
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pre-constructing a mortice-tenon joint based-layer to achieve an enhanced SEI on Li metal anode†
Kun Wang‡
a,
Chutao Wang‡a,
Sheng Liu‡a,
Congcong Dub,
Qingyi Zhenga,
Jiaqing Cuia,
Xinxin Yanga,
Yuxin Tang b,
Ruming Yuana,
Mingsen Zhenga,
Jingmin Fan*a and
Quanfeng Dong*a
b,
Ruming Yuana,
Mingsen Zhenga,
Jingmin Fan*a and
Quanfeng Dong*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces, Collaborative Innovation Center of Chemistry for Energy Materials (i-ChEM), Engineering Research Centre of Electrochemical Technologies of Ministry of Education, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: jmfan@xmu.edu.cn; qfdong@xmu.edu.cn
bQingyuan Innovation Laboratory, Quanzhou, 362801, China
First published on 25th January 2025
Abstract
For the efficient functioning of a lithium anode, it requires an ideal protective layer that has its own strength and strongly bonds with the substrate. There are many studies on the strength of such protective layers, but very few reports on their bond strength with substrates. Herein, a design strategy is proposedto pre-construct a based-layer, where a mortice-tenon joint will connect with the subsequent electrochemically active SEI that is set on a Li anode surface. Initially, a tightly bonded base layer was chemically formed via the reaction between 2-(fluorosulfonyl)difluoroacetate (DFSA) and lithium metal. Then, trimethylsilyl 2-(fluorosulphonyl)difluoroacetate (TSFSA), which has a similar molecular structure and same functional group as DFSA, was introduced to act as an SEI enhancer that can preferentially decompose over carbonate solvents under electrochemical conditions with the same components of the based-layer, which was thus strengthened to form an enhanced SEI (ESEI). The Li anode with ESEI achieved long cycling stability (≥ 2100 h) and a high average CE (99.2%) in carbonate electrolytes. Full cells with high cathode loading (20.5 mg cm−2) also achieved high cycling stability at low N/P ratios, demonstrating its great prospects for practical applications in high energy density Li-metal batteries.
Broader contextLithium metal batteries (LMBs) with high energy density are promising candidates for next-generation energy storage applications. However, the practical application of lithium metal anodes (LMAs) in carbonate electrolytes is hindered by poor reversibility, which is primarily caused by the instability of the solid electrolyte interphase (SEI). An ideal SEI must meet two determining factors: structural strength (ionic conductivity, mechanical stability, and low diffusion barrier) and a strong bond with the substrate. There are many studies on the former, but very few reports on the latter. Here, a design strategy is proposed to pre-construct a based-layer, where a mortise-tenon joint that connects with the subsequent electrochemically active SEI has been set on a LMA. The introduced SEI enhancer preferentially decomposed over carbonate solvents under electrochemical conditions with the same components as those of the based-layer, which was thus strengthened to form an enhanced SEI (ESEI). The ESEI achieved high lithium reversibility and extended cycling life. This research not only provides insights into addressing challenges associated with lithium dendrite growth in carbonate electrolytes but also contributes to the improvement of the practical full cells, demonstrating great potential for the application of high-energy-density LMBs. |
Introduction
Lithium metal anodes (LMAs) are considered the most ideal anode materials for electrochemical energy storage owing to their extremely high theoretical specific capacity (3860 mA h g−1) and low electrochemical potential (−3.04 V vs. standard hydrogen electrode).1–4 Li metal can further enhance the energy density of Li-ion batteries when paired with the current commercial Li-ion battery cathodes, such as Li iron phosphate (LFP) and LiNi0.8Co0.1Mn0.1O2 (NCM811).5–7 However, the high activity of Li metal in conventional carbonate electrolytes usually leads to the growth of Li dendrites and continuous reaction between the active Li and the electrolyte, resulting in a short cycle life of Li-metal batteries (LMBs), which severely limits the further application of LMBs.8–11Solid electrolyte interphase (SEI) acts as a protective layer between lithium and the electrolyte by terminating undesirable reactions and ensuring the uniform plating of lithium.12–14 However, the SEI obtained from the in situ reaction with lithium in conventional carbonate electrolytes is heterogeneous and mechanically unstable, which negatively impacts the structure and composition of SEI. Incorporating inorganic components into SEI can effectively balance the ionic conductivity, diffusion barriers, and surface energy at the lithium metal interface, thereby facilitating the smooth stripping of lithium. LiF is widely recognized as an effective way to improve the homogeneity of SEI and regulate the lithium-ion flux.15,16 However, monolayer SEI consisting only of small LiF particles has certain drawbacks, such as it is not compact, the connectivity between its particles is poor, and LiF exhibits low ionic conductivity (≈10−13–10−14 S cm−1).17 Other types of inorganic components (e.g., Li2S, Li3N, etc.) exhibit high ionic conductivity of ≈10−4–10−5 S cm−1,18 excellent lithium affinity, and effective dendrite inhibition. SEI containing these species exhibits high Li+ flux, thereby ensuring uniform striping of lithium. Therefore, this will further improve the compatibility of LMAs and enhance the electrochemical performance of LMBs.19
In recent years, most strategies to construct stable LMAs have been focused on electrolytes,20–22 additives,23–26 and artificial SEI designs.15,27–30 However, the interactions between the electrochemically formed SEI and the lithium metal via conventional electrolyte engineering are weak owing to the presence of natural passivation layers (LiOH, Li2CO3, and Li2O). The composition, homogeneity, and mechanical stability of SEI cannot be guaranteed, which will exacerbate SEI cracking and excessive depletion of the beneficial constituents during the cycling process.31 Although SEIs artificially constructed via a chemical reaction have a stronger binding force, SEIs that are pre-constructed exclusively via immersion and other methods are uncontrollable and face difficulty in resisting the structural damage caused by large volume changes.32,33 In addition, most of the artificial SEI design processes are cumbersome and increase the complexity of the process, which is unfavorable.34 Therefore, to achieve homogeneous and strongly mechanically stable SEI, it is necessary to adopt an incremental and controllable nanoscale treatment for the SEI composition and structure. However, there is currently no easy and efficient way to construct such ordered and controllable SEI at LMAs.
At least two basic requirements determine the performance of SEI: first, its own strength should be high, and second, it should have a strong bond with the substrate. There are several works on the former, but very few reports on the latter. In this study, we propose an enhanced SEI via pre-constructing a based-layer where the mortice and tenon joint that connects the subsequent SEI was set on a Li metal anode in carbonate electrolytes (Fig. 1). Specifically, the Li metal surface was pre-treated with 2-(fluorosulphonyl)difluoroacetic acid (DFSA) to in situ generate lithium fluosulfonyl difluoroacetate (DFSA–Li) as a tightly bonded based-layer, firstly. Then, trimethylsilyl 2-(fluorosulphonyl)difluoroacetate (TSFSA) which contains a similar component was introduced to act as an SEI enhancer to strengthen the mortice-tenon joint based-layer. During subsequent electrochemical reactions, TSFSA decomposes preferentially over carbonate solvents with the same components (LiF, Li2O, and Li2S), which can perfectly integrate with the based-layer to form enhanced SEI (ESEI). Under this strategy, LMAs achieved over 2100 hours of long cycling and an average coulombic efficiency of 99.2% in the carbonate electrolyte. Moreover, this strategy proved effective in carbonate electrolytes without FEC. Finally, this strategy demonstrated significantly improved rate performance, cycle stability, capacity retention, and capacity utilization in full cells and showed great prospects for practical applications in high-energy-density Li-metal batteries.
