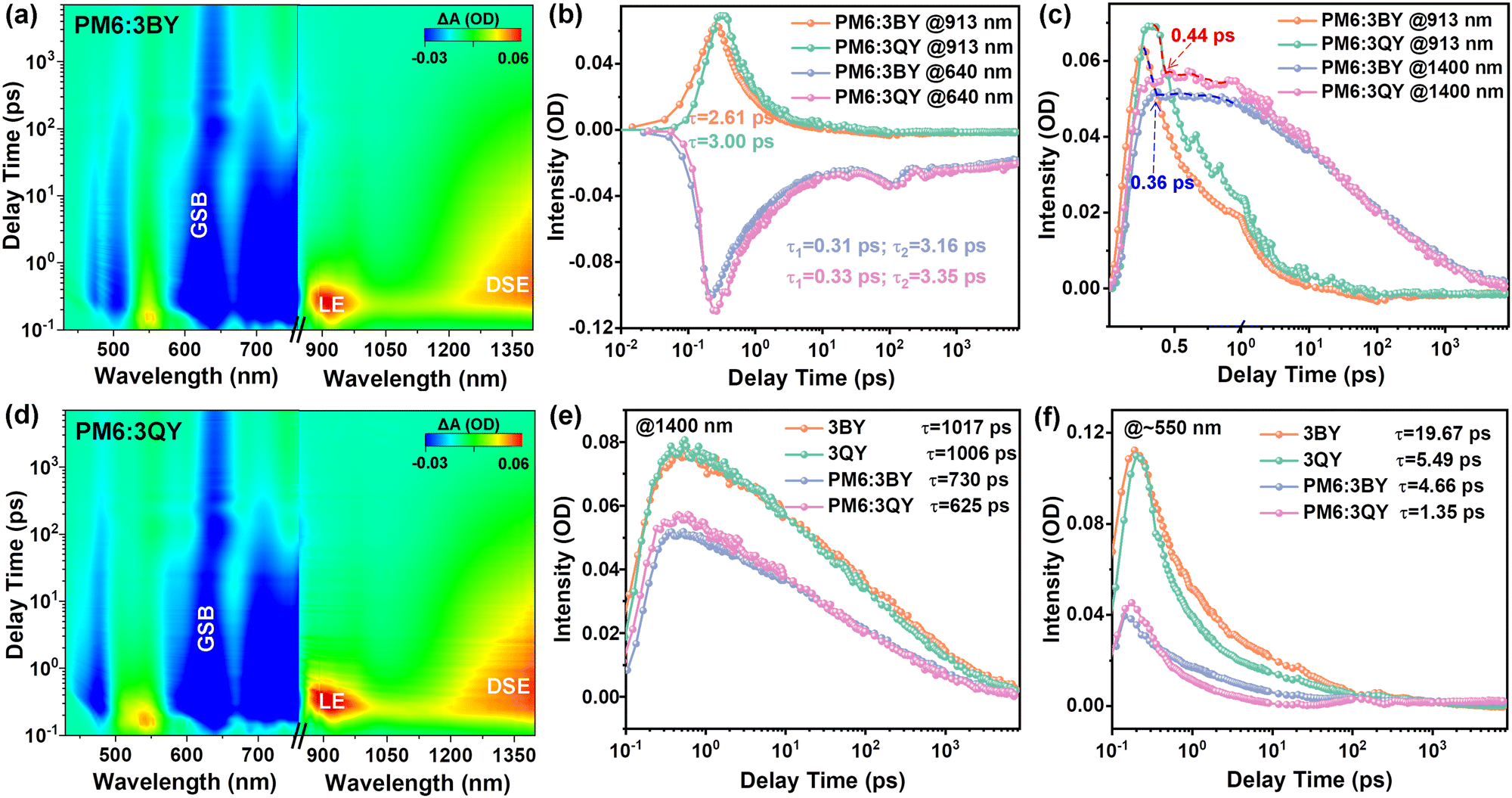 | ||
| Fig. 4 (a) and (d) 2D maps of the fs-TA spectra of PM6:3BY and PM6:3QY blend films in the range of 430–760 nm and 840–1400 nm with pump at 780 nm. (b) TA traces showing the hole-transfer kinetics. (c) TA traces of the conversion between LE and DSE excited species. (e) TA traces of the DSE species in neat acceptors and blend films. (f) TA traces of the excited species probed at ∼550 nm in neat acceptors and blend films. | ||
The morphology and aggregation properties of the active layer were subsequently investigated. As described in Fig. 5(a) and (d), similar fibrous phase-separated textures were observed for the two blends based on the obtained atomic force microscopy (AFM) images (Fig. S19, ESI†), with a smaller root-mean square roughness (RMS) of 1.80 nm for PM6:3QY than that of PM6:3BY (2.16 nm). From the transmission electron microscopy (TEM) images, we unexpectedly discovered larger domains in the PM6:3QY blend. Therefore, these microscopic results reveal the greater crystallinity of the PM6:3QY blend with nice fibrous phase-separations and a smooth surface, which favored the transportation and collection of free carriers more efficiently. GIWAXS studies were next carried out to explore the molecular crystallinity and orientations (Fig. 5(b), (c), and (e)). The two blends displayed alike (010) diffraction peaks in the OOP direction with a qz of ∼17 nm−1. However, PM6:3QY had a slightly greater CCL010 of 2.22 nm than that of PM6:3BY (2.00 nm) and hence a more expanded molecular aggregation. Because of the very close (010) diffraction locations of the acceptors (qz = 16.67, 16.86 nm−1) and PM6 (qz = 16.93 nm−1) (Fig. S20, ESI†), it was unreliable to distinguish the π–π packing behaviors of the donor and acceptor in their blends. Fortunately, the well-defined (100) diffraction plots in the IP direction provided a reliable platform to investigate the donor and acceptor separately. As given in Fig. S21 (ESI†), the (100) diffractions could be perfectly divided into two splitting components. Judging from the individual (100) diffractions of the neat acceptors (Fig. 2(d) and (e)) and PM6 (Fig. S20, ESI†), the two peaks at qxy ∼3.35 and 4.15 nm−1 could be classified as the donor and acceptor components, respectively. We further calculated the CCLs for each resolved profile and display the results in Fig. 5(f), with the data provided in Table S5 (ESI†). It is evident that in the respective blend, the 3QY component had a mildly greater CCL than that of 3BY. More interestingly, the CCL of the PM6 component in PM6:3QY delivered a marked increase over that in PM6:3BY. The more ordered molecular stacking could be expected to facilitate charge transport, supported by the greater charge mobilities of the PM6:3QY device obtained from the SCLC method (Fig. S22 and Table S2, ESI†).
