
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrolyte tailoring and interfacial engineering for safe and high-temperature lithium-ion batteries†
Chenyang
Shi
a,
Zhengguang
Li
g,
Mengran
Wang
 *acef,
Shu
Hong
h,
Bo
Hong
*acef,
Yaxuan
Fu
a,
Die
Liu
g,
Rui
Tan
*b,
Pingshan
Wang
g and
Yanqing
Lai
adef
*acef,
Shu
Hong
h,
Bo
Hong
*acef,
Yaxuan
Fu
a,
Die
Liu
g,
Rui
Tan
*b,
Pingshan
Wang
g and
Yanqing
Lai
adef
aSchool of Metallurgy and Environment, Central South University, Changsha 410083, Hunan, China. E-mail: mengranwang93@163.com; bop_hong@csu.edu.cn
bDepartment of Chemical Engineering, Swansea University, Swansea, SA1 8EN, UK. E-mail: rui.tan@swansea.ac.uk
cEngineering Research Centre of Advanced Battery Materials, The Ministry of Education, Changsha 410083, Hunan, China
dHunan Province Key Laboratory of Nonferrous Value-Added Metallurgy, Central South University, Changsha 410083, Hunan, China
eNational Energy Metal Resources and New Materials Key Laboratory, Changsha, 410083, Hunan, China
fNational Engineering Research Center of Advanced Energy Storage Materials, Changsha, 410083, Hunan, China
gSchool of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, Hunan, China
hTianjin Lishen Battery Joint-Stock Co., Ltd, Tianjin, 300000, China
First published on 13th February 2025
Abstract
The deployment of lithium-ion batteries, essential for military and space exploration applications, faces restrictions due to safety issues and performance degradation stemming from the uncontrollable side reactions between electrolytes and electrodes, particularly at high temperatures. Current research focuses on interfacial modification and non-flammable electrolyte development, which fails to simultaneously improve both safety and cyclic performance. This work introduces a synergistic approach by incorporating weakly polar methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (MDFSA) and non-flammable 2-(2,2,2-trifluoroethoxy)-1,3,2-dioxaphospholane 2-oxide (TFP) to achieve a localized high-concentration electrolyte (LHCE) that can stabilize both anode and cathode interfaces and thus improve the cycling life and safety of batteries, particularly at evaluated temperatures. As a result, the NCM811|Gr pouch cell with MDFSA-containing LHCE exhibits a high capacity retention rate of 79.6% at 60 °C after 1200 cycles due to the formation of thermally and structurally stable interfaces on the electrodes, outperforming pouch cells utilizing commercial carbonate-based (capacity retention: 23.7% after 125 cycles). Additionally, pouch cells in the charging state also exhibit commendable safety performance, indicating potential for practical applications.
Broader contextAchieving net-zero emissions by 2050 requires rapid advancement in renewable energy and efficient energy storage technologies. Lithium-ion batteries (LIBs) show great promise for grid-scale energy storage, especially with emerging battery chemistries based on layered transition metal oxides, which offer potential for higher energy and power densities. However, thermal issues are challenging the durability of LIBs, as demonstrated by frequent reports of battery failures and fire incidents. This highlights the need for breakthrough materials solutions to address these thermal issues effectively. This work introduces a synergistic approach by incorporating weakly polar methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (MDFSA) and non-flammable 2-(2,2,2-trifluoroethoxy)-1,3,2-dioxaphospholane 2-oxide (TFP) to achieve a localized high-concentration electrolyte (LHCE) that can stabilize both anode and cathode interfaces and thus improve the cycling life and safety of batteries, particularly at evaluated temperatures. As a result, the NCM811|Gr pouch cell with MDFSA-containing LHCE exhibits a high capacity retention rate of 79.6% at 60 °C after 1200 cycles due to the formation of thermally and structurally stable interfaces on the electrodes, outperforming pouch cells utilizing commercial carbonate-based electrolytes (capacity retention: 23.7% after 125 cycles). Additionally, pouch cells in the charging state also exhibit commendable safety performance, indicating potential for practical applications. |
Introduction
Currently, there is substantial demand for lithium-ion batteries (LIBs) with high safety and exceptional performance at high temperatures, particularly for military and space exploration applications.1,2 However, LIBs with commercial electrolytes often face poor adaptability, limited thermal stability and insufficient safety due to uncontrollable side reactions between the electrolyte and electrodes at elevated temperatures.3 Commercial electrolytes tend to form organic-rich and thermally unstable interfacial films on both cathode and anode electrodes, compromising interface stability and exacerbating side reactions at elevated temperatures.4,5 Specifically, when the temperature exceeds 60 °C, the decomposition of organic components in the solid electrolyte interphase (SEI) film on the graphite anode triggers uncontrollable side reactions between the electrolyte and the electrode, which deplete active materials and rapidly degrade electrochemical performance. On the cathode side, high temperatures increase the activity of the cathode, while the primarily organic cathode electrolyte interphase (CEI) film is insufficient to prevent side reactions between the electrode and electrolyte, leading to the dissolution of transition metals and the structural collapse of the cathode, resulting in a decline in electrochemical performance.6 Consequently, constructing stable and thermally resistant interfacial films is crucial for enhancing the electrochemical performance at high temperatures.7Several strategies have been employed to enhance the stability of the cathode and anode electrode interfaces at high temperatures, such as introducing functional additives,8 solvent optimization, and solvation structure design.9 Using high-concentration/localized high-concentration electrolytes10,11 based on solvation structure design to form inorganic-rich and thermally resistant interface films on the cathode and anode electrodes has proven to be an effective approach.12 Though effective, this approach struggles to simultaneously achieve the optimal construction of both anode and cathode interfaces, leading to suboptimal cycling life. Currently, thermally stable localized high-concentration electrolytes (LHCEs), typically formulated with diluents with strongly positive electrostatic potential (e.g., TTE), often contribute to the formation of rough and non-uniform interface films, which subsequently trigger side reactions.
Addressing these challenges requires selecting solvents from the molecular structure level and tuning the solvation structure to enhance electrode interface stability at high temperatures—an approach that is both important and highly demanding. A synergistic approach is proposed in this work, i.e., incorporating methyl 2,2-difluoro-2-(fluorosulfonyl)acetate (MDFSA) and non-flammable 2-(2,2,2-trifluoroethoxy)-1,3,2-dioxaphospholane 2-oxide (TFP) to modulate the localized high-concentration electrolyte solvation structure that favors the formation of thermally and structurally stable interfaces on electrodes. The active diluent MDFSA, upon decomposition at the anode, effectively increases the thermally stable inorganic components within the SEI film, since its decomposition temperature (>700 °C) is significantly higher than that of organic compounds (60 °C), thereby promoting the overall thermal stability of anodes. Meanwhile, the strongly positively charged TFP interacts with FSI− to form a stable CEI film on the cathode. This simultaneous modification thus effectively prevents electrode/electrolyte side reactions at evaluated temperatures. Therefore, the optimized electrolyte system forms a thin and uniform interfacial film predominantly composed of inorganic materials on the electrode surface. Furthermore, the decomposition temperature of the SEI film has increased from 60 °C to 250 °C, and the phase transition temperature of the cathode material has increased from 150 °C to 170 °C, indicating a significant improvement in the overall thermal stability of the battery. The NCM811|Gr pouch cell with MDFSA-based electrolyte exhibits a capacity retention of 79.6% at 60 °C after 1200 cycles, demonstrating superior performance compared to pouch cells using carbonate-based electrolytes (capacity retention: 23.7% after 125 cycles). This approach introduces a novel design strategy for developing high-temperature and non-flammable electrolytes for NCM811|Gr batteries.
Results and discussion
Thermally stable electrolyte preparation
In contrast to the commercial electrolyte, the localized high-concentration electrolyte, based on non-flammable cyclic phosphate ester (TFP) and inert diluent 1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether (TTE), can largely enhance safety. However, the inertness and strongly positive electrostatic potential of TTE prevent the electrolyte from forming a stable interface film on the electrode surface (Fig. 1a). This is attributed to the strongly positive electrostatic potential of TTE, which allows it to combine with anions and enter the Helmholtz layer on the cathode side; however, its high oxidation potential prevents it from participating in the formation of the CEI film, resulting in uneven deposition at the cathode interface. Given this, selecting the redox-active and weakly positive charge (i.e., weak polarity) solvent chemistries holds the premise for producing stable interfacial layers on both electrodes (Fig. 1a): (a) on the one hand, compared with inert solvent species, redox-active solvents prefer to undergo reduction on the surface of anodes and produce an inorganic-rich interface film with FSI− ions; (b) on the other hand, a strategic combination of solvents with varying polarities can increase the likelihood that strongly polar solvents, in conjunction with FSI−, will cooperatively form a stable CEI film on the cathode.13 To validate our strategy, we selected the redox-active and weakly positive electrostatic potential solvent MDFSA to replace the inert and strongly positive electrostatic potential TTE, a common solvent used in most non-flammable electrolytes.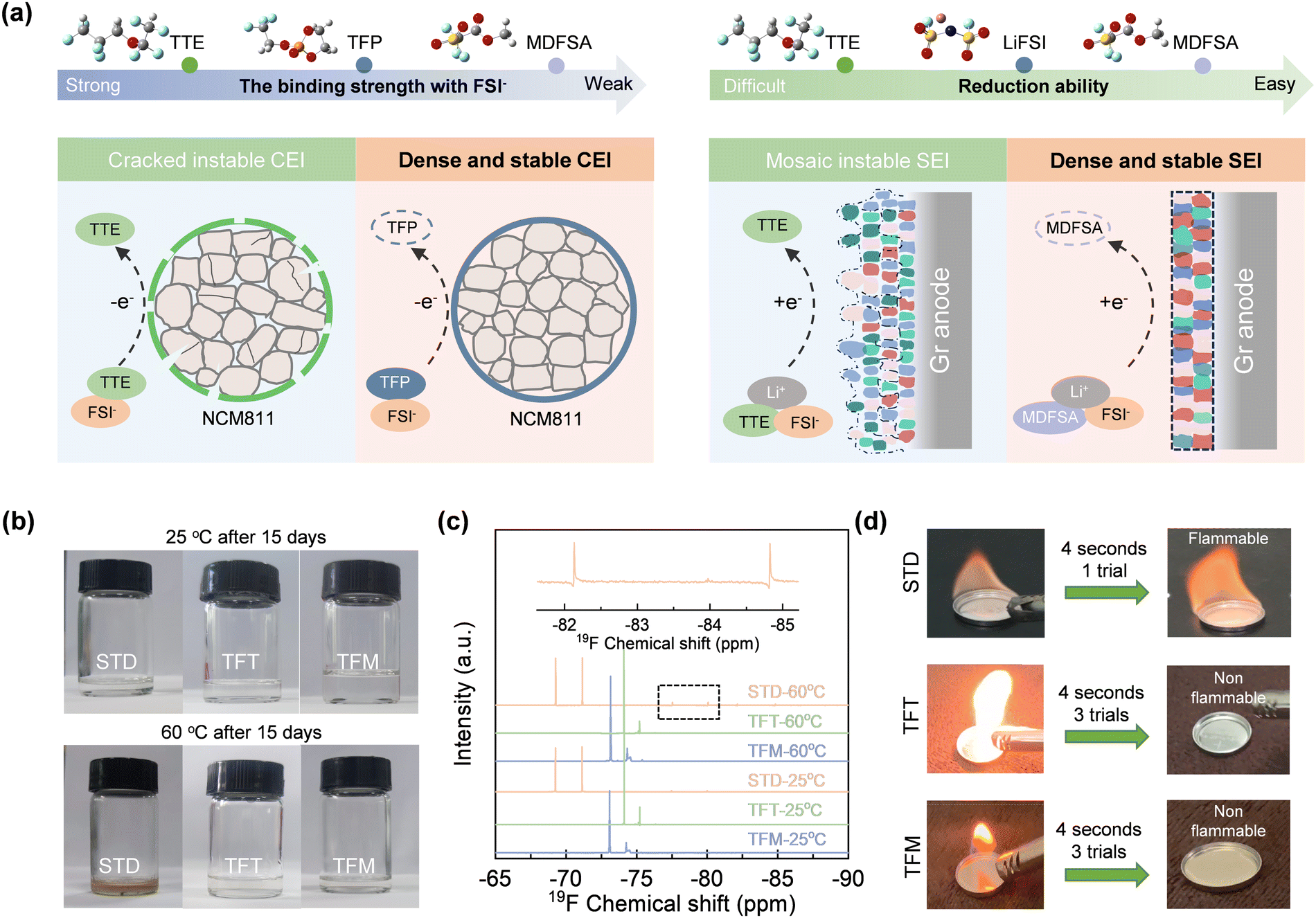 | ||
| Fig. 1 Design strategy and functional electrolyte tailoring. (a) Schematic diagram illustrating the influence of various electrolytes on the interface. (b) Photos of different electrolyte solutions after undergoing thermal treatment at 25 and 60 °C for 15 days. (c) 19F NMR spectroscopy of different electrolyte solutions at 25 and 60 °C. The insets are the enlarged curves at chemical shifts in the ranges of −86–83 ppm and −78–75 ppm. (d) Combustion tests of different electrolytes. STD: 1 M LiPF6 in EC (ethylene carbonate)/DMC (dimethyl carbonate)/EMC (ethyl methyl carbonate) = 1/1/1 (v/v/v); TFT: 1 M LiFSI in TFP (2-(2,2,2-trifluoroethoxy)-1,3,2-dioxaphospholane 2-oxide)/FEMC (methyl 2,2,2-trifluoroethyl carbonate)/TTE (1,1,2,2-tetrafluoroethyl 2,2,3,3-tetrafluoropropyl ether) = 2/6/2 (v/v/v); TFM: 1 M LiFSI TFP/FEMC/MDFSA (methyl 2,2-difluoro-2-(fluorosulfonyl)acetate) = 2/6/2 (v/v/v). | ||
Ignition and high-temperature storage tests were conducted to assess the safety and thermal stability of the electrolytes (TFM: MDFSA-involved bifunctional electrolyte; TFT: TTE involved non-flammable control; and STD: commercial control) at high temperatures. As illustrated in Fig. 1b, the electrolytes remained unchanged in color after a 15-day treatment at room temperature. However, when treated at 60 °C for the same period, the STD electrolyte turns noticeably yellow, indicating decomposition at high temperatures. In contrast, TFT and TFM electrolytes show no significant color change under entire room and high temperature conditions. Further analysis with nuclear magnetic resonance (NMR) confirms the composition of TFT and TFM electrolytes compared to the STD variant, highlighting their superior thermal stability. As shown in Fig. 1c, STD exhibits significant changes in its components at 60 °C compared to 25 °C. Specifically, two doublets at −77.5 to −78.6 ppm and −83.7 to −85.7 ppm, and one singlet at −190.1 ppm were observed. These peaks are possibly attributed to the generation of PO3F2−, PO2F2−, and HF, respectively, at 60 °C,14 suggesting the decomposition of LiPF6. In contrast, NMR spectra of the TFT and TFM electrolytes showed no changes, indicating their excellent stability at high temperatures. This stability highlights their potential suitability for high-temperature applications. Furthermore, both TFT and TFM electrolytes remained non-flammable after multiple ignition attempts, indicating a significant enhancement in safety compared to the STD electrolyte (Fig. 1d). Due to the high concentration of lithium salts and the strong polarity of the solvent, the viscosities of the TFT and TFM electrolytes increase to 99.8 and 89.1 mPa s, respectively, while their conductivities decrease to 1.3 and 1.9 mS cm−1. Given the Li-ion transference number of the TFM electrolyte measured as 0.55, the Li ion conductivity is calculated to be 1.05 mS cm−1.11,15 Moreover, the oxidation potential of the TFT and TFM electrolytes (5.2 V) shows a significant improvement compared to the STD electrolyte (4.5 V; Fig. S1, ESI†).
Rationales of selecting solvent chemistries
The properties of the interfacial layers formed on electrodes are determined by the interactions between solvent molecules and salt ions, as well as the inherent solvation structures. To investigate these solvent–ion interactions, density functional theory (DFT) and molecular dynamics (MD) simulations were employed to derive the HOMO and LUMO energy levels of each pair. As shown in Fig. 2a, the Li+–FSI− interaction (−2.85 eV) is significantly stronger than Li+–TFP (−2.41 eV) and Li+–FEMC (−1.64 eV), indicating that Li+ preferentially binds with FSI− to form a solvation structure involving anions (referring to the Debye–Hückel theory16). In contrast, the interactions between Li+ and TTE (−0.73 eV) or MDFSA (−1.28 eV) are considerably weaker, likely due to the molecular surface charge distribution (Fig. S2, ESI†). The solvation structures of TFT and TFM electrolytes were further characterized using Raman spectroscopy. As shown in Fig. S3 (ESI†), the peak at 730 cm−1 is attributed to contact ion pairs (CIPs), while the peaks at 744 cm−1 and 756 cm−1 correspond to ion-pair aggregates (AGG-I) and more coordinated ion-pair aggregates (AGG-II), respectively.17 This indicates a pronounced solvent structure involving anion participation in both TFT and TFM electrolytes. Moreover, their solvation structures are nearly identical, suggesting that MDFSA has minimal impact on the solvation structure.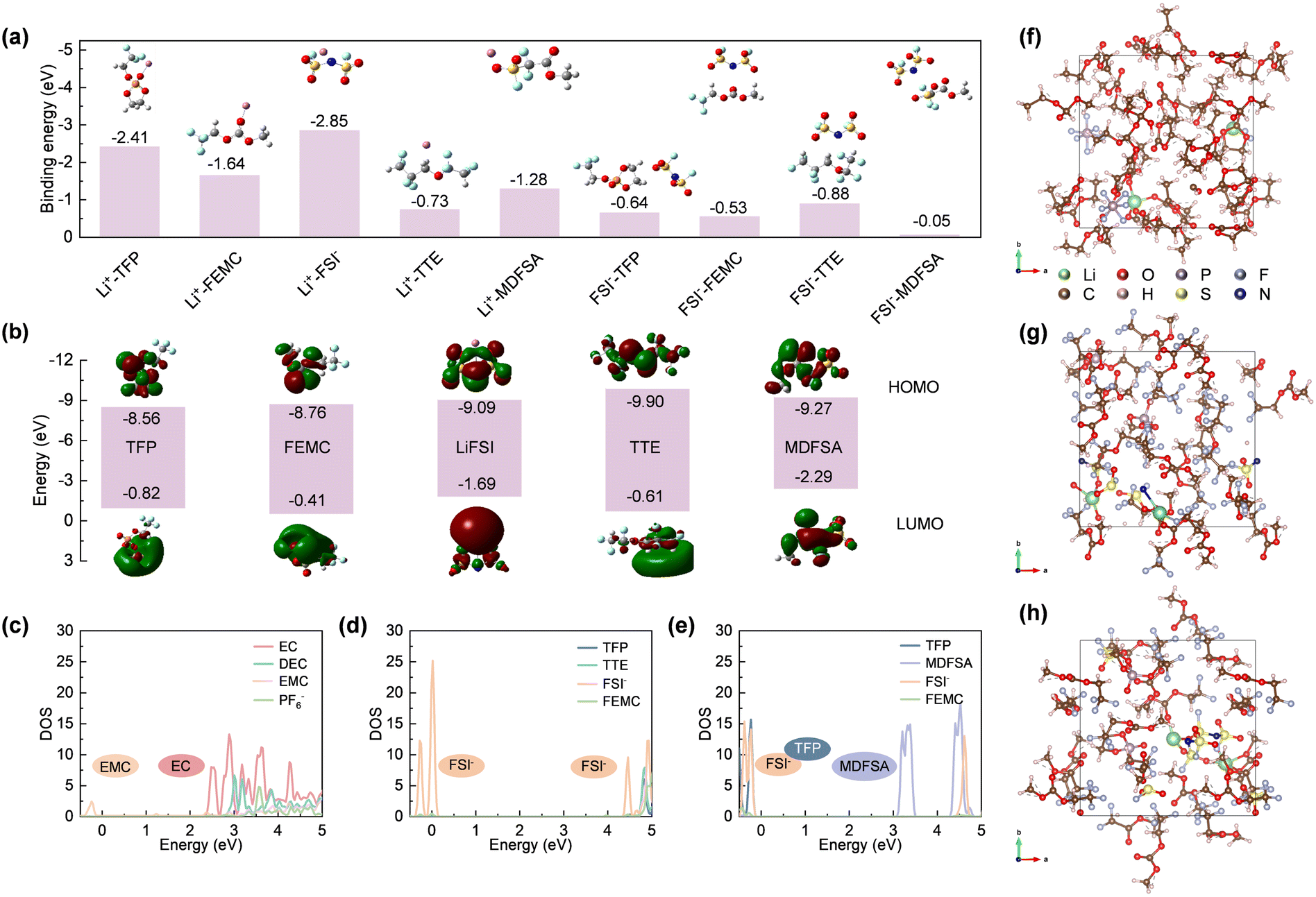 | ||
| Fig. 2 Validation of the selection of solvent molecules. (a) Interaction between FSI−, Li+ and different solvents. (b) Calculated HOMO and LUMO energy (eV) level diagrams of different solvents and corresponding salts. (c)–(h) Density of states (DOS) and corresponding snapshots obtained in quantum mechanical DFT-AIMD simulations of (c) and (f) STD, (d) and (g) TFT, and (e) and (h) TFM electrolytes. | ||
As shown in Fig. 2b, the LUMO level of LiFSI (−1.69 eV) is lower than that of TFP (−0.82 eV) and FEMC (−0.41 eV), indicating that LiFSI prefers to undergo reduction at the anode. In the presence of MDFSA with a much lower LUMO level (−2.29 eV), the reduction of electrolyte and the formation of the SEI may involve two species, LiFSI and MDFSA. Importantly, MDFSA rich in fluorine atoms can potentially enhance the formation of inorganic species LiF within the SEI. Regarding the films formed on cathodes, solvents with a strongly positive electrostatic potential preferentially associate with anions, accumulate on the cathode surface due to electric field forces, and subsequently decompose to form the CEI film.18 In TFT electrolytes, the excessively strongly positive electrostatic potential of TTE leads to preferential binding with FSI−. However, the low HOMO level of TTE hinders its decomposition on the cathode and formation of a CEI film, leading to uneven deposition.19 In contrast, the interaction between MDFSA and FSI− is significantly weaker than that of the TFP. This indicates that FSI− will preferentially bind with TFP, and the higher HOMO level of TFP will undergo ring-opening polymerization on the cathode surface, forming a stable CEI film in conjunction with FSI−.13
To further evaluate the impact of different electrolyte systems on the interface film, DOS calculations were performed for these systems. In Fig. 2c, the LUMO of the STD electrolyte aligns with EC indicating that the SEI film is primarily composed of organic compounds from EC decomposition. The LUMO of the TFT electrolyte is aligned with FSI−, indicating a predominance of inorganic substances (e.g., LiF) generated from FSI− decomposition (Fig. 2d). However, excessive usage of lithium salts may lead to rapid performance degradation. In the TFM electrolyte (Fig. 2e), MDFSA decomposes before FSI−, which might increase the inorganic content in the SEI film enhancing cycle stability.20 Furthermore, CV testing of Gr|Li batteries reveals that the reduction sequence of electrolyte components aligns with the theoretical calculations (Fig. S4, ESI†). Additionally, the HOMO of the STD electrolyte is aligned with the EMC which dominates the formation of the CEI film. The HOMO of the TFT electrolyte aligns with FSI−, and the stable film-forming component TFP does not contribute to the CEI film formation (Fig. 2d). This occurs because the strongly positively charged TTE preferentially binds with TFP and deposits on the cathode, while the low HOMO value of TTE prevents its involvement in the formation of the CEI film, leading to uneven deposition on the cathode, aligning with previous findings.19 In the TFM electrolyte, the HOMO is positioned at both TFP and FSI−, indicating cooperative formation of the CEI film (Fig. 2e). This is facilitated by the weakly positive charge from MDFSA, allowing FSI− to preferentially bind with TFP and deposit on the cathode. Theoretical calculations show that the introduction of MDFSA can enhance the formation of a stable interface film on the electrode surface.
Electrochemical performance validation
To validate the electrochemical performance, NCM811|Gr pouch cells with different electrolytes were tested at 60 °C (Fig. 3). As illustrated in Fig. 3a, the pouch cell with the STD electrolyte retains 23.7% of its initial capacity after 125 cycles. This degradation may be attributed to the decomposition of the interface film, and subsequent uncontrollable electrode/electrolyte side reactions. The pouch cell with the TFT electrolyte exhibits significant capacity degradation in the initial cycles with a capacity retention of 8.2% after 280 cycles, likely due to the uneven deposition of the CEI film and excessive usage of lithium salts. In contrast, the pouch cell with the TFM electrolyte demonstrates excellent electrochemical performance with a capacity of 132.1 mA h g−1 and a capacity retention rate of 79.6% over 1200 cycles at 60 °C, suggesting performance stabilization effects from MDFSA. This stable cycling performance outperforms that reported in previous literature10,14,21–37 (Fig. 3f).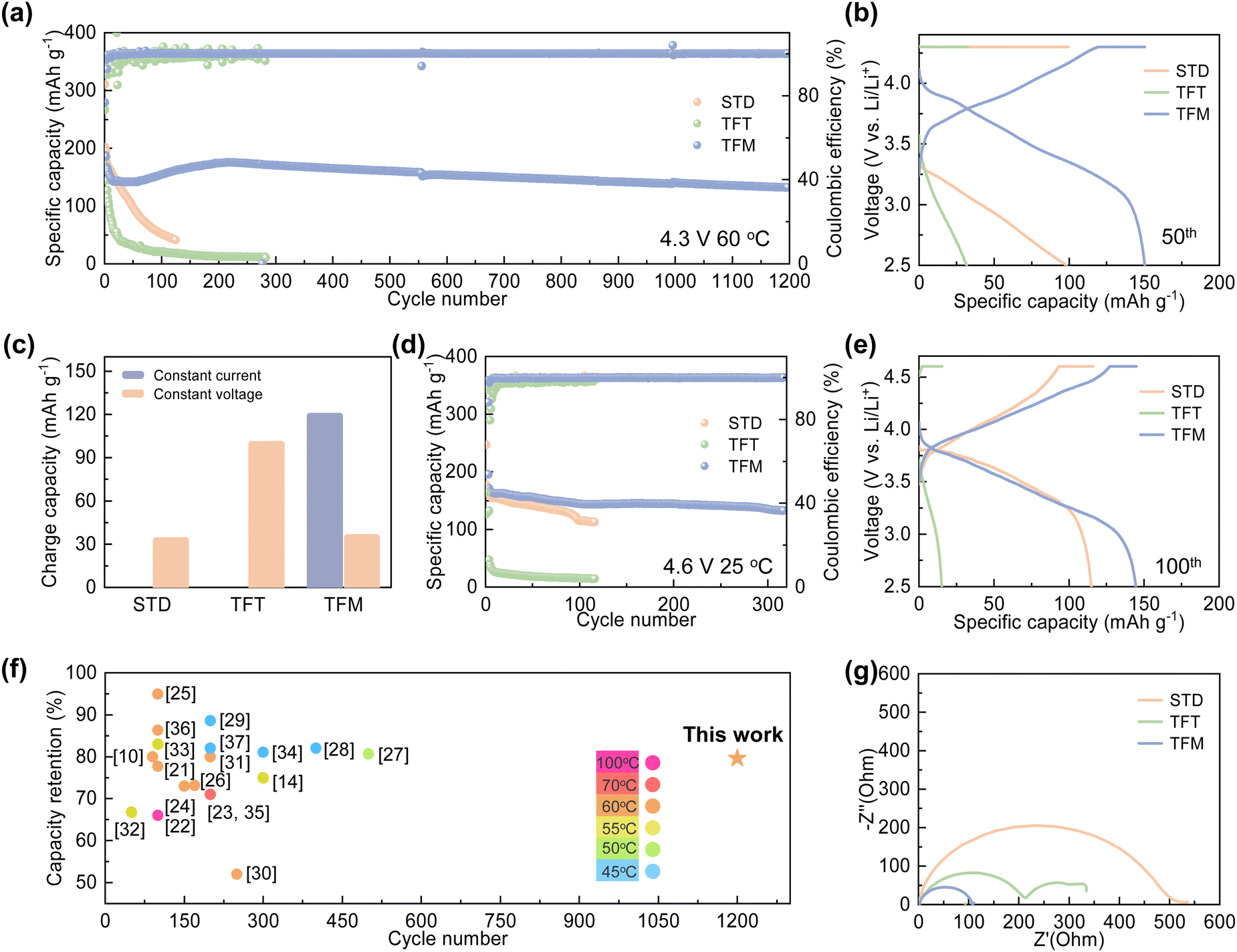 | ||
| Fig. 3 Electrochemical performance of NCM811|Gr pouch cells at 0.5C with different electrolytes at varied temperatures. (a) Performance of NCM811|Gr pouch cells at 0.5C with different electrolytes at 60 °C. (b) 50th charging–discharging curves. (c) Capacity of constant current charging and constant voltage charging. (d) Performance of NCM811|Gr pouch cells at 0.5C with different electrolytes at 25 °C at a 4.6 V charging cut-off voltage. (e) 100th charging–discharging curves. (f) Comparison of capacity retention with previous reports. (g) Impedance of pouch cells with different electrolytes after 100 cycles. | ||
From the 50th cycle, it is clear that the capacity of cells using STD and TFT electrolytes primarily relies on constant current charging (Fig. 3b and c), indicating a significant increase in internal impedance and consequent electrochemical performance deterioration. On the other hand, the cycling profiles of cells with TFM electrolyte show no notable changes. Impedance tests on pouch cells after varying cycles reveal that after 100 cycles, the impedance of cells with STD and TFT electrolytes increases significantly, a result of continuous solvent decomposition at the electrode surface which thickens the interface film and raises battery impedance (Fig. 3g and Fig. S5, ESI†). In contrast, cells with the TFM electrolyte exhibit stable impedance, suggesting a stable interface film at high temperatures. Additionally, impedance tests on pouch cells after various cycles reveal that the impedance of cells with STD and TFT electrolytes progressively increases with cycling, indicating ongoing side reactions at the battery interface under high-temperature conditions. In contrast, cells with TFM electrolyte exhibit minimal changes in impedance, highlight TFM's effectiveness in suppressing interfacial side reactions at high temperatures and improve the battery cycling stability (Fig. S6, ESI†). Moreover, when the cut-off voltage is raised to 4.6 V, the TFM-based pouch cells exhibit a capacity of 135.2 mA h g−1 after 300 cycles, with a capacity retention rate of 80% at room temperature, whereas the pouch cells with STD electrolyte maintain a capacity retention rate of 67% after 100 cycles (Fig. 3d). The charge–discharge curves show that the polarization of cells with the TFM electrolyte is significantly lower, compared to those with the STD and TFT electrolytes (Fig. 3e). Furthermore, the TFM electrolyte demonstrates excellent electrochemical performance at room temperature, with the cells exhibiting a capacity of 105.8 mA h g−1 after 1200 cycles. In comparison, cells with the STD electrolyte show a capacity of 100.1 mA h g−1 after 800 cycles, while cells with TFT electrolyte experience rapid decay due to unstable interface film formation (Fig. S7, ESI†). As shown in Fig. S8 (ESI†), TFM electrolyte exhibits excellent compatibility with the lithium metal anode. Improving the rate performance of pouch cells using TFM electrolyte will be the focus in the next phase of work.
Characterization of anode interfacial layers
Further physio-chemical characterization of the cycled anode was performed. As shown in Fig. S9a–c (ESI†), noticeable coatings appear on the graphite anode surfaces with STD and TFT electrolytes, indicating intense electrolyte decomposition. In contrast, the graphite anode with the TFM electrolyte displays distinctive particles on its surface without any coverage from side-product coatings, suggesting that the TFM electrolyte can maintain stability on the anode surface, effectively preventing the formation of disruptive side products (Fig. S9c, ESI†). Furthermore, the thickness of the SEI film on the graphite anode with STD and TFT electrolytes is 7–8 nm (Fig. 4a and b), while the SEI film thickness with the TFM electrolyte is ∼4 nm (Fig. 4c), suggesting that STD and TFT electrolytes undergo continuous and intense decomposition to produce a thicker SEI. Additionally, the graphite structure after cycling shows no significant changes, suggesting that the co-intercalation of phosphoric ester molecules does not cause cracking of the graphite structure (Fig. S9d–f, ESI†).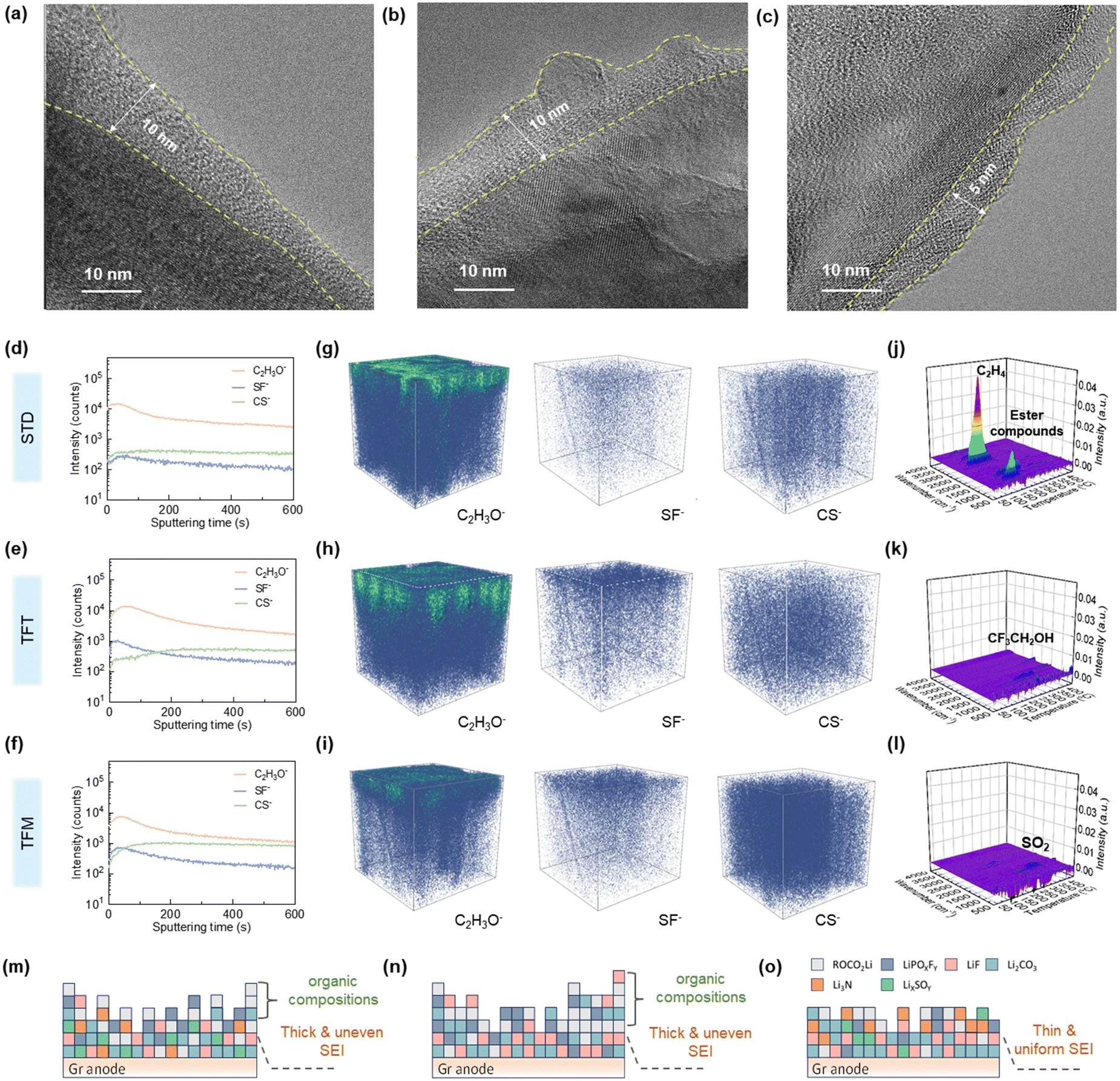 | ||
| Fig. 4 Characterization of anode interfacial layers. (a)–(c) TEM images of the cycled graphite anode using (a) STD, (b) TFT and (c) TFM electrolytes. (d)–(f) Normalized ToF-SIMS intensity depth profiles of surface and bulk fragments composed of the anode–electrolyte interphase with the (d) STD, (e) TFT and (f) TFM electrolytes. (g)–(i) 3D visualization of selected various secondary-ion fragments on the graphite anodes when being cycled with the (g) STD, (h) TFT and (i) TFM electrolytes given by the TOF-SIMS characterization. (j)–(l) Analysis of decomposition products at the anode after cycles with the (j) STD, (k) TFT and (l) TFM electrolytes by thermogravimetry-Fourier transform infrared reflection-Mass spectrometry (TG-FTIR-MS). (m)–(o) Schematic diagram of SEI films with (m) STD, (n) TFT and (o) TFM electrolytes. | ||
X-ray photoelectron spectroscopy (XPS) was employed to further analyze the composition of the SEI film (Fig. S10, ESI†). The main components on the surface of the anode with STD and TFT electrolytes include C–O (533.5 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (532 eV), and ROCO2Li (54.5 eV),38 confirming that under high-temperature conditions, the electrolyte forms an organic-based SEI film (Fig. S10a, b, d and e, ESI†). The content of inorganic substances in the SEI film with the TFT electrolyte is significantly higher than that with STD electrolyte, attributed to the FSI− decomposition. In contrast, the SEI film formed using TFM electrolyte primarily consists of Li2CO3 (55.5 eV) and LiF (56.5 eV), proving an inorganic-rich SEI that favors the thermal stability (Fig. S10c and f, ESI†). Moreover, elemental distribution reveals higher carbon content in SEI films formed with the STD electrolyte (Fig. S11, ESI†), indicating a greater organic content compared to those formed with TFT and TFM electrolytes. Additionally, the oxygen content proportion in the SEI film formed with the TFT electrolyte is significantly higher than that with the TFM electrolyte, indicating more extensive electrolyte decomposition at the anode. Furthermore, the SEI film formed with TFM electrolyte contains significantly higher levels of S, N, and F elements compared to that in the TFT electrolyte. This is attributed to the synergistic contribution of FSI− and MDFSA in forming the SEI film, which increases its inorganic content and enhances its stability at high temperatures (Fig. S12, ESI†).
O (532 eV), and ROCO2Li (54.5 eV),38 confirming that under high-temperature conditions, the electrolyte forms an organic-based SEI film (Fig. S10a, b, d and e, ESI†). The content of inorganic substances in the SEI film with the TFT electrolyte is significantly higher than that with STD electrolyte, attributed to the FSI− decomposition. In contrast, the SEI film formed using TFM electrolyte primarily consists of Li2CO3 (55.5 eV) and LiF (56.5 eV), proving an inorganic-rich SEI that favors the thermal stability (Fig. S10c and f, ESI†). Moreover, elemental distribution reveals higher carbon content in SEI films formed with the STD electrolyte (Fig. S11, ESI†), indicating a greater organic content compared to those formed with TFT and TFM electrolytes. Additionally, the oxygen content proportion in the SEI film formed with the TFT electrolyte is significantly higher than that with the TFM electrolyte, indicating more extensive electrolyte decomposition at the anode. Furthermore, the SEI film formed with TFM electrolyte contains significantly higher levels of S, N, and F elements compared to that in the TFT electrolyte. This is attributed to the synergistic contribution of FSI− and MDFSA in forming the SEI film, which increases its inorganic content and enhances its stability at high temperatures (Fig. S12, ESI†).
The roughness and thickness of the SEI have also been studied using electrochemical atomic force microscopy (AFM). As depicted in Fig. S13 (ESI†), the roughness of the graphite anode surface with TFM electrolyte is ∼53 nm, indicating that the decomposition reaction of the TFM electrolyte on the anode side is negligible and barely affects the structural characteristics of the graphite (Fig. S13d, ESI†). In contrast, the graphite surfaces in cells using STD and TFT electrolytes were more nonuniform, indicative of their roughness, about 113 and 130 nm, respectively (Fig. S13b and c, ESI†).
To investigate the distribution of different components, the cycled anode was analyzed using TOF-SIMS (Fig. 4d–i). The analysis included sputtering depth profiles (Fig. 4d–f) and 3D rendering images (Fig. 4g–i), focusing on fragments such as C2H3O− (indicative of organic products), SF− (indicative of FSI−), and CS− (indicative of MDFSA), facilitating the elucidation of the SEI formation process.39 The content of C2H3O− in the SEI film is significantly higher with STD and TFT electrolytes compared to the TFM electrolyte, and is distributed throughout the entire SEI film. This suggests that solvent decomposition occurs early in the cycling process, leading to SEI film decomposition and continued side reactions between the solvent and the anode at high temperatures. In contrast, C2H3O− in the SEI film with the TFM electrolyte is primarily concentrated in the outer layer, indicating the formation of an inorganically dominated SEI film in the initial cycles, which maintains stability at high temperatures. The decomposition product SF− derived from FSI− in TFT and TFM electrolytes is uniformly distributed across the SEI film, indicating that in localized high-concentration electrolyte systems, anions undergo reduction at the anode to predominantly form an SEI film composed of inorganic components.23 The concentration of SF− in the SEI film resulting from TFT decomposition exceeds that in TFM electrolyte. This is because the instability of the SEI film formed by TFT results in ongoing decomposition of FSI− at the anode. Moreover, the decomposition product CS− originating from MDFSA is evenly distributed within the SEI film, suggesting that MDFSA undergoes decomposition and contributes to the SEI film formation during the initial cycling stages to ensure the stability of the SEI film and prevents the subsequent decomposition of solvents.
Furthermore, TG-FTIR-MS was employed to further confirm the thermal stability of the SEI film through analysis of the anode after cycling. As shown in Fig. 4j, the graphite anode with the STD electrolyte decomposes at 60 °C, with the main decomposition products being C2H4 (1264 cm−1 and 3011 cm−1)40 and esters (1742 cm−1).41 This indicates that SEI films primarily composed of organic materials undergo significant decomposition at high temperatures (Fig. 4m). In contrast, the graphite anode with the TFT electrolyte decomposes only at temperatures approaching 200 °C, with the main decomposition product being 2,2,2-trifluoroethanol (1337 cm−1) (Fig. 4k).42 This indicates that the fluorinated solvent gradually decomposes at the anode during cycling (Fig. 4n). In contrast, the decomposition temperature of the graphite anode with the TFM electrolyte is further elevated to 250 °C, with the main decomposition product SO2 (1300 cm−1) (Fig. 4l).43 This proves that the SEI film formed with the TFM electrolyte maintains stability at high temperatures. These findings suggest that the SEI film, predominantly inorganic and formed through the synergistic interaction of FSI− and MDFSA, enhances the battery's electrochemical performance at high temperatures (Fig. 4o).
Characterization of cathode interfacial layers
Further physio-chemical characterization of the cycled cathode was also performed to investigate the impact of different electrolytes. According to Fig. 5a, c, e and Fig. S14 (ESI†), the cathode surfaces demonstrate thicker CEI films when utilizing STD and TFT electrolytes and the structural changes after cycling, transitioning from a layered structure to a spinel phase. Conversely, the CEI film with TFM electrolyte appears thinner and more uniform and the structure maintains a layered configuration. This aligns with the observation on the anode side and indicates the extensive side reactions when using STD and TFT electrolytes. The surfaces of the cathodes with STD and TFT electrolytes are more nonuniform with a roughness of 298 nm and 353 nm, respectively, while the surface of the cathode with TFM electrolyte is noticeably smoother (Ra = 237 nm), as shown in Fig. S15 (ESI†). These findings suggest that the utilization of STD and TFT electrolytes leads to a more intensive buildup of surface by-products resulting from side reactions at the cathode. Of great note, NCM811 cycled with the TFM electrolyte exhibits no mechanical fracturing (Fig. 5f) and effectively limits the dissolution of transition metals from the cathode (Fig. S16, ESI†), in agreement with the stable and consistently low resistance in the cycling. In comparison, STD and TFT electrolytes show significantly more and larger voids within NCM811 particles due to ongoing side effects like transition–metal dissolution (Fig. 5b and d). XRD analysis of the cathode before and after cycling further reveals minimal structural changes when using the TFM electrolyte. This further confirms that the CEI film formed with the TFM electrolyte effectively stabilizes the cathode structure (Fig. S17, ESI†).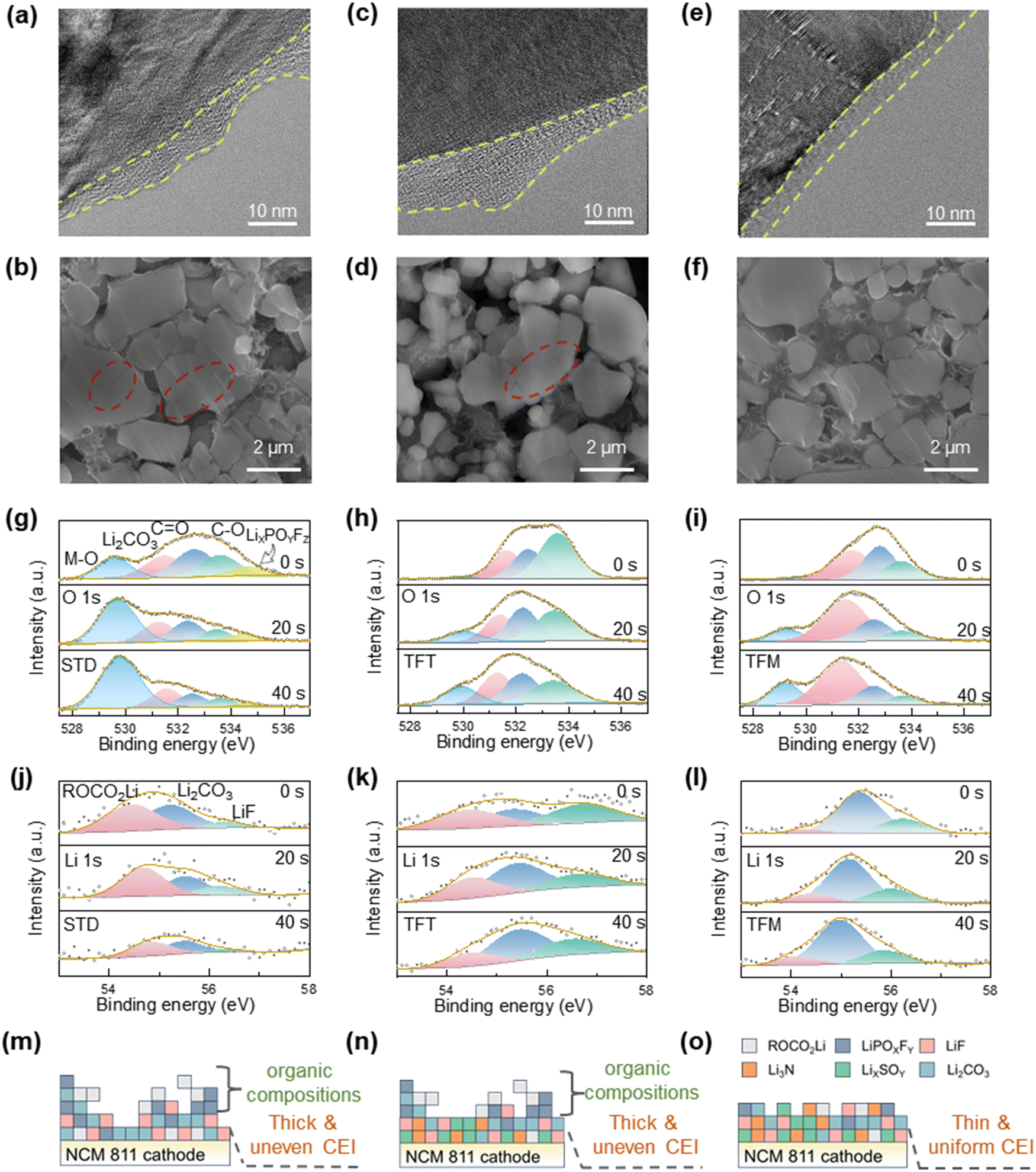 | ||
| Fig. 5 Characterization of cathode interfacial layers. (a)–(f) SEM of the cross-sections and TEM images of cycled NCM811 electrodes using (a) and (b) STD, (c) and (d) TFT and (e) and (f) TFM electrolytes. (g)–(l) XPS depth profiles of O 1s and Li 1s for NCM811 after cycles with the (g) and (j) STD, (h) and (k) TFT and (i) and (l) TFM electrolytes. The NCM811 cathode surface has been sputtered for 20 and 40 s to show the depth composition change of the CEI film. (m)–(o) Schematic diagram of CEI films with the (m) STD, (n) TFT and (o) TFM electrolytes. | ||
The composition of the CEI film was investigated by XPS. Analysis of the O 1s spectra (Fig. 5g–i) reveals that the M–O peak at 529.5 eV corresponds to transition metals within the cathode material. The predominance of M–O peaks with the STD electrolyte suggests that its CEI film is uneven, resulting in a cracked cathode. Additionally, due to the more preferential oxidation tendency of the EMC on the cathode side, the intensity of CO at 532.5 eV and C–O at 533.5 eV increases gradually with cycling. This results in the formation of an organic-dominated and uneven CEI film (Fig. 5m). With the TFT electrolyte, the uneven deposition of the CEI film induced by TTE leads to continuous solvent decomposition reactions, resulting in a significant increase in the content of C–O on the cathode surface with cycling (Fig. 5i). In contrast, on the cathode surface with the TFM electrolyte, the CEI film synergistically generated by FSI− and TFP effectively enhances the stability of the interface, resulting in the content of C–O and CO showing no significant increase with cycling. Additionally, the Li 1s spectrum reveals that the CEI film formed with the STD electrolyte is primarily composed of organic ROCO2Li, with a low content of LiF. This suggests that the CEI film is mainly derived from solvent decomposition at the cathode (Fig. 5j). As cycling progresses, the CEI film with the TFT electrolyte shows that ROCO2Li is the main component, attributed to the FEMC decomposition (Fig. 5k). In contrast, the composition of the CEI film with the TFM electrolyte remains stable throughout the cycling process without significant changes. The content of ROCO2Li generated from solvent decomposition is low, while inorganic Li2CO3 and LiF are the primary components. The F 1s spectrum reveals that LiF is the dominant component in the CEI film formed with TFM electrolyte, effectively enhancing the interface's stability at high-temperatures (Fig. S18, ESI†). Elemental distribution analysis (Fig. S19, ESI†) also shows that the CEI film formed with the STD electrolyte has a higher oxygen content, indicating a greater proportion of organic materials. Furthermore, the fluorine content in the CEI film formed with the TFM electrolyte is significantly higher than that in the TFT electrolyte, indicating a notable increase in inorganic materials.44,45 This also indicates that the CEI film is predominantly inorganic and exhibits excellent potential for high-temperature applications (Fig. 5o).
To further investigate the stability of the CEI film, in situ high-temperature XRD (HT-XRD) tests were conducted on the activated electrode in the charging state after three cycles, with the presence of the electrolyte. At 150 °C, the shift of the (003)L peak indicated that the layered structure (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) of the NCM811 material using STD and TFT electrolytes transformed into a disordered spinel phase (Fdm) (Fig. 6a). In contrast, structural changes in the NCM811 material using the TFM electrolyte occurred only at 170 °C. As the temperature continued to rise to 230 °C, NCM811 materials using STD and TFT electrolytes further transformed into a rock salt phase (Fmm)46,47 (Fig. 6b). In contrast, materials using the TFM electrolyte only exhibited changes at 270 °C indicating a significant improvement in the thermal stability of the cathode based on TFM electrolyte, ensuring the stability of the electrode structure over extended cycles at high temperatures (Fig. 6c).
m) of the NCM811 material using STD and TFT electrolytes transformed into a disordered spinel phase (Fdm) (Fig. 6a). In contrast, structural changes in the NCM811 material using the TFM electrolyte occurred only at 170 °C. As the temperature continued to rise to 230 °C, NCM811 materials using STD and TFT electrolytes further transformed into a rock salt phase (Fmm)46,47 (Fig. 6b). In contrast, materials using the TFM electrolyte only exhibited changes at 270 °C indicating a significant improvement in the thermal stability of the cathode based on TFM electrolyte, ensuring the stability of the electrode structure over extended cycles at high temperatures (Fig. 6c).
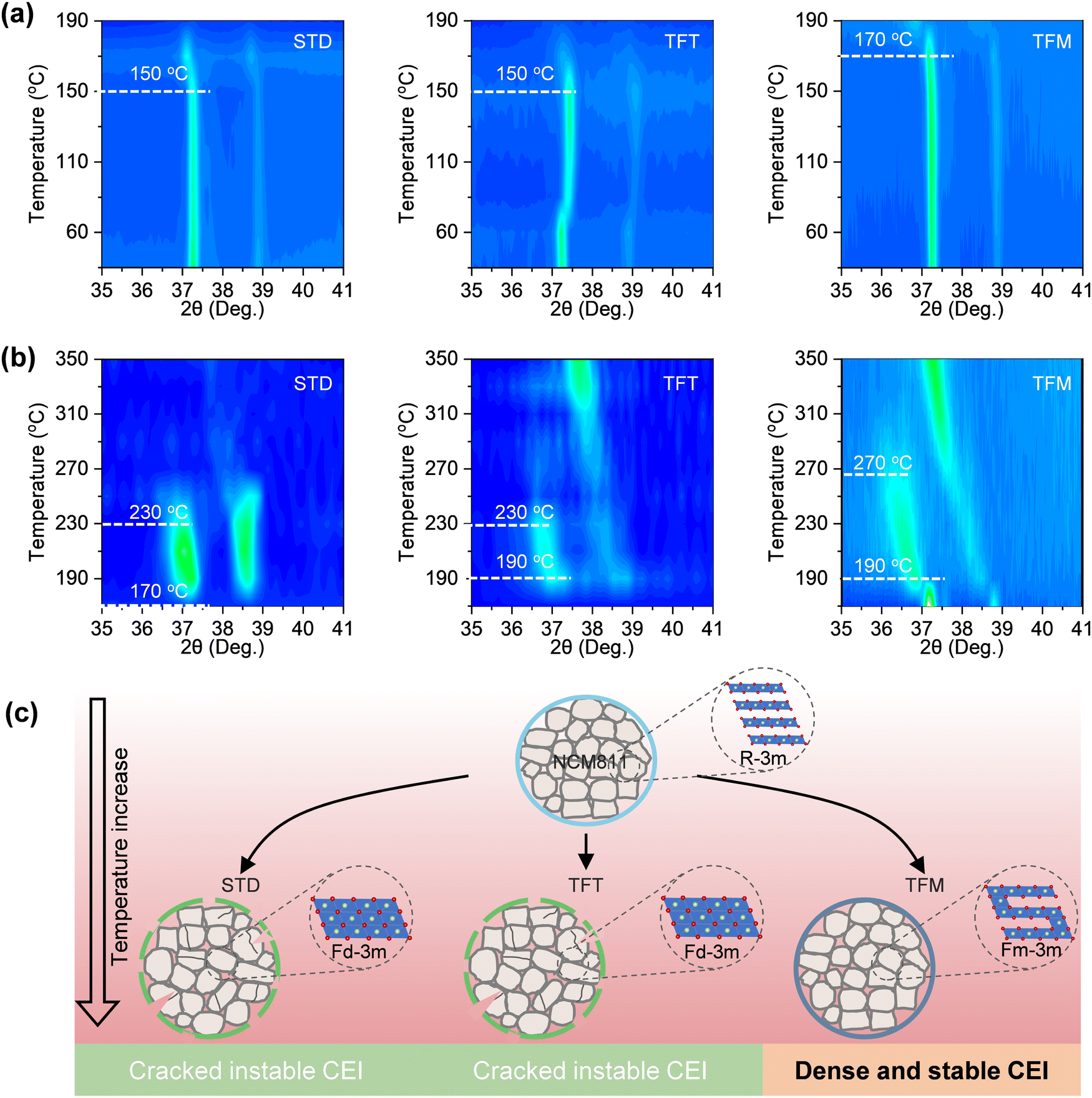 | ||
| Fig. 6 Stability characterization of CEI films at high temperature. (a) and (b) In situ HT-XRD patterns for the charged NCM811 cathode with different electrolytes at different temperatures. (c) Schematic diagram of the stability of the cathode at high temperature. | ||
The nail penetration test was conducted using 600 mA h Gr|NCM811 pouch cells with STD and TFM electrolytes to further assess the safety performance. In the initial stage, batteries with the STD electrolyte exhibit noticeable volume expansion, possibly due to the generation of a large amount of gas. Subsequently, the battery ruptures, releasing a significant amount of smoke and ejecting flames. As the combustion progresses, the battery gradually turns into charcoal as shown in Fig. S20a (ESI†). In stark contrast, batteries with TFM electrolyte show no structural changes throughout the puncture process and no combustion phenomenon, indicating superior safety performance of this electrolyte (Fig. S20b, ESI†).
Conclusions
This study stabilizes the electrode interface by electrolyte tailoring and interfacial engineering, enhancing both the safety and the electrochemical performance at high temperatures. By replacing the inert and strongly positive electrostatic potential of TTE with the active and weakly positive electrostatic potential of MDFSA, the inorganic content in the SEI films was increased, which also avoids the uneven deposition caused by TTE in the CEI films. Additionally, the feasibility of this approach was verified through theoretical calculations. On the anode side, the lower LUMO values of MDFSA and FSI− facilitate preferential reduction, leading to a notable increase in the inorganic content, thus ensuring the structural stability of the SEI film at high temperatures. On the cathode side, DOS analysis and experimental results reveal that TFP and FSI− can synergistically decompose to form a CEI film, fully leveraging the effect of TFP's ring-opening polymerization on enhancing the structural stability of the cathode, while preventing the uneven deposition caused by the low HOMO level of TTE. Therefore, based on this rational design, both electrode interfaces are stabilized, resulting in construction of batteries with long cycling life and high safety at high temperatures. Additionally, further optimization of the electrolyte components to enhance the battery's rate performance, along with refining other battery components to ensure inherent safety, is anticipated to expand the application scenarios of lithium-ion batteries.Data availability
The data are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 52034011 and 52101278), the Young Elite Scientists Sponsorship Program by CAST (2023QNRC001), the Fundamental Research Funds for Central Universities of the Central South University (2022ZZTS0405) and the Central South University Research Programme of Advanced Interdisciplinary Studies (2023QYJC005). R. T. acknowledges the RSC researcher collaboration grant (C23-8220221815) and the Royce Industrial Collaboration Grant (RICP-R4-100029).References
- L. Li, J. Yang, R. Tan, W. Shu, C. J. Low, Z. Zhang, Y. Zhao, C. Li, Y. Zhang, X. Li, H. Zhang, X. Zhao, Z. Kou, Y. Xiao, F. Verpoort, H. Wang, L. Mai and D. He, Nat. Chem. Eng., 2024, 1, 542–551 CrossRef.
- F. Li, Y. Lin, J. Liu, J. Chen, X. Wan, L. Zhao, L. Xi, Z. Li, H. Zhang, X. Xu, Z. Zhou, B. Su, M. Zhu and J. Liu, Energy Environ. Sci., 2025, 18, 1241–1254 RSC.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J.-G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer, Y. Du, O. Borodin, Y. Wang, X.-L. Wang, K. Xu, Z. Xu, C. Wang and Z. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS PubMed.
- Z. Sun, J. Zhao, M. Zhu and J. Liu, Adv. Energy Mater., 2023, 14, 2303498 CrossRef.
- Y. Yin, Y. Yang, D. Cheng, M. Mayer, J. Holoubek, W. Li, G. Raghavendran, A. Liu, B. Lu, D. M. Davies, Z. Chen, O. Borodin and Y. S. Meng, Nat. Energy, 2022, 7, 548–559 CrossRef CAS.
- Y. Zhu, W. Li, L. Zhang, W. Fang, Q. Ruan, J. Li, F. Zhang, H. Zhang, T. Quan and S. Zhang, Energy Environ. Sci., 2023, 16, 2825–2855 RSC.
- A. M. Haregewoin, A. S. Wotango and B.-J. Hwang, Energy Environ. Sci., 2016, 9, 1955–1988 RSC.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- F. A. Kreth, L. Köps, C. Leibing, S. Darlami Magar, M. Hermesdorf, K. Schutjajew, C. Neumann, D. Leistenschneider, A. Turchanin, M. Oschatz, J. L. Gómez Urbano and A. Balducci, Adv. Energy Mater., 2024, 14, 2303909 CrossRef CAS.
- G. A. Giffin, Nat. Commun., 2022, 13, 5250 CrossRef CAS PubMed.
- L. Chen, H. Wu, X. Ai, Y. Cao and Z. Chen, Battery Energy, 2022, 1, 20210006 CrossRef CAS.
- Q. Zheng, Y. Yamada, R. Shang, S. Ko, Y.-Y. Lee, K. Kim, E. Nakamura and A. Yamada, Nat. Energy, 2020, 5, 291–298 CrossRef CAS.
- C. Yang, M. Zheng, R. Qu, H. Zhang, L. Yin, W. Hu, J. Han, J. Lu and Y. You, Adv. Mater., 2023, 36, 2307220 CrossRef PubMed.
- R. Tan, A. Wang, C. Ye, J. Li, D. Liu, B. P. Darwich, L. Petit, Z. Fan, T. Wong, A. Alvarez-Fernandez, M. Furedi, S. Guldin, C. E. Breakwell, P. A. A. Klusener, A. R. Kucernak, K. E. Jelfs, N. B. McKeown and Q. Song, Adv. Sci., 2023, 10, 2206888 CrossRef CAS PubMed.
- G. M. Kontogeorgis, B. Maribo-Mogensen and K. Thomsen, Fluid Phase Equilib., 2018, 462, 130–152 CrossRef CAS.
- J. Chen, H. Lu, X. Kong, J. Liu, J. Liu, J. Yang, Y. Nuli and J. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317923 CrossRef CAS PubMed.
- D. Wu, C. Zhu, H. Wang, J. Huang, G. Jiang, Y. Yang, G. Yang, D. Tang and J. Ma, Angew. Chem., Int. Ed., 2024, 63, e202315608 CrossRef CAS PubMed.
- M. Fang, B. Du, X. Zhang, X. Dong, X. Yue and Z. Liang, Angew. Chem., Int. Ed., 2023, 63, e202316839 CrossRef PubMed.
- C. Zhu, C. Sun, R. Li, S. Weng, L. Fan, X. Wang, L. Chen, M. Noked and X. Fan, ACS Energy Lett., 2022, 7, 1338–1347 CrossRef CAS.
- Y. Zou, Z. Ma, G. Liu, Q. Li, D. Yin, X. Shi, Z. Cao, Z. Tian, H. Kim, Y. Guo, C. Sun, L. Cavallo, L. Wang, H. N. Alshareef, Y. K. Sun and J. Ming, Angew. Chem., Int. Ed., 2023, 62, e202216189 CrossRef CAS PubMed.
- J. Wang, Q. Zheng, M. Fang, S. Ko, Y. Yamada and A. Yamada, Adv. Sci., 2021, 8, 2101646 CrossRef CAS PubMed.
- L. Chen, J. Lu, Y. Wang, P. He, S. Huang, Y. Liu, Y. Wu, G. Cao, L. Wang, X. He, J. Qiu and H. Zhang, Energy Storage Mater., 2022, 49, 493–501 CrossRef.
- L. You, K. Duan, G. Zhang, W. Song, T. Yang, X. Song, S. Wang and J. Liu, J. Phys. Chem. C, 2019, 123, 5942–5950 CrossRef CAS.
- X. Zhang, L. Zou, Y. Xu, X. Cao, M. H. Engelhard, B. E. Matthews, L. Zhong, H. Wu, H. Jia, X. Ren, P. Gao, Z. Chen, Y. Qin, C. Kompella, B. W. Arey, J. Li, D. Wang, C. Wang, J. G. Zhang and W. Xu, Adv. Energy Mater., 2020, 10, 2000368 CrossRef CAS.
- J. Xu, J. Zhang, T. P. Pollard, Q. Li, S. Tan, S. Hou, H. Wan, F. Chen, H. He, E. Hu, K. Xu, X. Q. Yang, O. Borodin and C. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- X. Zhuang, S. Zhang, Z. Cui, B. Xie, T. Gong, X. Zhang, J. Li, R. Wu, S. Wang, L. Qiao, T. Liu, S. Dong, G. Xu, L. Huang and G. Cui, Angew. Chem., Int. Ed., 2023, 63, e202315710 CrossRef PubMed.
- K. An, Y. H. T. Tran, S. Kwak, J. Han and S. W. Song, Adv. Funct. Mater., 2021, 31, 2106102 CrossRef CAS.
- J.-G. Han, M.-Y. Jeong, K. Kim, C. Park, C. H. Sung, D. W. Bak, K. H. Kim, K.-M. Jeong and N.-S. Choi, J. Power Sources, 2020, 446, 227366 CrossRef CAS.
- J. W. Park, D. H. Park, S. Go, D.-H. Nam, J. Oh, Y.-K. Han and H. Lee, Energy Storage Mater., 2022, 50, 75–85 CrossRef.
- H. M. Jung, S.-H. Park, J. Jeon, Y. Choi, S. Yoon, J.-J. Cho, S. Oh, S. Kang, Y.-K. Han and H. Lee, J. Mater. Chem. A, 2013, 1, 11975–11981 RSC.
- X. Yan, C. Chen, X. Zhu, L. Pan, X. Zhao and L. Zhang, J. Power Sources, 2020, 461, 228099 CrossRef CAS.
- Z. Xie, J. He, Z. Xia, Q. Cai, Z. Tang, J. Cai, Y. Chen, X. Li, Y. Fan, L. Xing, Y. Shen and W. Li, J. Energy Chem., 2023, 86, 197–207 CrossRef CAS.
- G. J. Chung, Y. H. T. Tran, J. Han, K. Kim, Y. S. Lee and S.-W. Song, Chem. Eng. J., 2022, 446, 137288 CrossRef CAS.
- Q. Li, S. Jiao, L. Luo, M. S. Ding, J. Zheng, S. S. Cartmell, C.-M. Wang, K. Xu, J.-G. Zhang and W. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 18826–18835 CrossRef CAS PubMed.
- Y. Gu, S. Fang, L. Yang and S.-I. Hirano, J. Mater. Chem. A, 2021, 9, 15363–15372 RSC.
- Y. Qian, Y. Kang, S. Hu, Q. Shi, Q. Chen, X. Tang, Y. Xiao, H. Zhao, G. Luo, K. Xu and Y. Deng, ACS Appl. Mater. Interfaces, 2020, 12, 10443–10451 CrossRef CAS PubMed.
- C. Shi, X. Huang, J. Gu, Z. Huang, F. Liu, M. Wang, Q. Wang, B. Hong, Z. Zhang, J. Li and Y. Lai, J. Energy Chem., 2023, 87, 501–508 CrossRef CAS.
- X. Liu, J. Zhang, X. Yun, J. Li, H. Yu, L. Peng, Z. Xi, R. Wang, L. Yang, W. Xie, J. Chen and Q. Zhao, Angew. Chem., Int. Ed., 2024, e202406596 CAS.
- G. Hübner, G. Rauhut, H. Stoll and E. Roduner, Phys. Chem. Chem. Phys., 2002, 4, 3112–3121 RSC.
- S. Niu, Y. Zhou, H. Yu, C. Lu and K. Han, Energy Convers. Manage., 2017, 149, 495–504 CrossRef CAS.
- J. Vaynberg and L. M. Ng, Surf. Sci., 2005, 577, 188–199 CrossRef CAS.
- A. B. M. S. Y. Wang, O. Saur, J. C. Lavalley and B. A. Morrowa, Appl. Catal., B, 1998, 16, 279–290 CrossRef.
- Z. Sun, F. Li, J. Ding, Z. Lin, M. Xu, M. Zhu and J. Liu, ACS Energy Lett., 2023, 8, 2478–2487 CrossRef CAS.
- F. Li, Z. Liu, C. Liao, X. Xu, M. Zhu and J. Liu, ACS Energy Lett., 2023, 8, 4903–4914 CrossRef CAS.
- Z. Dai, H. Zhao, W. Chen, Q. Zhang, X. Song, G. He, Y. Zhao, X. Lu and Y. Bai, Adv. Funct. Mater., 2022, 32, 2206428 CrossRef CAS.
- X. Zhou, B. Zhang, P. Lyu, L. Xi, F. Li, Z. Ma, M. Zhu and J. Liu, Energy Environ. Sci., 2024, 17, 8174–8188 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05263c |
| This journal is © The Royal Society of Chemistry 2025 |