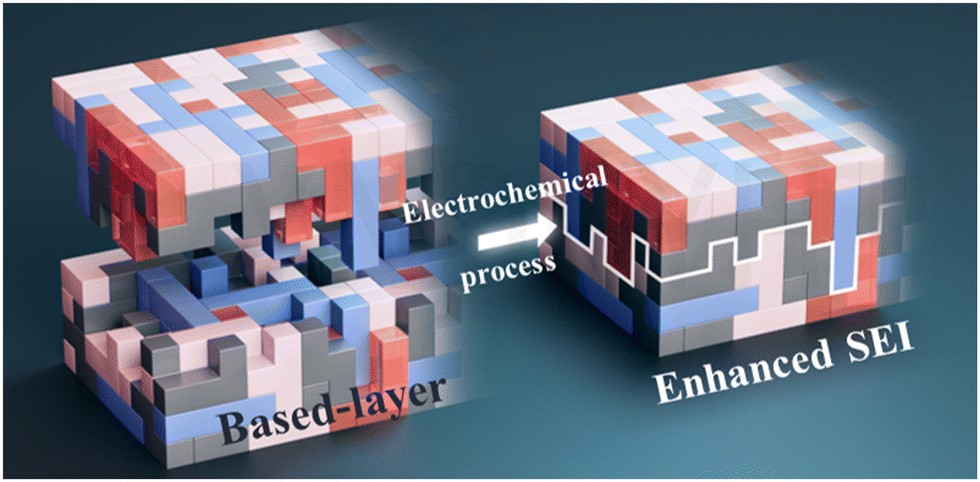 | ||
| Fig. 1 Schematic of the enhanced SEI (ESEI) fabrication process. | ||
Results and discussion
Design mechanism of the based-layer and SEI enhancer
The choice of sulfonyl fluoride group as an SEI enhancer is owing to its typically low reduction stability, and the elements F, O, and S within the group can help construct an inorganic SEI. Density-functional theory (DFT) calculations were used to calculate the LUMO and HOMO energy levels of the relevant molecules (Fig. 2(a) and Fig. S1, ESI†). The LUMO energy levels of sulfonyl fluoride group molecules (DFSA–Li for −1.529 eV and TSFSA for −1.541 eV) are lower than those of the salt (LiPF6 for −1.441 eV) and solvents (EC for 1.067 eV, EMC for 1.196 eV, and FEC for 0.517 eV), indicating that the sulfonyl fluoride group molecules are more readily reduced to participate in the construction of the inner SEI layer.35,36 Therefore, with the stepwise action of the based-layer (DFSA–Li) and SEI enhancer (TSFSA), the high LUMO energy levels of the undesirable solvents (EC and EMC) theoretically make it difficult for them to participate in the outer SEI construction, while the limited involvement of favorable solvent (FEC) and salts (LiPF6) in the outer SEI construction is acceptable as they act as F donors.37,38 The further reconstruction of DFSA–Li was studied using ab initio molecular dynamics (AIMD) simulations based on DFT. As shown in Fig. 2(b), DFSA–Li molecules rapidly release F atoms within 100 fs to form LiF and reconstruct within 500 fs, with O and S atoms, further reacting with Li atoms. Thus, the quick reconstruction kinetics of DFSA–Li molecules can fast construct the mortice-tenon joint-based layer that will be a foundation of the enhanced SEI. | ||
| Fig. 2 (a) LUMO energy level diagrams of the investigated salt (LiPF6), solvents (EC, EMC and FEC), DFSA–Li and TSFSA. (b) Ab initio molecular dynamics simulations with DFSA–Li on the Li (110) surface. Photographs of (c) bare-Li and (d) DFSA–Li. (e) FT-IR spectra of the based-layer, DFSA and bare-Li. XPS spectra of (f) C 1s, (g) F 1s and (h) S 2p for the based-layer. | ||
Construction and characteristics of the based-layer and SEI enhancer
The DFSA–Li based-layer on Li metal was constructed through a spontaneous chemical reaction between DFSA and the Li metal. In this reaction, the carboxylic acid group in DFSA underwent a displacement reaction with Li to form DFSA–Li. As the concentration of the reactants increased, the Li surface changed from a shiny metallic color to black and formed a film-like structure (Fig. 2(c), (d) and Fig. S2, S3, ESI†). X-ray diffraction (XRD) was used to characterize the crystalline structure of Li metal (Fig. S4, ESI†). The intensity of the characteristic peaks of the (211) and (220) planes decreased after the reaction, indicating that DFSA preferentially reacted with Li of these two planes and reduced the likelihood of Li dendrite growth.39–41 Scanning electron microscopy (SEM) images and elemental mapping images reveal the formation of the based-layer (Fig. S5, ESI†). The signals of F and S elements on the Li surface originate from the reaction products of DFSA. The thickness of the based-layer was difficult to observe on the cross-section views, indicating good compatibility and binding forces with Li metal (Fig. S6 and S7, ESI†). Therefore, DFSA–Li can be so tightly bound to Li that it can blend seamlessly with it. The Fourier Transform infrared spectroscopy (FT-IR) compared the functional groups on the Li surface (Fig. 2(e)). The peak corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration on the surface of Li shifted from 1773.1 cm−1 (DFSA) to 1673.2 cm−1 (DFSA–Li) after the reaction, indicating the formation of lithium carboxylate salts. X-ray photoelectron spectroscopy (XPS) was used to study the chemical composition of the based-layer (Fig. 2(f)–(h)). The high-resolution C 1s spectrum shows peaks at 291.1 eV and 289.9 eV assigned to C–F and OC–O, revealing the retention of the DFSA structure.42 Peaks corresponding to LiF and Li–S in the F 1s and S 2p spectra indicate that DFSA–Li has begun to react with Li to form the desired inorganic SEI components. These results further confirm the formation of the based-layer on the Li metal surface after DFSA treatment.
O vibration on the surface of Li shifted from 1773.1 cm−1 (DFSA) to 1673.2 cm−1 (DFSA–Li) after the reaction, indicating the formation of lithium carboxylate salts. X-ray photoelectron spectroscopy (XPS) was used to study the chemical composition of the based-layer (Fig. 2(f)–(h)). The high-resolution C 1s spectrum shows peaks at 291.1 eV and 289.9 eV assigned to C–F and OC–O, revealing the retention of the DFSA structure.42 Peaks corresponding to LiF and Li–S in the F 1s and S 2p spectra indicate that DFSA–Li has begun to react with Li to form the desired inorganic SEI components. These results further confirm the formation of the based-layer on the Li metal surface after DFSA treatment.
It is not enough to have only the based-layer for an ideal SEI as its film-like structure cannot completely prevent the penetration of undesirable solvents. Next, we designed to further construct enhanced SEI through electrochemical in situ decomposition of the mortise-tenon joint. In order to achieve good bonding and compatibility with the based-layer, the selection of the decomposed components is of great significance. The trimethylsilyl 2-(fluorosulphonyl)difluoroacetate (TSFSA), which has a similar molecular structure and the same functional group as DFSA, was chosen as a functional molecule to achieve this aim. A solution of 1 M LiPF6 in FEC/EMC (BE) was selected as the basic electrolyte for the study due to its good fundamental performance in carbonate electrolytes.25 We assembled Li||Cu cells for cyclic voltammetry (CV) tests to verify the role of TSFSA in the subsequent electrochemical progressive reactions (Fig. 3(a) and Fig. S8–S9, ESI†). Compared to BE, TSFSA showed reduction peaks at about 2.0 V and 1.5 V, preferentially decomposing them compared to the other components in the electrolyte, which contributed to the strengthening of the based-layer. Moreover, the electrolyte of TSFSA also provided a higher capacity during the stripping phase of Li (forward scan of CV), exhibiting a larger peak area. In addition, CE tests and plating overpotential curves of Li||Cu cells containing the TSFSA electrolyte at a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2 also confirmed the high efficiency of the SEI formed from the electrochemical decomposition of unique sulfonyl fluoride groups as described in Fig. S10 and S11 (ESI†). The SEI incrementally induced by TSFSA achieved a higher CE, long-cycle stability, and lower plating overpotential. The dosage of DFSA and TSFSA was also optimized. The amounts of DFSA and TSFSA were also optimized in order to further select the appropriate reaction concentration for the formed ESEI. The ability to construct a based-layer with different DFSA reaction concentrations (1 vol% to 6 vol%) was evaluated using Li||Li symmetric cells (Fig. S12, ESI†). The cell performance of DFSA-treated Li anode exhibited significant improvement. Specifically, a DFSA reaction concentration of 4 vol% provided the best based-layer, which has the lowest overpotential and the longest cycling stability for Li||Li cells. Li||Cu cells at this concentration also exhibited the lowest nucleation and growth overpotentials (Fig. S13, ESI†). Fig. S14 (ESI†) also shows the CE of different TSFSA concentrations. The electrolyte with 2 vol% TSFSA demonstrated higher cycle stability (over 180 cycles) and CE. Therefore, the optimal DFSA reaction concentration and TSFSA concentration were determined, and the gradual modulation and construction of these two enhancers will ultimately result in an excellent ESEI.
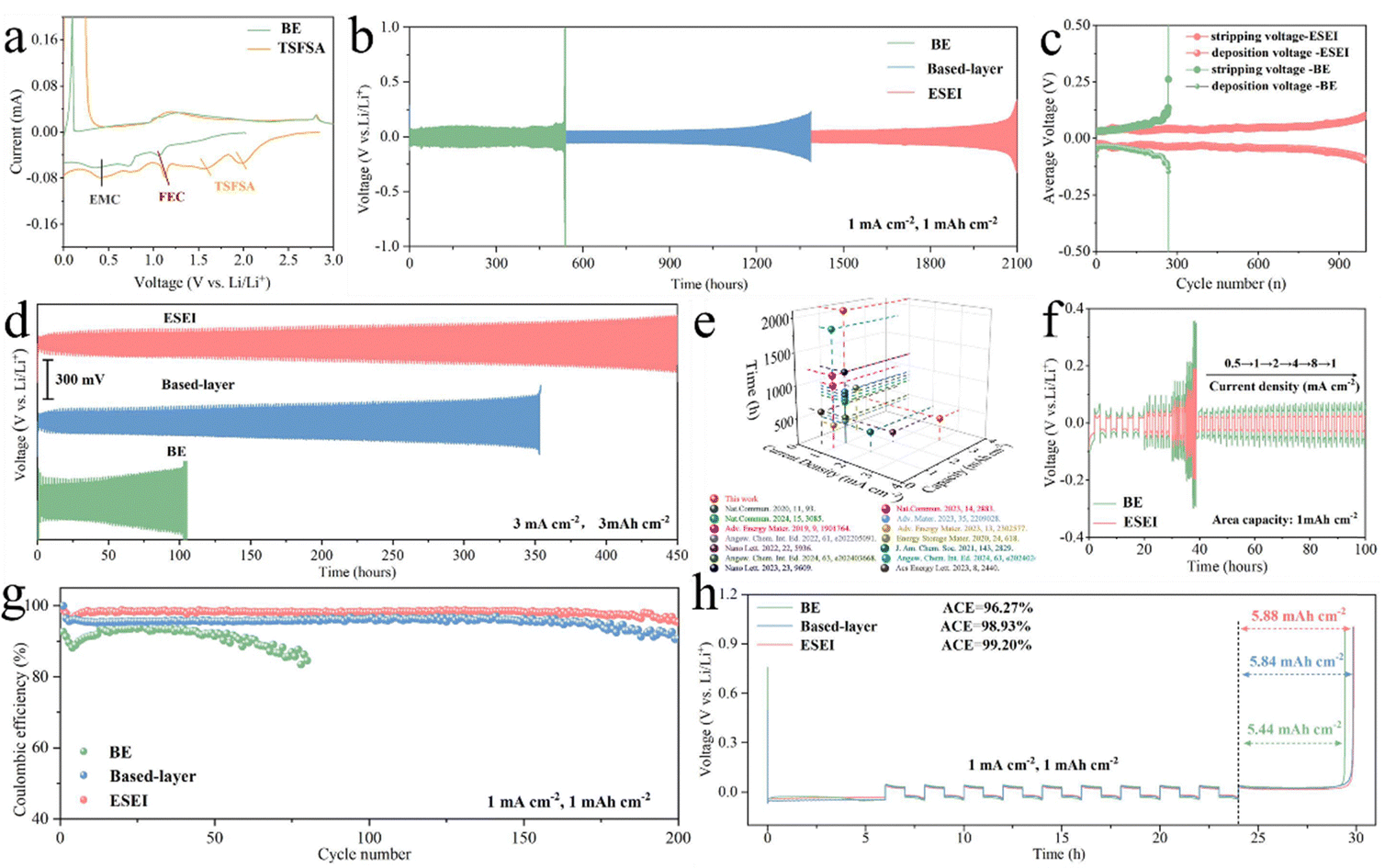 | ||
Fig. 3 (a) CV profiles of the first cycle Li||Cu cells in BE and TSFSA electrolytes at 1 mV s−1. (b) Charge–discharge profiles of Li||Li symmetric cells for ESEI, based-layer and BE of 1 mA cm−2 with 1 mA h cm−2. (c) Comparative voltage profiles of the symmetrical cells for ESEI and BE of 1 mA cm−2 with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) mA h cm−2. (d) Charge–discharge profiles of Li||Li symmetric cells for ESEI, based-layer and BE of 3 mA cm−2 with 3mA h cm−2. (e) Comparison of the electrochemical performance with previous studies using carbonate electrolytes. See Table S1 (ESI†) for details. (f) Voltage profiles of Li||Li symmetric cells at various current densities (0.5–8 mA cm−2) with a fixed capacity of 1 mA h cm−2. (g) CE in different Li||Cu cells of 1 mA cm−2 with 1 mA h cm−2. (h) Voltage–time curves to calculate the average CE of Li–Cu cells of 1 mA cm−2 with 1 mA h cm−2. Electrolyte: 1 M LiPF6 in FEC/EMC (v/v = 3:7). mA h cm−2. (d) Charge–discharge profiles of Li||Li symmetric cells for ESEI, based-layer and BE of 3 mA cm−2 with 3mA h cm−2. (e) Comparison of the electrochemical performance with previous studies using carbonate electrolytes. See Table S1 (ESI†) for details. (f) Voltage profiles of Li||Li symmetric cells at various current densities (0.5–8 mA cm−2) with a fixed capacity of 1 mA h cm−2. (g) CE in different Li||Cu cells of 1 mA cm−2 with 1 mA h cm−2. (h) Voltage–time curves to calculate the average CE of Li–Cu cells of 1 mA cm−2 with 1 mA h cm−2. Electrolyte: 1 M LiPF6 in FEC/EMC (v/v = 3:7). | ||
Plating morphologies and electrochemical performance of the ESEI
The long-term cycle stability of Li||Li symmetric cells was used to verify the effectiveness of ESEI. At a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2, the cell with a conventional carbonate electrolyte failed after 530 h of cycling. Although the reliance on based-layer significantly improves the cycling stability of the battery (from 530 h to 1380 h), it is still insufficient to achieve the goal of long-cycle LMAs. Impressively, after the electrochemical incremental reaction process, ESEI enhanced the stability of Li plating/stripping, increasing the cycle life to 2100 h (Fig. 3(b)). On one hand, the based-layer and SEI enhancer (TSFSA) dominated the construction of the SEI and prevented the involvement of the solvents. On the other hand, the uniform and efficient SEI blocked the formation of dead Li species, which helped maintain the stable overpotential during cycling and avoided rapid battery failure (Fig. 3(c)).43 ESEI also achieved better cycle stability at higher current densities and capacities (Fig. 3(d)). Compared to BE (100 h) and based-layer (350 h), ESEI showed excellent adaptability at high current densities with a cycling time of more than 450 h at 3 mA cm−2 and 3 mA h cm−2. Further combined with previous literature reports, it was found that Li||Li symmetric cells exhibited longer cycle life than other reported carbonate electrolyte cells, confirming the feasibility of the ESEI design (Fig. 3(e) and Table S1, ESI†). When increasing the current density (from 0.5 mA cm−2 to 8 mA cm−2), the symmetric cells with ESEI exhibited lower Li plating/stripping overpotential, demonstrating the excellent interfacial performance provided by the uniform and efficient SEI.44 In contrast, bare Li anodes in BE electrolyte showed higher overpotential, and the overpotential continued to increase even when the current density was returned to 0.5 mA cm−2 (Fig. 3(f) and Fig. S15, ESI†).Compared to the severe excess of lithium in Li||Li symmetric cells, the CE of Li||Cu half-cells testing can more intuitively reflect the reversibility of Li metal. At 1 mA cm−2 and 1 mA h cm−2, the ESEI anode exhibited the highest CE and cycle stability (98.1%, 200 cycles), showing significant improvement compared to BE (91.0%, 80 cycles) and based-layer (95.45%, 200 cycles) (Fig. 3(g) and Fig. S16, ESI†). Further increasing the current density to 2 and 3 mA cm−2, the half-cell with ESEI still exhibits higher coulombic efficiency improvement and longer cycle life compared to the BE anode (Fig. S17, ESI†). Furthermore, an average CE of up to 99.2% was obtained from the Aurbach method indicating that the ESEI suppresses excessive consumption of the active Li (Fig. 3(h)).45 To demonstrate the general applicability of this strategy to highly reactive carbonate electrolytes, stability and CE tests were conducted in commercial electrolytes. At a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2, Li||Li symmetric cells and Li||Cu half cells achieved 800 hours of cycle stability (BE ≤ 150 h) and an average CE of 98.53% (0.5 mA cm−2) as shown in Fig. S18 and S19 (ESI†), significantly improving the reversibility of Li anodes in FEC-free carbonate electrolytes. These results indicate that this mortise-tenon joint enhancement of SEI is designed to have excellent LMAs compatibility, which is inextricably linked to the structure and composition of the ESEI, the modulation of lithium plating morphology by the ESEI, and the excellent Li+ transfer kinetics.
In order to investigate the wettability of different Li metal anode SEIs in carbonate electrolytes, contact angle tests were performed (Fig. S20, ESI†). Bare Li exhibited the poorest wettability in the BE electrolyte (34.2°). The construction of the based-layer on the Li surface improved wettability (14.47°), likely due to the stronger affinity between the lithium organic carboxylate and F functional polar groups in DFSA–Li and FEC. When TSFSA was introduced into the electrolyte, the pre-ESEI achieved optimal electrolyte interface wettability (10.71°). The improved wettability improved the distribution of Li+ at the anode interface, reduced interfacial polarization and facilitated the subsequent electrochemical construction of the ESEI.46 This would promote the modulation of Li+ plating morphology by ESEI. Thus, SEM and AFM images can visually observe the microstructure of the different Li anodes after cycling, confirming the advantages of the ESEI. As shown in Fig. S21 (ESI†), the anode surface in BE exhibited severe cracking after the 10th stripping in the Li||Li symmetric cells, allowing the electrolyte to quickly enter these cracks and form new weak SEI. The based-layer constructed a first protective film, resulting in very few cracks during cycling. However, the ESEI showed no cracks after 10 cycles, demonstrating that the TSFSA can strengthen the original based-layer to form the so-called ESEI. High-resolution atomic force microscopy (AFM) was used to compare the surface roughness of the metallic lithium anode at this stage. The significant surface undulations and a high root-mean-square roughness (Rq = 718.0 nm) indicated the uneven surface of BE. Although the based-layer effectively mitigated this issue (Rq = 217 nm), the uneven surface still resulted in high localized currents, promoting the dendritic growth on the lithium surface. In contrast, the enhanced SEI greatly improved the surface smoothness (Rq = 30.1 nm), effectively preventing the high local current (Fig. S21 and Table S2, ESI†). The heterogeneous Young's modulus distribution in BE is also in stark contrast to the based-layer and ESEI, indicating that the SEI in BE undergoes repeated fracture-regeneration cycles, leading to continuous consumption of active lithium and electrolyte decomposition. The uniform Young's modulus distribution of ESEI demonstrates the stability of the SEI, laying a solid foundation for subsequent stable cycling (Fig. S22, ESI†). After 50 cycles, the anode surface in BE turned from a shiny metallic color to black, and the Li metal surface became loose and rough due to the formation of porous Li dendrites and dead Li. With the improvement of the based-layer, the blackened areas and Li dendrites on the anode surface were reduced, but loose gaps remained. In stark contrast, the incrementally formed ESEI anode surface retained a shiny metallic color without noticeable Li dendrites or cracks, indicating uniform Li plating/stripping (Fig. 4(a) and Fig. S23, ESI†).
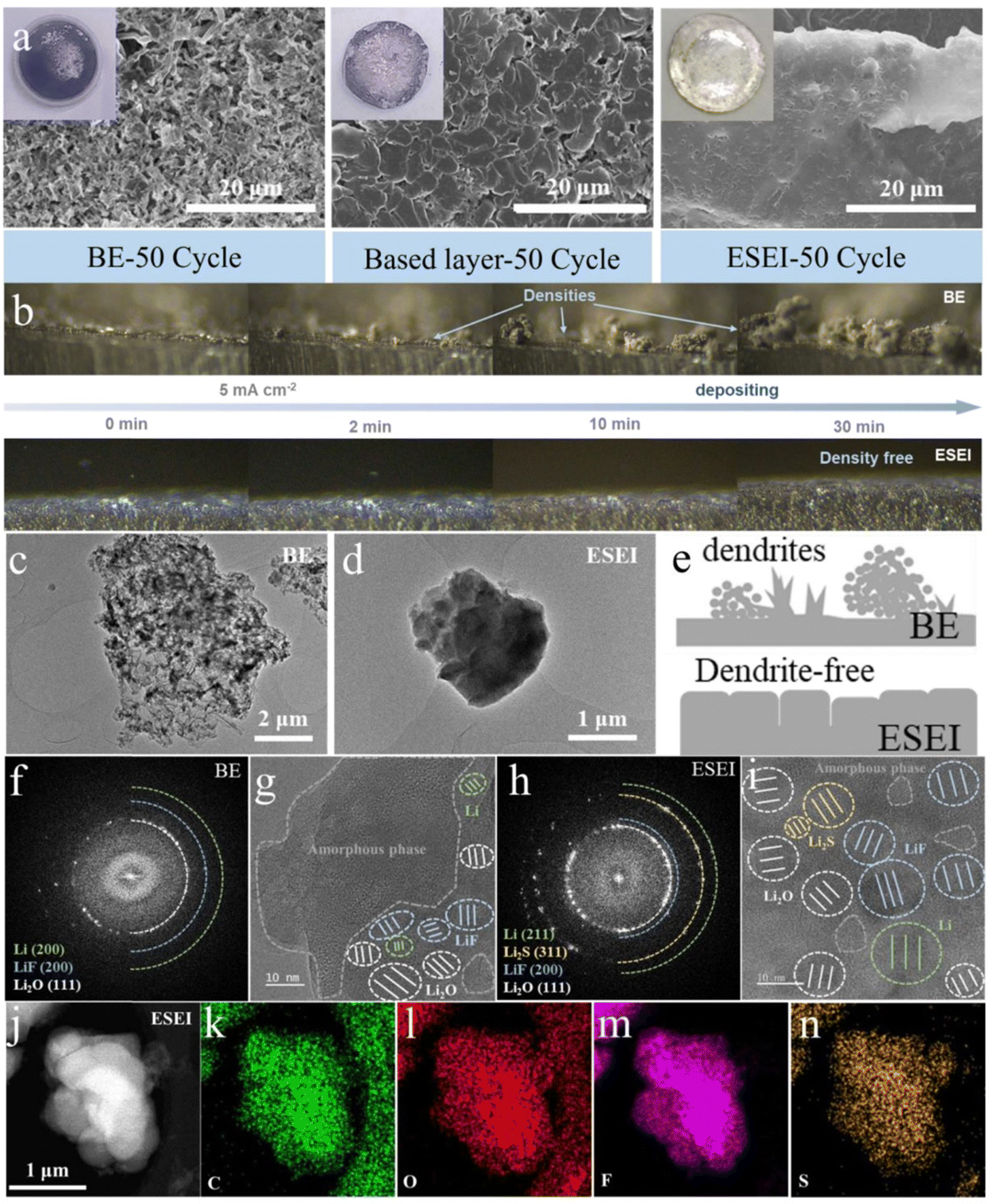 | ||
| Fig. 4 (a) Contact angle measurements of BE, based-layer and ESEI. (b) SEM images of BE, based-layer and ESEI after 50 cycles. The top left corner is photographs of the corresponding Li anode. (b) In situ optical microscope observation of the Li striping process for BE and ESEI at a current density of 2 mA cm−2. Cryo-TEM images of (c) BE and (d) ESEI in Li||Li symmetric cell after 5 cycles. (e) Schematic of plating behavior of Li metal in different SEI. (f) SAED pattern of BE. (g) HRTEM image of BE. (h) SAED pattern of ESEI. (i) HRTEM image of ESEI. (j)–(n) HAADF-STEM image and elemental mapping of ESEI. | ||
To visually observe the effects of the enhanced SEI on Li striping behavior, in situ electrochemical optical microscopy studies were conducted using the optical dual-electrode cells assembled with quartz and polytetrafluoroethylene (Fig. S25, ESI†). As shown in Fig. 4(b), the optical images were captured at 0 min, 2 min, 10 min and 30 min under a current density of 2 mA cm−2, and dynamic videos are also included in Movies 1–2 (ESI†). BE exhibited fast dendrite growth within the first 2 minutes of plating, with extensive disordered growth as the striping continued. In contrast, the surface of ESEI showed no significant dendrite growth within 30 minutes and exhibited a smooth and dense morphology with uniformly large plating thicknesses throughout the striping process. These results indicate that the enhanced SEI has a strong affinity for lithium metal and exhibits excellent plating/stripping stability under harsh conditions.
Structure and components characterization of ESEI
To correlate the structure–activity relationship of the advanced LMA with ESEI, we conducted an analysis of the structure and composition of both BE and ESEI.First, owing to the electron beam sensitivity of Li metal and SEI, we used cryogenic transmission electron microscopy (Cryo-TEM) to analyze the morphology and composition of the interface. Given the unique method of ESEI construction, thin layers of Li surface after five cycles were scraped off and dispersed onto a copper grid. A morphology comparison showed that post-cycle BE presented a rigid appearance with multiple spikes, while the Li metal induced by ESEI exhibited a flexible appearance with smooth particles, which was consistent with the results from the above SEM images (Fig. 4(c)–(e)). Regarding internal composition, the SEI films of BE contained a relatively high amount of amorphous organic components and a lower amount of beneficial inorganic components such as LiF, whereas ESEI was primarily composed of inorganic components, which were uniformly distributed (Fig. 4(f)–(i) and Fig. S26, S27, ESI†). HAADF-STEM and elemental mapping further highlighted the compositional differences in SEI between the two samples. Specifically, the contents of C and O elements in BE far exceeded that of F, and the distribution of F was highly uneven, which is detrimental to uniform Li plating. In contrast, the uniform distribution of C, O, F and S elements in ESEI also confirmed the high content of inorganic components (Fig. 4(j)–(n) and Fig. S28, ESI†), which can enable the effective migration of Li+ and achieve rapid and uniform plating.
Regrettably, due to the limitations of the sample preparation method, it was difficult to observe the thickness of the SEI using Cryo-TEM. To further analyze the composition of the SEI at different depths, the samples were analyzed by XPS after five cycles at a current density of 1 mA cm−2. As shown in Fig. 5(a) and (b), common species are present in all systems at C 1s, including C–C (284.8 eV), C–O (286.2 eV), CO (289.3 eV), and OC–O (289.9 eV). For the conventional SEI formed from the BE electrolyte, the C 1s spectra show a high proportion of C–F (291.1 eV) and C–O species with increasing sputtering depth, suggesting that a large number of organic phases will be present in the SEI formed from the massive decomposition of FEC and EMC solvents. Interestingly, ESEI only exhibited weak P–F (687.7 eV) at 0 nm in the F 1s spectrum and then disappeared at 20 nm, replaced by the presence of a large amount of LiF (684.8 eV), demonstrating that the 0–20 nm region consisted of an organic–inorganic hybrid phase and the inner layer was the inorganic phase.47 The O 1s spectrum showed SO species, confirming the continuous decomposition of the sulfonyl fluoride groups to create a superior SEI.42 Meanwhile, compared to the SEI of the BE electrolyte, the ESEI contains more abundant Li2O (528.1 eV) species. The increasing proportion of the Li2O species, which have recently been recognized as a key factor in enhancing CE, is beneficial for achieving high-performance LMBs.17,48 The S 2p spectrum displayed similar characteristics to those of F and O, S mainly existing as Li2S species present throughout the ESEI, whereas the undecomposed sulfonyl fluoride groups are mainly concentrated in the outer layer of the ESEI, which will unite the other components as a resilient organic outer layer (Fig. S29, ESI†). The distribution of species at different sputtering depths was further investigated (Fig. 5(c) and (d)). The C elemental content in ESEI decreased dramatically (from 19.1% to 5.8%), inhibiting the unfavorable decomposition of the free solvent. The elemental F content increased significantly (from 12.1% to 30.8%) under the combined effect of based-layer and TSFSA. Thus, ESEI consists of an organic–inorganic hybrid outer layer, enriched with inorganic phase inner layers at grain boundaries. The inorganic components, such as LiF, Li2O, and Li2S, of the SEI not only enhance its own mechanical strength and inhibit the penetration of dendritic dendrites but also increase the Li+ transport flux and promote highly reversible Li plating/stripping. Time-of-flight secondary ion mass spectrometry (TOF-SIMs) was used to further study the spatial composition of the SEI. As shown in Fig. 5(e), (f) and Fig. S30–S32 (ESI†), the 3D reconstructed sputtering images indicated that the organic phase fragments C2H3O− and C2F− in ESEI were sparsely distributed only at the top, whereas the inorganic phase fragments LiF2− and S− were densely and uniformly distributed. In sharp contrast, the SEI derived from BE was significantly thicker than that derived from ESEI, with both organic and inorganic phases uniformly distributed throughout the entire SEI. This type of SEI cannot provide effective buffering during Li plating/stripping, leading to SEI rupture and further degradation of the electrode.49 Additionally, such a thick SEI is unfavorable for Li-ion transport and increases polarization. Therefore, the ESEI can induce the formation of a thin and dense SEI with an outer organic–inorganic hybrid phase and inner inorganic structure. Specifically, the external organic polymer can provide good volumetric buffering during cycling, while the internal LiF and Li2O regulate the flux of Li ions, inducing uniform lithium striping. Li2S enhances the ionic conductivity of the SEI layer, facilitating the adhesion of SEI components.
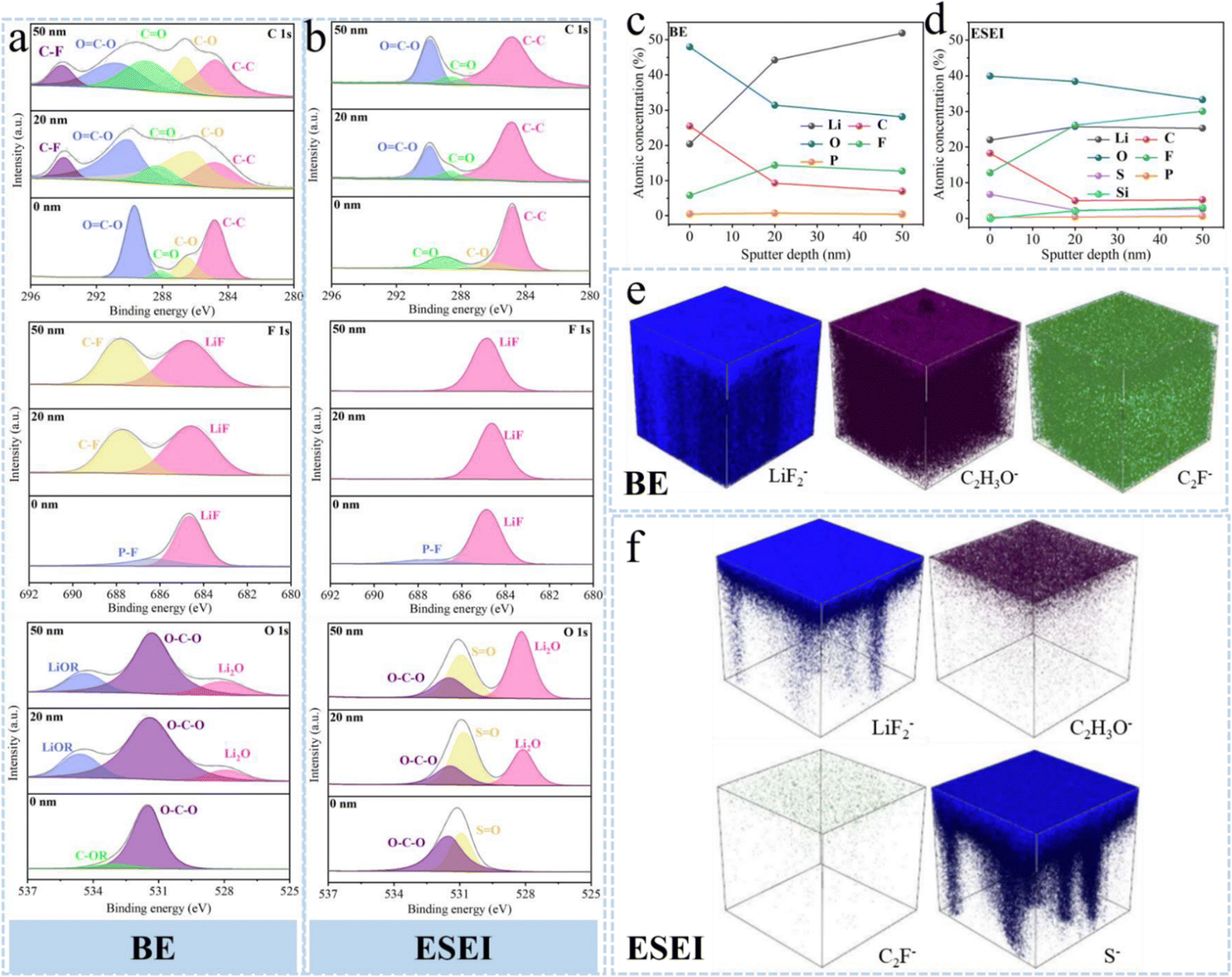 | ||
| Fig. 5 XPS depth characterization of C 1s, F 1s and O 1s spectra for (a) BE and (b) ESEI after 5 cycles. Elemental ratios at different depths for (c) BE and (d) ESEI. 3D TOF-SIMS sputtering images of selected secondary ion fragments for (e) BE of LiF2−, C2H3O− and C2F− and (f) ESEI of LiF2−, C2H3O−, C2F− and S−. | ||
Dynamics characterization of the ESEI
Electrochemical impedance spectroscopy (EIS) measurements were conducted to further characterize the interface features of the electrodes, and highlight the advantages of ESEI for Li+ transport. In all three cells, BE exhibited the highest Rint (including SEI and charge transfer impedance) and the fresh electrodes started to get damaged. During cycling, the Rint of all cells decreased, while that of ESEI exhibited the lowest Rint after 50 cycles (Fig. 6(a)–(c) and Fig. S33, ESI†). This phenomenon was attributed to the functions of the sulfonyl fluoride group at the electrode–electrolyte interface, while the high ionic conductivity of Li2S in ESEI accelerated the transport of Li+.50 Given that the interfacial properties have thus changed, the reaction kinetics were improved as well. The fitting results of the exchange current density also supported this conclusion. The j0 values obtained from the Tafel equations indicate the speed of charge transfer processes on the electrodes. The j0 value of ESEI (1.262 mA cm−2) was higher than that of BE (0.446 mA cm−2) and based-layer (0.877 mA cm−2), implying that the ESEI had faster charge transfer kinetics and higher stability of plating/stripping (Fig. 6(d)). EIS measurements conducted at different temperatures were used to study the effect of Li plating activation energy and electrochemical kinetics at the electrode/electrolyte interface. Interestingly, the activation energy (Ea) of pre-ESEI (60.11 kJ mol−1) was significantly higher than that of BE (48.00 kJ mol−1) in the unactivated state (Fig. S34, ESI†). After two cycles of activation, the electrochemical reactions completed the construction of the ESEI. At this point, the Ea of BE increased to 61.36 kJ mol−1 due to the slow Li+ transfer kinetics resulting from the poor SEI of BE. Although the construction of pre-ESEI was affected due to the initial reaction, the final ESEI formed through the incremental construction exhibited a lower activation energy (56.45 kJ mol−1), which showed a lower Li+ diffusion energy barrier (Fig. 6(e), (f), Fig. S35, S36 and Tables S3–S6, ESI†). The change in the activation energy reflects the influence of SEI on the energy barrier for Li plating. This demonstrated the strong affinity of the SEI enhanced by ESEI for Li metal.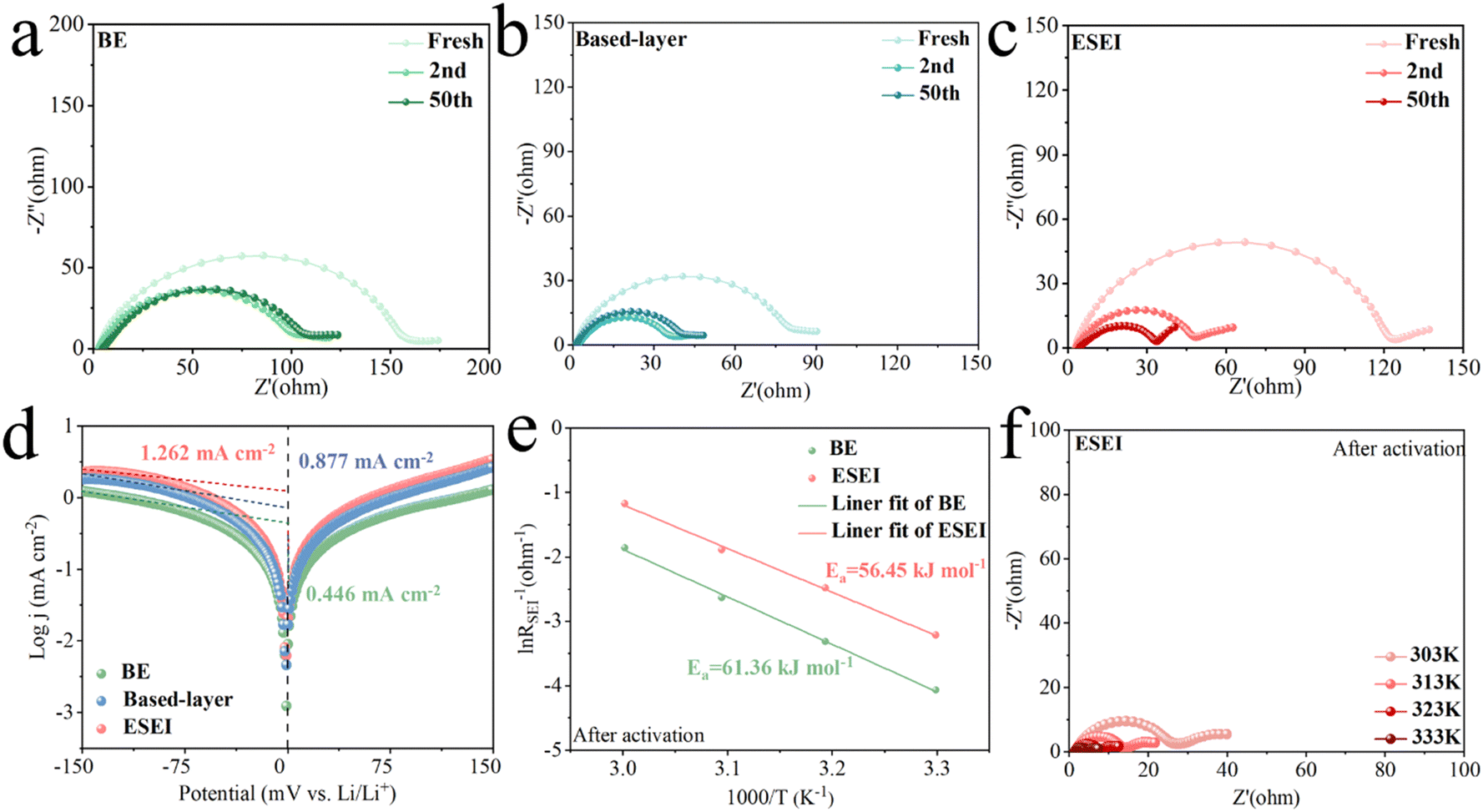 | ||
| Fig. 6 Nyquist plots of Li||Li symmetric cells at various cycles for (a) BE, (b) based-layer and (c) ESEI. (d) Tafel plots and exchange current densities of Li||Li symmetric cells for BE, based-layer and ESEI. (e) Arrhenius behavior and corresponding activation energy for Li+ diffusion through the activated SEI film derived from RSEI in Nyquist plots at various temperatures of Li||Li symmetric cells with BE and ESEI after 2 cycles. (f) EIS plots of the Li||Li symmetric cells with ESEI at different temperatures after 2 cycles. | ||
Electrochemical performance of the high-loading full cells
Based on the excellence of ESEI in LMAs, Li||LFP full cells were assembled using a highly loaded LFP (20.5 mg cm−2) as the cathode to evaluate the effectiveness of ESEI in real lithium metal batteries (LMBs) systems. Fig. 7(a) shows the cycling performance of the full battery at different rates. The Li||LFP cells with ESEI can be discharged at higher rates with specific capacities of 155.1, 154.5, and 147.4 mA h g−1 at 0.2 C, 0.5 C, and 1 C, respectively, which are higher than those provided by the BE and the based-layer at the same rates. Notably, at the high current density of 4 C (13.94 mA cm−2), BE only delivered a specific capacity of 7.7 mA h g−1, the cell basically fails at such a high current density. Due to the reinforcement of the LMAs by the based-layer a specific capacity of 106.4 mA h g−1 was achieved. Finally, with the help of ESEI, the Li||LFP cells exhibit a capacity of 111.9 mA h g−1. In addition, ESEI exhibits a narrow voltage gap on the capacity–voltage curve proving the fast reaction kinetics of ESEI. The advanced component regulation in ESEI led to the highest discharge plateau and lowest charge plateau, consistent with the symmetric cell and impedance test results (Fig. 7(b)–(d)). To validate this hypothesis, different scan rate CV tests were conducted on full cells after two activation cycles (Fig. S37a–c, ESI†). BE exhibited a split reduction peak at higher scan rates (>0.1 mV s−1) after activation, indicating that the BE anode SEI is unsatisfactory for high-loading full cells. The lithium-ion diffusion coefficients of BE, based-layer and ESEI were fitted using the Randles–Sevcik equation. Fig. S37d and e (ESI†) showed the highest lithium-ion diffusion coefficient of ESEI, indicating that Li2S in the SEI enhanced ionic conductivity, enabling the anode to achieve rapid ion transport under high-current conditions.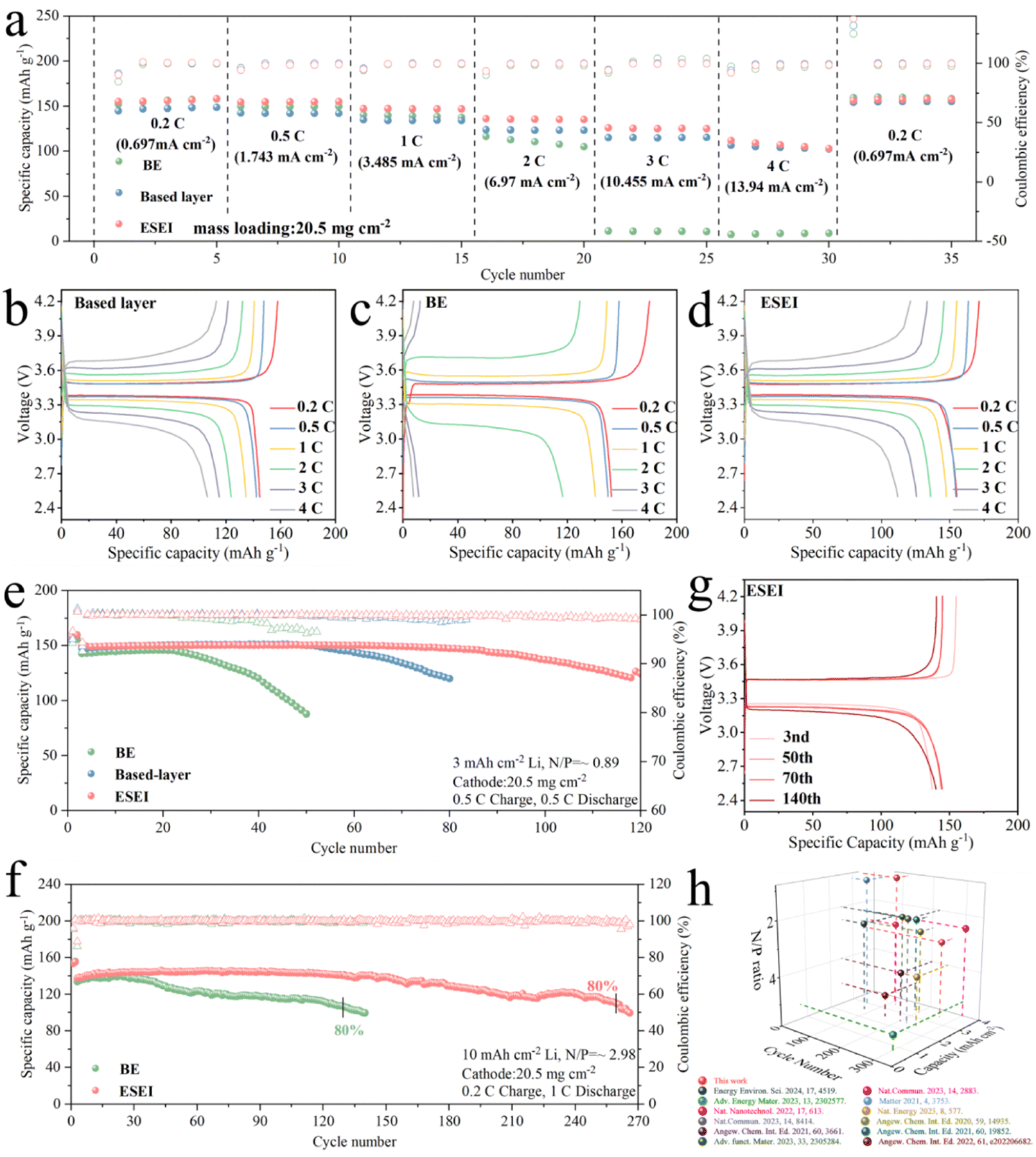 | ||
| Fig. 7 (a) Rate performance of Li||LFP full cells with BE, based-layer and ESEI from 0.2 to 4 C. Charge–discharge profiles of Li||LFP full cells with (b) BE, (c) based-layer and (d) ESEI at different rates. (e) Long-term cycling performance of Li||LFP full coin cells with BE, based-layer and ESEI in a 1 M LiPF6 in FEC/EMC (v/v = 3:7) electrolyte; conditions: 3 mA h cm−2 deposited Li, high area loading LPF (3.485 mA h cm−2, 20.5mgcm−2). The cells were activated at 0.1 C for 2 cycles, then charged at 0.5 C and discharged at 0.5 C in subsequent cycles. (f) Long-term cycling performance of Li||LFP full cells with BE and ESEI in a 1 M LiPF6 in FEC/EMC (v/v = 3:7) electrolyte; conditions: 10 mA h cm−2 deposited Li, high area loading LPF (3.485 mA h cm−2, 20.5mgcm−2). The cells were activated at 0.1 C for 2 cycles, then charged at 0.2 C and discharged at 1 C in subsequent cycles. (g) Charge–discharge profiles of Li||LFP full cells with ESEI under a N/P of 2.98. (h) Comparing the long-cycling performance of our work with previous studies in carbonate electrolyte full cells. See Table S7 (ESI†) for details. | ||
In order to evaluate the electrochemical performance of LMBs under real and more severe conditions, Li||LFP cells under low N/P ratio conditions were further matched to test the long-cycle performance to fully reflect the true results of the lithium anode. First, electrolytes containing TSFSA were used to deposit 3 mA h cm−2 of Li on a Cu foil with a corresponding N/P ratio of 0.89. As expected, Li||LPF full cells assembled with ESEI maintained stable cycling for over 115 cycles at extremely low N/P ratios with a capacity retention of 80% (Fig. 7(e)). In contrast, Li||LFP full cells of BE only maintained stable cycling for about 20 cycles before experiencing a rapid capacity drop due to Li depletion. The full cells assembled with the based-layer exhibited 80% capacity retention after 80 cycles, demonstrating the enhancer's positive effect on enhancing the SEI during the electrochemical reaction. Further increasing the capacity to 10 mA h cm−2, with a corresponding N/P ratio of 2.98, the same trend was observed (Fig. 7(f)). Under the influence of ESEI, Li||LFP full cells achieved stable cycling for over 260 cycles (with 80% capacity retention). BE maintained 80% capacity for only about 130 cycles. During cycling, ESEI effectively maintained interface stability with a slight increase in overpotential after 140 cycles, while BE exhibited a significant increase in overpotential and depleted Li metal during cycling (Fig. 7(g) and Fig. S38, ESI†). Fig. S39 (ESI†) shows the corresponding Nyquist plots after 50 cycles. The significantly increased Rct indicated severe interfacial degradation in BE during cycling. The smallest Rct of ESEI demonstrated improved charge transfer kinetics, attributed to the enhanced stability of the SEI through electrochemical reactions. This stability allowed rapid ion diffusion and suppressed dendrite formation. CV test results further reveal the smaller polarization change of ESEI after 50 cycles, confirming that ESEI prevented parasitic reactions between Li and the electrolyte, enabling uniform, dendrite-free Li deposition (Fig. S40, ESI†).
Using a commercialized electrolyte without FEC matched with a high-loading LFP cathode, the practicality of the based-layer was further studied (Fig. S38, ESI†). Even with a thicker Li anode (300 μm), full cell assembled with BE only maintained 80% capacity for 50 cycles. Encouragingly, ESEI increased the cycle stability of the full cell by more than three times (160 cycles). The capacity–voltage curves of full cells demonstrated that ESEI could stabilize the Li metal interface even without FEC, enhancing battery reversibility. In particular, the performance of the full cells for ESEI is better than those of the previously reported full cells in carbonate electrolytes (Fig. 7(h) and Table S7, ESI†).
To investigate the feasibility of ESEI in high-energy-density batteries, the LSV curves of the electrolyte containing TSFSA were first tested. TSFSA exhibited better oxidation stability in FEC-riched electrolytes under 5 V conditions (Fig. S42, ESI†), indicating its applicability to high-voltage full cells. The assembled Li||NCM811 full cell featured cathodes with high mass-loading (26.0 mg cm−2) and was charged to 4.6 V to achieve higher capacity (6.4 mA h cm−2). After 70 cycles, ESEI demonstrated higher capacity (246.5 mA h g−1 at 0.1 C and 218.9 mA h g−1 at 1 C), excellent cycling performance and lower polarization compared to the rapid capacity decay observed in cells composed of BE and NCM811 (Fig. S43, ESI†). Therefore, this SEI-enhancing strategy has a good application prospect for further improving the energy density of LMBs.
Conclusions
In summary, we proposed a strategy to build an ESEI on a lithium metal anode by chemically pre-constructing a mortice-tenon joint based-layer, and then further forming it through in situ electrochemical reactions with SEI enhancer. The DFSA–Li as the based-layer was first obtained via the reaction between DFSA and Li, which firmly bonds with the Li metal, thereby providing an embedded base on Li metal. Meanwhile, the TSFSA functional molecule in the electrolyte acted as an SEI enhancer, preferentially decomposing over the carbonate solvent to form the same components as those of the SEI, thus achieving a perfect combination between the based-layer and the subsequent electrochemically decomposed productions, resulting in a final ideal SEI, namely, ESEI. This enhanced SEI achieved long-cycle stability of the Li metal anode in carbonate electrolyte for over 2100 hours at a current density of 1 mA cm−2 and a high average CE of 99.2%. The full cells achieved higher cycling stability when paired with a high-loading LPF cathode (20.5 mg cm−2). Thus, this strategy holds promise for realizing the next generation of high energy density Li-metal batteries.Author contributions
K. W., C. W. and Q. D. conceived of the idea and designed the experiments. K. W., S. L., Q. Z., J. C., and X. Y performed the cell test, electrochemical measurements, and characterizations. R. Y. conducted the theoretical calculation. C. D. performed cryo-TEM. K. W. and C. W. recorded and analysed the XPS and TOF-SIMs. All authors discussed and analysed the data. K. W. wrote the manuscript. Y. T., M. Z., J. F., and Q. D. revised the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the NSFC projects (22179112, 22072117, 22021001). We are also grateful for Professor Yan Jiawei's group at Xiamen University for their assistance in AFM testing.References
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- B. Liu, J.-G. Zhang and W. Xu, Joule, 2018, 2, 833–845 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- C. Yang, K. Fu, Y. Zhang, E. Hitz and L. Hu, Adv. Mater., 2017, 29, 1701169 CrossRef PubMed.
- S. Li, M. Jiang, Y. Xie, H. Xu, J. Jia and J. Li, Adv. Mater., 2018, 30, 1706375 CrossRef PubMed.
- R. Wang, W. Cui, F. Chu and F. Wu, J. Energy Chem., 2020, 48, 145–159 CrossRef.
- Y. Zhang, T.-T. Zuo, J. Popovic, K. Lim, Y.-X. Yin, J. Maier and Y.-G. Guo, Mater. Today, 2020, 33, 56–74 CrossRef CAS.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. Xu, C. Wang, B. Horstmann, R. Amine, C.-C. Su, J. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036–1052 CrossRef CAS.
- A. Hu, F. Li, W. Chen, T. Lei, Y. Li, Y. Fan, M. He, F. Wang, M. Zhou, Y. Hu, Y. Yan, B. Chen, J. Zhu, J. Long, X. Wang and J. Xiong, Adv. Energy Mater., 2022, 12, 2202432 CrossRef CAS.
- H. Wang, Z. Yu, X. Kong, S. C. Kim, D. T. Boyle, J. Qin, Z. Bao and Y. Cui, Joule, 2022, 6, 588–616 CrossRef CAS.
- X.-R. Chen, B.-C. Zhao, C. Yan and Q. Zhang, Adv. Mater., 2021, 33, 2004128 CrossRef CAS PubMed.
- Y. Han, B. Liu, Z. Xiao, W. Zhang, X. Wang, G. Pan, Y. Xia, X. Xia and J. Tu, InfoMat, 2021, 3, 155–174 CrossRef CAS.
- P. Zou, Y. Sui, H. Zhan, C. Wang, H. L. Xin, H.-M. Cheng, F. Kang and C. Yang, Chem. Rev., 2021, 121, 5986–6056 CrossRef CAS PubMed.
- R. Pathak, K. Chen, A. Gurung, K. M. Reza, B. Bahrami, J. Pokharel, A. Baniya, W. He, F. Wu, Y. Zhou, K. Xu and Q. Qiao, Nat. Commun., 2020, 11, 93 CrossRef CAS PubMed.
- B. Jagger and M. Pasta, Joule, 2023, 7, 2228–2244 CrossRef CAS.
- H. Zeng, K. Yu, J. Li, M. Yuan, J. Wang, Q. Wang, A. Lai, Y. Jiang, X. Yan, G. Zhang, H. Xu, J. Wang, W. Huang, C. Wang, Y. Deng and S.-S. Chi, ACS Nano, 2024, 18, 1969–1981 CrossRef CAS PubMed.
- S. Ni, M. Zhang, C. Li, R. Gao, J. Sheng, X. Wu and G. Zhou, Adv. Mater., 2023, 35, e2209028 CrossRef PubMed.
- H. Chen, A. Pei, D. Lin, J. Xie, A. Yang, J. Xu, K. Lin, J. Wang, H. Wang, F. Shi, D. Boyle and Y. Cui, Adv. Energy Mater., 2019, 9, 1900858 CrossRef.
- X. Zhang, P. Xu, J. Duan, X. Lin, J. Sun, W. Shi, H. Xu, W. Dou, Q. Zheng, R. Yuan, J. Wang, Y. Zhang, S. Yu, Z. Chen, M. Zheng, J.-F. Gohy, Q. Dong and A. Vlad, Nat. Commun., 2024, 15, 536 CrossRef CAS PubMed.
- J.-n Duan, Q. Hou, R.-m Yuan, J. Fan, M. Zheng and Q. Dong, J. Mater. Chem. A, 2023, 11, 548–558 RSC.
- T. D. Pham, A. Bin Faheem, J. Kim, K. Kwak and K.-K. Lee, Adv. Funct. Mater., 2023, 33, 2305284 CrossRef CAS.
- Y.-H. Tan, G.-X. Lu, J.-H. Zheng, F. Zhou, M. Chen, T. Ma, L.-L. Lu, Y.-H. Song, Y. Guan, J. Wang, Z. Liang, W.-S. Xu, Y. Zhang, X. Tao and H.-B. Yao, Adv. Mater., 2021, 33, 2102134 CrossRef CAS PubMed.
- X. Cui, J. Cheng, C. Li, Z. Sun, K. Li, Y. Wang, X. Fan, S. Tang, X. Lin, R. Yuan, B. Mao, M. Zheng and Q. Dong, Energy Environ. Mater., 2023, 6, 12393 CrossRef.
- S. Li, W. Zhang, Q. Wu, L. Fan, X. Wang, X. Wang, Z. Shen, Y. He and Y. Lu, Angew. Chem., Int. Ed., 2020, 59, 14935–14941 CrossRef CAS PubMed.
- A. Muthulatha, T. Bhanupriya, S. Srikanth and S. Pramodhini, Res. Dermatol., 2021, 27, 423–428 Search PubMed.
- S. Stalin, P. Chen, G. Li, Y. Deng, Z. Rouse, Y. Cheng, Z. Zhang, P. Biswal, S. Jin, S. P. Baker, R. Yang and L. A. Archer, Matter, 2021, 4, 3753–3773 CrossRef CAS.
- Y. Xie, Y. Huang, Y. Zhang, T. Wu, S. Liu, M. Sun, B. Lee, Z. Lin, H. Chen, P. Dai, Z. Huang, J. Yang, C. Shi, D. Wu, L. Huang, Y. Hua, C. Wang and S. Sun, Nat. Commun., 2023, 14, 2883 CrossRef CAS PubMed.
- J. Pokharel, A. Cresce, B. Pant, M. Y. Yang, A. Gurung, W. He, A. Baniya, B. S. Lamsal, Z. Yang, S. Gent, X. Xian, Y. Cao, W. A. Goddard, K. Xu and Y. Zhou, Nat. Commun., 2024, 15, 3085 CrossRef CAS PubMed.
- Y. Tan, Z. Sun, Y. Liu, H. Yan, C. Wang, J. Fan, M. Zheng and Q. Dong, Adv. Energy Mater., 2024, 14, 2304153 CrossRef CAS.
- H. Zhang, G. Gebresilassie Eshetu, X. Judez, C. Li, L. M. Rodriguez-Martinez and M. Armand, Angew. Chem., Int. Ed., 2018, 57, 15002–15027 CrossRef CAS PubMed.
- U. S. Meda, L. Lal, M. Sushantha and P. Garg, J. Energy Storage, 2022, 47, 103564 CrossRef.
- R. Xu, Y. Xiao, R. Zhang, X.-B. Cheng, C.-Z. Zhao, X.-Q. Zhang, C. Yan, Q. Zhang and J.-Q. Huang, Adv. Mater., 2019, 31, 1808392 CrossRef PubMed.
- R. Xu, X.-Q. Zhang, X.-B. Cheng, H.-J. Peng, C.-Z. Zhao, C. Yan and J.-Q. Huang, Adv. Funct. Mater., 2018, 28, 1705838 CrossRef.
- Y. Gao, G. Wu, W. Fang, Z. Qin, T. Zhang, J. Yan, Y. Zhong, N. Zhang and G. Chen, Angew. Chem., Int. Ed., 2024, 63, e202403668 CrossRef CAS PubMed.
- S. Kim, S. O. Park, M.-Y. Lee, J.-A. Lee, I. Kristanto, T. K. Lee, D. Hwang, J. Kim, T.-U. Wi, H.-W. Lee, S. K. Kwak and N.-S. Choi, Energy Storage Mater., 2022, 45, 1–13 CrossRef.
- T. Hou, G. Yang, N. N. Rajput, J. Self, S.-W. Park, J. Nanda and K. A. Persson, Nano Energy, 2019, 64, 103881 CrossRef CAS.
- Y. Liu, J. Wu and Y. Yang, J. Electrochem. Soc., 2020, 167, 160535 CrossRef.
- Z. Sun, Y. Wang, S. Shen, X. Li, X. Hu, M. Hu, Y. Su, S. Ding and C. Xiao, Angew. Chem., Int. Ed., 2023, 62, e202309622 CrossRef CAS PubMed.
- X. Hu, Y. Gao, B. Zhang, L. Shi and Q. Li, Ecomat, 2022, 4, e12264 CrossRef CAS.
- Q. Zhao, Y. Deng, N. W. Utomo, J. Zheng, P. Biswal, J. Yin and L. A. Archer, Nat. Commun., 2021, 12, 6034 CrossRef CAS PubMed.
- D.-Y. Han, I. K. Han, H. Y. Jang, S. Kim, J. Y. Kwon, J. Park, S. Back, S. Park and J. Ryu, Energy Storage Mater., 2024, 65, 103176 CrossRef.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao, F. Wei, J.-G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef PubMed.
- N.-W. Li, Y.-X. Yin, C.-P. Yang and Y.-G. Guo, Adv. Mater., 2016, 28, 1853–1858 CrossRef CAS PubMed.
- B. D. Adams, J. Zheng, X. Ren, W. Xu and J.-G. Zhang, Adv. Energy Mater., 2018, 8, 1702097 CrossRef.
- W. Zhang, H. L. Zhuang, L. Fan, L. Gao and Y. Lu, Sci. Adv., 2018, 4, eaar4410 CrossRef PubMed.
- Y.-X. Zhan, Z.-Y. Liu, Y.-Y. Geng, P. Shi, N. Yao, C.-B. Jin, B.-Q. Li, G. Ye, X.-Q. Zhang and J.-Q. Huang, Energy Storage Mater., 2023, 60, 102799 CrossRef.
- G. M. Hobold, C. Wang, K. Steinberg, Y. Li and B. M. Gallant, Nat. Energy, 2024, 9, 580–591 CrossRef CAS.
- M. Tian, L. Ben, H. Yu, Z. Song, Y. Yan, W. Zhao, M. Armand, H. Zhang, Z.-B. Zhou and X. Huang, J. Am. Chem. Soc., 2022, 144, 15100–15110 CrossRef CAS PubMed.
- R. Zhang, B. Chen, C. Shi, J. Sha, L. Ma, E. Liu and N. Zhao, Small, 2023, 19, 2208095 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee04617j |
| ‡ K. W., C. W., S. L. contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2025 |