 | ||
| Fig. 5 (a) and (d) AFM (left) and TEM images (right) of the blend films. (b) and (e) GIWAXS patterns and (c) line-cut profiles. (f) CCLs of PM6 and the acceptor in their blends resolved from the (100) diffractions in IP azimuth. | ||
In situ absorption spectra were further combined to elucidate the evolutions of the donor and acceptor in the two blend films.51 As Fig. 6(a) and (b) show, the two systems had similar 2D-absorption appearances with comparable solution-to-film transformation durations of ∼150 ms. However, by extracting the time-dependent absorption shifts of the donor and acceptor components separately, an amazing disparity was observed. In terms of the PM6 component in the two blend solutions (stage I), a conspicuous bathochromic shift was afforded when it was blended with 3QY (Fig. 6(c) and Fig. S23, ESI†). This result signifies the greater pre-aggregation of PM6 when it interacts with 3QY molecules in solution (I), of which the preferred pre-aggregation can be partially preserved during the transformation stage (II), with remaining bathochromic shift occurring in the post-film stage (III) (Fig. 6(c)). In addition, both the 3BY and 3QY components displayed comparable maximum absorptions in their respective blend solutions (Fig. 6(d) and Fig. S23, ESI†). This suggests the similar pre-aggregation behaviors of the two oligomers and is logical because of their infirm intermolecular entanglement compared to the polymer PM6. Meanwhile, the two acceptors revealed very close transformation times, which were different from the kinetics of the neat acceptors (Fig. 2(g)–(i)) and should be ascribed to the dilute effect of PM6. However, it is worth noting that the acceptors received enhanced but different aggregations at the post-film stage. In particular, 3QY had enlarged shifts after the transformation stage compared to that of 3BY, supporting the stronger crystallinity of 3QY in the blend film (Fig. 5(f)). This improvement should be partly attributed to the stronger inherent self-aggregation and crystallinity of the 3QY molecule than that of 3BY as suggested by the GIWAXS results (Fig. 2(f)). Additionally, the moderately limited intermolecular interactions between the polymer donor and 3QY (discussed below) should be another factor, which would suppress the excessive blending and thus promote the homogeneous self-aggregation process.
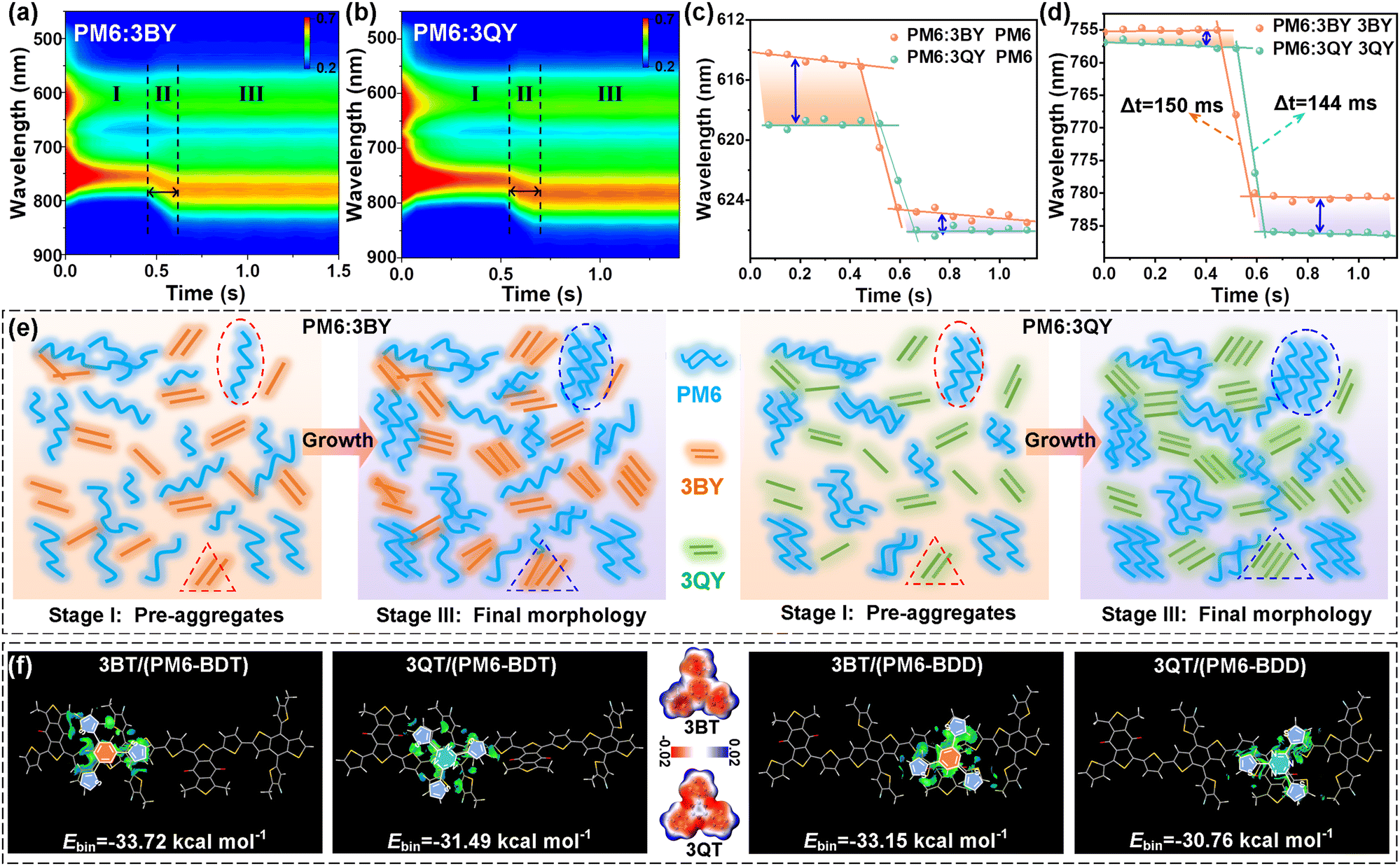 | ||
| Fig. 6 (a) and (b) 2D in situ absorption patterns of PM6:3BY and PM6:3QY blends and (c) and (d) relevant maximum peak shifts of the donor and acceptor in the blends. (e) Schematic of the film-formation kinetics, including solution-stage pre-aggregation, domain growth, and aggregates in the post-film stage. (f) Optimized molecular configurations and IGMH isosurfaces of the PM6 dimer and the core segments (3BT, 3QT) of the oligomers. PM6-BDT and PM6-BDD stand for the different interaction regions of the PM6 skeleton. | ||
Supported by the above results, Fig. 6(e) visibly depicts the pre-aggregation in the solutions and the final phase-separation behaviors in the post-film stage of the two blend systems. As for stage I, the PM6 segments had a stronger pre-aggregation attribute in the PM6:3QY solution compared to that in PM6:3BY. However, the corresponding 3QY and 3BY acceptors showed similar pre-aggregation properties in the two blend solutions. When it comes to stage III, the homologous pre-aggregation propensity of PM6 was well-inherited, affording more orderly interchain stacking and relatively larger domains of the PM6 component in the PM6:3QY film. The 3QY component, meanwhile, received an increased crystallinity over that of 3BY in their respective blend films. The preferable film-formation kinetics of the PM6:3QY system resulted in its unique morphology, that is, an increased but appropriate phase-separation with fine fibrous textures and a smooth surface (Fig. 5), favoring charge generation, transport, and collection while alleviating energy losses. As we previously suggested, intermolecular interaction inside a heterojunction is one crucial factor to drive the film-forming procedure and final morphologies.4,62,63 Thus, figuring out the interactions could be helpful to interpret the distinctive phase-separations, particularly the dramatically different pre-aggregation behaviors of the PM6 component in the two blends. However, the largely extended molecular structures of the star-shaped acceptors with complex spatial configurations make it unrealistic to complete simulations. Considering the only difference of the two acceptors, it is reasonable to employ the two central units, namely 3BT and 3QT, and these should be equivalent to evaluating the interactions between the donor and acceptor segments. In view of the differential electronic atmosphere of the PM6 π-skeleton, we progressively put the 3BT or 3QT unit in three separate regions of the PM6 dimer, including the BDT region (labeled as PM6-BDD), BDD region (labeled as PM6-BDD), and the transition area between BDT and BDD, for the subsequent calculations. The optimized packing configurations and calculated Hirshfeld partition (IGMH) isosurfaces are provided in Fig. S24 (ESI†) and Fig. 6(f). From the perspective of the molecular electrostatic potential (ESP), as given in Fig. 6(f), the 3QT segment may favor interactions with PM6 because of the more positive ESP distributions of 3QT owing to the electron-deficient triazine. This would enlarge the ESP difference with electronegative PM6 (Fig. S25, ESI†) and improve their electrostatic interactions. Beyond our expectation, all the results suggested greater intermolecular binding energies (Ebin) between PM6 and 3BY than those between PM6 and 3QY. For instance, when interacting with PM6-BDT, 3BT/PM6-BDT afforded a larger Ebin (33.72 kcal mol−1) than that of 3QT/PM6-BDT (31.49 kcal mol−1), and the same trends for the other two packing modes (Fig. S24, ESI†). The strong interactions between PM6 and 3BT should be attributed to the more relaxed configuration of the 3BT moiety, leaving greater molecular freedom to interact with the PM6 backbone (Fig. S26, ESI†). In other words, the multiple N⋯H intramolecular conformational locks of 3QT confine the molecule rotary and reduce the orbital coupling and global interactions with PM6 (Fig. S26, ESI†). Similar results could be mirrored between the oligomer acceptors and donor in their blend film. The relatively stronger interaction between 3BY and PM6 would increase the opportunity to form heterojunctions and reduce phase-separations. Likewise, the limited interactions between 3QY and PM6 would force more prominent self-aggregations of the homogeneous molecules, which should be one contributor to the stronger pre-aggregation of the PM6 component in the PM6:3QY solution. Collectively assisted by the greater crystallinity of 3QY, moderately expanded phase-separations with superior exciton/charge properties were realized for the PM6:3QY system, affording it with an outstanding photovoltaic performance.
Conclusion
To conclude, we report two new three-arm star-shaped oligomer acceptors, namely 3BY and 3QY, featuring non-fused cores to bridge the acceptor arms. Notably, 3QY carrying a triazine center endows multisite intramolecular non-covalent interactions, resulting in an accelerated self-assembly and greater molecular ordering. Meanwhile, the relatively stiffer molecular configuration of 3QY modulated the interactions with the donor segment and refined the film-forming kinetics of the blend systems, affording excellent heterojunction textures and charge-transfer property for the PM6:3QY system. These merits collectively account for the outstanding PCE up to 19.27% in the PM6:3QY solar cell, which represents a new record efficiency for oligomer acceptors so far. Moreover, the large-size star-shaped oligomers with notable Tgs contributed to decent long-term device stability under continuous thermal annealing. In summary, this study provides a promising approach to design star-shaped acceptors by employing non-fused cores and discloses the structure–performance relationship modulated by non-covalent interactions. These findings should provide guidance for future research on high-performance and long-term stable OSCs.Author contributions
C. S. conceived the idea, synthesized the oligomer acceptors, and tested the basic properties. J. W. fabricated the solar cells, measured the device performance and exciton/charge kinetics. F. B. carried out theoretical calculation. H. J. measured the in situ absorption spectra. C. Y. conducted the GIWAXS measurements. Y. L., J. C., and X. B. supervised the study. Y. L., C. S. and X. B. prepared and revised the manuscript. All the authors participated in the result analysis.Data availability
The data that supports the finding of this study are available in the ESI† of this article or from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by National Natural Science Foundation of China (62305350 and 22375213), the Youth Innovation Promotion Association CAS (2021211), Shandong Energy Institute (SEIS202108, SEII202111), QIBEBT/SEI/QNESL (S202305), China Postdoctoral Science Foundation (2022M723260, 2023M743636, 2024T170962), Shandong Postdoctoral Innovative Talents Support Plan (SDBX2022031, SDBX2023031) for financial support. The authors appreciate the beamline BL16B1 of the Shanghai Synchrotron Radiation Facility for providing beam time. The authors also appreciate Dr Ren from Xingyuan Aurora (Suzhou) photoelectric Technology Co. Ltd for the support of fs-TA measurements.References
- H. Yao and J. Hou, Angew. Chem., Int. Ed., 2022, 61, e202209021 CrossRef CAS
.
- J. Yi, G. Zhang, H. Yu and H. Yan, Nat. Rev. Mater., 2024, 9, 46–62 CrossRef CAS
- Y. Li, L. Yu, L. Chen, C. Han, H. Jiang, Z. Liu, N. Zheng, J. Wang, M. Sun, R. Yang and X. Bao, Innovation, 2021, 2, 100090 Search PubMed
- C. Han, J. Wang, S. Zhang, L. Chen, F. Bi, J. Wang, C. Yang, P. Wang, Y. Li and X. Bao, Adv. Mater., 2023, 35, 2208986 CrossRef CAS
- J. Wang, Y. Li, C. Han, L. Chen, F. Bi, Z. Hu, C. Yang, X. Bao and J. Chu, Energy Environ. Sci., 2024, 17, 4216–4227 RSC
- X. Wang, J. Wang, P. Wang, C. Han, F. Bi, J. Wang, N. Zheng, C. Sun, Y. Li and X. Bao, Adv. Mater., 2023, 35, 2305652 CrossRef CAS
- C. Chen, L. Wang, W. Xia, K. Qiu, C. Guo, Z. Gan, J. Zhou, Y. Sun, D. Liu and W. Li, Nat. Commun., 2024, 15, 6865 CrossRef CAS PubMed
- S. Guan, Y. Li, C. Xu, N. Yin, C. Xu, C. Wang, M. Wang, Y. Xu, Q. Chen and D. Wang, Adv. Mater., 2024, 2400342 CrossRef CAS
- Y. Sun, L. Wang, C. Guo, J. Xiao, C. Liu, C. Chen, W. Xia, Z. Gan, J. Cheng and J. Zhou, J. Am. Chem. Soc., 2024, 146, 12011–12019 CrossRef CAS PubMed
- P. Ding, D. Yang, S. Yang and Z. Ge, Chem. Soc. Rev., 2024, 53, 2350–2387 RSC
- Y. Jiang, X. Dong, L. Sun, T. Liu, F. Qin, C. Xie, P. Jiang, L. Hu, X. Lu and X. Zhou, Nat. Energy, 2022, 7, 352–359 CrossRef CAS
- Y. Liang, D. Zhang, Z. Wu, T. Jia, L. Lüer, H. Tang, L. Hong, J. Zhang, K. Zhang and C. J. Brabec, Nat. Energy, 2022, 7, 1180–1190 CrossRef CAS
- S. Li, C.-Z. Li, M. Shi and H. Chen, ACS Energy Lett., 2020, 5, 1554–1567 CrossRef CAS
- C. Guan, C. Xiao, X. Liu, Z. Hu, R. Wang, C. Wang, C. Xie, Z. Cai and W. Li, Angew. Chem., Int. Ed., 2023, 135, e202312357 CrossRef
- K. Zhang, Z. Jiang, J. Qiao, P. Lu, C. Qin, H. Yin, X. Du, W. Qin and X. Hao, Energy Environ. Sci., 2023, 16, 3350–3362 RSC
- K. An, W. Zhong, F. Peng, W. Deng, Y. Shang, H. Quan, H. Qiu, C. Wang, F. Liu and H. Wu, Nat. Commun., 2023, 14, 2688 CrossRef CAS PubMed
- Y. Yang, J. Wang, Y. Zu, Q. Liao, S. Zhang, Z. Zheng, B. Xu and J. Hou, Joule, 2023, 7, 545–557 CrossRef CAS
- X. Lai, S. Chen, X. Gu, H. Lai, Y. Wang, Y. Zhu, H. Wang, J. Qu, A. K. K. Kyaw and H. Xia, Nat. Commun., 2023, 14, 3571 CrossRef PubMed
- C. Sun, J.-W. Lee, C. Lee, D. Lee, S. Cho, S.-K. Kwon, B. J. Kim and Y.-H. Kim, Joule, 2023, 7, 416–430 CrossRef CAS
- C. Zhang, J. Song, J. Xue, S. Wang, Z. Ge, Y. Man, W. Ma and Y. Sun, Angew. Chem., Int. Ed., 2023, 135, e202308595 CrossRef
- J.-W. Lee, C. Sun, C. Lee, Z. Tan, T. N.-L. Phan, H. Jeon, D. Jeong, S.-K. Kwon, Y.-H. Kim and B. J. Kim, ACS Energy Lett., 2023, 8, 1344–1353 CrossRef CAS
- C. Sun, J. W. Lee, Z. Tan, T. N. L. Phan, D. Han, H. G. Lee, S. Lee, S. K. Kwon, B. J. Kim and Y. H. Kim, Adv. Energy Mater., 2023, 13, 2301283 CrossRef CAS
- X. Gu, X. Zhang and H. Huang, Angew. Chem., Int. Ed., 2023, 135, e202308496 CrossRef
- C. Zhang, J. Song, L. Ye, X. Li, M. H. Jee, H. Y. Woo and Y. Sun, Angew. Chem., 2024, 136, e202316295 CrossRef
- Y. Li, F. Qi, B. Fan, K. K. Liu, J. Yu, Y. Fu, X. Liu, Z. Wang, S. Zhang and G. Lu, Adv. Mater., 2024, 2313393 CrossRef CAS
- X. Liu, Z. Zhang, C. Wang, C. Zhang, S. Liang, H. Fang, B. Wang, Z. Tang, C. Xiao and W. Li, Angew. Chem., Int. Ed., 2024, 136, e202316039 CrossRef
- J. Song, C. Zhang, C. Li, J. Qiao, J. Yu, J. Gao, X. Wang, X. Hao, Z. Tang and G. Lu, Angew. Chem., Int. Ed., 2024, e202404297 CAS
- J. Liu, X. Duan, J. Song, C. Liu, J. Gao, M. H. Jee, Z. Tang, H. Y. Woo and Y. Sun, Energy Environ. Sci., 2024, 17, 3641–3650 RSC
- P. Tan, H. Chen, H. Wang, X. Lai, Y. Zhu, X. Shen, M. Pu, H. Lai, S. Zhang and W. Ma, Adv. Funct. Mater., 2024, 34, 2305608 CrossRef CAS
- F. Qi, Y. Li, R. Zhang, F. R. Lin, K. Liu, Q. Fan and A. K. Y. Jen, Angew. Chem., Int. Ed., 2023, 135, e202303066 CrossRef
- J. Wu, Z. Ling, L. R. Franco, S. Y. Jeong, Z. Genene, J. Mena, S. Chen, C. Chen, C. M. Araujo and C. F. Marchiori, Angew. Chem., Int. Ed., 2023, 62, e202302888 CrossRef CAS
- X. Chen, X. Zhang, X. Xiao, Z. Wang and J. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202216010 CrossRef CAS
- M. Lv, Q. Wang, J. Zhang, Y. Wang, Z. G. Zhang, T. Wang, H. Zhang, K. Lu, Z. Wei and D. Deng, Adv. Mater., 2024, 36, 2310046 CrossRef CAS PubMed
- Y. Li, L. Mei, Z. Ge, C. Liu, J. Song, Y. Man, J. Gao, J. Zhang, Z. Tang and X. K. Chen, Adv. Mater., 2024, 2403890 CrossRef CAS
- J. W. Lee, C. Sun, H. Jeon, T. H. Q. Nguyen, T. N. L. Phan, X. Bao, Y. H. Kim and B. J. Kim, Adv. Funct. Mater., 2024, 2404569 CrossRef CAS
- W. Liu, X. Xu, S. He, R. Sun, Q. Chen, J. Min, Z. Zhang, J. Yuan, Y. Li and Y. Zou, Macromolecules, 2023, 56, 8623–8631 CrossRef CAS
- X. Liao, Q. Xie, Y. Guo, Q. He, Z. Chen, N. Yu, P. Zhu, Y. Cui, Z. Ma and X. Xu, Energy Environ. Sci., 2022, 15, 384–394 RSC
- J. W. Lee, C. Sun, J. Lee, D. J. Kim, W. J. Kang, S. Lee, D. Kim, J. Park, T. N. L. Phan and Z. Tan, Adv. Energy Mater., 2024, 14, 2303872 CrossRef CAS
- C. Wang, X. Ma, Y.-F. Shen, D. Deng, H. Zhang, T. Wang, J. Zhang, J. Li, R. Wang and L. Zhang, Joule, 2023, 7, 2386–2401 CrossRef CAS
- Y. Kervella, J. M. A. Castán, Y. A. Avalos-Quiroz, A. Khodr, Q. Eynaud, T. Koganezawa, N. Yoshimoto, O. Margeat, A. Rivaton and A. J. Riquelme, J. Mater. Chem. C, 2023, 11, 8161–8169 RSC
- X.-M. Huang, N. Chen, D.-N. Ye, A.-G. Zhong, H. Liu, Z. Li and S.-Y. Liu, Sol. RRL, 2023, 7, 2300143 CrossRef CAS
- A. Rafiq, R. Hussain, M. U. Khan, M. Y. Mehboob, M. Khalid, Shehnaz, M. M. Alam, M. Imran and K. Ayub, Energy Technol., 2022, 10, 2100751 CrossRef CAS
- M. Khalid, U. H. Ishaque, M. A. Asghar, M. Adeel, M. M. Alam, M. Imran, R. Baby, A. A. Braga, M. F. ur Rehman and M. S. Akram, Mater. Chem. Phys., 2023, 299, 127528 CrossRef CAS
- H. Li, Y. Wang, L. Yu, C. Liu, C. Zhou, S. Sun, M. Li, Y. Tao, G. Xie and H. Xu, Chem. Eng. J., 2021, 425, 131487 CrossRef CAS
- N. Rani Kumar and A. R. Agrawal, ChemistryOpen, 2023, 12, e202200203 CrossRef CAS
- R. A. Marcus, Angew. Chem., Int. Ed. Engl., 1993, 32, 1111–1121 CrossRef
- G. Zhou, M. Zhang, Z. Chen, J. Zhang, L. Zhan, S. Li, L. Zhu, Z. Wang, X. Zhu and H. Chen, ACS Energy Lett., 2021, 6, 2971–2981 CrossRef CAS
- R. Shivhare, G. J. Moore, A. Hofacker, S. Hutsch, Y. Zhong, M. Hambsch, T. Erdmann, A. Kiriy, S. C. Mannsfeld and F. Ortmann, Adv. Mater., 2022, 34, 2101784 CrossRef CAS
- S. Liu, J. Yuan, W. Deng, M. Luo, Y. Xie, Q. Liang, Y. Zou, Z. He, H. Wu and Y. Cao, Nat. Photonics, 2020, 14, 300–305 CrossRef CAS
- N. Zhao, C. Yang, F. Bian, D. Guo and X. Ouyang, J. Appl. Crystallogr., 2022, 55, 195–203 CrossRef CAS
- Y. F. Shen, H. Zhang, J. Zhang, C. Tian, Y. Shi, D. Qiu, Z. Zhang, K. Lu and Z. Wei, Adv. Mater., 2023, 35, 2209030 CrossRef CAS
- R. Ma, X. Jiang, J. Fu, T. Zhu, C. Yan, K. Wu, P. Müller-Buschbaum and G. Li, Energy Environ. Sci., 2023, 16, 2316–2326 RSC
- Y. Cheng, Q. Mao, C. Zhou, X. Huang, J. Liu, J. Deng, Z. Sun, S. Jeong, Y. Cho and Y. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202308267 CrossRef
- D. He, F. Zhao, C. Wang and Y. Lin, Adv. Funct. Mater., 2022, 32, 2111855 CrossRef CAS
- L. J. A. Koster, V. Mihailetchi, H. Xie and P. W. Blom, Appl. Phys. Lett., 2005, 87, 203502 CrossRef
- L. J. A. Koster, V. D. Mihailetchi, R. Ramaker, H. Xie and P. W. Blom, Org. Optoelectron. Photonics II, 2006, 6192, 457–467 Search PubMed
- C. M. Proctor, M. Kuik and T.-Q. Nguyen, Prog. Polym. Sci., 2013, 38, 1941–1960 CrossRef CAS
- J. Vollbrecht and V. V. Brus, Energies, 2021, 14, 4800 CrossRef CAS
- L. Zhang, Z. Zhang, D. Deng, H. Zhou, J. Zhang and Z. Wei, Adv. Sci., 2022, 9, 2202513 CrossRef
- Z. Chen, C. He, P. Ran, X. Chen, Y. Zhang, C. Zhang, R. Lai, Y. M. Yang, H. Chen and H. Zhu, Energy Environ. Sci., 2023, 16, 3373–3380 RSC
- K. Jiang, J. Zhang, C. Zhong, F. R. Lin, F. Qi, Q. Li, Z. Peng, W. Kaminsky, S.-H. Jang and J. Yu, Nat. Energy, 2022, 7, 1076–1086 CrossRef
- P. Wang, F. Bi, H. Jiang, X. Wang, C. Cui, Y. Li and X. Bao, Adv. Funct. Mater., 2024, 34, 2411862 CrossRef
- X. Wang, J. Wang, P. Wang, C. Han, F. Bi, J. Wang, N. Zheng, C. Sun, Y. Li and X. Bao, Adv. Mater., 2023, 35, 2305652 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04149f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |