
Iron clusters and single atom sites cooperatively promote bifunctional oxygen reaction activity in ultra-stable flexible zinc–air batteries†
Qi
Liu‡
a,
Panzhe
Qiao‡
cde,
Di
Shen
b,
Ying
Xie
 b,
Baoluo
Wang
b,
Tianyu
Han
b,
Hongtu
Shi
b,
Lei
Wang
*b and
Honggang
Fu
*ab
b,
Baoluo
Wang
b,
Tianyu
Han
b,
Hongtu
Shi
b,
Lei
Wang
*b and
Honggang
Fu
*ab
aKey Laboratory of Superlight Materials and Surface Technology of the Ministry of Education of the People's Republic of China, Harbin Engineering University, Harbin, 150001, China. E-mail: fuhg@vip.sina.com; fuhg@hlju.edu.cn
bKey Laboratory of Functional Inorganic Material Chemistry, Ministry of Education of the People's Republic of China, Heilongjiang University, Harbin, 150080, China. E-mail: wanglei0525@hlju.edu.cn
cShanghai Synchrotron Radiation Facility, Shanghai Advanced Research Institute, Chinese Academy of Sciences, Shanghai, 201210, China
dShanghai Institute of Applied Physics, Chinese Academy of Science, Shanghai, 201210, China
eUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 5th February 2025
Abstract
Enhancing the bifunctional activity of electrocatalysts for oxygen reduction/evolution reactions (ORR/OER), along with improving water retention in gel-polymer electrolytes, is essential for developing high-performance flexible zinc–air batteries (FZABs). Herein, we synthesize a structure that combines Fe single atom sites with an FeN4 configuration and clusters of four coordinated Fe atoms anchored on worm-like polypyrrole (FeSA/FeAC@PPy/CC) using an electrochemical deposition strategy. It shows a promoted bifunctional ORR/OER activity with a small potential gap of 0.694 V. Theoretical calculations indicate that Fe single atom sites lower the energy barrier of the rate-determining step for both the ORR and OER, while Fe clusters optimize the energy barriers associated with oxygen-containing intermediates. The interaction between Fe single atom sites and clusters shifts the d-band center of the metal closer to the Fermi level, leading to electron depletion at the Fe centers. This adjustment triggers a rearrangement of the orbital electrons and enhances the adsorption interaction with oxygen orbitals, thereby improving both the ORR and OER activities. Additionally, a water-locking hydrogen bonding network electrolyte composed of polyacrylamide and ethylene glycol is utilized to enhance low-temperature tolerance. Thus, the assembled FeSA/FeAC@PPy/CC-based FZAB demonstrates ultra-stable operation for 210 h at 25 °C and 167 h at −40 °C.
Broader contextFlexible zinc–air batteries (FZABs) are emerging as a promising class of energy storage devices. However, their development is primarily hindered by the sluggish kinetics of air-cathode reactions and the inadequate low-temperature tolerance and water retention of alkaline gel-polymer electrolytes (GPEs). Transition metal-based single atom site catalysts (SACs) are renowned for their superior electrochemical activity in the oxygen reduction reaction (ORR). Nevertheless, the strong adsorption of oxygen-containing intermediates can limit the oxygen evolution reaction (OER), thereby influencing the charging voltage of zinc–air batteries (ZABs). It is thus crucial to balance the adsorption strength of these intermediates for both the ORR and OER to optimize their functionality in ZABs. To address this challenge, we have developed a composite structure consisting of FeN4 single atom sites and Fe clusters on worm-like polypyrrole (FeSA/FeAC@PPy/CC), which demonstrates excellent bifunctional oxygen activity. Moreover, we employed a water-locking hydrogen bonding network electrolyte comprised of ethylene glycol and polyacrylamide to improve low-temperature tolerance. Consequently, the FeSA/FeAC@PPy/CC-based FZAB exhibited ultra-stable performance at −40 °C. This study provides critical insights into constructing highly efficient and stable low-temperature FZABs. |
Introduction
With the advent of the carbon-neutral development paradigm, there has been a noticeable shift among researchers towards environmentally friendly energy storage solutions.1,2 Zinc–air batteries (ZABs) have garnered significant attention for their high energy density, abundant zinc reserves, and environmentally benign nature, however, the slow kinetics of the air-cathode reaction remains a crucial bottleneck for improving battery performance.3–5 The high cost and limited sources of precious metals necessitate the exploration of cost-effective bifunctional oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) electrocatalysts to optimize the discharging and charging processes.6,7 Furthermore, flexible zinc–air batteries (FZABs) face particular challenges related to gel-polymer electrolytes (GPEs) and their sensitivity to temperature fluctuations, particularly at low temperatures.8,9 Therefore, engineering effective bifunctional ORR/OER catalysts and enhanced low-temperature resilience of GPEs maybe an effective strategy to address these problems.Transition metal based single-atom site catalysts (SACs) exhibit high utilization of metal atoms and provide plentiful active sites, which are consistently situated at the optimal position on the ORR volcano plot for maximum activity.10,11 In particular, Fe–N–C catalysts guarantee superior ORR performance due to having the lowest kinetic barriers and fewer free energy changes during the *O to *OOH transition.12,13 Guided by the Sabatier principle, Fe-based SACs exhibit a strong surface bonding capability on the left side of the volcano plot, enhancing the adsorption of reactive species and benefiting the ORR process.14 However, the strong adsorption strength can lead to high desorption barriers for reverse reactions, namely the OER, thereby impacting the optimal performance of ZABs.15 Previously, we discovered that SACs undergo restructuring to form specific cluster structures during the electrocatalytic reaction process, which further enhances the catalytic activity.16–18 Additionally, clusters not only provide high active surface areas and robust electrochemical stability, but also allow for tunable compositional and structural characteristics.19,20 Theoretically, the coexistence of single atom sites and clusters disrupts the typical symmetric charge distribution of traditional Fe SACs, enabling effective control over the coordination environment of the metal centers. Consequently, the design of Fe-based catalysts featuring both single atom sites and clusters may effectively modulate the reaction barriers of the ORR/OER, potentially enhancing electrocatalytic activity. Electrochemical deposition is an effective strategy to synthesize uniform structural catalysts, while conductive polymers (such as polypyrrole and polyaniline) possess numerous nitrogen-containing species that could anchor metal species.21,22 Therefore, utilizing conductive polymers as carriers to anchor metal species through a coordination strategy is a worthwhile endeavor. Additionally, a crucial challenge for FZABs is their pronounced susceptibility to low temperatures, due to the common GPE monomers being limited in functionality to just a few hours under conditions below 0 °C.23–25 Alternatively, incorporating organic molecules with highly polar functional groups such as −OH, −COOH, −SO3, and −NH2 can establish a more robust hydrogen bond network with H2O molecules. This could prevent the freezing of water molecules below 0 °C and promote the low-temperature performance of FZABs.26,27
In this work, a two-step electrodeposition strategy is employed to anchor Fe single atom sites and clusters coexisting on worm-like polypyrrole (FeSA/FeAC@PPy/CC) as a bifunctional oxygen reaction electrocatalyst. This kind of unique three-dimensional structure can provide more active sites, greatly shortening the diffusion paths of mass transfer. Furthermore, Fe single atom sites reduce the energy barrier of the rate-determining step for both the ORR and OER, while Fe clusters average the energy barriers of oxygen-containing intermediates. The collaboration between Fe single atom sites and clusters shifts the d-band center of the metal closer to the Fermi level, thereby improving the bifunctional oxygen reaction activity. The assembled aqueous ZAB by using FeSA/FeAC@PPy/CC as the air-cathode shows a small initial charging–discharging potential gap of 0.77 V at 10 mA cm−2 for stably operating for 437 h. More importantly, we introduced ethylene glycol (EG) into polyacrylamide (PAM) to develop a novel PAM/EG GPE, which was used to construct a FZAB. The FZAB demonstrates a stable cycling performance over a wide-temperature range from a high temperature of 40 °C to a low temperature of −40 °C. Our work proves the great application potential of non-precious based electrocatalysts in wide-temperature ZABs.
Results and discussion
Pretreated carbon cloth (CC) with a smooth surface is used to synthesize FeSA/FeAC@PPy/CC (Fig. S1 and S2, ESI†). An illustration of FeSA/FeAC@PPy/CC synthesis by a two-step electrochemical deposition strategy is depicted in Fig. 1a. Uniform polypyrrole was firstly grown on the CC substrate at a constant potential of 0.9 V (PPy/CC). The amount of PPy deposited on a CC substrate can be controlled by adjusting the deposition voltages (Fig. S3, ESI†). At constant potentials of 0.8, 0.9 and 1.0 V, the amounts of PPy deposited onto the obtained PPy/CC-0.8, PPy/CC, and PPy/CC-1.0 were determined to be 1.68, 2.15, and 2.69 mg cm−2, respectively, using a gravimetric method. This demonstrated that the PPy loading increases with the increase in preparation voltage. Additionally, the thickness of the deposited PPy gradually increases with escalating potential from the top view of the carbon fibers in the CC (Fig. S4, ESI†). Then, the Fe species were deposited onto the PPy/CC support by cyclic voltammetry (CV) at a potential range from −0.6 to 0.2 V for 20 cycles to prepare FeSA/FeAC@PPy/CC, which showed a three-dimensional (3D) worm-like structure with a diameter of approximately ≈45 nm (Fig. 1b). | ||
| Fig. 1 (a) Schematic illustration for the synthesis of FeSA/FeAC@PPy/CC. (b) SEM, (c) TEM, (d) HRTEM, (e) and (f) elemental mapping and (g) HAADF-STEM images of iron clusters (red circles) coupled with iron single atoms (blue circles) of FeSA/FeAC@PPy/CC. | ||
Transmission electron microscopy (TEM) reveals an absence of distinct lattice fringes, suggesting that the Fe did not form nanoparticles (Fig. 1c and d).28 As shown in Fig. 1e and f, the Fe, N, O and C elements are uniformly dispersed in FeSA/FeAC@PPy/CC. The presence of oxygen originates from the unavoidable surface oxidation when the sample is exposed to air. The aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images are presented in Fig. 1g; the bright spots represent the significantly higher electron density of Fe compared to the non-metallic elements C and N, indicating that Fe is dispersed on atomic scale. Furthermore, both individual and more highly concentrated bright spots are distinctly observed, confirming the coexistence of Fe single atoms and Fe clusters in the FeSA/FeAC@PPy/CC system. The Fe loading in FeSA/FeAC@PPy/CC is approximately 0.92 wt% as determined by an inductively coupled plasma optical emission spectrometry (ICP-OES) test (Table S1, ESI†). Moreover, the untreated CC without treatment is not favorable for the growth of PPy (Fig. S5, ESI†).
The X-ray diffraction (XRD) pattern of FeSA/FeAC@PPy/CC only shows two characteristic peaks at 26.5° and 44.7°, corresponding to the C (002) and C (100) crystal planes of PPy/CC, respectively (Fig. 2a). The absence of additional metal peaks suggests that no significant formation of Fe-based compounds occurs in FeSA/FeAC@PPy/CC, implying that Fe may exist in atomic-scale entities. Due to the small particle size and low concentration, its crystalline phases are undetectable. X-ray photoelectron spectroscopy (XPS) could provide comprehensive analyses of the specific bonding structures and states in FeSA/FeAC@PPy/CC. The wide spectrum of FeSA/FeAC@PPy/CC reveals the presence of Fe, O, N and C elements, consistent with TEM elemental mapping results (Fig. S6, ESI†). As shown in Fig. 2b, the Fe 2p XPS spectrum of FeSA/FeAC@PPy/CC displays the doublet peaks of Fe0 (706.7/721.2 eV), Fe2+ (709.2/722.6 eV), and Fe3+ (711.6/724.0 eV) and satellite peaks (717.0/729.2 eV).29 The presence of Fe0 demonstrated the existence of Fe–Fe bonds in FeSA/FeAC@PPy/CC, and the presence of multiple valence states maybe enhance the ORR/OER reactions. As shown in Fig. 2c, the pyridinic-N (398.6 eV), Fe–N (399.4 eV), graphitic-N (401.1 eV), and oxidized N (403.2 eV) exist in the N 1s spectrum.30 The pyridinic-N with lone pair electrons can be oxidized for promoting the OER, while graphitic-N increases the number of conjugated π electrons to enhance oxygen adsorption for promoting the ORR.31 The presence of Fe–N bonds indicates the coordination between Fe atoms and nitrogen-containing species in the PPy.
 | ||
| Fig. 2 (a) XRD patterns of FeSA/FeAC@PPy/CC and PPy/CC. (b) Fe 2p and (c) N 1s XPS spectra of FeSA/FeAC@PPy/CC. (d) The normalized Fe K-edge XANES and (e) FT-EXAFS spectra of Fe foil, Fe2O3, FePc and FeSA/FeAC@PPy/CC. (f) FT-EXAFS fitting curves of the Fe K-edge for FeSA/FeAC@PPy/CC, while the inset is the structural model. (g) WT count plots of the Fe K-edge at K-space for Fe foil, Fe2O3, FePc and FeSA/FeAC@PPy/CC. | ||
X-ray absorption fine structure (XAFS), including X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS), utilized to further determine the coordination structures of Fe species. The Fe K-edge XANES spectra reveals that the peak position of FeSA/FeAC@PPy/CC was located between the Fe foil and Fe2O3, approaching that of Fe phthalocyanine (FePc), suggesting the Fe in FeSA/FeAC@PPy/CC is around the +2 valence state (Fig. 2d). As shown in Fig. 2e, the EXAFS spectrum of FeSA/FeAC@PPy/CC exhibits two main peaks. In detail, the peak at 1.5 Å in R-space coincide with the Fe–N peak position in FePc, slightly offset from the Fe–O peak position in Fe2O3. The peak at 2.6 Å in R-space alignes with the Fe–Fe bond. However, the Fe–Fe bond position in the synthetic FeSA/FeAC@PPy/CC aligns with that in Fe2O3 and is significantly different from the 2.2 Å position observed in Fe foil. This is attributed to the Fe clusters in FeSA/FeAC@PPy/CC being in nanoscale, in contrast to the larger size of standard bulk Fe foil. Moreover, the Fe foil contains only Fe–Fe coordination bonds, while the FeSA/FeAC@PPy/CC includes nitrogen atoms surrounding the iron, resulting in electron loss, similar to the behavior observed in Fe2O3. Consequently, the length of the Fe–Fe bond in FeSA/FeAC@PPy/CC is longer than that in the Fe foil, approaching the bond length in Fe2O3. Previous studies also indicated that Fe–Fe bonds in Fe clusters coordinated with non-precious metal atoms are significantly stretched compared to those in Fe foil.32,33 Moreover, the Fe–N and Fe–Fe coordination numbers are respectively 3.78 and 2.76, suggesting the presence of the Fe4–FeN4 configuration (Fig. 2f and Table S2, ESI†). Additionally, wavelet transform (WT) analyses reveal that the maximum intensities at approximately 5.0 and 6.5 Å−1 in K-space correspond to Fe–N and Fe–Fe (Fig. 2g), respectively, further identifying the presence of Fe–Fe and Fe–N coordination in FeSA/FeAC@PPy/CC. To study the formation mechanism of FeSA/FeAC@PPy/CC, we further performed constant potential deposition of iron species on PPy/CC substrates every 0.1 V within a potential range from +0.2 V to −0.6 V, with each potential held for 10 min. All the synthetic samples were characterized using XAFS as shown in Fig. S7 (ESI†). FT-EXAFS fitting analysis in Table S3 (ESI†) indicated that only Fe–N bonds were detected at the potentials of 0.2 V and 0.1 V, and the coordination number increased from 3 to 4, suggesting the formation of Fe atom sites. As the potential decreased to −0.2 V, Fe–Fe bonds began to appear, implying the formation of Fe clusters. In the potential range of −0.3 V to −0.6 V, both Fe–Fe and Fe–N bonds coexists. Based on the above analysis, we can infer that in the electrodeposition process, Fe monomers are initially formed, followed by the gradual appearance of Fe clusters, eventually resulting in structures that contain both FeN4 configurations and Fe4 clusters.
For further researching the influence of electrochemical deposition conditions on the structure of catalysts, the Fe@PPy/CC-10 and Fe@PPy/CC-30 were also prepared via 10 and 30 CV cycles, respectively. The Fe loadings for Fe@PPy/CC-10 and Fe@PPy/CC-30 are approximately 0.56 wt% and 1.22 wt% (Table S1, ESI†), respectively. TEM and XRD analyses show no obvious nanoparticles, suggesting that Fe is present as atomically dispersed species (Fig. S8–S10a, ESI†). Additionally, wide-scan XPS confirms the presence of Fe, O, N and C in both Fe@PPy/CC-10 and Fe@PPy/CC-30 (Fig. S10b, ESI†). The Fe 2p spectra exhibit peaks corresponding to Fe0, Fe2+ and Fe3+, along with satellite peaks (Fig. S10c, ESI†). Moreover, the N 1s spectra in Fig. S10d (ESI†) reveal the presence of pyridinic-N, Fe–N, graphitic-N, and oxidized N in both samples. Notably, the content of Fe–N in Fe@PPy/CC-10 is higher than in Fe@PPy/CC-30, suggesting a potentially greater proportion of Fe–N coordination in Fe@PPy/CC-10. The N contents for the catalysts Fe@PPy/CC-10, FeSA/FeAC@PPy/CC, and Fe@PPy/CC-30 are 10.6 at%, 11.0 at%, and 12.1 at%, respectively (Fig. S11 and Table S4, ESI†). The high N content ensures superior electrical conductivity and provides numerous sites for anchoring metals. The presence of Fe–N peaks in all three catalysts confirms the successful loading of Fe. Notably, the FeSA/FeAC@PPy/CC exhibits a high proportion of pyridinic-N and graphitic-N. The XANES spectra indicate that both Fe@PPy/CC-10 and Fe@PPy/CC-30 exhibit oxidation states close to that of FePc, approximately at +2 valence (Fig. S12a, ESI†). The FT-EXAFS spectra show that both Fe@PPy/CC-10 and Fe@PPy/CC-30 exhibit Fe–N and Fe–Fe bonds (Fig. S12b, ESI†), and further fitting results are presented in Table S2 (ESI†). Based on the coordination configuration and Fe loading results, the ratio between single atom and clusters in Fe@PPy/CC-10 and Fe@PPy/CC-30 is calculated to be 1.14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1.05:1, respectively (Table S5, ESI†). It can be concluded that the content of single atoms and clusters could be adjusted by tuning the electrodeposition conditions. To investigate the impact of different PPy loadings on CC on the subsequent iron deposition for forming the FeN4 structure, we utilized PPy/CC-0.8 and PPy/CC-1.0 as substrates for iron species electrodeposition, designated as FeSA/FeAC@PPy/CC-0.8 and FeSA/FeAC@PPy/CC-1.0, respectively. FT-EXAFS spectra fitting analyses revealed that the coordination numbers for Fe–N and Fe–Fe bonds in FeSA/FeAC@PPy/CC-0.8 are approximately 3 and 10, respectively, whereas they are 4 and 6 for FeSA/FeAC@PPy/CC-1.0 (Fig. S13 and Table S2, ESI†). The results indicate that the amount of PPy loaded on the CC significantly influences the state of the deposited iron species, suggesting that an optimal loading of PPy facilitates the formation of both FeN4 single atoms and Fe4 clusters.
:1 and 1.05:1, respectively (Table S5, ESI†). It can be concluded that the content of single atoms and clusters could be adjusted by tuning the electrodeposition conditions. To investigate the impact of different PPy loadings on CC on the subsequent iron deposition for forming the FeN4 structure, we utilized PPy/CC-0.8 and PPy/CC-1.0 as substrates for iron species electrodeposition, designated as FeSA/FeAC@PPy/CC-0.8 and FeSA/FeAC@PPy/CC-1.0, respectively. FT-EXAFS spectra fitting analyses revealed that the coordination numbers for Fe–N and Fe–Fe bonds in FeSA/FeAC@PPy/CC-0.8 are approximately 3 and 10, respectively, whereas they are 4 and 6 for FeSA/FeAC@PPy/CC-1.0 (Fig. S13 and Table S2, ESI†). The results indicate that the amount of PPy loaded on the CC significantly influences the state of the deposited iron species, suggesting that an optimal loading of PPy facilitates the formation of both FeN4 single atoms and Fe4 clusters.
The OER performances were conducted using a three-electrode system in 1.0 M KOH electrolyte. The linear sweep voltammetry (LSV) curves in Fig. 3a reveal that FeSA/FeAC@PPy/CC exhibits an overpotential of 294 mV at 20 mA cm−2, which is much lower than that of RuO2/CC of 375 mV. The RuO2/CC electrode was prepared by a drop coating method, which is associated with non-uniformity compared to self-supporting electrodes synthesized from an in situ grown strategy. Additionally, the presence of Nafion may negatively impact mass transport. Thus, the OER performance of RuO2/CC may be inferior to that of other rotating disk electrode tests. Even at a high current density of 50 mA cm−2, FeSA/FeAC@PPy/CC displays a modest overpotential of 333 mV, indicating its superior OER performance (Fig. 3b). The FeSA/FeAC@PPy/CC exhibits enhanced OER activity, comparable to that of previously reported atomic dispersion catalysts (Table S6, ESI†). The XPS analysis presented in Fig. 2c revealed that FeSA/FeAC@PPy/CC contains a high proportion of pyridinic-N. The lone pair electrons in pyridinic-N can be oxidized, thereby promoting the OER.
 | ||
| Fig. 3 Electrochemical performances. (a) OER polarization curves, (b) the corresponding overpotentials at 20 and 50 mA cm−2, (c) Tafel plots and (d) linear plots of scan rates versus current density of FeSA/FeAC@PPy/CC and RuO2/CC tested in 1.0 M KOH electrolyte. (e) Durability of FeSA/FeAC@PPy/CC for the OER after 5000 CV cycles, the inset is the chronopotentiometry curve at 20 mA cm−2. (f) ORR polarization curves and (g) Tafel plots of FeSA/FeAC@PPy/CC and Pt/C tested in O2-saturated 0.1 M KOH electrolyte. (h) LSV curves at different rotation rates and calculated K–L plots (inset) at different potentials for FeSA/FeAC@PPy/CC. (i) Stability of FeSA/FeAC@PPy/CC for 10000 CV cycles. | ||
Tafel slope is an important indicator of reaction kinetics that can be analysed using LSV curves. We selected the potential corresponding to a current density of 10 mA cm−2 as the onset potential and extended it by 60 mV to establish the endpoint for the Tafel slope. As shown in Fig. 3c, the FeSA/FeAC@PPy/CC exhibited a smaller Tafel slope of 91.9 mV dec−1 compared to RuO2/CC of 102.6 mV dec−1. The electrochemistry active surface area (ECSA) was further evaluated by fitting the electrical double layer capacitance (Cdl) from CV at different rotational speeds. As shown in Fig. 3d and Fig. S14 (ESI†), FeSA/FeAC@PPy/CC displayed a higher Cdl of 193.4 mF cm−2 than that of RuO2/CC (18.7 mF cm−2). It has been demonstrated that FeSA/FeAC@PPy/CC exhibits a significantly larger ECSA. The unique 3D structure of FeSA/FeAC@PPy/CC facilitates mass transfer and electron transport, while the coexistence of Fe single atom sites and Fe clusters provides numerous active sites. Additionally, the self-supporting electrode can be utilized directly without binders, thereby maximizing the retention of active sites. Consequently, FeSA/FeAC@PPy/CC exhibits a larger ECSA, indicating enhanced electrocatalytic activity.
Meanwhile, the in situ electrodeposition for constructing self-supported electrodes was also beneficial to the electron and mass transfer, thus promoting the electrocatalytic activity. As indicated by the electrochemical impedance spectra (EIS) depicted in Fig. S15 (ESI†), FeSA/FeAC@PPy/CC exhibits the smallest semicircle and the lowest charge transfer resistance (Rct), effectively reducing the energy required to overcome charge transfer during the process, indicating superior charge transfer kinetics at the interface.34 Stability is a crucial indicator for evaluating the electrocatalytic performance of catalysts. As shown in Fig. 3e, FeSA/FeAC@PPy/CC demonstrates a potential shift of only 6 mV at a current density of 20 mA cm−2 after 5000 CV cycles, indicating excellent stability. Additionally, constant potential curves were further employed to validate the OER stability of FeSA/FeAC@PPy/CC. At a current density of 20 mA cm−2 corresponding to a potential of 1.53 V, FeSA/FeAC@PPy/CC demonstrates stable operation for 100 h, further confirming its excellent durability. After the stability test, the 3D worm-like structure of FeSA/FeAC@PPy/CC remains intact, with no visible metal nanoparticles detected (Fig. S16a–c, ESI†). Elemental mapping further confirms the uniform distribution of Fe, N, O and C, as shown in Fig. S16d and e (ESI†). The HAADF-STEM image reveals the coexistence of atomically dispersed Fe single atoms and Fe clusters (Fig. S16f, ESI†). Furthermore, the FeSA/FeAC@PPy/CC after the OER stability test was characterized using XAFS analysis (Fig. S17 and Table S2, ESI†). The coordination numbers of the Fe–N and Fe–Fe bonds remains unchanged, demonstrating the structural stability of FeSA/FeAC@PPy/CC. The strong coupling between single atom sites and clusters, along with the improvement of the symmetric coordination structure in SACs, is a crucial factor contributing to the exceptional stability of FeSA/FeAC@PPy/CC.
The ORR tests were evaluated in O2-saturated 0.1 M KOH electrolyte. To investigate the intrinsic activity of the catalyst, we adopted a rotating disk electrode (RDE) for the ORR test, which could ensure adequate contact between the catalyst and oxygen molecules to enhance the reaction kinetics. Therefore, we scraped the catalyst from the self-supporting material to prepare catalyst ink and subsequently spreaded it on the surface of RDE for the ORR test. As shown in Fig. 3f, the FeSA/FeAC@PPy/CC catalyst exhibited an onset potential (Eonset) of 0.97 V and half-wave potential (E1/2) of 0.83 V, which could be comparable to the commercial Pt/C (0.99 V, 0.85 V). Additionally, FeSA/FeAC@PPy/CC exhibits a larger limiting current density (JL) of 5.5 mA cm−2, which demonstrates its outstanding proton transport capacity originated from a higher ECSA. The Tafel slope was depicted in Fig. 3g, where the potential range from 10 mV to 70 mV below the Eonset is designated as the strong polarization region for calculation. The small Tafel slope of 75.0 mV dec−1 indicates the rapid ORR reaction kinetics of FeSA/FeAC@PPy/CC. The LSV curves at various rotation rates along with the corresponding approximately linear Koutecky–Levich (K–L) plots, suggesting first-order reaction kinetics of FeSA/FeAC@PPy/CC towards the ORR (Fig. 3h). As a comparison, the electrocatalytic performance on the catalyst composed of solely FeN4 single atoms prepared under a constant potential deposition condition of 0.1 V was also tested. As shown in Fig. S18 (ESI†), it displays much lower OER and ORR activities than those of FeSA/FeAC@PPy/CC. Therefore, the synergy between single atom sites and clusters could effectively promote the bifunctional activity.
To study the impact of iron loading on electrocatalytic performance, the Fe@PPy/CC-10 and Fe@PPy/CC-30 were also tested for comparison. As illustrated in Fig. S19a and b (ESI†), the OER activities of Fe@PPy/CC-10 and Fe@PPy/CC-30 indicates overpotentials of 342 and 330 mV at a current density of 20 mA cm−2, respectively, and overpotentials of 387 and 374 mV at a higher current density of 50 mA cm−2. The Tafel slopes for Fe@PPy/CC-10 and Fe@PPy/CC-30 are measured at 127.7 and 115.7 mV dec−1, respectively (Fig. S19c, ESI†). As for the ORR catalyst, Fe@PPy/CC-10 showed an Eonset of 0.95 V and E1/2 of 0.79 V, with a relatively low Tafel slope of 86.4 mV dec−1, slightly exceeding that of Fe@PPy/CC-30 (0.93 V, 0.77 V, 96.2 mV dec−1) (Fig. S20, ESI†). Moreover, the Cdl values of Fe@PPy/CC-10 and Fe@PPy/CC-30 are about 122.1 and 146.2 mF cm−2 (Fig. S21, ESI†), highlighting the significant ECSA of self-supporting electrodes. However, the electrocatalytic performances of both catalysts, in terms of OER, ORR and ECSA, are found to be inferior to that of FeSA/FeAC@PPy/CC, indicating that optimal and moderate Fe content is crucial for achieving superior electrocatalytic performance.
The calculated electron transfer number (n) is about 4 at different potentials, indicating that FeSA/FeAC@PPy/CC primarily conducts a four-electron transfer process, similar to Pt/C (Fig. S22a and b, ESI†). As illustrated in Fig. 3i, FeSA/FeAC@PPy/CC exhibits an E1/2 loss of only 5 mV after 10000 CV cycles, significantly lower than that of Pt/C of 21 mV (Fig. S22c, ESI†), implying the superior ORR stability of FeSA/FeAC@PPy/CC. The 3D worm-like structure in FeSA/FeAC@PPy/CC facilitates the exposure of active sites, endowing a large ECSA and accelerating mass transport processes to enhance the electrochemical reaction rate for the ORR and OER. The in situ growth strategy via electrodeposition avoids the use of organic conductive agents and binders during the test, which prevents the blocking of active sites and electrolyte poisoning. Furthermore, this growth method can eliminate stability issues caused by catalyst detachment and structural degradation for long-term electrochemical testing.
Density functional theory (DFT) calculations were further employed to elucidate the intrinsic electrocatalytic behavior of oxygen reactions on FeSA/FeAC@PPy/CC. The Fe4–FeN4 model was built based on the results of EXAFS fitting. We explored all potential adsorption active sites for oxygen-containing intermediates (Fig. 4a and Table S7, ESI†), and the two models of the FeN4 and Fe clusters were also studied for comparison (Fig. S23 and S24, ESI†). As shown in Fig. 4b, the adsorption energies of O2 on Fe4–FeN4, FeN4 and Fe clusters are calculated to be −4.57, −4.29 and −8.57 eV (Table S8, ESI†), respectively, indicating a spontaneous process for O2 adsorption. Notably, the moderate adsorption energy of Fe4–FeN4 suggests that desorption occurs easily, thereby facilitating the ORR process. The surface adsorption of oxygen intermediates is predominantly influenced by the electronic coupling between atomic orbitals and adsorbates.35 It is widely accepted that metallic species serve as the primary active sites for the adsorption of reactive species because they typically occupy advantageous positions in the volcano plot for the ORR and OER.36,37 Therefore, we selected the Fe site as the adsorption site for our calculations. At U = 0 V and pH = 13, the Fe4–FeN4, FeN4, and Fe cluster models exhibit a downhill energy barrier for the spontaneous reactions *O2 → *OOH → *O → *OH, while the *OH → OH− process showed an uphill energy barrier, signifying an endothermic reaction (Fig. 4c). Thus, the rate-determining step (RDS) for the Fe4–FeN4, FeN4, and Fe cluster models corresponds to the *OH → OH− reaction, with energy barriers of 1.96, 1.64, and 4.01 eV, respectively. Moreover, the RDS for all three models remains consistent at U = 1.23 V (Fig. 4d). In the Fe4–FeN4 model, the presence of Fe single atom sites facilitates the desorption of *OH, effectively reducing the energy barrier of the RDS. In contrast, the Fe cluster in the Fe4–FeN4 model averages the energy barriers of the intermediates, thereby enhancing the overall ORR activity.
 | ||
| Fig. 4 DFT calculations. (a) Side and top views of the oxygen-containing intermediates adsorbed on Fe4–FeN4 models at the Fe site of FeN4. (b) O2 absorption energy values on the Fe sites of Fe4–FeN4, FeN4 and Fe cluster models. Free energy diagrams of intermediate species in the (c) and (d) ORR and (e) and (f) OER processes for Fe4–FeN4, FeN4 and Fe cluster models at U = 0 V and U = 1.23 V. (g) DOS of Fe4–FeN4, FeN4, and Fe cluster models. Differential charge density of (h) Fe4–FeN4, (i) FeN4 and (j) Fe cluster models. | ||
In the OER, Fe4–FeN4 and FeN4 undergo a non-spontaneous reaction pathway of *OH → *O → *OOH → *O2, as depicted in Fig. 4e and f. At pH = 14, U = 0 V and 1.23 V, the RDS for Fe4–FeN4, FeN4 and Fe cluster models is consistently the transition from *OOH to *O2, while the reduced energy barrier of Fe4–FeN4 suggests its superior catalytic performance in redox reactions. As observed in the ORR process, the incorporation of FeN4 in Fe4–FeN4 is beneficial for lowering the energy barrier of RDS during the OER. To further elucidate the primary active site, we have supplemented the DFT calculation data about the free energy diagrams of intermediate species at various Fe sites on the Fe4–FeN4 model (Fig. S25, ESI†). The results indicate that the energy barriers for both the ORR and OER at the different Fe4 sites are significantly higher than those at the FeN4 site in the Fe4–FeN4 model, particularly for the RDS step (Fig. 4c–f). This finding suggests that the oxygen reactions are more favorable at the FeN4 site. Notably, the energy barrier values for different oxygen intermediates (*OOH, *O, and *OH) considerably vary on the FeN4 site, whereas the energy barrier values appear more uniform across the Fe4 sites. Upon introducing the Fe4 site to the FeN4 model, the Fe4–FeN4 model exhibits reduced energy barriers for the oxygen intermediates, especially averaging the energy barriers, potentially enhancing the transition of these intermediates. Additionally, the Fe cluster demonstrates a higher O2 adsorption value (Fig. 4b), which may hinder subsequent desorption and reactions. Based on the above analyses, we propose that while the FeN4 sites serve as the primary active sites for the ORR and OER, the Fe4 sites may play a role in averaging the energy barriers of the intermediates. Thus, the synergy between Fe single atom sites and clusters enhances the bifunctional oxygen reaction activity of the Fe4–FeN4 model.
The DOS value at the Fermi level can qualitatively indicate the electronic conductivity of a catalyst.38 The peak values at the Fermi level for Fe4–FeN4, FeN4, and Fe cluster models are 25.52, 11.60, and 18.95, respectively. This suggests that Fe4–FeN4 exhibits a higher charge transfer rate. In predicting the adsorption strength of metals with reaction intermediates, the d-band centers (Ed) of Fe4–FeN4, FeN4 and Fe cluster models relative to the Fermi level are calculated to be −2.77, −3.82 and −3.07 eV, respectively. The proximity of the d-band center to the Fermi level in the Fe4–FeN4 model suggests that the interaction between Fe single atom sites and clusters significantly enhance the electronic occupancy of the Fe atoms. This facilitates electron migration in the outer metal d-orbitals, thereby enhancing the catalytic capabilities and improving ORR/OER activity.39 As the differential charge density displayed in Fig. 4h–j, the charge transfer quantities for Fe in Fe4–FeN4, FeN4 and Fe cluster models are 0.21 e−, 0.08 e− and 0.18 e−, respectively. Due to a higher proportion of Fe atoms, there is a propensity to transfer charge to surrounding electrons in Fe4–FeN4, resulting in a significant charge transfer. As orbital splitting occurs only in the presence of a ligand field, we primarily consider the influence of Fe clusters on the d orbitals charge distribution of Fe in the Fe4–FeN4 model. The electron filling is related to the oxidation state of the metal with the d orbital electron configuration of Fe being 3d64s2, which includes a total of eight electrons. According to the aforementioned XAFS analysis, the Fe in FeSA/FeAC@PPy/CC is primarily in the +2 oxidation state, and the arrangement of the remaining six electrons in the d orbitals should be considered. The differential charge density, shown in Fig. 4h and i, indicates that the introduction of Fe clusters leads to the central metal in FeN4 losing partial electrons, thereby reducing the electron density at the Fe sites and further enhancing the ligand N attraction to the surrounding metal electrons, resulting in the delocalization of charge on the Fe atoms and ultimately affecting the d band center. Fig. S26a (ESI†) shows that in the Fe4–FeN4 model, the d band center of FeN4 shifts to a lower energy compared to pure FeN4, effectively modulating the adsorption strength of oxygen-containing intermediates and enhancing the OER activity. Furthermore, the characteristic of the d band center being distant from the Fermi level leads to a change in the distribution of d orbital electrons, allowing electrons to transition to higher energy levels, which in turn causes the rearrangement of 3d orbital electrons in Fe (Fig. S26b, ESI†).33,40 This electron redistribution significantly enhances the interaction with oxygen orbitals, improving the electrocatalytic activity for oxygen reactions.
Inspired by the excellent bifunctional ORR/OER activity, the FeSA/FeAC@PPy/CC was utilized as a self-supporting air-cathode to assemble an aqueous ZAB (Fig. 5a). The battery used the mixture of Pt/C and RuO2 as the air-cathode was also tested. Generally, the performance of the ORR is related to the discharging voltage of ZAB, while the OER performance correlates with the charging voltage. Consequently, strong ORR activity is associated with a high discharging voltage, whereas robust OER activity corresponds to a low charging voltage. As shown in Fig. 5b, the FeSA/FeAC@PPy/CC-based ZAB demonstrates a high open-circuit voltage of approximately 1.46 V, surpassing that of the Pt/C + RuO2-based ZAB (1.41 V). The power density was calculated based on discharge voltages at varying discharge currents ranging from 0 to 400 mA cm−2. The FeSA/FeAC@PPy/CC-based ZAB achieved a peak power density of 205.1 mW cm−2, which was about 1.53 times that of the Pt/C + RuO2-based ZAB (Fig. 5c). Additionally, it was observed that the FeSA/FeAC@PPy/CC-based ZAB still maintained a discharge voltage of 0.4 V even under a high discharge current of 400 mA cm−2, while it approached 0 V for Pt/C + RuO2-based ZAB. This implied the exceptional high-current tolerance of the FeSA/FeAC@PPy/CC. Furthermore, the FeSA/FeAC@PPy/CC-based ZAB exhibited higher discharge voltage plateaus across all tested current densities of 2, 5, 10, and 20 mA cm−2 compared to the Pt/C + RuO2-based ZAB (Fig. 5d). When the measurement returns from a current density of 20 mA cm−2 back to 2 mA cm−2, the FeSA/FeAC@PPy/CC-based ZAB essentially maintained its initial discharge voltage, demonstrating its good stability. Specific capacity tests calculated by the consumption of zinc mass for the FeSA/FeAC@PPy/CC-based ZAB was 758 mAh gZn−1 at 10 mA cm−2, approximately 92.4% of the theoretical capacity, surpassing that of the Pt/C + RuO2-based ZAB (707 mAh gZn−1) (Fig. 5e). As illustrated in Fig. 5f, three FeSA/FeAC@PPy/CC-based ZABs connect in series could power a 4 V LED display, proving its potential for practical application.
 | ||
| Fig. 5 Aqueous rechargeable ZAB performances. (a) Scheme of aqueous ZAB. (b) Open-circuit voltages. (c) Discharging curves and the corresponding power density plots. (d) Discharging curves at 2–20 mA cm−2. (e) Specific discharging capacities at 10 mA cm−2 for FeSA/FeAC@PPy/CC and Pt/C + RuO2-based ZABs. (f) Photo of three FeSA/FeAC@PPy/CC-based ZABs driving a 4 V LED viewing screen. (g) Long-term galvanostatic charging–discharging plots of FeSA/FeAC@PPy/CC and Pt/C + RuO2-based ZAB at a current density of 10 mA cm−2. (h) Voltage efficiency and round-trip efficiency of FeSA/FeAC@PPy/CC at 1, 100, 200, 300, and 400 cycles of (g). | ||
Long-term stability was further conducted, which involved cycles of charging for 30 min and discharging for 30 min. The voltage efficiency was calculated based on the ratio of discharging to charging voltages, and the round-trip efficiency was determined by comparing the integral areas of discharging and charging curves. Initial tests conducted at a current density of 5 mA cm−2 demonstrates that the FeSA/FeAC@PPy/CC-based ZAB could cycle stably for 400 h with an initial charging–discharging voltage difference of 0.72 V, outperforming the Pt/C + RuO2-based ZAB (0.78 V, 215 h) (Fig. S27, ESI†). The FeSA/FeAC@PPy/CC-based ZAB exhibits charging–discharging voltage differences of 0.69 V, 0.69 V, 0.71 V, and 0.73 V at 100, 200, 300, and 400 h, respectively, demonstrating its superior cycling performance. At a higher current density of 10 mA cm−2, the initial charging voltage of the FeSA/FeAC@PPy/CC-based ZAB is 1.98 V and the discharging voltage is 1.21 V (Fig. 5g), resulting in a voltage difference of 0.77 V, much lower than the Pt/C + RuO2 of 0.87 V. Even after 100 h, 200 h, 300 h and 400 h tests, the FeSA/FeAC@PPy/CC-based ZAB is able to maintain charging–discharging voltage differences of 0.79 V, 0.81 V, 0.83 and 0.86 V, respectively (Fig. S28, ESI†). After 400 h of cycling, the FeSA/FeAC@PPy/CC-based ZAB exhibited only a 3.0% decrease in voltage efficiency and a 3.4% reduction in round-trip efficiency (Fig. 5h). It is noteworthy that the ZAB featuring FeSA/FeAC@PPy/CC achieved the longest stable operation time of 437 h.
The performance of the FeSA/FeAC@PPy/CC-based ZAB in terms of high current density was further evaluated. As depicted in Fig. S29 (ESI†), the FeSA/FeAC@PPy/CC-based ZAB exhibits a high and stable discharge voltage platform at the current range from 30 mA cm−2 to 80 mA cm−2, sustaining a discharge voltage of 0.88 V even at the upper limit of 80 mA cm−2. The charging–discharging cycling performance of the aqueous ZABs under high current density is illustrated in Fig. S30 (ESI†). The FeSA/FeAC@PPy/CC-based ZAB demonstrates excellent cycling capabilities, enduring 392 h at 20 mA cm−2, 364 h at 40 mA cm−2, and 290 h at 60 mA cm−2, with respective charging–discharging voltage gaps of 0.92 V, 1.06 V, and 1.26 V. This further underscores the superior performance of aqueous ZABs under high current density. Therefore, the high power density and remarkable durability ensure that the FeSA/FeAC@PPy/CC-based ZAB exhibits significant potential for commercial applications. Furthermore, the FeSA/FeAC@PPy/CC-based ZAB demonstrates performances comparable to those of other reported atomic dispersion catalysts (Table S6, ESI†).
The FeSA/FeAC@PPy/CC was further used to construct a sandwich-type FZAB (Fig. 6a). The significant challenge faced by the FZAB is the rapid dehydration of the GPEs at room temperature and gradual freezing at low temperatures.24 Hence, enhancing the water retention and low-temperature resistance of the GPEs is crucial for the commercialization of FZABs. For the synthesis of GPEs, ethylene glycol (EG) was introduced to polyacrylamide (PAM) to develop a novel GPE as PAM/EG. Based on the relationship between the volume fraction of EG in aqueous solution and the freezing point, we incorporated an EG aqueous solution with a volume fraction of 55% into the PAM/EG system (Fig. S31, ESI†). As shown in Fig. S32a (ESI†), the addition of EG significantly enhances the ionic conductivity and accelerates proton transfer. DFT calculations indicate that PAM/EG exhibits the highest adsorption energy for H2O molecules, suggesting that the incorporation of EG considerably strengthens the interaction between PAM and H2O molecules, resulting in a more robust intermolecular hydrogen bonding network (Fig. S32b and S33, ESI†). Consequently, the energy required to overcome intermolecular forces for H2O loss and freezing is increased, leading to the anticipation that PAM/EG will possess excellent water retention and antifreeze capabilities. Relatively, the needle-like structures of PAM/EG could produce a certain level of porosity between the molecules (Fig. S34, ESI†), resulting in its excellent absorption capacities of 154.3 g g−1 and 13.7 g g−1 in deionized water and alkaline electrolyte (Fig. S32c, ESI†), respectively. As depicted in Fig. 6b, PAM/EG exhibits superior water retention capabilities (75.4 wt% retention after seven days), further indicating the long-term stability.
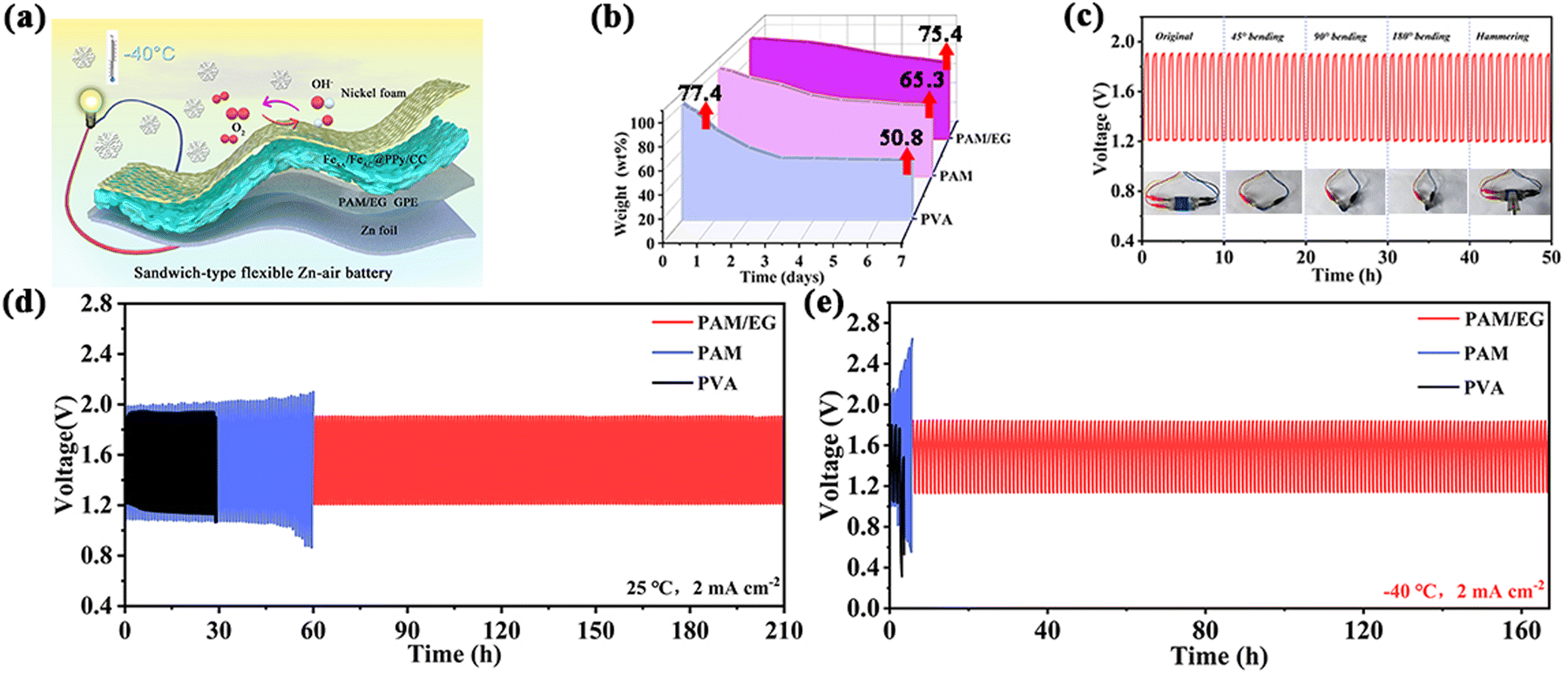 | ||
| Fig. 6 (a) Illustration of sandwich-type FZABs. (b) Liquid retention capability of PAM/EG, PAM and PVA. (c) Cycling stability of FeSA/FeAC@PPy/CC-based FZABs under different bending states and hammering at 2 mA cm−2. Galvanostatic cycling curves with 30 min discharging and 30 min charging per cycle under (d) 25 °C and (e) −40 °C at 2 mA cm−2. | ||
Due to the inability to test electrocatalytic performances at temperatures below 0 °C, we assessed the low-temperature performance by adjusting the electrolyte temperature to 15 °C, 0 °C, and 5 °C (Fig. S35, ESI†). As illustrated in Fig. S36 (ESI†), the potential difference (ΔE) values between the OER potential at 20 mA cm−2 and the ORR potential at E1/2 for FeSA/FeAC@PPy/CC are approximately 0.73 V, 0.77 V and 0.81 V at 15 °C, 10 °C and 5 °C, respectively. The results indicate the favorable low-temperature oxygen reaction activity of FeSA/FeAC@PPy/CC. Subsequently, the sandwich-type FZAB was assembled by using FeSA/FeAC@PPy/CC as the air-cathode, and zinc plate as the anode. At shown in Fig. 6c, the charging–discharging curves of FeSA/FeAC@PPy/CC-based FZABs show no significant fluctuations at any bending and folding due to the superior mechanical performance of PAM/EG GPE (Fig. S37, ESI†).
To evaluate the mechanical properties of PAM/EG, PAM, and PVA GPEs, the tensile stress–strain curves were further tested. As shown in Fig. S38a (ESI†), PAM/EG exhibits a tensile strength of 2.2 MPa and a maximum elongation at break of 877.8%, significantly higher than those of PAM (1.3 MPa, 531.9%) and PVA (0.6 MPa, 239.3%). Additionally, compressive strength tests revealed that PAM/EG can withstand a maximum pressure of 1.5 MPa with a maximum compression strain of 64.9%, outperforming PAM (1.1 MPa, 50.1%) and PVA (0.5 MPa, 39.1%) (Fig. S38b, ESI†). These results indicate that PAM has superior mechanical strength compared to PVA due to its chemically cross-linked molecular network formed through polymerization, which is more robust than the physically cross-linked structure of PVA. The exceptional mechanical properties of PAM/EG can be attributed to the introduction of EG, which forms a hydrogen bond network that enhances molecular density and intermolecular forces, thereby increasing the material rigidity and load-bearing capacity. At 25 °C, the FeSA/FeAC@PPy/CC-based FZABs exhibit an open circuit voltage of 1.49 V, a power density of 98.8 mW cm−2, and a specific capacity of 722.7 mAh g−1 (Fig. S39a–c, ESI†). As shown in Fig. 6d, the FZABs assembled with PAM/EG demonstrates an extended lifetime of 210 h, markedly surpassing that of FZABs assembled with PAM (60 h) and PVA (29 h).
Under low-temperature conditions of −40 °C, they shows an open circuit voltage of 1.41 V, a power density of 30.2 mW cm−2, and a specific capacity of 570.2 mAh g−1 (Fig. S39a–c, ESI†). Furthermore, they displays outstanding rate performances across current densities ranging from 1 to 5 mA cm−2 at both room and low temperatures, indicating excellent stability (Fig. S39d, ESI†). Long-term cycle tests were also performed for FZABs at a low temperature of −40 °C. As displayed in Fig. 6e, the FZAB assembled with PAM/EG exhibits a discharging voltage of 1.14 V and a charging voltage of 1.82 V at 2 mA cm−2 and could stably operate for 167 h, attributed to its exceptional to low temperature resistance capabilities (Fig. S40, S41 and S42, ESI†).
The high-temperature resistance of the GPEs at 40 °C is illustrated in Fig. S43 (ESI†). After one day, the water retention rates of PAM/EG, PAM, and PVA are 93.4, 65.1, and 47.4 wt%, respectively. Moreover, PAM/EG still maintains 68.1 wt% water retention rates after five days, demonstrating its superior high-temperature water retention capability. This enhanced performance is attributed to the introduction of EG, which significantly strengthens the interaction between H2O molecules through hydrogen bonding, effectively reducing water evaporation. Additionally, FZABs assembled with FeSA/FeAC@PPy/CC as the air cathode were evaluated using the three electrolytes at 40 °C. As depicted in Fig. S44 (ESI†), the FZAB featuring the PAM/EG electrolyte exhibits an initial discharging voltage of 1.28 V and a charging voltage of 1.71 V at a current density of 2 mA cm−2. It is consistently cycled for 126 h, significantly surpassing the performance of batteries with PAM (25 h) and PVA (12 h) electrolytes. The results demonstrate the enhanced performance and stability of the PAM/EG electrolyte under high-temperature conditions.
As illustrated in Fig. S45 (ESI†), the FeSA/FeAC@PPy/CC-based FZABs display stable performance across a range of operating temperatures from −40 to 40 °C, further highlighting their exceptional charge–discharge recovery capabilities. The performance of the Pt/C + RuO2-based FZAB at room and low temperatures was further evaluated. As shown in Fig. S46 (ESI†), it could stably operate for 160 hours and 104 hours at 25 °C and −40 °C, respectively, which are significantly lower than the operation times of the FeSA/FeAC@PPy/CC-based FZAB. It also exhibits a much higher potential gap than that of the FeSA/FeAC@PPy/CC-based FZAB. The performance of the FeSA/FeAC@PPy/CC-based FZAB also surpasses most of the reported FZABs (Table S9 and S10, ESI†). Subsequently, we investigated the zinc corrosion potential and hydrogen evolution reaction (HER) suppression effect of PAM/EG GPE. As shown in Fig. S47a (ESI†), the FZAB assembled with the PAM/EG electrolyte exhibits a corrosion potential of −1.10 V, which is more positive than the −1.22 V value observed in PAM. This indicates that the zinc anode demonstrates higher corrosion resistance in the PAM/EG electrolyte. Furthermore, the HER potential for the PAM/EG electrolyte is −0.19 V at 5 mA cm−2, whereas that for the PAM electrolyte was −0.06 V (Fig. S47b, ESI†). It indicates that the addition of EG significantly suppresses an undesirable HER activity, thereby preventing the formation of zinc dendrites and enhancing the stability of the zinc/electrolyte interface. Consequently, the FZABs assembled with the PAM/EG electrolyte show exceptional cycling stability under wide-temperature conditions. Due to the FeSA/FeAC@PPy/CC-based FZAB having exceptional low-temperature resistance and excellent water retention capabilities, PAM/EG effectively ensures the efficiency of the catalyst for assembling FZABs, demonstrating its prospects for widespread future applications.
Conclusion
We have successfully designed a bifunctional oxygen reaction electrocatalyst of FeSA/FeAC@PPy/CC characterized by the coexistence of Fe single atom sites and clusters. It exhibits good bifunctional electrocatalytic activity with a potential of 1.524 V at 20 mA cm−2 for the OER and an E1/2 of 0.83 V for the ORR, as well as high-performance aqueous ZABs. The enhanced activity is attributed to the Fe single atom sites reducing the energy barrier of the RDS, and the Fe clusters could balance the energy barriers of intermediates, thus optimizing the overall reaction process. Then, a novel PAM/EG GPE was constructed for assembling FZABs by using FeSA/FeAC@PPy/CC as the air-cathode. The FZAB shows exceptionally long lifespans of 210 h at room temperature and 167 h at a low temperature of −40 °C, which is ascribed to the introduction of EG significantly promoting the interaction with H2O molecules, thereby significantly improving the water retention and low-temperature resistance of GPEs. Our work provides new insight for constructing highly-efficient and long cycling-time FZABs.Data availability
The data that support the findings of this study are available upon reasonable request from the corresponding author.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We gratefully acknowledge the support of this research by the National Key R&D Program of China (2023YFA1507204), the National Natural Science Foundation of China (U20A20250, 22179034), and the Natural Science Foundation of Heilongjiang Province (ZD2023B002).Notes and references
- W. Fang, W. Guo, R. Lu, Y. Yan, X. Liu, D. Wu, F. M. Li, Y. Zhou, C. He, C. Xia, H. Niu, S. Wang, Y. Liu, Y. Mao, C. Zhang, B. You, Y. Pang, L. Duan, X. Yang, F. Song, T. Zhai, G. Wang, X. Guo, B. Tan, T. Yao, Z. Wang and B. Y. Xia, Nature, 2024, 626, 86–91 CrossRef CAS PubMed.
- P. Bonnick, M. Redko, C. Wang, M. Frey, M. Jones, S. Wang, G. Allred, C. Ling, R. Sugiura and J. Muldoon, ACS Energy Lett., 2023, 8, 4251–4258 CrossRef CAS.
- C.-X. Zhao, J.-N. Liu, J. Wang, D. Ren, J. Yu, X. Chen, B.-Q. Li and Q. Zhang, Adv. Mater., 2021, 33, 2008606 CrossRef CAS.
- T. Zhou, N. Zhang, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 1132–1153 RSC.
- L. Tang, H. Peng, J. Kang, H. Chen, M. Zhang, Y. Liu, D. H. Kim, Y. Liu and Z. Lin, Chem. Soc. Rev., 2024, 53, 4877–4925 RSC.
- L. Huo, M. Lv, M. Li, X. Ni, J. Guan, J. Liu, S. Mei, Y. Yang, M. Zhu, Q. Feng, P. Geng, J. Hou, N. Huang, W. Liu, X. Y. Kong, Y. Zheng and L. Ye, Adv. Mater., 2024, 36, 2312868 CrossRef CAS PubMed.
- H. Lei, L. Ma, Q. Wan, Z. Huangfu, S. Tan, Z. Wang and W. Mai, Nano Energy, 2022, 104, 107941 CrossRef CAS.
- M. Jiao, L. Dai, H.-R. Ren, M. Zhang, X. Xiao, B. Wang, J. Yang, B. Liu, G. Zhou and H.-M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202301114 CrossRef CAS.
- C. Gu, X.-Q. Xie, Y. Liang, J. Li, H. Wang, K. Wang, J. Liu, M. Wang, Y. Zhang, M. Li, H. Kong and C.-S. Liu, Energy Environ. Sci., 2021, 14, 4451–4462 RSC.
- X. Zheng, P. Li, S. Dou, W. Sun, H. Pan, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 2809–2858 RSC.
- L. Lyu, X. Hu, S. Lee, W. Fan, G. Kim, J. Zhang, Z. Zhou and Y.-M. Kang, J. Am. Chem. Soc., 2024, 146, 4803–4813 CrossRef CAS.
- H. Adabi, A. Shakouri, N. U. Hassan, J. R. Varcoe, B. Zulevi, A. Serov, J. R. Regalbuto and W. E. Mustain, Nat. Energy, 2021, 6, 834–843 CrossRef CAS.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer III, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- M. Xiao, Y. Chen, J. Zhu, H. Zhang, X. Zhao, L. Gao, X. Wang, J. Zhao, J. Ge, Z. Jiang, S. Chen, C. Liu and W. Xing, J. Am. Chem. Soc., 2019, 141, 17763–17770 CrossRef CAS PubMed.
- H. Zheng, D. Deng, X. Zheng, Y. Chen, Y. Bai, M. Liu, J. Jiang, H. Zheng, Y. Wang, J. Wang, P. Yang, Y. Xiong, X. Xiong and Y. Lei, Nano Lett., 2024, 24, 4672–4681 CrossRef CAS PubMed.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS.
- G. Xing, M. Tong, P. Yu, L. Wang, G. Zhang, C. Tian and H. Fu, Angew. Chem., Int. Ed., 2022, 61, e202211098 CrossRef CAS.
- M. Tong, F. Sun, G. Xing, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2023, 62, e202314933 CrossRef CAS PubMed.
- X. Guo, J. Shi, M. Li, J. Zhang, X. Zheng, Y. Liu, B. Xi, X. An, Z. Duan, Q. Fan, F. Gao and S. Xiong, Angew. Chem., Int. Ed., 2023, 62, e202314124 CrossRef CAS PubMed.
- X. Zheng, X. Cao, Z. Sun, K. Zeng, J. Yan, P. Strasser, X. Chen, S. Sun and R. Yang, Appl. Catal., B, 2020, 272, 118967 CrossRef CAS.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- X. Wang, F. Liu, H. Qin, J. Li, X. Chen, K. Liu, T. Zhao, W. Yang, M. Yu, G. Fan and F. Cheng, Angew. Chem., Int. Ed., 2024, 63, e202409628 CAS.
- Y. Zhang, H. Qin, M. Alfred, H. Ke, Y. Cai, Q. Wang, F. Huang, B. Liu, P. Lv and Q. Wei, Energy Storage Mater., 2021, 42, 88–96 CrossRef.
- X. Fan, R. Zhang, S. Sui, X. Liu, J. Liu, C. Shi, N. Zhao, C. Zhong and W. Hu, Angew. Chem., Int. Ed., 2023, 62, e202302640 CrossRef CAS PubMed.
- Q. Liu, H. Shi, T. Han, L. Wang and H. Fu, Energy Storage Mater., 2024, 67, 103255 CrossRef.
- Q. Wang, Q. Feng, Y. Lei, S. Tang, L. Xu, Y. Xiong, G. Fang, Y. Wang, P. Yang, J. Liu, W. Liu and X. Xiong, Nat. Commun., 2022, 13, 3689 CrossRef CAS.
- F. Chen, D. Zhou, J. Wang, T. Li, X. Zhou, T. Gan, S. Handschuh-Wang and X. Zhou, Angew. Chem., Int. Ed., 2018, 57, 6568–6571 CrossRef CAS PubMed.
- Z. An, P. Yang, D. Duan, J. Li, T. Wan, Y. Kong, S. Caratzoulas, S. Xiang, J. Liu, L. Huang, A. I. Frenkel, Y.-Y. Jiang, R. Long, Z. Li and D. G. Vlachos, Nat. Commun., 2023, 14, 6666 CrossRef CAS PubMed.
- X. Wan, Q. Liu, J. Liu, S. Liu, X. Liu, L. Zheng, J. Shang, R. Yu and J. Shui, Nat. Commun., 2022, 13, 2963 Search PubMed.
- Z. Zhu, H. Yin, Y. Wang, C.-H. Chuang, L. Xing, M. Dong, Y.-R. Lu, G. Casillas-Garcia, Y. Zheng, S. Chen, Y. Dou, P. Liu, Q. Cheng and H. Zhao, Adv. Mater., 2020, 32, 2004670 CrossRef CAS.
- Y. Liu, S. Roy, S. Sarkar, J. Xu, Y. Zhao and J. Zhang, Carbon Energy, 2021, 3, 795–826 CrossRef CAS.
- J. Chen, C. Qiu, L. Zhang, B. Wang, P. Zhao, Y. Zhao, H. Wang, G. Yang, A. Sun, J. Fan, Q. Xv and O. J. Rojas, Energy Environ. Sci., 2024, 17, 4746–4757 RSC.
- H. Zhang, H.-C. Chen, S. Feizpoor, L. Li, X. Zhang, X. Xu, Z. Zhuang, Z. Li, W. Hu, R. Snyders, D. Wang and C. Wang, Adv. Mater., 2024, 36, 2400523 CrossRef CAS PubMed.
- P. Chen, W. Zhou, Z. Xiao, S. Li, H. Chen, Y. Wang, Z. Wang, W. Xi, X. Xia and S. Xie, Energy Storage Mater., 2020, 33, 298–308 CrossRef.
- T. He, Y. Chen, Q. Liu, B. Lu, X. Song, H. Liu, M. Liu, Y.-N. Liu, Y. Zhang, X. Ouyang and S. Chen, Angew. Chem., Int. Ed., 2022, 61, e202201007 CrossRef CAS.
- J. Liu, J. Xiao, B. Luo, E. Tian and G. I. N. Waterhouse, Chem. Eng. J., 2022, 427, 132038 CrossRef CAS.
- Y. Wei, H. Xia, H. Lan, D. Xue, B. Zhao, Y. Yu, Y. Hu and J.-N. Zhang, Adv. Energy Mater., 2024, 14, 2303011 CrossRef CAS.
- K. Wang, H. Du, S. He, L. Liu, K. Yang, J. Sun, Y. Liu, Z. Du, L. Xie, W. Ai and W. Huang, Adv. Mater., 2021, 33, 2005587 CrossRef CAS PubMed.
- S. Sun, X. Zhou, B. Cong, W. Hong and G. Chen, ACS Catal., 2020, 10, 9086–9097 CrossRef CAS.
- Y. Zhao, Z. Shen, J. Huo, X. Cao, P. Ou, J. Qu, X. Nie, J. Zhang, M. Wu, G. Wang and H. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308349 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05508j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |