
Corrosion of metallic anodes in aqueous batteries
Xuejin
Li†
*a,
Pengyun
Liu†
a,
Cuiping
Han
 b,
Tonghui
Cai
a,
Yongpeng
Cui
a,
Wei
Xing
*a and
Chunyi
Zhi
*c
b,
Tonghui
Cai
a,
Yongpeng
Cui
a,
Wei
Xing
*a and
Chunyi
Zhi
*c
aState Key Laboratory of Heavy Oil Processing, Shandong Key Laboratory of Intelligent Energy Materials, School of Materials Science and Engineering, China University of Petroleum, Qingdao 266580, China. E-mail: lxjupc@upc.edu.cn; xingwei@upc.edu.cn
bFaculty of Materials Science and Energy Engineering/Institute of Technology for Carbon Neutrality, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen, 518055, China
cDepartment of Materials Science and Engineering, City University of Hong Kong, Hong Kong 999077, P. R. China. E-mail: cy.zhi@cityu.edu.hk
First published on 6th February 2025
Abstract
Aqueous metal batteries offer high energy density and excellent compatibility with various cathode materials, and hence, they are attracting significant attention. However, the corrosion of metallic anodes seriously deteriorates the battery performance in terms of capacity loss, retarded reaction kinetics, and shortened cycling life. In this review, the fundamental corrosion mechanisms of metallic anodes in aqueous electrolytes and the key influencing factors are comprehensively discussed and summarized. Subsequently, recent achievements in corrosion inhibition are summarized and categorized to evaluate the advantages and disadvantages of various strategies. Furthermore, advanced characterization techniques used in corrosion mechanism analyses and protection assessments are elucidated. Finally, the current challenges in addressing the corrosion issue and potential developments in this field are presented.
 Xuejin Li | Xuejin Li is currently an Associate Professor in the Department of Materials Chemistry at the China University of Petroleum (East China). He received his PhD degree from the China University of Petroleum under the co-supervision of Prof. Zifeng Yan and Prof. Wei Xing. He then moved to the Institute of Physics-SongShan Lake Materials Laboratory as a Postdoctoral Fellow. His research interests focus on aqueous electrolyte batteries and hydrogen oxidation reaction electrocatalysis. |
 Pengyun Liu | Pengyun Liu is currently an Associate Professor in the Department of Materials Chemistry at the China University of Petroleum (East China). She received her PhD degree from the Curtin University under the co-supervision of Prof. Zongping Shao and Prof. Moses Tade. Her current research interests mainly focus on perovskite materials and devices, including perovskite photovoltaic devices and photo-rechargeable energy storage devices. |
 Wei Xing | Wei Xing received his PhD in chemical engineering from the China University of Petroleum in 2005 under the co-supervision of Prof. Zifeng Yan and Prof. G. Q. Max Lu. After seven years of research and teaching in Shandong University of Technology, he joined the China University of Petroleum as a Full Professor from December 2012. His research interests cover porous materials for energy storage, CO2 capture, and heterogeneous catalysis. |
 Chunyi Zhi | Chunyi Zhi is currently a Professor in the Department of Materials Science and Engineering at the City University of Hong Kong. He received his PhD degree in Physics from the Institute of Physics, Chinese Academy of Sciences. He then moved to the National Institute for Materials Science (NIMS) in Japan as a Postdoctoral fellow, and he later became an ICYS Research Fellow, Researcher (faculty) and Senior Researcher (permanent). His research focuses on wearable/flexible energy storage devices and aqueous electrolyte batteries. |
Broader contextMetallic anodes, including Zn, Al, and Mg, are widely applied in aqueous batteries owing to their high energy density and excellent compatibility with various cathode materials. However, they typically suffer from severe corrosion in aqueous electrolytes, and their corrosion behaviors are complicated. Corrosion processes of metallic anodes are of different types owing to their various properties. Even for the same metal, the corrosion behaviors are distinct in different environments and can be affected by pH, electrolyte ions, and dissolved oxygen concentration. Understanding the corrosion mechanisms of metallic anodes in various electrolyte environments is crucial in achieving effective corrosion inhibition methods and obtaining the desired battery performance. Currently, almost no review has specifically addressed, analyzed, or summarized the corrosion issue of metallic anodes in aqueous metal batteries (AMBs). Presenting a review in this research frontier is very necessary to provide insights on recent advances and potential guidance for improving the overall performance of AMBs. This review comprehensively introduced the corrosion behaviors of various metallic anodes, a variety of corrosion prevention tactics, and sophisticated characterization methods. We also discussed the challenges and future perspectives of this field to offer insights into further development of AMBs. |
1. Introduction
Aqueous metal batteries (AMBs), which directly adopt metals as anodes (such as Zn, Al, and Mg), exhibit superior advantages not only in the field of large-scale energy storage but also in wearable and biocompatible applications.1,2 Electrochemistry on the anodic side is based on the reversible deposition-dissolution of metals (Fig. 1a). The employment of metallic anodes endow AMBs with higher energy densities than “rocking chair”-type metal-ion batteries that store metal ions in anode hosts.3,4 In addition, metallic anodes provide more choices for matching cathode materials, such as air electrodes and sulfur electrodes, that have higher theoretical capacity than intercalation-type cathode materials.5,6 However, they typically suffer from serious corrosion issues in aqueous electrolytes, which are normally accompanied by the production of hydrogen and inert corrosion products, resulting in capacity deterioration and unsatisfied lifespan.7,8 | ||
| Fig. 1 (a) Illustration of the metal battery configuration in aqueous electrolytes; (b) illustration of metal corrosion in aqueous electrolytes; illustrations of (c) general corrosion, (d) pitting corrosion, (e) intergranular corrosion or crevice corrosion, and (f) galvanic corrosion with corresponding morphologies or potential demonstrations. | ||
Electrochemical corrosion of metal is a redox reaction (Fig. 1b) and can be expressed by the following equation:
| M + nY → Mn+ + nY− | (1.1) |
The above-mentioned redox reactions are irreversible and result in continuous metal dissolution. Even worse, the energy produced via these process cannot be utilized for the battery.11,12 Another problem is that the corrosion products, such as inert metal oxides/hydroxides and H2, are critically detrimental to battery performance, resulting in capacity loss, retarded reaction kinetics, and short lifespan.13 Moreover, the HER and ORR will fatally increase the pH of the electrolyte, which in turn may negatively influence the battery performance.
According to the corrosion morphology, corrosion can be divided into general corrosion and localized corrosion. General corrosion suggests that the corrosion proceeds in a relatively uniform manner over the entire surface of metal anodes (Fig. 1c). Damage to metals caused by general corrosion is unitary, and the derivative harm to metal is unlikely. However, in most cases, metallic anodes suffer from severe localized corrosion, which is more intense in some specific regions than in an adjacent area.8,14,15 When active species in electrolytes, such as Cl−, dissolved oxygen, or some oxidants, are selectively absorbed on the defective area of metals, corrosion on the area is more likely to happen and form the pitting nucleus. Under suitable potential, pitting corrosion is accelerated due to the enrichment of active species in the pits and develops to the inner side of metals (Fig. 1d). Corrosions are promoted in the pits due to the formation of oxygen-concentration cells and occluded cells. Corrosion may also occur along the grain boundaries because of the uneven electrochemical properties between grain crystals and grain boundaries (Fig. 1e). The boundaries have more defects and aggressive anions prefer to absorb on these regions, leading to more serious corrosion. If metallic impurities exist in the metal anode, a galvanic-type corrosion cell will be formed, and this results in galvanic corrosion of metals at the metal/impurities interface (Fig. 1f). It is demonstrated that the surface potentials between matrix metals and impurities or boundaries are different (Fig. 1e and f),16,17 which will facilitate the corrosion at interfaces. Damages to the metal electrode by localized corrosion are more serious than those that appear outwardly. Apart from the mass loss of metals, the current distribution on the metallic anode will be apparently affected by localized corrosion, resulting in uneven deposition and dissolution of metal during the charge–discharge process.18 To this end, the corrosion inhibition of metallic anodes in aqueous electrolytes is particularly important for improving the efficiency and lifespan of AMBs.
Metallic anodes including Zn, Al, and Mg are widely applied in aqueous metal–air batteries, which typically select alkaline electrolytes. The self-corrosion issue of these anodes has recently gained great interest from industry and researchers. Strategies such as using corrosion inhibitors and protective coating on anodes have been proposed to solve the corrosion issue in metal–air batteries.19–22 Compared with the process in metal–air batteries, less information is available on corrosion issues in other types of AMBs, which still requires great efforts in this intriguing area. Corrosion processes of metallic anodes are different due to their various properties. Even for the same metal, the corrosion behaviors are distinct under different environments and can be affected by pH, electrolyte ions, and dissolved oxygen concentration. Understanding the corrosion mechanisms of metallic anodes in various electrolyte environments is crucial in achieving effective corrosion inhibition methods and obtaining the desired battery performance.
Currently, almost no review has specifically addressed, analyzed, or summarized the corrosion issue of metallic anodes in AMBs. Although several reviews related to Zn anodes have been reported, they mainly focus on Zn dendrite issues.3,23–25 A recent review related to Zn anode corrosion has been reported, which briefly introduced the corrosion mechanism, corrosion inhibition, and methods for Zn corrosion analyses.26 However, the study and discussion of other metal anodes such as Al, Mg, and Fe are not included in this publication. The corrosion reaction process of metal anodes is complicated and variable in a complex electrochemical reaction environment, and an accurate understanding of the corrosion mechanism is still lacking. Presenting a review in this research frontier is very necessary to provide recent advances and potential guidance for improving the overall performance of AMBs. This review article first introduces the corrosion behaviors of various metallic anodes including Zn, Al, Mg, and Fe. The mechanisms and corrosion products in different electrolyte systems, as well as potential damages to the battery, will be discussed in this review. Following that, we will detail various strategies that inhibit corrosion, with corresponding advancements highlighted. Then, the review summarizes advanced characterization approaches as measurement criteria for the metallic anode corrosion issue. The final section discusses the challenges and perspectives of this field, so that it presents insights for further development of AMBs.
2. Corrosion mechanism
Corrosion behaviors including corrosion product, corrosion rates, and side reactions are different for various metal anodes such as Zn,7,27,28 Al,21,29,30 Mg,31 and Fe32 in aqueous electrolytes due to their respective characteristics. Even for the same metal, its corrosion behaviors vary with the environment it is in. This section will summarize and discuss the corrosion mechanisms, influencing factors, and corrosion products of Zn, Al, Mg, and Fe anodes.2.1 Corrosion behavior of Zn anodes
Aqueous Zn batteries including Zn-ion, Zn–air, and alkaline Zn–Ni are promising candidate systems for next-generation energy storage.33–37 From a thermodynamic point of view, Zn has a lower standard electrode potential (−0.763 V vs. SHE) than HERs in all pH ranges (Fig. 2a), which can theoretically predict that Zn is intended to be corroded in aqueous electrolytes. Zn's corrosion behaviors will be discussed in this section. | ||
| Fig. 2 (a) Pourbaix diagram of Zn–H2O at 25 °C. (b) Fraction Zn species at different pH values. (c) Dissolution process illustration of Zn. Reproduced from ref. 38 with permission from Elsevier, copyright 2020. (d) Various Zn corrosion modes at different pH values. (e) Illustrations of cathodic reactions and rate-determining steps of Zn corrosion. (f) Pourbaix diagram with the consideration of kinetics factors for 10−4 mol L−1 Zn2+. Reproduced from ref. 39 with permission from Elsevier, copyright 1991. | ||
In acidic electrolyte environments, the corrosion process of Zn can be expressed by the following equations:
| Anodic reaction: Zn–2e− → Zn2+ | (2.1) |
| Cathodic reaction: H+ + 2e− → H2 | (2.2) |
Thermodynamically, the most stable Zn species formed in an acidic environment is Zn2+ (Fig. 2a). Zn corrosion proceeds under the cathodic control and the corrosion rate is determined by the cathodic HER kinetics.41 The chemical kinetics of HERs controls the overall corrosion rate of Zn, when the pH value is smaller than 4.42 When the pH increases to the range of 3–5, the transport of H+ is slow and the corrosion rate of Zn is controlled by H+ diffusion or a mixed kinetic.43 Corrosion behaviors in acidic environments are illustrated in Fig. 2d. It should be noted that local pH at the corrosion interface may increase with continuous corrosion reactions and give rise to a different corrosion mechanism.
| Cathodic reaction: 2H2O + 2e− → 2OH− + H2 | (2.3) |
| O2 + 2H2O + 4e− → 4OH− | (2.4) |
ORR kinetics are activation controlled and are also independent of the solution pH due to the high irreversibility of ORRs. Dissolved oxygen plays a vital role in the cathode depolarization process and will accelerate Zn corrosion. Anodic reactions are relatively complicated in this pH region, which can be expressed as follows:41
| Anodic reaction: Zn–2e− → Zn2+ | (2.1) |
| Zn2+ + 2OH− → Zn(OH)2 | (2.5) |
| Zn(OH)2 → ZnO + H2O | (2.6) |
| Zn(OH)2 + 2OH− → Zn(OH)42− | (2.7) |
It is expected that Zn2+ will react with OH− to form a Zn(OH)2 precipitate (eqn (2.5)), parts of which can be dehydrated to form ZnO (eqn (2.6)). The local concentration of OH− at very active corrosion sites may be high enough to form zincate ions (eqn (2.7)). Notably, the precipitate formed at this stage is porous and pseudo-passive, which will not reduce the corrosion rate (Fig. 2d). Anions including SO42−, Cl−, and ClO4− in the electrolytes may also participate in the corrosion reaction. Cl− may react with Zn2+ or Zn(OH)2 and form zinc chloride hydroxide according to the following reactions:44
| 5Zn2+ + 8OH− + 2Cl− + H2O → Zn5(OH)8Cl2·H2O | (2.8) |
| 5Zn(OH) + 2Cl− + H2O → Zn5(OH)8Cl2·H2O + 2OH− | (2.9) |
ClO4− ions can accelerate the dissolution of Zn due to their adsorption on the metal surface and subsequent participation in the active dissolution:
| Zn + 2ClO4− → Zn(ClO4)2(aq) + 2e− | (2.10) |
In the presence of SO42−, a zinc hydroxide sulfate intermediate is formed and the intermediate is then hydrated through the following reaction:45
| Zn(OH)2 + 3Zn2+ + 4OH− + SO42− + 5H2O → Zn4(OH)6SO4·5H2O | (2.11) |
Compared with Zn4(OH)6SO4·5H2O, Zn5(OH)8Cl2·H2O is more porous due to the aggressive nature of Cl−. The tendency for these ions to be absorbed and to form ion pairs or complexes with dissolving Zn2+ ions follows the sequence of ClO4− < SO42− < Cl−.46 Thus, the rate of active dissolution of Zn increases in the order of ClO4− < SO42− < Cl−. The electrolytes in zinc-ion batteries are typically mild acid or neutral (such as ZnSO4, pH = 3–6), in which the corrosion of the Zn anode follows the mechanism discussed above.
| Zn + H2O → ZnO + 2H+ + 2e− | (2.12) |
| Zn + H2O + H+ → Zn(OH)+ + 2H+ + 2e− | (2.13) |
Although ZnO is formed, it is pseudo-passive and cannot reduce the corrosion of Zn, which is similar to the condition in mild electrolytes. With continuous corrosion, a passive film is formed. However, the produced H+ will simultaneously destabilize the passive film dynamically. The presence of the pseudo-passive layer is a net result of the passive layer formation and destabilization processes. Between pH 11 and 13, zinc oxides or zinc hydroxy complexes will be formed, and anode reaction kinetics will control the corrosion rate in this range. At pH 11, the metal hydrolysis reaction is limited, and as-produced H+ is getting less. Thus, small domains of passivity are formed at pH 11. At pH 12, a compact ZnO passive film can be formed by an adsorption model or a nucleation-growth model, which can be expressed as follows:47,48
| Zn + OH− → ZnOHads + e− | (2.14) |
| ZnOHads → ZnO + H+ + e− | (2.15) |
| Zn + 2OH− → ZnO + H2O + 2e− | (2.16) |
This compact film is called type III oxide. It is thin but plays the most important role in inhibiting further corrosion. When the pH increases to 13–14, the Zn(OH)3− complex will be generated via the following reactions:41
| ZnOHads + OH− → Zn(OH)2 + e− | (2.17) |
| Zn(OH)2 + OH− → Zn(OH)3− | (2.18) |
With continuous corrosion, local supersaturation of these complexes will impede OH− from transporting to the metal interface, thereby hindering further formation of the Zn(OH)3− complex. Then, Zn(OH)2 can gradually build upon the surface. Meanwhile, ZnO could also be formed by the dissolution of Zn(OH)3− or Zn(OH)42− as follows:41,47
| Zn(OH)3− + H+ → ZnO + H2O | (2.19) |
| Zn(OH)42− → ZnO + H2O + 2OH− | (2.20) |
As a result, a porous passivation film is produced and is called type I oxide. During this process, the dissolution of Zn(OH)2 is the rate-determining step, and further corrosion reaction must occur across a porous layer. Once the local OH− concentration drops to an extent, the dissolution of Zn(OH)2 to zinc hydroxy complexes is stopped, and a well-defined passive layer is formed between the metal surface and the type I layer, which is described as type II oxide. Although type III oxide is produced in much smaller quantities than type I or II, it plays a more important role in making Zn transit to the passive state.47 The Zn corrosion process in alkaline electrolytes is summarized and illustrated in Fig. 2d. The alkaline zinc batteries or zinc–air batteries typically select a high-concentration KOH solution as an electrolyte (pH > 13), suggesting that the Zn corrosion in these battery systems follows eqn (2.17)–(2.20).
The cathodic reactions and rate-determining steps are summarized and displayed in Fig. 2e. The HER is the only cathodic reaction in acidic electrolytes, and its chemical kinetics determine the overall corrosion rate. With the increase in pH to 4–6, H+ ion diffusion or mixed kinetics control the corrosion rate. The ORR may also proceed due to the local drop of H+ concentration. In the pH range of 7–10, the ORR is the main cathodic reaction, which is also diffusion-controlled and independent of pH. Thus, the corrosion rates do not vary significantly in this range. With the further increase in pH, the passive film is gradually formed, and the corrosion rate is determined by the anode reaction kinetics. Taking the kinetic factors into consideration, the Pourbaix diagram can be expressed in Fig. 2f.
Pitting corrosion is one of the most common corrosion types in Zn batteries. It will not occur in acidic electrolytes due to the clear surface of Zn. One important precondition for pitting corrosion is the presence of passivation films. Some researchers hold the view that pitting corrosion is caused by the specific absorption of aggressive halide ions (Cl−, Br−, and I−) on the surface of the passivation film.53,54 Once the halide ions penetrate the passivation film under pitting potential, corrosion pits emerge and local corrosion occurs. Local acidification theory is also widely accepted for interpreting the pitting behavior.54,55 Zn hydrolysis (eqn (2.12) and (2.15)) increases the H+ concentration locally and destabilizes the passivation film, paving the way for pitting corrosion. Besides, pitting corrosion easily takes place in the Zn/electrolyte/air triple-phase boundary due to the large amount of dissolved oxygen. Corrosion rates and corrosion potentials in pits and surrounding pseudo-passive regions are different. Once there exists the corrosion potential difference, a galvanic corrosion cell may be formed, in which Zn in pits acts as the anode and Zn under the pseudo-passivation film as the cathode. The formation of galvanic corrosion will accelerate the corrosion rate in the anode side.
2.2 Corrosion behavior of the Al anode
Aqueous Al batteries mainly include primary Al–air batteries and recently emerging aqueous Al-ion batteries, which normally employ highly alkaline or acidic electrolytes.56–59 It is known that Al is amphoteric and is reactive in both acidic and alkaline environments. As illustrated in the Pourbaix diagram of Al–H2O (Fig. 3a), the stable domain of Al is far below that of water, implying that Al strongly tends to be corroded with the decomposition of water. Based on the pH values, the dissolving species varies from Al3+ ions to Al2O3 and AlO2−. In this section, Al's corrosion mechanisms will be summarized and discussed. | ||
| Fig. 3 (a) Pourbaix diagram of Al–H2O at 25 °C. Illustration of the corrosion mechanism of the Al anode in (b) acidic electrolytes, (c) neutral electrolytes, and (d) alkaline electrolytes. | ||
| Al2O3 + 6H+ → 2Al3+ + 3H2O | (2.21) |
| AlOX → Alaq3+ + VAl(OX)3− | (2.22) |
| Al + 3H+ → Al3+ + 3/2H2 | (2.23) |
In the presence of anions such as Cl−, SO42−, and NO3−, the corrosion process gets more complicated due to the participation of these anions. Compared with other anions, Cl− ions are more aggressive and can accelerate the corrosion rate remarkably. On the one hand, Cl− ions assist in the dissolution of Al2O3 films initially and reduce the induction period. The absorbed Cl− ions react with Al2O3 as follows:61
| Al3+(in Al2O3) + 4Cl− → AlCl4− | (2.24) |
On the other hand, Cl− ions can directly participate in the dissolution reaction of Al. In the induction period, Cl− ions penetrate the oxide layer and form complexes, which leads to pitting corrosion and destroys the oxide film repair kinetics. The reaction can be described as follows:61,62
| Al + nCl− → AlCl(n−3)− + 3e− | (2.25) |
After total removal of the oxide film, Cl− ions are specifically adsorbed onto the metal surface and replace the adsorption of water molecules, facilitating the transfer of Al from the metallic phase to the solutions in the form of a complex with Cl− (as described in eqn (2.25)). The corrosion reactions of the Al anode in the recently developed aqueous Al-ion batteries, which typically use AlCl3 as the electrolyte, can be explained by eqn (2.23)–(2.25).
Al corrosion in the acidic electrolytes containing SO42− and NO3− is much slower and the corrosion mechanism is somewhat uncertain. The reduced corrosion rate in SO42-containing solutions may be caused by the less aggressiveness of SO42− than that of Cl−. It is also reported that the presence of SO42− ions in Cl−-containing electrolytes can retard the corrosion reactions due to the stronger absorptive affinity of SO42−.63,64 However, at higher concentrations, SO42− ions can attack the bare metal surface at the pre-existing pit bottoms. Another point of view suggests that a passive film containing Al(OH)SO4 and Al2(SO4)3·18H2O is formed during exposure to a sulfate-containing environment.65 This kind of passivation film might contribute to the low corrosion rate in electrolytes containing SO42−.
It is generally believed that NO3− ions play an inverse role with H+ and help develop and sustain a passive film, thus decreasing the corrosion rate. However, the reaction pathways are still not clear as numerous species have been found to be possible products, including Al(NO3)3, NO, NH3, N2, and NO2−.66–69 Some researchers also argue that an increase in HNO3 concentration would promote Al dissolution. The researchers stated that soluble complex ions were formed by complexation reactions between NO3− ions and hydrated Al3+ ions and, thus, increased the Al dissolution rate.61,70 From the above discussions, it is speculated that the Al dissolution behavior is more likely affected by the nature of anions rather than by the pH value.
| Al3+ + OH− → Al(OH)2+ | (2.26) |
| Al3+ + 2OH− → Al(OH)2+ | (2.27) |
| Al3+ + 3OH− → Al(OH)3 | (2.28) |
At this stage, the corrosion rate is determined by the nucleation and growth of aluminum hydroxide. The soluble Al(OH)2+ and Al(OH)2+ species diffuse outward to electrolyte solutions and water penetrates to the Al/Al(OH)3 interface, respectively. As the reactions continue, the hydroxide layer thickens and densifies gradually, playing a passivating effect on the corrosion process and slowing down the corrosion rate. Finally, the transport of these soluble species is the rate-determining step. Corrosion mechanisms of Al in mild electrolytes are illustrated in Fig. 3c.
Hydrogen produced from water reduction can diffuse through both Al and Al2O3 films. Once the generation rate of H2 is higher than the diffusion rate, the hydrogen will nucleate and grow gas bubbles at the Al/Al2O3 interface. Eventually, the bubbles can generate sufficient pressure to blow holes through the passivation film and initiate pitting corrosion.71
| Al(OH)3 + OH− → Al(OH)4− | (2.29) |
The diffusion of OH− ions and Al(OH)4− to/away from the film/electrolyte interface is the rate-controlling step.74 It should be noted that the direct ejection of Al3+ ions from oxide film to electrolyte can never occur in alkaline electrolytes due to the thermodynamic instability of Al3+ ions in alkaline solutions. Meanwhile, the dissolution of the Al2O3 film occurs at the film/electrolyte interface according to the following equation:
| Al2O3 + OH− + H2O → Al(OH)4− | (2.30) |
In highly alkaline electrolytes, the oxide film on the surface of Al is totally dissolved and OH− from electrolytes can directly attack the bare Al surface according to the following reaction:
| Al + 4OH− → Al(OH)4− | (2.31) |
Meanwhile, the influences of OH− and Al(OH)4− ion diffusions on the corrosion rate are not significant in highly alkaline environments due to the high concentration of OH− and large solubility of Al(OH)4− ions. Eqn (2.31) clearly defines the corrosion of the Al anode in the Al–air battery as a result of the electrolyte's excessive alkalinity.
2.3 Corrosion behavior of the Mg anode
Mg metal aqueous batteries mainly refer to primary Mg–MnO2 and Mg–air batteries with neutral electrolytes. Rechargeable Mg–air batteries are still far from being practical because of the low energy conversion efficiency and low high-rate stability of Mg. Mg anode suffers from more severe self-corrosion than Al and Zn due to its more reactive nature. As illustrated in the Mg–H2O Pourbaix diagram (Fig. 4a–c), Mg's almost no stable domain in aqueous electrolytes in the whole pH range due to its much lower equilibrium potential than hydrogen evolution potential. Compared with those of Al and Zn, the corrosion behaviors of Mg in aqueous electrolytes are more complicated. This section will discuss and summarize the corrosion mechanisms of Mg and their influencing factors.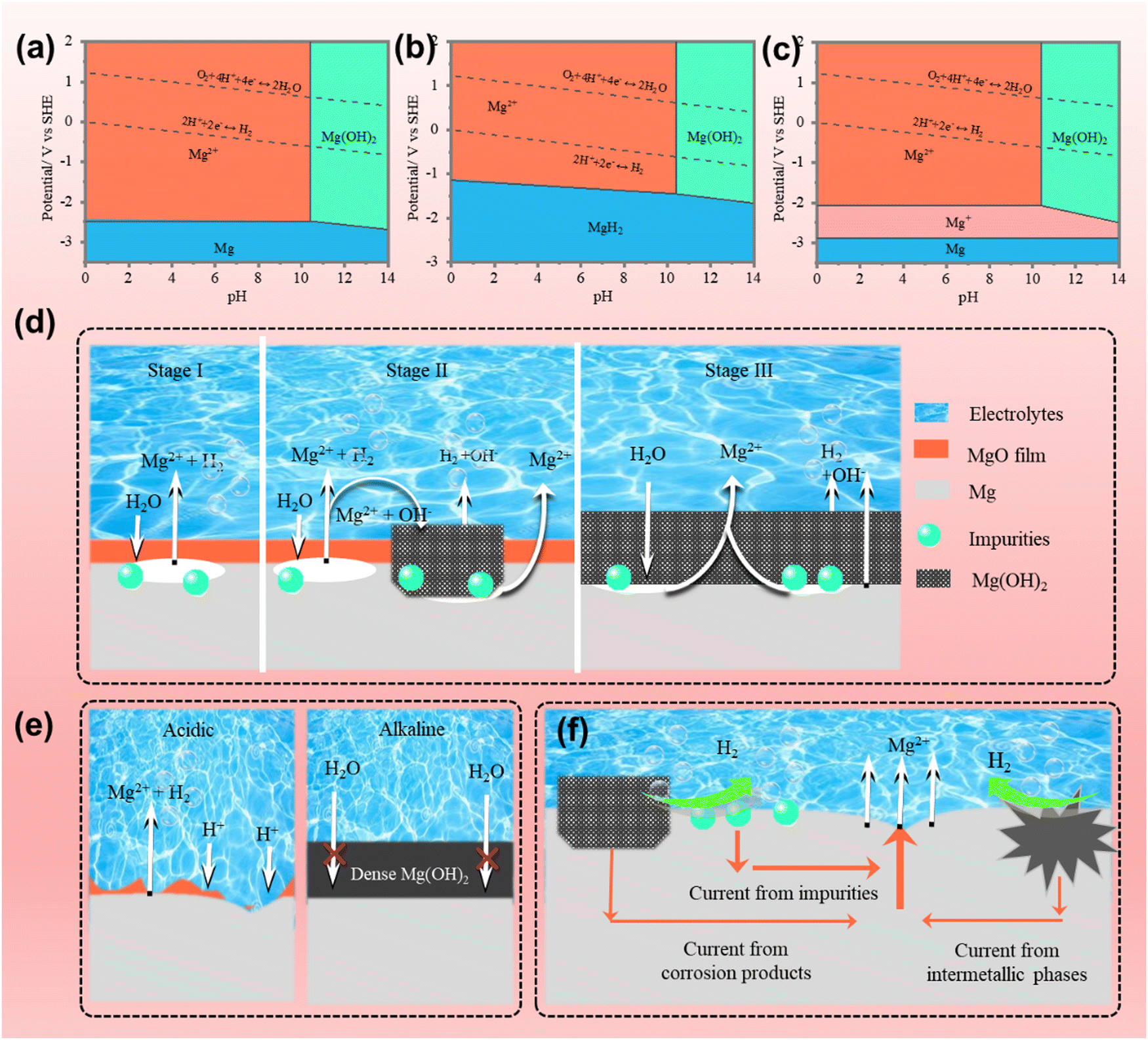 | ||
| Fig. 4 Pourbaix diagrams of Mg–H2O at 25 °C (a) without Mg+ and MgH2, (b) in the presence of MgH2, and (c) in the presence of Mg+. Corrosion mechanism of Mg (d) in neutral electrolytes, and (e) in acidic and alkaline electrolytes. (f) Galvanic corrosion mechanism of Mg. | ||
Until now, several corrosion mechanism hypotheses have been proposed, including the magnesium hydride intermediate model, univalent Mg-based model, film-based model, and metal spalling model. However, these models are controversial and cannot individually interpret all the corrosion behaviors. The magnesium hydride intermediate model suggests that MgH2 exists on the Mg surface (Fig. 4b) and plays a vital role in the corrosion process.81,82 Mg is first reduced to MgH2, which can be further oxidized to Mg(OH)2 (in neutral or alkaline environments) or Mg2+ (in acidic environments). These processes can be expressed as follows:
| Mg + 2H+ + 2e− → MgH2 | (2.32) |
| MgH2 + 2H2O → Mg(OH)2 + 2H2 | (2.33) |
| MgH2 + 2H+ → Mg2+ + 2H2 | (2.34) |
Mg can also be dissolved directly in the uncovered regions by the following reaction:
| Mg–2e− → Mg2+ | (2.35) |
The formation of MgH2 is a cathodic reaction, so the NDE cannot be reasonably explained. The univalent Mg-based model indicated that the dissolution of Mg initiates with the formation of Mg+ (Fig. 4c) as follows:83,84
| Mg–e− → Mg+ | (2.36) |
Mg+ ions are not stable and will be further oxidized to Mg2+via the following reactions:
| 2Mg + 2H+ → 2Mg2+ + H2 | (2.37) |
| 2Mg+ + 2H2O → 2Mg2+ + 2OH− + H2 | (2.38) |
The NDE can be understood via the above-mentioned oxidization processes. However, this mechanism cannot interpret the dissolution hysteresis phenomenon in neutral or alkaline environments. Besides, Mg+ species have never been experimentally detected, suggesting the irrationality of this mechanism.
Due to the uneven corrosion behaviors, the spalling mechanism infers that Mg dissolves as minute particles or flakes to spall off.87 Further corrosion of these metallic Mg particles would result in the generation of H2 with no current flow. Though this mechanism can explain the NDE, Mg's relatively positive corrosion potential cannot still be thoroughly explained.
| Mg + H2O → Mg2+ + 2OH− + H2 | (2.39) |
Since Mg–MnO2 or Mg–air batteries typically use a neutral NaCl electrolyte, the corrosion reaction mechanism of the Mg anode can be described by eqn (2.39).
Mg(OH)2 is stable in aqueous electrolytes at a pH higher than 10.5 and serves as a protective barrier to further corrosion (Fig. 4e). In acidic environments, the MgO/Mg(OH)2 film is highly soluble, giving rise to a rapid corrosion reaction as follows:96,97
| MgO + 2H+ → Mg2+ + H2O | (2.40) |
| Mg + 2H+ → Mg2+ + H2 | (2.41) |
The Mg matrix in Mg alloys exhibits an analogous corrosion mechanism with pure Mg. The HER is the main cathodic reaction of Mg corrosion due to its much higher equilibrium potential than that of Mg/Mg2+. Though dissolved oxygen influences corrosion behavior, the effect is quite weak, which is not typically considered.
2.4 Corrosion behaviors of Fe and other metallic anodes
Fe-based aqueous batteries mainly include alkaline Fe–Ni batteries,100 Fe–air batteries,101 and recently reported Fe–S batteries, Fe–I2 batteries, as well as Fe-ion batteries with weakly acidic FeSO4 electrolytes.102–104 The Fe anode is prone to be corroded in acidic electrolytes due to its lower equilibrium potential than hydrogen evolution potentials, which can be described as follows with the HER as a cathodic reaction:105| Fe + 2H+ → Fe2+ + H2 | (2.42) |
In the presence of dissolved oxygen, the corrosion process can also proceed through the following reaction with ORR as a cathodic reaction:
| Fe + 2H+ + O2 → Fe2+ + H2O | (2.43) |
When the pH value of electrolytes is higher than 6, the ORR is the main cathodic reaction. In the absence of dissolved oxygen, the Fe corrosion rate is relatively low. The corrosion reaction is accompanied by water reduction and hydrogen evolution, which can be described as follows:
| Fe + 2H2O → Fe2+ + 2OH− + H2 | (2.44) |
Fe2+ ions are soluble in neutral electrolytes and Fe(OH)2 may be formed on the surface locally due to the increase in OH− concentrations in the corrosion area. In alkaline electrolytes, the Fe2+ ions generated from corrosion precipitate on the surface with OH− from electrolytes, which results in the passivation of Fe and protects Fe from further corrosion. It should be noted that the passive film is hardly formed in the presence of aggressive ions (such as Cl− and Br−).
From the above-mentioned discussions, Fe corrosion is mainly affected by the pH of electrolytes, dissolved oxygen, and anions in the electrolytes. Besides, the charge/discharge states of alkaline Fe-based batteries may influence corrosion behaviors. Fe is known to be transformed into Fe(OH)2, Fe3O4, and Fe(OH)3 during the discharge process. These discharge products have inhibition on the corrosion of Fe. However, this passivation would also result in a decrease in battery kinetics.
Metallic Li and Na are so reactive with water that they cannot be utilized directly as anodes in aqueous electrolytes. The reactions are described as follows:
| 2Li + H2O → 2LiOH + H2 | (2.45) |
| 2Na + 2H2O → 2NaOH + H2 | (2.46) |
Li and Na metals must be protected with coating layers to prevent them from corrosion by water in aqueous Li metal and Na metal batteries.
2.5 Influencing factors and derived problems
 | ||
| Fig. 5 Influencing factors and derived problems of metallic anode corrosion. | ||
Corrosion behaviors under various working conditions including temperature, charge/discharge states, and charge/discharge rates are different. For example, at low current densities, OH− can efficiently transport across the porous type I ZnO passivation layer and inhibit the formation of the compact type II ZnO layer.7 Besides, the morphology and surface products will change with continuous charge/discharge process, leading to a variation in corrosion behaviors. Higher operating temperatures are reported to promote the corrosion rate except in the electrolytes with sulfates and nitrates.61,73
The properties of metallic anodes such as specific surface area, porosity, and particle size also influence corrosion behaviors. Anodes with sufficient porosity and high specific surface area will cause enhanced corrosion susceptibility due to the difficult formation of a passivation layer and improved corrosion interface.7,110 Pitting corrosions or galvanic corrosions prefer to occur at grain boundaries or cracks due to the microstructural or compositional differences,76,111 suggesting that a more uniform surface or phase structure is preferable for corrosion inhibition. The elemental composition of metallic anodes also affects the corrosion process. Elements with a higher HER overpotential in the anodes, such as In, Sn, and Bi, will hinder the parasitic HER and, thus, reduce the corrosion rates. Moreover, the intermetallic phases in the anodes formed in the alloying process will also influence the corrosion process. Especially for Mg alloys, the second phase can serve as a cathode and enhance the dissolution of Mg at the α-Mg phase/secondary phase interface due to its more noble electrochemical activity than the Mg matrix.112 However, the continuous distribution of the second phase can suppress further Mg corrosion propagation. In addition, impurities in the metallic anodes may promote metal dissolutions around the impurities through the galvanic corrosion process.
Thus, the effects of the electrolyte composition and the metal anode itself on the battery's electrochemical performance and corrosion of the metal anode must be thoroughly taken into account when manufacturing aqueous metal batteries. Some factors need particular consideration. The starting point is to choose a metal anode that has a low specific surface area, good structural uniformity, and minimal impurities. The anode's processing technology must meet strict specifications to guarantee that there are as few defects and grain boundaries as feasible. It is necessary to eliminate contaminants from the metal anode, particularly those that contain metal elements with a lower hydrogen evolution overpotential. Furthermore, choosing the right electrolyte is crucial. On the premise of ensuring that electrochemistry can operate, neutral or weakly acidic and weakly alkaline electrolytes should be selected as far as possible. In addition, electrolyte salts that include caustic anions should be avoided when choosing electrolyte salts. Ultimately, the oxygen content should be minimized during battery assembly; for instance, the electrolyte's dissolved oxygen can be evacuated through an inert environment.
3. Corrosion inhibition
3.1 Artificial interface engineering
The artificial interface on the metallic anode surface plays an essential role in corrosion inhibition by protecting the metallic anode from direct exposure to the aqueous electrolytes. A stable metal/electrolyte interface can be established, thereby increasing the cycling stability of metallic anodes. Typically, this interface serves as a physical barrier and confines the transport of OH−, H+, H2O, and other aggressive anions to metal surfaces (Fig. 6a). Surface chemistry at the interface such as charge property and hydrophobicity may also be changed and prohibit the accessibility of specific ions and water molecules (Fig. 6b).114 Besides, introducing metallic-based interfaces with higher HER overpotentials would suppress the HER at the interface (Fig. 6c). According to the above-discussed corrosion mechanism, five crucial factors, namely reactivity, structural stability, electrical conductivity, ionic conductivity, and hydrophilicity, should be taken into consideration to construct robust interfaces (Fig. 6d). A more reactive property than the metallic anode is preferred for reactivity if the protective layer is conductive. Otherwise, a galvanic cell will be formed with the layer as a cathode, accelerating metallic anode corrosion. As for the structural stability, the coating layer should be robust in the electrolytes and keep working during the battery cycling process. From the aspect of electrical conductivity, a semiconductor or insulator is desirable because it will not form a galvanic cell with the sheltered metal anode. Besides, the metal ions will not plate on the outer protective film surface.3 For ionic conductivity and hydrophilicity, a good metal ion conductor with superior hydrophilicity is beneficial for the metal ion transport during the charge/discharge process, which enhances the reaction kinetics.38 In conclusion, there are several requirements that the perfect artificial interface layer should fulfill (Fig. 6e). It should be light and not drastically lower the energy density of the battery. Excellent chemical and electrochemical stability is another requirement for the perfect interface coating to guarantee its continued functionality throughout the battery's extended charge and discharge cycles. To guarantee that the electrolyte ions may penetrate the interface layer and uniformly deposit on the metal negative electrode's surface, it should also possess superior ion conductivity and insulation. Furthermore, it must have strong mechanical properties to prevent breaking or falling off while the battery is operating. Recent advancements in corrosion inhibition by various artificial interfaces are summarized in Fig. 6f and Table 1. | ||
| Fig. 6 Schematic of corrosion inhibition mechanisms for interface engineering: (a) physical barrier, (b) surface modification, and (c) chemical inhibition. (d) Properties of carbon coating layers, metallic compounds, and organic polymers. (e) Requirements for an ideal interfacial protection layer. (f) Corrosion inhibition performance for various artificial interfaces. Reproduced from ref. 49 and 115–124 with permission from John Wiley & Sons, Elsevier, and the Royal Society of Chemistry. | ||
| Anode | Electrolyte | Coating layer | Corrosion inhibition efficiency | Battery performance | Ref. | |
|---|---|---|---|---|---|---|
| 1 | Zn | 3 M Zn(SO3CF3)2 | TiO2 | Suppressed HER | Enhanced cycling stability | 115 |
| 2 | Zn | 9 M KOH | Al2O3 | 78.6% | Enhanced cycling stability | 116 |
| 3 | Zn | 6 M KOH | Porous carbon | — | Reduced capacity but enhanced stability | 117 |
| 4 | Zn | 7 M KOH | Polyaniline | 85% | Enhanced stability | 118 |
| 5 | Zn | 6 M KOH | SiO2 | 40% | — | 125 |
| 6 | Zn | 2 M ZnSO4 | MOF | — | Enhanced cycling stability | 49 |
| 7 | Zn | 2 M ZnSO4 | ZrO2 | Higher Ecorr | Enhanced cycling stability | 119 |
| 8 | Zn | 2 M ZnSO4 | Indium-based interface layer | Lower Icorr | Enhanced cycling stability | 120 |
| 9 | Zn | 2 M ZnSO4/0.1 M MnSO4 | Al2Si2O5(OH)4, | 56% | Enhanced cycling stability | 121 |
| 10 | Zn | 2 M ZnSO4 | PVDF@TiO2 | Reduced corrosion reaction | Reduced capacity but enhanced stability | 122 |
| 11 | Zn | 2 M ZnSO4/0.1 M MnSO4 | Polyamide | Higher Ecorr and lower Icorr | 60-fold enhancement in running lifetime | 123 |
| 12 | Zn | 3 M Zn(SO3CF3)2 | Al2O3 | 40% | Enhanced cycling stability | 124 |
| 13 | Zn | 6 M KOH | Neodymium film | 90.9% | — | 126 |
| 14 | Zn | 2 M ZnSO4 | Zn2SiO4/CNT | lower Icorr and more positive Ecorr | Extended lifespan | 127 |
| 15 | Zn | 2 M ZnSO4 | ZnSn alloy interphase | Lower Icorr | 250% improvement in cycling life | 128 |
| 16 | Zn | 2 M ZnSO4 | Graphene acid/carbon nanofiber | Lower Icorr | Ultra-low self-discharge behavior | 129 |
| 17 | Zn | 2 M ZnSO4 | Hydroxyapatite | Higher Ecorr and lower Icorr | 10 times longer lifespan | 130 |
| 18 | Zn | 2 M ZnSO4 | Cellulose | 61.9% | Improved ICE and cycling stability | 131 |
| 19 | Zn | 2 M ZnSO4 | Dopamine-functionalized polypyrrole | Higher corrosion voltage and reduced H2 generation | 5 and 2.5 times higher capacity retentions | 132 |
| 20 | Zn | 2 M ZnSO4 | ZnNb2O6 | Lower Icorr | Longer-term stable cycling | 133 |
| 21 | Al | PAA–KOH | Prussian blue | 81.2% | Increased anode efficiency and energy density | 134 |
| 22 | Al | 4 M KOH | LDH-PVA-acetal | 81.0% | Improved capacity and cycling stability | 30 |
 | ||
| Fig. 7 (a) Diagram of the modification route and the electric field distributions of zinc and Zn–G anodes. Reproduced from ref. 135 with permission from John Wiley & Sons, copyright 2021. (b) Schematic of the synthesis of HsGDY and concentration field simulation after a constant diffusion duration of 5 s for Zn@HsGDY symmetric cell. Reproduced from ref. 138 with permission from John Wiley & Sons, copyright 2020. (c) Preparation diagram of zinc anode coated with a surface functional graphene layer. Reproduced from ref. 139 with permission from Elsevier, copyright 2023. | ||
The hydrophobicity of the carbon layer should be taken into consideration. Carbon is advantageous in blocking water molecules due to its inherent hydrophobicity. Superhydrophobic surfaces have been proven to be qualified for corrosion inhibition of metals.140,141 A superhydrophobic carbon black film prepared by electrospray is reported to protect stainless steel from corrosion in a sulfuric acid solution, exhibiting positive corrosion inhibition.142 This work suggests that the hydrophobicity depends on the thickness of the carbon layer, and the porous carbon layers gradually lose the hydrophobicity in aqueous electrolytes because of the distant interaction of water vapor with the metal surface. Notwithstanding the effective protectivity, the hydrophobic surface would suppress the ion transport across the electrode/electrolyte interface, resulting in an inferior battery performance.
Apart from the role of the carbon layer as a physical barrier, the chemical properties of carbon influence corrosion protection. Functional groups decorated on the carbon can tune the accessibility of ions from electrolytes. A graphene film with abundant oxygen-containing groups was selected to protect Zn from corrosion (Fig. 7c), by which the aggressive anionic species were prohibited from accessing the metal surface.139 Chemical modification of the carbon layer should have advantages in inhibiting specific ions from electrolytes, but related research is still limited.
Although the carbon layer has been proven effective in corrosion inhibition, several drawbacks remain. Carbon is typically electrically conductive and may cause metal ion plating or hydrogen evolution outside the layer. Moreover, the hydrophobic nature of carbon will enlarge the ion interface transport resistance and thus increase the overpotential or voltage polarization of the metallic anode. A trade-off between corrosion protection and battery kinetic should be taken into account under this condition.
 | ||
| Fig. 8 Timeline of metallic compound-based interfacial layer strategies for metallic anode protection from 2020 to 2024. All insets were obtained from the literature.119,127,146–153 | ||
Preparing thin and compact layers with good protectivity and ion conductivity may help break the confrontation. In contrast with the conventional doctor blade coating method, sol–gel method, and other solution methods, atomic layer deposition (ALD) is a promising thin-film deposition technique, which enables precise control of the thickness of the film with good protectivity. For instance, an ultrathin ALD TiOx layer (Fig. 9a) has been reported to suppress the corrosion of the Al current collector.154 The ALD TiOx thickness can be precisely controlled (from a deposition rate of ∼0.02–0.04 nm cycle−1) and the Ti-oxidation state was easily tuned by adjusting the TiCl4 and H2O precursor pulse lengths. As a result, the corrosion resistance of Al was improved by orders of magnitude. ALD Al2O3 was also reported to increase the corrosion resistance of the Zn anode.115,116,124 Although the ALD Al2O3-protected Zn anode exhibited a higher overpotential initially due to the insulating nature of Al2O3, it outperformed bare Zn during the long-term cycling process in terms of lower resistance and smoother surface. Moreover, ALD Al2O3 layers gave rise to improved surface wettability, suggesting that this strategy enables both promising corrosion inhibition and fast reaction kinetics.
 | ||
| Fig. 9 (a) Illustration of a single ALD cycle with a TiOx coating of thickness of ∼0.04 nm. Reproduced from ref. 154 with permission from the American Chemical Society, copyright 2024. (b) Schematic of the artificial ZnS layer preparation. Reproduced from ref. 155 with permission from John Wiley & Sons, copyright 2020. (c) Schematic of the procedure for fabricating the Zn@ZSO composite foil. Reproduced from ref. 156 with permission from John Wiley & Sons, copyright 2022. | ||
Beyond the ALD method, the in situ formation of the artificial interface on the metal surface is another effective strategy to control surface chemistry. A precisely controllable ZnS interface can be formed by treating Zn anodes with sulfur vapor (Fig. 9c), which serves as a physical barrier to inhibit corrosion and modifies the charge distribution at the interface.155 The unbalanced charge distribution improved Zn2+ diffusion at the interface and adhesion of the ZnS layer to the metal surface, leading to both superior corrosion inhibition and fast ion transport kinetics. An “etching-nucleation-growth” strategy was also developed to generate a tightly arranged ultrathin ZnSiO3 nanosheet array. The ZnSiO3 is nucleated in the holes generated by the initial etching of Zn2+ ions, which gives the in situ-formed ZnSiO3 strong adhesion to the Zn anode. The porosity and morphology of the protective layer can be regulated effectively by the pH and reaction time.
Some specific metal compounds such as indium-based species, Al2O3, and Bi2O3 serve as not merely physical barriers but also chemical corrosion inhibitors owing to their higher hydrogen evolution overpotential. Among various metal compounds, indium-based species such as In, In2O3, and In(OH)3 manifest the best effect on suppressing hydrogen evolution because of the good chemical inactivity of indium.28,120,143,157,158 A metallic indium layer will suppress HERs and by-product formation, give rise to uniform nucleation of Zn, and reduce Zn dendrites (Fig. 10). Benefiting from the promising corrosion inhibition effect, the cell performed well in long-term cycling without swelling.143 Even in alkaline electrolytes, this strategy is proven to be effective. In2O3 and In(OH)3 may be formed in alkaline electrolytes, which are insulative and can promote metal plating underneath the coating film.120
 | ||
| Fig. 10 (a) Schematic of the stripping/plating behaviors of bare Zn and Zn/In anodes. (b) Thickness change of the symmetric cells. (c) Gas evolution characterizations of bare Zn and Zn/In anodes in ZnSO4 electrolytes. (d) XRD patterns of the bare Zn and Zn/In anodes before and after cycling. Reproduced from ref. 143 with permission from John Wiley & Sons, copyright 2020. (e) Digital images and SEM images of Zn anodes with/without an indium layer before and after plating. Reproduced from ref. 120 with permission from Elsevier, copyright 2020. | ||
As to metallic compound-based protective layers, lower thickness, compact structure, and good ionic conductivity are preferred to provide desirable protection and avoid sacrificing the battery performance. While they are advantageous in mechanical strength and insulation (Fig. 6d), cracks may be formed during long-term cycling. Besides, their chemical stability in various electrolytes should be taken into consideration.
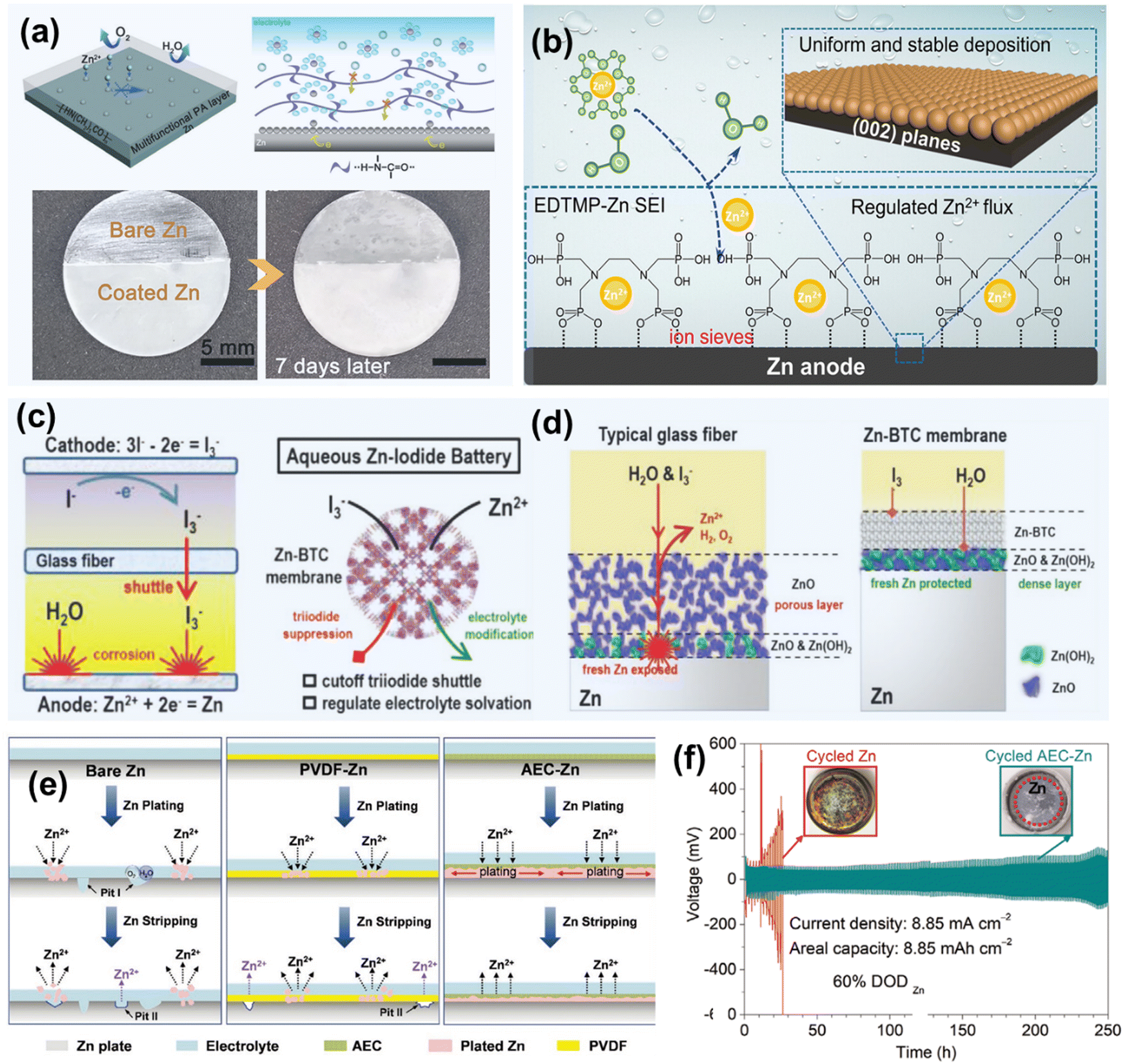 | ||
| Fig. 11 (a) Schematic of a PA layer for corrosion inhibition, and optical images of Zn electrodes with/without the PA layer. Reproduced from ref. 123 with permission from the Royal Society of Chemistry, copyright 2019. (b) Schematic illustration of the Zn deposition behavior on EDTMP15-Zn@Zn. Reproduced from ref. 163 with permission from John Wiley & Sons, copyright 2024. (c) Schematic of Zn–I2 batteries with a glass fiber separator and a MOF-based separator. (d) Component illustration at the electrolyte/electrode interfaces after corrosion process. Reproduced from ref. 49 with permission from John Wiley & Sons, copyright 2020. (e) Schematic of Zn plating/stripping processes for bare Zn, PVDF-Zn, and anti-corrosion elastic constraint-protected Zn electrode. (f) Comparison of the cycling stability and optical images of the cells after cycling. Reproduced from ref. 122 with permission from John Wiley & Sons, copyright 2020. | ||
3.2 Alloying engineering
Alloying is an exceedingly employed strategy to tune the physicochemical properties such as mechanical strength and corrosion resistance. A multitude of Zn-, Al-, and Mg-based alloys have been reported and great advancements in corrosion inhibition have already been achieved.112,128,171–180 The corrosion inhibition effect significantly depends on the type and doping amount of alloying elements. This section will summarize and discuss the corrosion inhibition effects and mechanisms of various alloying elements.Generally, the HER is suppressed on metals such as Pb, Cd, In, Hg, Bi, and Sn. It is anticipated that introducing these metallic elements into the base metal may give rise to increased HER overpotentials, thereby reducing the corrosion rates (Fig. 12a). For instance, Zn–Bi alloys were proven to present a much lower corrosion rate than pure Zn anode in KOH electrolytes.181,182 It is also reported that Al–Zn, Al–In, and Al–Sn alloys exhibit a lower corrosion potential than the pure Al anode in alkaline electrolytes due to their higher HER overpotentials.177,183,184 Nonetheless, the superfluous addition of these alloying elements will accelerate the corrosion reactions of the matrix metals. Once the content of alloying elements exceeds its solubility in parent metals, the segregative phases will be formed, which may act as the cathodic sides and accelerate the corrosion reactions at the grain boundaries.185
 | ||
| Fig. 12 Schematic of corrosion inhibition mechanisms of alloying elements: (a) increasing HER overpotential, (b) sacrificial anode protection, and (c) formation of the protective layer. (d) Exhibition of mostly reported HER catalysts in the periodic table. Reproduced from ref. 186 with permission from the American Chemical Society, copyright 2020. (e) Corrosion inhibition efficiency of common alloying elements in Zn-, Al-, and Mg-alloys. Reproduced from ref. 8, 171, 172, 185 and 187–189. (f) Schematic of the electrochemical impacts of alloying elements in Mg alloys; Cs refers to solid solubility. Reproduced from ref. 190 with permission from Elsevier, copyright 2017. (g) Schematic of the grain boundary engineering, EBSD IPF mapping of Zn–Ti alloy, and SEM-EDS mapping of Zn–Ti alloy. Reproduced from ref. 177 with permission from Nature Publishing Group, copyright 2023. | ||
Some more chemically reactive elements were also employed as sacrificial agents to protect parent metals from corrosion (Fig. 12b). In this aspect, a galvanic cell will be assembled with an alloying agent as the anode, by which the base metals are protected.172 Take Mg as an example: a small amount of Mg can protect the Al matrix from corrosion due to both the increased HER overpotential and sacrificial anodic protection.172 However, this strategy is not suitable for Mg metals due to its relatively reactive nature and low equilibrium potential.
In addition, the alloying elements may influence the corrosion products and affect the formation of passivation films (Fig. 12c). According to reports, the Zn addition to Al anode promoted the formation of two-type Zn oxidation films, including a porous Zn(OH)2 layer and a protective ZnO layer.191 These layers were more protective than the Al(OH)3 layer in alkaline electrolytes and suppressed the Al corrosion. However, this protective layer will also reduce the charge/discharge efficiency due to its passivation effect. Adding Al into the Mg anode led to the formation of MgxAl(OH)2x+2Cl in the NaCl electrolyte, promoting the peel-off of Mg(OH)2 products and thereby reducing the NDE.192 A low concentration of alloying agents cannot affect the passivation film. In contrast, a large amount of alloying agents will promote the formation of the secondary phase and accelerate the corrosion reactions.
Fig. 12d exhibits the most reported single-atom catalysts for HERs, suggesting that these elements have lower HER overpotentials. Therefore, the other metallic elements such as Bi, Hg, and In in the periodic table can potentially suppress the HER and can be alloyed into metallic anodes for corrosion inhibition. Although these elements have already been confirmed to be effective in corrosion inhibitions (Fig. 12e), there is no criterion for the amount of alloying elements. The effects of alloying elements vary a lot in different research studies. A standard evaluation methodology should be established to examine the corrosion inhibition efficiency of various alloying elements accurately.
Notably, the alloying engineering in Mg anodes is more complicated due to the reactive nature of Mg. Most of the elements manifest limited solubility in crystalline Mg and give rather minimal changes to the corrosion potential of Mg alloys. On the one hand, alloying elements with a higher HER overpotential may be beneficial in slowing down Mg corrosion. On the other hand, the presence of a secondary phase will accelerate the dissolution of neighboring Mg. Nearly all intermetallic particles, except Mg2Ca, are thought to have more noble corrosive potentials than pure Mg, suggesting that they will act as cathodic sides of the galvanic cells and promote Mg dissolution.193 The effect of alloying engineering on Mg anode is the net result of the above-mentioned aspects, which the polarization curves can illustrate. From the dynamic polarization curves of various binary Mg-alloys, the Ecorr value shifts to more positive values and the cathodic current densities increase for all alloys.194 The electrochemical impacts of alloying elements in Mg have been summarized in a previous report (Fig. 12f).190 Alloying metal impurities will enrich the surface of the Mg matrix during the corrosion process, acting as a local cathode and enhancing the cathodic HER kinetics. Though some reports state that alloying elements such as Al, Zn, Ca, and In positively affect Mg corrosion inhibition, the improvements should be ascribed to the refined grain or secondary phase morphologies rather than to the chemistry effect.98,190,195Fig. 12f also demonstrates that only specific elements such as As, Sb, and Ge from groups 14 and 15 can reduce the cathodic reaction kinetics due to their electrocatalytic poisoning effect toward the HER.196,197
The secondary phase formed by alloying elements and base metals plays a vital role in corrosion inhibition. Grain boundaries (GBs) are important structural elements that link grains in polycrystalline metals and are more reactive than crystallographic planes respectively. As a result, significant intergranular corrosion sometimes results from localized corrosion that starts at GBs with high corrosion susceptibility and subsequently spreads to the deep or inside grains. Take Mg–Ca alloy as an example: corrosion preferentially occurred along the GBs due to the presence of Mg2Ca.98 As the secondary Mg2Ca is not continuous, the dissolution of Mg developed to the interiors of the Mg matrix, leading to shallow but spacious cracks. Once the cracks connect, the Mg grains detach from the bulk anode, resulting in low utilization. Tuning the grain boundary property or the bulk phase structure is an effective strategy to reduce corrosion.177,198–200 For example, constructing a Zn–Ti dual-phase alloy can alter the GBs of metallic Zn by forming Ti-containing intermetallic compounds that are thermodynamically stabilized at GBs (Fig. 12g). The HER-induced corrosion was efficiently suppressed regardless of aging or extended cycling. Larger grain sizes and fewer grain boundaries in the metallic electrode typically present better corrosion resistance.199 Through aging treatment or rolling processing, the separated phase distribution, grain boundary, and grain size can be modified.76,111,177,199
Alloying engineering has been proven to be an effective strategy for suppressing the corrosion of metals, and recent achievements in alloying engineering of metallic anodes are summarized in Table 2. The proportion of alloying elements is determinative of the corrosion inhibition efficiency. Though great advances have been achieved, insights into the mechanistic aspect of corrosion inhibition require more research, especially in the presence of multiple alloying elements. A standard evolution methodology should be established to examine the corrosion inhibition efficiency of various alloying elements accurately.
| Anode | Electrolyte | Alloying elements | Corrosion inhibition efficiency | Battery performance | Ref. | |
|---|---|---|---|---|---|---|
| 1 | Zn | 7 M KOH | 2% Bi | 91.5% | Slightly reduced capacity | 187 |
| 2 | Zn | 6 M KOH | 1.5% Bi | 53.8% | Lower cut-off voltage, enhanced capacity | 171 |
| 3 | Zn | 3 M ZnSO4/0.3 M MnSO4 | Cu | 83.7% | Prolonged cycling life | 8 |
| 4 | Zn | 7 M KOH | 3% Bi | 33.2% | — | 201 |
| 5 | Zn | 7 M KOH | 5% Ni | 86.3% | — | 202 |
| 6 | Al | 4 M KOH | 1% Mg | 89.2% | Enhanced Al utilization and capacity | 172 |
| 7 | Al | 4 M KOH | 1% Mg/0.9% Sn | 82.7% | Enhanced Al utilization and capacity | 172 |
| 8 | Al | 7 M KOH | 1% Mg/1% Zn/0.1% Bi/0.1% In | 37.3% | Higher cell potential and Al utilization | 203 |
| 9 | Al | 4 M NaOH | 0.05% In | 29.2% | Enhanced capacity and anodic efficiency | 204 |
| 10 | Al | 0.6 M NaCl | 0.6% Sn | 62.7% | — | 188 |
| 11 | Al | 1 M NaOH | Cu | Higher corrosion resistance | Higher discharge potential and Al efficiency | 205 |
| 12 | Mg | 3.5% NaCl | 9% Al/2.5% Pb | Higher Ecorr and lower Icorr | Enhanced discharge potential and Al utilization efficiency | 192 |
| 13 | Mg | 0.1 M NaCl | 1% Zn | More serve corrosion | — | 112 |
| 14 | Mg | 0.1 M NaCl | 0.3% Ge | Suppress HER | — | 112 |
| 15 | Mg | 0.1 M NaCl | Bi, Ge, Pb, Sb, Sn | Improved corrosion resistance | — | 196 |
| 16 | Mg | 3.5% NaCl | 0.1% Ca | 25.1% | Higher cell voltage and energy density | 185 |
| 17 | Mg | 0.6 M NaCl | 3–9% Al | Decreased self-corrosion | Higher operating voltage and capacity | 206 |
| 18 | Mg | 0.6 M NaCl | 3–9% Al | Lower Icorr | Higher Mg utilization | 195 |
| 19 | Mg | 0.1 M NaCl | 0.37% As | An order of magnitude lower H2 evolution | — | 197 |
| 20 | Mg | 3.5% NaCl | 1–3% Sn | Reduced Icorr and H2 evolution | — | 207 |
| 21 | Mg | 0.6 M NaCl | 1%, 5%, 10% Sn | 77%, 85%, 95% | — | 189 |
3.3 Electrolyte engineering
Electrolyte engineering refers to employing additives, designing gel or solid-state electrolytes, and developing hybrid electrolytes, which aim to suppress the cathodic HER. These approaches give rise to reduced HER active centers or sluggish HER kinetics, thereby decreasing the self-corrosion of metallic anodes.ΔGads = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(55.5Kads) ln(55.5Kads) | (3.1) |
 | ||
| Fig. 13 (a) Molecular structures of commonly used organic inhibitors. (b) Schematic of the corrosion inhibition mechanism of organic inhibitors. (c) HOMO energy level of different molecules. Reproduced from ref. 212 with permission from Elsevier, copyright 2024. (d) Channel size illustration of CuC7 and CuC15 with different alkyl chain lengths. Reproduced from ref. 213 with permission from the Royal Society of Chemistry, copyright 2024. | ||
Organic inhibitors may either dynamically generate an absorption layer on the surface or produce a protective layer with metal ions (Fig. 13b).214,215 Take SDBS as an example: it is mainly composed of hydrophilic groups and hydrophobic alkyl chains.216 The highest occupied molecular orbital exclusively distributes in the sulfonate anion region, which exhibits a negative electrostatic potential and helps to interact with the metal surface (Fig. 13c).212,217 While the polar groups help inhibitors to adsorb onto the metal surface, the non-polar part (such as the hydrocarbon chain) is forced towards the aqueous solution side due to its electrophilic reactivity. These tails repel the water and aggressive molecules away from the surface and thus decrease their corrosion effects.
Efficiency in inhibiting corrosion is primarily dependent on the completeness of the protective or absorption layer, which is closely tied to inhibitor concentrations and intrinsic characteristics. Increasing the inhibitor concentration to the critical micelle concentration value gradually increases, and full coverage can finally be achieved.218 The adsorption of inhibitors on the metal surface typically obeys either Langmuir isotherm or Freundlich isotherm,209,219 which can be expressed as follows:
 | (3.2) |
 | (3.3) |
 | (3.4) |
 | (3.5) |
 are the charge transfer resistances of the metallic anodes in electrolytes with and without inhibitors, respectively. Icorr and
are the charge transfer resistances of the metallic anodes in electrolytes with and without inhibitors, respectively. Icorr and  are the corrosion current densities of the metallic anodes in electrolytes with and without inhibitors. Besides, the molecules containing the carbonyl group, carboxyl group, amino group, benzene rings, heterocyclic ring, or more heteroatoms should present higher corrosion inhibition efficiency due to the improved absorption interaction (Fig. 13a).39,219,220 Moreover, the alkyl chain length significantly influences the corrosion efficiency. Too short an alkyl chain may make it hard to form an intact film, while a too-long alkyl chain might result in a loose adsorbed layer.221 The film formed by a longer alkyl chain presents a smaller channel size and higher water molecular/Zn2+ ion insertion energies (Fig. 13d).213 Lastly, the effectiveness of corrosion inhibition also depends on the inhibitor's capacity to participate in the solvation shell. Solvated water rather than free water is more likely to be the origin of H2 evolution and “parasitic” side reactions.222 By substituting solvated water molecules, certain organic compounds can participate in the solvation structure of Zn ions, lowering the possibility of corrosion at the Zn electrode/electrolyte interface.214,223–228
are the corrosion current densities of the metallic anodes in electrolytes with and without inhibitors. Besides, the molecules containing the carbonyl group, carboxyl group, amino group, benzene rings, heterocyclic ring, or more heteroatoms should present higher corrosion inhibition efficiency due to the improved absorption interaction (Fig. 13a).39,219,220 Moreover, the alkyl chain length significantly influences the corrosion efficiency. Too short an alkyl chain may make it hard to form an intact film, while a too-long alkyl chain might result in a loose adsorbed layer.221 The film formed by a longer alkyl chain presents a smaller channel size and higher water molecular/Zn2+ ion insertion energies (Fig. 13d).213 Lastly, the effectiveness of corrosion inhibition also depends on the inhibitor's capacity to participate in the solvation shell. Solvated water rather than free water is more likely to be the origin of H2 evolution and “parasitic” side reactions.222 By substituting solvated water molecules, certain organic compounds can participate in the solvation structure of Zn ions, lowering the possibility of corrosion at the Zn electrode/electrolyte interface.214,223–228
Combining different inhibitors gives rise to a higher corrosion inhibition efficiency, thanks to their synergistic effect.220,229 On the one hand, the branched and linear structures in organic inhibitors result in a more integrated protective layer. Inhibitors with a benzene ring or a heterocyclic ring exhibit strong adhesion on the metal surface (Fig. 14a). However, the as-formed absorption layer or protective layer may be loose and incomplete due to the steric effect of a highly branched structure. Introducing linear organic inhibitors such as PEG into an imidazole-containing system contributed to an intact protection film, improving the corrosion inhibition efficiency.220 On the other hand, organic and inorganic composite inhibitors may present better corrosion protection performance. Some inorganic corrosion inhibitors such as ZnO and K2SnO3 endow in situ deposition of Zn or Sn layers on the anode surface.56,230,231 Together with physical barrier actions, the increased HER overpotential by this kind of in situ formed metallic layers contributes to an enhanced corrosion inhibition efficiency. The inhibitors may also have significant influences on the solvation shell structure. For example, the radius of the hydrated zinc complex (4.30 Å) is approximately five times as large as that of the bare zinc cation itself (0.83 Å) (Fig. 14b). The hydration shells are mainly occupied by the coordinating water molecules. By selecting redox-inactive cations with appropriate coulombic repulsion energy and solvation rigidity (Fig. 14c), the solvation shell structure has been modified, leading to markedly improved cyclability.223
 | ||
| Fig. 14 (a) Schematic of the absorption for different inhibitors with a linear alkyl chain or a heterocyclic ring. Effect of key parameters of redox-inactive cationic additives on tip blocking: (b) physical dimensions of a hydrated Zn ion and scheme of the tip effect. (c) Illustration of the charge and the water exchange rate modifications by redox-inactive cations. Reproduced from ref. 223 with permission from John Wiley & Sons, copyright 2022. (d) Corrosion inhibition efficiencies of various corrosion inhibitors for Zn-, Al-, and Mg-based anodes. | ||
Recent advances in corrosion inhibitors for AMBs are summarized in Fig. 14d and Table 3. Though various inhibitors have already achieved remarkable corrosion inhibitions, further efforts need to be made to clarify their mechanisms. Especially for inorganic inhibitors, it is yet unknown how to restrict active centers for cathodic HERs or anodic dissolution, which impedes the development of effective corrosion inhibitors in the future. More emphasis should be paid to the adsorption characteristics of inorganic inhibitors, which impact the corrosion inhibitor efficiency. Since corrosion inhibitors may alter metallic anodes' reversible stripping/plating behaviors, their impact on battery kinetics should also attract greater attention.
| Anode | Electrolyte | Corrosion inhibitor | Corrosion inhibition efficiency | Battery performance | Ref. | |
|---|---|---|---|---|---|---|
| 1 | Zn | 7 M KOH | DTAB | 80.2% | — | 232 |
| 2 | Zn | 6 M KOH | OED | 85.5% | Both improved capacity and stability | 233 |
| 3 | Zn | 1 M Li2SO4/2 M ZnSO4 | PEG200 | 72.4% | 32% improvement in capacity | 167 |
| 4 | Zn | 9.5 M KOH | PEG | 88.9% | — | 218 |
| 5 | Zn | 8.5 M KOH | PEG600 | 82% | — | 234 |
| 6 | Zn | 26% NH4Cl | SDBS | 89.7% | — | 217 |
| 7 | Zn | 3 M KOH | PEG 600 and Tween 20 | 89% | 33% improvement in capacity | 235 |
| 8 | Zn | 6 M KOH | PEI | 55.2% | Improved stability | 236 |
| 9 | Zn | 7 M KOH | PNE | 98.1% | Improved capacity | 211 |
| 10 | Zn | 3 M KOH | IMZ and PEG600 | 79.9% | — | 220 |
| 11 | Zn | 26% NH4Cl | HEC | 92.07% | — | 209 |
| 12 | Zn | 7 M KOH | SDS | 77.5% | 24% improvement in capacity | 237 |
| 13 | Zn | ZnCl2/NH4Cl | AE | 60–80% | — | 219 |
| 14 | Zn | 6 M KOH | BAT | 76.9% | 31.9% improvement in capacity | 238 |
| 15 | Zn | 6 M KOH | BTA and SDBS | 88.5% | 34.3% improvement of capacity | 238 |
| 16 | Zn | 2 M ZnSO4 | Sodium anthraquinone-2-sulfonate | Reduced corrosion | Longer cycle life | 239 |
| 17 | Zn | 1 M ZnSO4 | 1,2-Butanediol | Lower Icorr | 5–20 fold zinc cyclability enhancement | 240 |
| 18 | Zn | 6 M KOH | CMC | 43.5% | — | 241 |
| 19 | Zn | 2 M ZnSO4 | Trehalose | Lower Icorr and higher Ecorr | two times enhancement in cycle stability | 242 |
| 20 | Zn | 2 M ZnSO4 | Zinc pyrrolidone carboxylate | More positive Ecorr | Enhanced cycle stability | 243 |
| 21 | Zn | 2 M ZnSO4 | Nitrilotriacetic acid | Much lower Icorr | regulate the Zn deposition | 244 |
| 22 | Zn | 2 M ZnSO4 | Imidazo[1,2-b]pyridazine | lower Icorr | Better reversibility | 245 |
| 23 | Zn | 2 M ZnSO4 | Pyrimidine | lower Icorr | Accelerating the desolvation process | 246 |
| 24 | Zn | 1 M Zn(OAc)2 | Ethylene glycol | 15 times lower corrosion rate | Higher capacity retention | 247 |
| 25 | Zn | 3 M Zn(OTf)2 | Urea/LiOAc | Far less Icorr | highly reversible plating/stripping | 229 |
| 26 | Zn | 2 M ZnSO4 | Zinc acetylacetonate | Lower Icorr and higher Ecorr | 10 times longer cyclability | 248 |
| 27 | Zn | 2 M ZnSO4 | Ammonium hydroxide | Lower HER potential | much better cyclic stability | 249 |
| 28 | Zn | 2 M ZnSO4 | Sc3+ additive | Higher corrosion resistance | 5–8 fold cycle life | 250 |
| 29 | Zn | 2 M ZnSO4 | Sodium 3,3′-dithiodipropane sulfonate | Lower Icorr and higher Ecorr | Extended cycle life | 251 |
| 30 | Zn | 2 M ZnSO4 | C5SeCN | Reduced Icorr | Improved Zn utilization | 252 |
| 31 | Zn | 2 M ZnSO4 | Protonated triglycine | Increased Ecorr | Prolonged operation life | 253 |
| 32 | Al | 4 M NaOH | 6-thioguanine | 36.56% | Enhanced Al utilization: from 38.5% to 74.6% | 254 |
| 33 | Al | 4 M NaOH | N-9 | 92.8% | 15–25% improvement of capacity | 255 |
| 34 | Al | 4 M NaOH | [Allmim][NTf2] | 95.1% | 48.5% improvements in capacity | 256 |
| 35 | Al | 4 M KOH | APG and K2SnO3 | 94.1% | 250% improvements in capacity | 56 |
| 36 | Al | 4 M NaOH | Na2SnO3 and casein | 83.1% | 89.3% improvements in capacity | 230 |
| 37 | Al | 4 M NaOH | L-Cysteine/ZnO | 85% | Enhanced Al utilization: from 7.4% to 19.9% | 257 |
| 38 | Al | 5 M KOH | PEG di-acid and ZnO | 66% | 57.3% improvements in capacity | 258 |
| 39 | Al | 5 M KOH | Flax straw extract | 62% | 23.5% improvements in capacity | 259 |
| 40 | Al | 4 M NaOH | C12BBr | 56.3% | 96% improvements in capacity | 260 |
| 41 | Al | 4 M NaOH | L-Aspartic/CaO | 74.5% | — | 261 |
| 42 | Al | 4 M NaOH | 8-HQ/ZnO | 71.5% | Enhanced Al utilization: from 56.2% to 70.3% | 262 |
| 43 | Mg | 3.5% NaCl | DG | 94% | Enhanced Mg utilization: from 39.4% to 89.7% | 263 |
| 44 | Mg | 3.5% NaCl | Na3PO4 and SDBS | 95.2% | Enhanced Mg utilization: from 44.1% to 49.1% | 264 |
| 45 | Mg | 2 M MgSO4/2 M Mg(NO3)2 | NaF–Na3PO4 | 98.8% | 57% improvement in capacity | 208 |
| 46 | Mg | 0.6 M NaCl | Na3PO4·12H2O | 95.6% | 25% decrease of capacity | 265 |
| 47 | Mg | 0.6 M NaCl | NaVO3 | 72.2% | 6.7% improvement in capacity | 265 |
| 58 | Al | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M LiTFSI M LiTFSI |
Hydrolyzation type-anodic additive | Three orders lower Icorr | Improved cycling stability and initial efficiency | 266 |
 | ||
| Fig. 15 (a) Synthetic procedure of the PANa-cellulose hydrogel electrolyte and illustration of entrapped OH− ions and water in the hydrogel via hydrogen bonding. Reproduced from ref. 274 with permission from John Wiley & Sons, copyright 2019. (b) Design principle of PAM-based hydrogel electrolyte and its structural illustration. Reproduced from ref. 275 with permission from John Wiley & Sons, copyright 2019. (c) Schematic of the solvation sheath in different electrolytes and illustrations of reduced water reduction reaction and Al anode corrosion in the WIS electrolyte. Reproduced from ref. 286 with permission from Elsevier, copyright 2020. | ||
Gel electrolytes prepared by direct gelatinization of aqueous electrolytes were also reported, which can be doped with corrosion inhibitors such as lignin, pyrazole, Pb2+, and polyethylene glycol.287–289 The combination of water immobilization and corrosion inhibitors gives rise to good corrosion inhibition efficiency.
Water-in-salt (WIS) electrolytes contain more excellent dissolved salt than water, in which all water molecules are a part of ion solvation shells and no free water is present. This type of super-concentrated electrolyte has been applied in various aqueous batteries due to its expanded electrochemical stability window.290–292 As the water molecules are all immobilized by an electrolyte salt, the corrosion reaction of metallic anode and cathodic HERs in WIS electrolytes is suppressed. For example, a WIS electrolyte containing 16 M potassium acetate (KOAc) and 4 M KOH was developed to suppress Al corrosion in Al–air batteries.286 With the gradual increase in the amount of KOAc, acetate anions cooperated with the sheaths of K+, and more water molecules were involved in binding interactions in solvation sheaths (Fig. 15c). Fewer water molecules adsorbed onto the Al anode surface due to the greatly reduced free water. Therefore, the HER was significantly inhibited and Al anode corrosion was obviously reduced.
3.4 Other strategies
Some other promising strategies, ranging from tuning the exposure crystal plane to rationally designing the battery configurations, have also been developed to address the corrosion issue of metallic anode. Recently, some researchers have proposed that the corrosion behavior of metallic anodes is highly related to their crystallographic planes. Zn corrosion is mainly prone to occur in the (100) plane, while it is not likely to happen on the (002) plane.2,159,293,294 Electrodeposition of Zn in the presence of various additives, such as indium sulfate, tin oxide, and boric acid, will give rise to different surface textures and crystallographic orientations. Among different additives, boric acid helps to produce Zn with the dominance of the (002) surface, which has a higher resistance to dendrite formation and corrosion. Similar phenomena were also detected in the Al anode, which suggested that the (001) plane has good corrosion resistance and the (110) plane is more sensitive.295,296 Experimental results confirmed that the hydrogen evolution rate and corrosion current density follow the order of (001) single crystal < (111) single crystal < (110) single crystal < polycrystalline. Therefore, tuning the crystallographic planes might be an effective way to suppress both dendrite formation and corrosion.Rational battery configuration may also help to remit the corrosion issue of metallic anodes. Displacing electrolytes from the electrode with a non-conductive oil during battery standby was developed in a primary Al–air battery to suppress the open-circuit corrosion.297 However, this battery design still has self-corrosion during the discharging process. A magnesium-moisture battery without additional electrolytes was proposed, in which metallic Mg served as the anode and a semi-conducting polyaniline (PANI) foam served as the cathode (Fig. 16a). Trace-absorbed water causes Mg corrosion, which generates Mg2+ ions and electrons. Because of the much lower electronic conductivity than the ionic conductivity of PANI, the electrons have to run into an external circuit due to the depolarization process.298 Although the discharge current is not high, this battery configuration prevented the energy loss caused by corrosion.
 | ||
| Fig. 16 (a) Schematic of the Mg-PANI foam battery. Reproduced from ref. 298 with permission from John Wiley & Sons, copyright 2015. (b) Schematic of a device combining electrical and chemical storage (1-Al anode; 2-aqueous alkaline electrolyte; 3-introduction of inert gas; 4-H2 collection; 5-ion-exchange membrane; and 6-cathode); Reproduced from ref. 299 with permission from Elsevier, copyright 2016. (c) Schematic of the aqueous Zn/Al-CO2 systems. Reproduced from ref. 300 with permission from John Wiley & Sons, copyright 2019. | ||
Another approach to raising the efficiency of the metallic anode is collecting the hydrogen produced by the parasitic cathodic reactions during corrosion. Devices combined with electrical energy and chemical energy (H2 storage) storage systems were developed (Fig. 16b and c).299,301 Apart from the normal function of the battery, the hydrogen produced during the corrosion can be stored. To effectively collect hydrogen, it should be mentioned that the anode compartment needs to be deoxygenated. A recently reported Zn/Al–CO2 system may also provide insights into this tactic, which adopted HER as the cathodic reaction to achieve both electrons and H2 collection.300 Similarly, the hydrogen produced in the corrosion process was also anticipated to be utilized by this battery system. Although this strategy reduces the side effects caused by corrosion, it is unsuitable for practical battery systems due to their complicated configurations.
The aforementioned review and discussion lead us to the conclusion that the anti-corrosion strategy of the metal anode can be macroscopically divided into two parts: electrode design and electrolyte design. The interfacial protective coating, which has multifunctional characteristics and is customizable, generally prevents the electrolyte from corroding to the metal anode through the physical barrier effect. However, the corrosion inhibition performance of the electrode is heavily dependent on the microstructural characteristics and inherent physicochemical properties of the coating layer, which undoubtedly increases the difficulty and cost of preparation. The alloy anode is easy to prepare on a large scale and has good stability, but alloying engineering requires strict control of the alloy ratio and process to ensure the anode's electrochemical performance. There are still issues with alloy element compatibility and precise phase composition management. Moreover, the introduced inactive competent inevitably decreases the energy density of the battery. The most popular approach in electrolyte design is the introduction of corrosion inhibitors. This approach offers special benefits such easy large-scale preparation, cheapness, and simple operation. However, while choosing additives, it is important to consider if they might cause problems with other battery parts (such as the separator and cathode), which could impact the battery's overall charge/discharge performance. Special attention must be paid to how corrosion inhibitors affect the battery's charge/discharge kinetics in order to prevent lowering the power characteristics.
4. Characterization techniques
Advanced techniques to characterize metallic anode corrosion and the as-proposed protective approaches play a vital role in understanding the corrosion processes and evaluating the corrosion inhibition performance. Electrochemical characterizations, including polarization curve method, electrochemical impedance spectroscopy (EIS), electrochemical noise (EN), and scanning electrochemical microscopy (SECM), make prominent contributions to corrosion mechanism determination. The formation of byproducts and passivation layers, which have huge impacts on further corrosion and battery performance, can be assessed by various spectroscopic techniques including X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy. A series of microscopy techniques can visually monitor the morphological evolution of the metallic anode during the corrosion process and long-term battery running.4.1 Electrochemical techniques
 | (4.1) |
However, the Tafel plots are easily affected by the passivation process or other redox reactions on the electrode surface, thereby increasing the difficulty of corrosion current measurement. Under this circumstance, linear polarization near Ecorr provides more accurate information. Corrosion current can be calculated by the slope of the linear polarization curve according to the Stern-Geary equation as follows:303
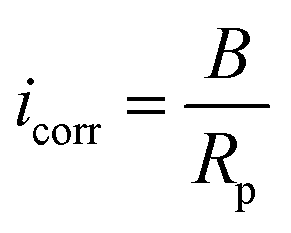 | (4.2) |
The metal dissolution rate increases during the anodic polarization and the as-produced byproducts may cause electrode passivation. Then, the corrosion current reduces due to the protection of the passivation film until the occurrence of pitting corrosion (Fig. 17a). Therefore, polarization curves can reveal the overall corrosion process and play a significant role in the mechanism study.11,240,304,305 Corrosion protection performance can also be easily evaluated by polarization curves. For instance, more positive corrosion potential and smaller corrosion current were observed for a Cu alloyed Zn anode, demonstrating good corrosion inhibition efficiency.8 Another example is that the N9 surfactant caused a marked decrease in cathodic current and slightly impeded the anodic curve, inferring that this corrosion inhibitor is a cathodic inhibitor.255
 | ||
| Fig. 17 (a) Schematic of polarization curves. Reproduced from ref. 302 with permission from Elsevier, copyright 2018. (b) In situ Nyquist plots of the Mg electrode in Na2SO4 electrolytes. Reproduced from ref. 306 with permission from Elsevier, copyright 2019. (c) Bode phase and Bode |Z| plots of the Mg–0.6%Si–2%Zn alloys. (d) Equivalent electrical circuit. Reproduced from ref. 307 with permission from Elsevier, copyright 2024. | ||
In summary, the polarization curve method is a fundamental technique to obtain corrosion reaction kinetics and describe the overall corrosion behaviors. However, one thing that should be particularly noted is that Ecorr cannot estimate the corrosion tendency individually. The positive shift of corrosion potential does not always suggest corrosion inhibition behavior, which should be evaluated together with corrosion current.
To better understand and quantify the EIS plots, the data are typically fitted using corresponding equivalent electrical circuits, which ordinarily include the components of resistance, capacitance, and constant phase element (CPE). In the equivalent electrical circuit (Fig. 17d), RΩ, Rt, and RL are the solution resistance, combined resistance of migration along the porous layer, and inductor resistance, respectively. CPE is described by its parameter Q and its exponent α, to account for the heterogeneities presented on the surfaces. The corrosion tendency, protective coating efficiency, and electrolyte/electrode interface modification can be evaluated by the variations of these parameters.310,311
 | ||
| Fig. 18 (a) Typical EN signals of Q235 steel including potential noise, current noise, noise resistance, and standard deviation. Reproduced from ref. 315 with permission from Maney Publishing, copyright 2017. (b) Localization Index of EN signals in different electrolytes. Reproduced from ref. 314 with permission from Elsevier, copyright 2019. (c) PSD curves for the electrochemical potential noise and electrochemical current noise signals obtained from the Al 6061-T6 alloy in a NaCl electrolyte. (d) Schematic of the information that may be obtained from an EDP. Reproduced from ref. 316 with permission from Taylor & Francis, copyright 2019. (e) ST analysis from the electrochemical current noise signals. (f) Comparison between the ST and wavelet transform using the current signal. Reproduced from ref. 317 with permission from Elsevier, copyright 2019. | ||
The power spectral density (PSD) of the EN signals can be obtained by fast Fourier transform or maximum entropy method, which provides characteristic information on corrosion behaviors in the frequency domain (Fig. 18c). If the PSD spectrum presents a white noise feature, the electrode is inferred to be dominated by uniform corrosion. It is reported that the average value of the PSD in a certain frequency range of 10–250 MHz corresponds well with the uniform corrosion rate.318 A roll-off slope of −20 dB per decade or less demonstrates the localized corrosion.319
Another method to analyze EN signals is wavelet transform, which can decompose the EN into wavelet coefficients of different scales. Events in the time domain, such as diffusion, activation, or mixed controlled processes, are converted into wavelet crystals with respective crystal energy. The energy distribution plot (EDP), referring to the plot of accumulated energy vs. the crystal name, provides detailed information about corrosion mechanisms. The position of the maximum relative energy in the EDP corresponds to the dominant corrosion type. Typically, short-time-scale crystals (D2 and D3 crystals) are associated with activation-controlled pitting corrosion. In contrast, medium-time-scale crystals (D4, D5, and D6) and long-time-scale crystals (D7 and D8) should be related to mixed-controlled localized corrosion and diffusion-controlled general corrosion, respectively (Fig. 18d).316,319,320
Furthermore, the Stockwell transform (ST) and Shannon Energy have also been presented as effective proposals to identify the corrosion type at different temperatures (Fig. 18e and f). ST analyses give rise to a series of amplitude scales and establish the relationship between EN signals and the corrosion types. Typically, the amplitude values from potential signals of 1 × 10−6–1 × 10−5 represent the material passivation, while the values of 1 × 10−5–1 × 10−4 are considered mixed corrosion. In the range of 1 × 10−4–1 × 10−3, the corrosion type is suggested as localized corrosion and uniform corrosion exhibits higher amplitudes. As to ST analyses from current signals, the amplitudes of 1 × 10−9–1 × 10−8, 1 × 10−8–1 × 10−7, and 1 × 10−7–1 × 10−6 imply the types of passivation, mixed corrosion, and localized corrosion, respectively.317 With this promising technique, it is possible to obtain corrosion information at any temperature in terms of corrosion type, corrosion rate, and occurrence time.
 | ||
| Fig. 19 (a) Current density distributions on Mg surface exposed to 2 M NaCl electrolyte at different time points. Reproduced from ref. 80 with permission from Elsevier, copyright 2015. (b) Schematic showing the mapping of NiO film breakdown on Ni at individual locations via SECM and map of breakdown potential (Ebreak) on different grains. Reproduced from ref. 326 with permission from the American Chemical Society, copyright 2022. (c) Schematic of the coaxial electrochemical cell and Non-interpolated SECM image of the scanned area covering the SEI-free (left) and SEI-covered (right) areas. Reproduced from ref. 329 with permission from Wiley, copyright 2022. | ||
The tip collection mode of SECM provides opportunities to detect electrochemical hydrogen evolution.322,330,331 It is reported that a platinum electrode enables the oxidation of hydrogen evolved from the metallic surface and can be used to identify the corrosion sites.332,333 The hydrogen evolution rate is related to the detection current and gives rise to a visualized corrosion distribution on the surface (Fig. 20a). In addition, the local pH distributions in the corroded region can be monitored in the potentiometric mode with a pH-sensitive antimony microelectrode tip (Fig. 20b and c),334,335 thereby leading to a better understanding of corrosion mechanisms. Through SECM equipped with a pH-sensitive microelectrode, the pH distribution and variation with time in the vicinity of a corroding surface can be visualized, which is of interest to understanding the corrosion behaviors on a microscale. For example, the pH distribution above an iron sample was presented.336 The spatial distribution of pH values from the surface into the bulk electrolyte can be observed from the inspection of this image. A relatively homogeneous pH distribution was observed over the Zn sample, suggesting its different corrosion mechanism from that of the iron sample. Moreover, the dissolved oxygen concentration near the metallic surface can be revealed. As shown in Fig. 20d, the oxygen can be intercepted by the SECM tip, which can be reduced at special potentials and give rise to corresponding current signals. In summary, SECM is a powerful tool for detecting the spatial distribution of corrosion reactions in real time, leading to a better understanding of the complicated corrosion mechanisms.
 | ||
| Fig. 20 (a) Schematic of H2 detection by SECM and the corresponding current distribution caused by H2 fluxes. Reproduced from ref. 337 with permission from Wiley, copyright 2022. (b) pH distribution in a plane perpendicular to the surface of iron after immersion in 0.1 M NaCl for 22 h. (c) pH distribution in a plane parallel to the surface of Zn after immersion in 0.1 M NaCl for 7 h. Reproduced from ref. 336 with permission from Elsevier, copyright 2011. (d) Illustration of the processes occurring at the electrode-electrolyte interface during SECM measurement; Reproduced from ref. 338 with permission from the American Chemical Society, copyright 2024. | ||
4.2 Spectroscopy characterization
Understanding the crystal and chemical structures of corrosion products on the metallic anode surface is crucial to interpreting the corrosion mechanism and evaluating its influence on the battery performance. Generally, the insoluble corrosion products will lead to the passivation of the metallic anode, which will suppress further corrosion and simultaneously hinder the battery kinetics. In this section, applications of advanced spectroscopic techniques in the field of corrosion inhibition, including XRD, Fourier transform infrared (FTIR) spectroscopy, Raman spectroscopy, and XPS, will be discussed.In situ FTIR spectroscopy analysis is a powerful tool to assess the gradual formation of corrosion products. The evolution of Zn surface chemistry in a NaCl solution was detected from the in situ FTIR spectra.344 With the increasing soaking time, the Zn–Cl peak intensity was enhanced, inferring that chloride species were gradually formed on the Zn surface during the corrosion process. In another case, the gradual appearance of Al3+-complexation was demonstrated by increasing the exposure time of the Al electrode in electrolytes containing a humic acid inhibitor, which would serve as a protective film on the Al anode.259 The characteristic absorption of hydroxy groups in metal hydroxides can also be detected to indicate the corrosion behavior. For instance, shallow Zn(OH)2 was formed on the surface of the MOF-protected Zn anode, demonstrating the inferior corrosion behavior of MOF-based Zn than that of bare Zn (Fig. 21a).49 From the above description, it can be inferred that FTIR spectroscopy plays a crucial role in the research related to the surface/interface modification engineering of metallic anodes.
 | ||
| Fig. 21 (a) Raman and FTIR spectra with glass fiber and MOF-based membrane. Reproduced from ref. 49 with permission from John Wiley & Sons, copyright 2020. (b) Raman mapping of the Mg electrode after immersion in 0.1 M NaCl electrolytes. Reproduced from ref. 345 with permission from the Institute of Physics, copyright 2017. (c) Raman spectra ZnSO4 electrolytes (with different concentrations and electrolytes in MOF-based film channels) and (d) Raman spectroscopy of separator harvested from Zn–I2 batteries after 1 h aging for GF separator and Zn-BTC membrane. | ||
On the other hand, Raman can probe the electrolyte composition at the electrode/electrolyte interface to evaluate its influence on the corrosion behaviors.160 Raman spectra of the electrolytes suggested that the hydroxy stretching feature corresponding to free water molecules was suppressed at higher electrolyte concentration levels.65 With the increase in salt concentration, the free water was transformed into solvating water, leading to corrosion inhibition. Another work also proposed this observation that evaluated the variation in Zn–OH2, Zn−OSO32−, SO42−, and HOH–OH2 stretching vibrations by Raman analysis.49 The results indicated that the electrolyte solvation sheath was enhanced and free water molecule was minimized in the protective film (Fig. 21c). Besides, the corrosive species in the electrode/electrolyte interface can be monitored as well. For instance, with the assistance of Raman analysis, the shuttling of corrosive I3− ions toward the Zn surface was confirmed (Fig. 21d) and the protection of MOF-based film is proposed.49
4.3 Morphology characterization
The morphology evolution of metallic anode surfaces, such as passivation film formation and corrosion pits, provides direct evidence for corrosion behaviors. The validity of corrosion inhibition strategies can be visually characterized by morphological information. In this section, four typical techniques, namely atomic force microscopy (AFM), optical microscopy, scanning electron microscopy (SEM), and X-ray computed tomography (CT) will be discussed.mV higher than that of the BCC phase in the AlCoCrFeNi2.1 eutectic high-entropy alloy (EHEA) (Fig. 22c).354 A micro-galvanic corrosion cell was established between FCC and BCC phases after EHEA was immersed in an acidic electrolyte, resulting in the selective dissolution of BCC phase at the FCC/BCC interface (Fig. 22d). These in situ AFM techniques have also underlined relations between local corrosion and structural defects, suggesting that pitting corrosion occurs at grain boundaries or defects.355 For example, the micro-topography of the SAC305 alloy demonstrated that the local corrosion appears on the eutectic phase (area 1) and β-Sn (area 2) sites (Fig. 22e).356
 | ||
| Fig. 22 AFM topography (left) image and Volta potential map (right) of a bare AA2024-T3 sample (a) before and (b) after the corrosion experiment in a 0.1 M NaClO4 electrolyte solution. Reproduced from ref. 353 with permission from Wiley, copyright 2022. (c) Surface topography and Volta potential maps/line profiles of EHEA in 0.5 MH2SO4 after 5min of immersion. (d) Schematic of the corrosion mechanism of EHEA in a sulfuric acid solution. Reproduced from ref. 354 with permission from Elsevier, copyright 2022. (e) Micro-topography of SAC305 alloy after immersion in 0.1 wt% FeCl3; Reproduced from ref. 356 with permission from Elsevier, copyright 2021. | ||
Moreover, in situ AFM allows quantitative probing of the local morphology variation in real space and real time at the nanoscale level. Combining in situ AFM with other ex situ characterizations that allow the identification of the corrosion products, more accurate surface evolution over time can be obtained. AFM can monitor in situ variations in height, amplitude, and phase angle on the surface and is typically employed to assess the corrosion product formation.357 For instance, in situ AFM was applied to monitor the initial nucleation and dissolution of Zn in the 5.0 M ZnCl2 electrolyte.358 The variation in height intuitively demonstrated the plating and striping process of Zn (Fig. 23a and b).
 | ||
| Fig. 23 In situ AFM images of (a) nucleation and early growth and (b) dissolution of Zn in the 5.0 M ZnCl2 electrolyte, captured during the galvanostatic charge–discharge process. Reproduced from ref. 358 with permission from the American Chemical Society, copyright 2021. (c) In situ EC-AFM topography measurement of immersed AA2024 with the application of chronoamperometric pulses. Reproduced from ref. 359 with permission from Springer Nature, copyright 2021. (d) Comparison between the tapping-mode AFM and contact-mode AFM images of inhibitor films formed on mica. Reproduced from ref. 360 with permission from NACE International, copyright 2021. | ||
In addition, in situ electrochemical AFM (EC-AFM) has been developed to depict the morphological variation under electrochemical control directly.251 By tuning the applied potential, the corrosion behaviors during the charge/discharge process may be revealed. Using in situ EC-AFM, a relationship between the microstructure of Al alloy and localized corrosion was elaborated, giving rise to guidance for the rational design of Al alloys.361 This type of in situ EC-AFM analysis can also clarify the dynamics of the corrosion process. For example, the topography measurement of a 0.5 M NaCl electrolyte-immersed AA2024 alloy with the application of chrono-amperometric pluses was conducted.359 The roughness, skewness, and kurtosis varied regularly with the pulses (Fig. 23c), illustrating the formation process of an amorphous oxide/oxyhydroxide layer.
Apart from the contact mode, tapping mode is another AFM imaging mode that allows the detection of samples' mechanical properties by phase imaging. By phase image, the difference between the amorphous matrix and α-Al can be distinguished, which provides in-depth observation of the earliest stages of passive film formation in Al metals.362 Tapping mode AFM has multiple advantages in the imaging of soft surfaces or weak adsorption of corrosion inhibitor molecules. For example, researchers found that tapping mode topography made it easier to distinguish the inhibitor adsorption film that formed on the mica substrate (Fig. 23d).360 Tapping mode AFM also provides a phase image that can differentiate between areas with different properties regardless of their topographical nature. Sometimes the phase images can provide detailed surface structure, while topography images are quite blurry.
In summary, in situ AFM is a powerful technique to monitor the diminutive morphological changes, which provides opportunities for resolving the early stage of the corrosion process.
 | ||
| Fig. 24 (a) Operando optical microscopic images of the electrode/electrolyte interface for bare Zn and indium-protected Zn. Reproduced from ref. 120 with permission from Elsevier, copyright 2020. (b) In situ optical images of bare Zn and protected Zn surfaces at a current density of 10 mA cm−2. Reproduced from ref. 122 with permission from John Wiley & Sons, copyright 2020. (c) In situ CT of Zn anodes undergoing aging. Reproduced from ref. 27 with permission from Cell Press, copyright 2023. | ||
5. Summary and prospect
This review article introduced the corrosion behaviors of metallic anodes in aqueous electrolytes. Corrosion mechanisms, possibly generated corrosion products, and influencing factors were discussed in depth. Various strategies to inhibit metallic anode corrosion, including interface engineering, alloying engineering, and electrolyte designing, were summarized. In addition, recent developments in advanced techniques to monitor corrosion processes in the past few years were presented. Research achievements in the field of corrosion inhibition for metallic anodes can be briefly summarized as follows:(1) The corrosion of metallic anodes results in problems including low utilization of metallic anodes, electrolyte depletion, passivation issues, and H2 generation. Electrons generated by the metal corrosion are consumed by the cathodic HER rather than run through an external circuit to the cathode of batteries, leading to an irreversible energy loss. The cathodic HER causes the reduction of water and H2 evolution, which will cause an increase in electrolyte concentration and battery internal pressure. Besides, the corrosion products may cause the passivation of metallic anode. Although they might be protective for further corrosion reactions, the battery kinetics is seriously affected.
(2) Zn anodes are thermodynamically unstable in aqueous electrolytes, and are inclined to be corroded with parasitic HERs or ORRs. In highly acidic electrolytes, Zn2+ is the main corrosion product accompanied by the reduction of H+. In the pH range of 4–11, water reduction and metal hydrolysis greatly influence the local pH values and corrosion product formation. Non-protective ZnO or Zn(OH)2 fractions are formed on the metal surface in this pH range, which is a net result of the passive layer formation caused by OH− and the destabilization process promoted by H+. With the continuous increase in pH, three types of ZnO/Zn(OH)2 are generated that can protect Zn from further corrosion but limit the battery kinetics. In highly alkaline electrolytes, zincate is the leading corrosion product. Impurities in Zn anodes, dissolved oxygen, and anions in the electrolytes, such as Cl−, ClO4−, SO42−, and CO32−, can participate in the corrosion reactions and make differences in the corrosion behaviors.
(3) Al anode corrosion in acidic electrolytes is similar to that of Zn. In mildly acidic or alkaline electrolytes, the compact Al2O3 layer can protect Al from corrosion and suppress the reversible plating/stripping of Al at the same time. In highly alkaline electrolytes, Al corrosion proceeds with the continuous formation of Al(OH)3 in the metal/passivation layer interface and the dissolution of Al(OH)3 in the passivation/electrolyte interface. The passivation layer on the surface plays an important role in the corrosion process. Similarly, impurities in Al anodes, dissolved oxygen, and aggressive anions can accelerate the corrosion reaction of Al, which should be mainly considered.
(4) Corrosion mechanisms of Mg are controversial and the most convincing one demonstrates that Mg corrosion is initiated with the local breakdown of the MgO film and the NDE is ascribed to the formation of Mg(OH)2 product. In neutral and alkaline electrolytes, Mg(OH)2 is the only corrosion product, which would serve as cathodic sites to accelerate the HER. However, when the pH value exceeds 10.5, Mg(OH)2 formed on the surface will passivate Mg from dissolution. It is worth noting that dissolved oxygen in electrolytes does not influence Mg corrosion much due to the high HER rates. Most of the metallic impurities are more noble than Mg and will accelerate Mg dissolution.
(5) Artificial interfaces including carbonaceous layers, metallic compounds, and organic polymers can protect metallic anodes from direct exposure to aqueous electrolytes, thereby reducing corrosion reactions. Apart from acting as physical barriers, some interfaces such as indium-based compounds also serve as chemical corrosion inhibitors, which can increase the HER overpotential. The abundant surface chemistry of carbon layers or organic polymers is also important in preventing aggressive anions from getting close to the metal surface. Though effective in corrosion inhibition, the coating layer will cause sluggish ion transport between the metal anode and electrolytes, resulting in inferior battery kinetics and large polarization. The corrosion inhibition efficiency and battery performance are heavily dependent on the microstructural characteristics of the coating layer, which undoubtedly increases the difficulty and cost of preparation. Most of the sophisticated synthetic techniques published in the literature are not suited for large-scale preparation, which restricts their practical applicability.
(6) Alloying engineering is another effective strategy to improve the corrosion resistance of metallic anodes. On the one hand, alloying elements or as-formed intermetallic phases with higher HER overpotentials are beneficial to reducing the cathodic HER. On the other hand, some alloying elements help to form protective corrosion layers (such as La(OH)3 and Al2O3) during the corrosion process. However, the formed secondary phase will accelerate the corrosion reaction if it presents a more noble electrochemical activity than the protected metal. Almost all Mg intermetallic phases can enhance the dissolution of Mg due to the formation of galvanic cells. Although the alloy anode is easy to prepare on a large scale, alloying engineering requires strict control of the alloy ratio and phase distribution to ensure the anode's electrochemical performance. There are still issues with alloy element compatibility and precise phase composition management during large-scale preparation.
(7) Electrolyte engineering significantly impacts the electrochemistry at the metallic anode/electrolyte interface. Metal corrosion behavior in aqueous electrolytes highly depends on the pH value, which should be considered when designing electrolytes. Reducing the contents of free water molecules through high-concentration electrolytes or gel electrolytes greatly diminishes the interaction between metal and water, thus protecting metallic anodes from corrosion. The corrosion inhibitor strategy offers special benefits such as easy large-scale preparation, cheapness, and simple operation. All that is required for this method is the addition of a very small quantity of an appropriate corrosion inhibitor to the electrolyte. Corrosion inhibitors suppress metal corrosion in various ways, including protective layer formation, surface chemistry modification, and adjustment of electrolyte pH. Although the addition of corrosion inhibitors may deteriorate the battery kinetics, it is the most facile and cost-effective strategy to suppress metal corrosion.
(8) The change of electrochemical signals or surface morphology can monitor corrosion behaviors. The polarization curve method, EIS, and EN technique are frequently employed to understand corrosion mechanisms and get information on corrosion tendency and rate. Some advanced techniques such as in situ SECM, in situ AFM, and operando optical microscopy allow for observing the spatial distribution of corrosion reaction in real time by monitoring surface currents and morphology evolutions. Besides, conventional characterization methods such as Raman, SEM, and FTIR can help distinguish corrosion products.
Besides these achievements discussed in this review, many challenges remain (Fig. 25). The challenges that should be addressed and potential further research directions are summarized as follows:
 | ||
| Fig. 25 Challenges and potential research directions toward the corrosion issue of metallic anodes. | ||
1. Fundamental corrosion mechanisms of metallic anodes in aqueous electrolytes should be further elucidated. Behaviors at the initial stage of the corrosion reaction are still unclear, and the formation of corrosion products essentially speculates on the existing mechanism. Besides, the corrosion behavior monitoring during the charge/discharge process of the battery deserves more effort, which should consider the reversible stripping/plating of the metal anode. Moreover, the corrosion mechanism of Mg is still controversial and advanced in situ techniques should be employed to classify the Mg corrosion behaviors.
2. Artificial interface engineering to protect metallic anodes from corrosion suffers from the main penalty of deteriorating battery kinetics. Although water molecules and aggressive anions can be kept out of the anode surface efficiently, sluggish metal ion migration across the coating layer results in large polarization and inferior rate capability. Besides, the structural integrity of the protective interfaces in large-scale production is still challenging, which may lead to serious pitting corrosion. The in situ formation of protective film or artificial solid electrolyte interface during the charge/discharge process may be a promising direction, which reduces the assembly cost and maintains the durability of the protective layer. High chemical/electrochemical stability, high integrity without fractures, high-speed electrolyte ion conductivity, a strong capacity to bond with the metal anode, and lightweight characteristics are all necessary for the perfect interface protection layer.
3. In alloying engineering, there is no standard criterion for choosing alloying elements and their corresponding amounts. A comprehensive examination of relationships between alloying elements and their protection efficiency is urgently needed to tackle this challenge. Meanwhile, the influences of alloying elements on the grain size, intermetallic phase distribution, and exposed crystalline facet should be considered in further research.
4. As to electrolyte engineering, developing new electrolyte systems with mild acidity/alkalinity and less aggressive anions is a potential direction to gain excellent corrosion inhibition. Despite various additives' great corrosion inhibition efficiency, the surface chemistry variation caused by organic additives lacks in-depth understanding. Further research on organic additives' adsorption/desorption kinetics and corresponding surface properties evolutions is urgently needed. A rational combination of different additives may be a potential research direction to improve the corrosion inhibition efficiency. Besides, hydrogel electrolytes with less free water molecules have tremendous application potential in suppressing metallic anode corrosion. Moreover, organic/aqueous hybrid electrolyte design is another attractive research direction in this field, as the reduced protons in the hybrid electrolytes may hinder the corrosion of metallic anodes.
5. Apart from the electrolyte pH values, local pH values play a crucial role in affecting the corrosion behaviors, which are seldom examined. Advanced microelectrochemical techniques such as scanning vibrating electrode techniques deserve more investigations, which can give rise to new mechanistic insights into the highly localized corrosion processes. Employing more in situ techniques such as AFM, SECM, and EN to study the metallic anode's corrosion should be a potential research trend.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review. All the figures and tables were reproduced from references with permissions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22109178, 52277229, 52202337, 22371153), Natural Science Foundation of Shandong Province (ZR2023MB051, ZR2021QB085), National Key Research and Development of China (2022YFA1503402), the Fundamental Research Funds for the Central Universities (2462023QNXZ015, 24CX07003A), and the Young Taishan Scholars Program of Shandong Province (tsqn202211082, tsqn202312224).References
- X. Cai, X. Wang, Z. Bie, Z. Jiao, Y. Li, W. Yan, H. J. Fan and W. Song, Adv. Mater., 2024, 36, 2306734 CrossRef CAS PubMed.
- Z. Chen, Y. Wang, Q. Wu, C. Wang, Q. He, T. Hu, X. Han, J. Chen, Y. Zhang, J. Chen, L. Yang, X. Wang, Y. Ma and J. Zhao, Adv. Mater., 2024, 36, 2411004 CrossRef CAS PubMed.
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed.
- W. Zhou, S. Ding, D. Zhao and D. Chao, Joule, 2023, 7, 1104–1107 CrossRef CAS.
- Y. Zhao, D. Wang, X. Li, Q. Yang, Y. Guo, F. Mo, Q. Li, C. Peng, H. Li and C. Zhi, Adv. Mater., 2020, 32, 2003070 CrossRef CAS PubMed.
- B. Ge, L. Hu, X. Yu, L. Wang, C. Fernandez, N. Yang, Q. Liang and Q.-H. Yang, Adv. Mater., 2024, 36, 2400937 CrossRef CAS PubMed.
- Z. Zhao, X. Fan, J. Ding, W. Hu, C. Zhong and J. Lu, ACS Energy Lett., 2019, 4, 2259–2270 CrossRef CAS.
- Z. Cai, Y. Ou, J. Wang, R. Xiao, L. Fu, Z. Yuan, R. Zhan and Y. Sun, Energy Storage Mater., 2020, 27, 205–211 CrossRef.
- Y. Liu, Q. Sun, W. Li, K. R. Adair, J. Li and X. Sun, Green Energy Environ., 2017, 2, 246–277 CrossRef.
- A. R. Mainar, O. Leonet, M. Bengoechea, I. Boyano, I. de Meatza, A. Kvasha, A. Guerfi and J. Alberto Blazquez, Int. J. Energy Res., 2016, 40, 1032–1049 CrossRef CAS.
- G. Chen, Y. Kang, H. Yang, M. Zhang, J. Yang, Z. Lv, Q. Wu, P. Lin, Y. Yang and J. Zhao, Adv. Funct. Mater., 2023, 33, 2300656 CrossRef CAS.
- Y.-H. Lee, Y. Jeoun, J. H. Kim, J. Shim, K.-S. Ahn, S.-H. Yu and Y.-E. Sung, Adv. Funct. Mater., 2024, 34, 2310884 CrossRef CAS.
- A. Kolesnikov, M. Kolek, J. F. Dohmann, F. Horsthemke, M. Börner, P. Bieker, M. Winter and M. C. Stan, Adv. Energy Mater., 2020, 10, 2000017 CrossRef CAS.
- W. Xiong, D. Yang, T. K. A. Hoang, M. Ahmed, J. Zhi, X. Qiu and P. Chen, Energy Storage Mater., 2018, 15, 131–138 CrossRef.
- C. Y. Chen, K. Matsumoto, K. Kubota, R. Hagiwara and Q. Xu, Adv. Energy Mater., 2019, 9, 1900196 CrossRef.
- C. Cai, R. Song, L. Wang and J. Li, Appl. Surf. Sci., 2018, 462, 243–254 CrossRef CAS.
- K. Yasakau, Corros. Mater. Degrad., 2020, 1, 345–372 CrossRef.
- W. Yuan, X. Nie, G. Ma, M. Liu, Y. Wang, S. Shen and N. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218386 CrossRef CAS PubMed.
- Y. Li, X. Fan, X. Liu, S. Qu, J. Liu, J. Ding, X. Han, Y. Deng, W. Hu and C. Zhong, J. Mater. Chem. A, 2019, 7, 25449–25457 RSC.
- P. Katsoufis, V. Mylona, C. Politis, G. Avgouropoulos and P. Lianos, J. Power Sources, 2020, 450, 227624 CrossRef CAS.
- C. Lv, Y. Zhang, J. Ma, Y. Zhu, D. Huang, Y. Li, H. Wang and Y. Tang, J. Mater. Chem. A, 2022, 10, 9506–9514 RSC.
- C. Lv, Y. Zhu, Y. Li, Y. Zhang, J. Kuang, Y. Tang, H. Li and H. Wang, Energy Storage Mater., 2023, 59, 102756 CrossRef.
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- J. Sun, F. Kang, D. Yan, T. Ding, Y. Wang, X. Zhou and Q. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202406511 CrossRef CAS PubMed.
- G. Wang, Q.-K. Zhang, X.-Q. Zhang, J. Lu, C. Pei, D. Min, J.-Q. Huang and H. S. Park, Adv. Energy Mater., 2025, 15, 2304557 CrossRef CAS.
- Z. Cai, J. Wang and Y. Sun, eScience, 2023, 3, 100093 CrossRef.
- S. D. Pu, B. Hu, Z. Li, Y. Yuan, C. Gong, Z. Ning, C. Chau, S. Yang, S. Zhang, L. Pi, Y. T. Tang, J. Yue, T. J. Marrow, X. Gao, P. G. Bruce and A. W. Robertson, Joule, 2023, 7, 366–379 CrossRef CAS.
- C. Liu, Z. Luo, W. Deng, W. Wei, L. Chen, A. Pan, J. Ma, C. Wang, L. Zhu, L. Xie, X.-Y. Cao, J. Hu, G. Zou, H. Hou and X. Ji, ACS Energy Lett., 2021, 6, 675–683 CrossRef CAS.
- Y. Wang, H. Y. H. Kwok, W. Pan, H. Zhang, X. Lu and D. Y. C. Leung, Appl. Energy, 2019, 251, 113342 CrossRef CAS.
- A. P. Sinha, T. S. Thomas and D. Mandal, Energy Storage Mater., 2023, 63, 102988 CrossRef.
- J. Gao, X. Li, Q. Liu, H. Fan, S. Gao, Y. Song and E. Wang, Chem. Eng. J, 2023, 464, 142655 CrossRef CAS.
- J. Wang, L. Cui, S. Li, T. Pu, X. Fang, S. Kang and X. Zhang, New J. Chem., 2020, 44, 1624–1631 RSC.
- H. Li, L. Ma, C. Han, Z. Wang, Z. Liu, Z. Tang and C. Zhi, Nano Energy, 2019, 62, 550–587 CrossRef CAS.
- X. Li, Y. Tang, J. Zhu, H. Lv, L. Zhao, W. Wang, C. Zhi and H. Li, Small, 2020, 16, 2001935 CrossRef CAS PubMed.
- Z. Liu, Y. Huang, Y. Huang, Q. Yang, X. Li, Z. Huang and C. Zhi, Chem. Soc. Rev., 2020, 49, 180–232 RSC.
- Y. Tang, X. Li, H. Lv, D. Xie, W. Wang, C. Zhi and H. Li, Adv. Energy Mater., 2020, 10, 2000892 CrossRef CAS.
- X. Li, Y. Tang, H. Lv, W. Wang, F. Mo, G. Liang, C. Zhi and H. Li, Nanoscale, 2019, 11, 17992–18008 RSC.
- C. Han, W. Li, H. K. Liu, S. Dou and J. Wang, Nano Energy, 2020, 74, 104880 CrossRef CAS.
- K. Wippermann, J. W. Schultze, R. Kessel and J. Penninger, Corros. Sci., 1991, 32, 205–230 CrossRef CAS.
- K. G. Boto and L. F. G. Williams, J. Electroanal. Chem. Interfacial Electrochem., 1977, 77, 1–20 CrossRef CAS.
- S. Thomas, N. Birbilis, M. S. Venkatraman and I. S. Cole, Corrosion, 2012, 68, 9 CrossRef.
- Z. Zembura and L. Burzynska, Corros. Sci., 1977, 17, 871–878 CrossRef CAS.
- M. Gmytryk and J. Sedzimir, Corros. Sci., 1967, 7, 683–695 CrossRef.
- M. Mouanga, P. Berçot and J. Y. Rauch, Corros. Sci., 2010, 52, 3984–3992 CrossRef CAS.
- C. Qiao, L. Shen, L. Hao, X. Mu, J. Dong, W. Ke, J. Liu and B. Liu, J. Mater. Sci. Technol., 2019, 35, 2345–2356 CrossRef CAS.
- L. M. Baugh, Electrochim. Acta, 1979, 24, 669–677 CrossRef CAS.
- M. Mokaddem, P. Volovitch and K. Ogle, Electrochim. Acta, 2010, 55, 7867–7875 CrossRef CAS.
- L. M. Baugh and A. Higginson, Electrochim. Acta, 1985, 30, 1163–1172 CrossRef CAS.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, P. He and H. Zhou, Adv. Mater., 2020, 32, 2004240 CrossRef CAS PubMed.
- J.-H. Lee, Y. Byun, G. H. Jeong, C. Choi, J. Kwen, R. Kim, I. H. Kim, S. O. Kim and H.-T. Kim, Adv. Mater., 2019, 31, 1904524 CrossRef CAS PubMed.
- B. Evanko, S. J. Yoo, J. Lipton, S.-E. Chun, M. Moskovits, X. Ji, S. W. Boettcher and G. D. Stucky, Energy Environ. Sci., 2018, 11, 2865–2875 RSC.
- L. Gao, Z. Li, Y. Zou, S. Yin, P. Peng, Y. Shao and X. Liang, iScience, 2020, 23, 101348 CrossRef CAS PubMed.
- S. M. A. E. Haleem, Br. Corros. J., 1976, 11, 215–218 CrossRef.
- X. G. Zhang, Corrosion and Electrochemistry of Zinc, Springer Science & Business Media, New York, 2013 Search PubMed.
- A. D. Keitelman, S. M. Gravano and J. R. Galvele, Corros. Sci., 1984, 24, 535–545 CrossRef CAS.
- S. Wu, Q. Zhang, D. Sun, J. Luan, H. Shi, S. Hu, Y. Tang and H. Wang, Chem. Eng. J, 2020, 383, 123162 CrossRef CAS.
- C. Wu, S. Gu, Q. Zhang, Y. Bai, M. Li, Y. Yuan, H. Wang, X. Liu, Y. Yuan, N. Zhu, F. Wu, H. Li, L. Gu and J. Lu, Nat. Commun., 2019, 10, 73 CrossRef CAS PubMed.
- Y. Wang, T. Wu, Y. Lu, W. Zhang and Z. Li, Angew. Chem., Int. Ed., 2024, e202416032 Search PubMed n/a.
- X. Yang, H. Gu, L. Chai, S. Chen, W. Zhang, H. Y. Yang and Z. Li, Nano Lett., 2024, 24, 8542–8549 CrossRef CAS PubMed.
- S.-M. Moon and S.-I. Pyun, Electrochim. Acta, 1999, 44, 2445–2454 CrossRef CAS.
- I. Boukerche, S. Djerad, L. Benmansour, L. Tifouti and K. Saleh, Corros. Sci., 2014, 78, 343–352 CrossRef CAS.
- R. Ambat and E. S. Dwarakadasa, J. Appl. Electrochem., 1994, 24, 911–916 CrossRef CAS.
- J. Flis and L. Kowalczyk, J. Appl. Electrochem., 1995, 25, 501–507 CrossRef CAS.
- W.-J. Lee and S.-I. Pyun, Electrochim. Acta, 2000, 45, 1901–1910 CrossRef CAS.
- S. Li and B. C. Church, Appl. Surf. Sci., 2018, 440, 861–872 CrossRef CAS.
- D. N. D. Singh, J. Electrochem. Soc., 1982, 129, 1869–1874 CrossRef CAS.
- G. O. H. Whillock and S. E. Worthington, in Shreir's Corrosion, ed. B. Cottis, M. Graham, R. Lindsay, S. Lyon, T. Richardson, D. Scantlebury and H. Stott, Elsevier, Oxford, 2010, vol. 2, pp. 1250–1269 Search PubMed.
- R. G. Wymer and R. E. Blanco, Ind. Eng. Chem., 1957, 49, 59–61 CrossRef CAS.
- G. T. Burstein and R. M. Organ, Corros. Sci., 2005, 47, 2932–2955 CrossRef CAS.
- K. F. Khaled, Corros. Sci., 2010, 52, 2905–2916 CrossRef CAS.
- B. C. Bunker, G. C. Nelson, K. R. Zavadil, J. C. Barbour and J. P. Sullivan, J. Phys. Chem. B, 2002, 106, 4705–4713 CrossRef CAS.
- R. S. Alwitt, J. Electrochem. Soc., 1974, 121, 1322 CrossRef CAS.
- S. S. Razavi-Tousi and J. A. Szpunar, Electrochim. Acta, 2014, 127, 95–105 CrossRef CAS.
- S.-I. Pyun and S.-M. Moon, J. Solid State Electrochem., 2000, 4, 267–272 CrossRef CAS.
- M. Jingling, W. Jiuba, Z. Hongxi and L. Quanan, J. Power Sources, 2015, 293, 592–598 CrossRef CAS.
- Y. Liu, Q. Pan, H. Li, Z. Huang, J. Ye and M. Li, J. Alloys Compd., 2019, 792, 32–45 CrossRef CAS.
- P. W. Beetz, Philos. Mag., 1866, 32, 269–278 Search PubMed.
- M. Curioni, F. Scenini, T. Monetta and F. Bellucci, Electrochim. Acta, 2015, 166, 372–384 CrossRef CAS.
- P. Gore, S. Fajardo, N. Birbilis, G. S. Frankel and V. S. Raja, Electrochim. Acta, 2019, 293, 199–210 CrossRef CAS.
- S. Thomas, N. V. Medhekar, G. S. Frankel and N. Birbilis, Curr. Opin. Solid State Mater. Sci., 2015, 19, 85–94 CrossRef CAS.
- G. G. Perrault, J. Electroanal. Chem. Interfacial Electrochem., 1970, 27, 47–58 CrossRef CAS.
- E. Gulbrandsen, J. Taftø and A. Olsen, Corros. Sci., 1993, 34, 1423–1440 CrossRef CAS.
- G. L. Song and A. Atrens, Adv. Eng. Mater., 1999, 1, 11–33 CrossRef CAS.
- R. L. Petty, A. W. Davidson and J. Kleinberg, J. Am. Chem. Soc., 1954, 76, 363–366 CrossRef CAS.
- P. F. King, J. Electrochem. Soc., 1966, 113, 536 CrossRef CAS.
- M. E. Straumanis and Y. N. Wang, J. Electrochem. Soc., 1955, 102, 304 CrossRef CAS.
- G. R. Hoey and M. Cohen, J. Electrochem. Soc., 1958, 105, 245 CrossRef CAS.
- M. Taheri, J. R. Kish, N. Birbilis, M. Danaie, E. A. McNally and J. R. McDermid, Electrochim. Acta, 2014, 116, 396–403 CrossRef CAS.
- M. Danaie, R. M. Asmussen, P. Jakupi, D. W. Shoesmith and G. A. Botton, Corros. Sci., 2013, 77, 151–163 CrossRef CAS.
- N. T. Kirkland, G. Williams and N. Birbilis, Corros. Sci., 2012, 65, 5–9 CrossRef CAS.
- G. S. Frankel, A. Samaniego and N. Birbilis, Corros. Sci., 2013, 70, 104–111 CrossRef CAS.
- L. Rossrucker, K. J. J. Mayrhofer, G. S. Frankel and N. Birbilis, J. Electrochem. Soc., 2014, 161, C115–C119 CrossRef CAS.
- M. Curioni, Electrochim. Acta, 2014, 120, 284–292 CrossRef CAS.
- N. Birbilis, A. D. King, S. Thomas, G. S. Frankel and J. R. Scully, Electrochim. Acta, 2014, 132, 277–283 CrossRef CAS.
- K. A. Unocic, H. H. Elsentriecy, M. P. Brady, H. M. Meyer, G. L. Song, M. Fayek, R. A. Meisner and B. Davis, J. Electrochem. Soc., 2014, 161, C302–C311 CrossRef CAS.
- J. Liang, P. B. Srinivasan, C. Blawert and W. Dietzel, Corros. Sci., 2010, 52, 540–547 CrossRef CAS.
- H. Wu, Z. Shi, X. Zhang, A. M. Qasim, S. Xiao, F. Zhang, Z. Wu, G. Wu, K. Ding and P. K. Chu, Appl. Surf. Sci., 2019, 478, 150–161 CrossRef CAS.
- M. Deng, D. Höche, S. V. Lamaka, L. Wang and M. L. Zheludkevich, Corros. Sci., 2019, 153, 225–235 CrossRef CAS.
- N. Wang, R. Wang, Y. Feng, W. Xiong, J. Zhang and M. Deng, Corros. Sci., 2016, 112, 13–24 CrossRef CAS.
- D. Lei, D. C. Lee, A. Magasinski, E. Zhao, D. Steingart and G. Yushin, ACS Appl. Mater. Inter., 2016, 8, 2088–2096 CrossRef CAS PubMed.
- H. Weinrich, J. Come, H. Tempel, H. Kungl, R.-A. Eichel and N. Balke, Nano Energy, 2017, 41, 706–716 CrossRef CAS.
- C. Bai, H. Jin, Z. Gong, X. Liu and Z. Yuan, Energy Storage Mater., 2020, 28, 247–254 CrossRef.
- X. Wu, A. Markir, Y. Xu, C. Zhang, D. P. Leonard, W. Shin and X. Ji, Adv. Funct. Mater., 2019, 29, 1900911 CrossRef.
- X. Wu, A. Markir, Y. Xu, E. C. Hu, K. T. Dai, C. Zhang, W. Shin, D. P. Leonard, K. I. Kim and X. Ji, Adv. Energy Mater., 2019, 9, 1902422 CrossRef CAS.
- D. Enning and J. Garrelfs, Appl. Environ. Microbiol., 2014, 80, 1226 CrossRef PubMed.
- E. E. Oguzie, B. N. Okolue, E. E. Ebenso, G. N. Onuoha and A. I. Onuchukwu, Mater. Chem. Phys., 2004, 87, 394–401 CrossRef CAS.
- R. C. Zeng, Y. Hu, S. K. Guan, H. Z. Cui and E. H. Han, Corros. Sci., 2014, 86, 171–182 CrossRef CAS.
- J. K. Lin, C. L. Hsia and J. Y. Uan, Scr. Mater., 2007, 56, 927–930 CrossRef CAS.
- J. Xu, J. Hu and S. Hu, Chem. Res. Chin. Univ., 2019, 35, 641–646 CrossRef CAS.
- P. Pei, K. Wang and Z. Ma, Appl. Energy, 2014, 128, 315–324 CrossRef CAS.
- X. Yin, Int. J. Electrochem. Sci., 2017, 12, 4150–4163 CrossRef CAS.
- R. L. Liu, Z. R. Zeng, J. R. Scully, G. Williams and N. Birbilis, Corros. Sci., 2018, 140, 18–29 CrossRef CAS.
- H. Jia, Z. Wang, B. Tawiah, Y. Wang, C.-Y. Chan, B. Fei and F. Pan, Nano Energy, 2020, 70, 104523 CrossRef CAS.
- M. Zhu, Q. Ran, H. Huang, Y. Xie, M. Zhong, G. Lu, F.-Q. Bai, X.-Y. Lang, X. Jia and D. Chao, Nano-Micro Lett., 2022, 14, 219 CrossRef CAS PubMed.
- K. Zhao, C. Wang, Y. Yu, M. Yan, Q. Wei, P. He, Y. Dong, Z. Zhang, X. Wang and L. Mai, Adv. Mater. Interfaces, 2018, 5, 1800848 CrossRef.
- K. Wongrujipairoj, L. Poolnapol, A. Arpornwichanop, S. Suren and S. Kheawhom, Phys. Status Solidi B, 2017, 254, 1600442 CrossRef.
- H. Wei, X. Hu, X. Zhang, Z. Yu, T. Zhou, Y. Liu, Y. Liu, Y. Wang, J. Xie, L. Sun, M. Liang and P. Jiang, Energy Technol., 2019, 7, 1800912 CrossRef.
- Y. N. Jo, S. H. Kang, K. Prasanna, S. W. Eom and C. W. Lee, Appl. Surf. Sci., 2017, 422, 406–412 CrossRef CAS.
- P. Liang, J. Yi, X. Liu, K. Wu, Z. Wang, J. Cui, Y. Liu, Y. Wang, Y. Xia and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908528 CrossRef CAS.
- K. Hu, X. Guan, R. Lv, G. Li, Z. Hu, L. Ren, A. Wang, X. Liu and J. Luo, Chem. Eng. J, 2020, 396, 125363 CrossRef CAS.
- C. Deng, X. Xie, J. Han, Y. Tang, J. Gao, C. Liu, X. Shi, J. Zhou and S. Liang, Adv. Funct. Mater., 2020, 30, 2000599 CrossRef CAS.
- R. Zhao, Y. Yang, G. Liu, R. Zhu, J. Huang, Z. Chen, Z. Gao, X. Chen and L. Qie, Adv. Funct. Mater., 2021, 31, 2001867 CrossRef CAS.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- H. He, H. Tong, X. Song, X. Song and J. Liu, J. Mater. Chem. A, 2020, 8, 7836–7846 RSC.
- M. Schmid, U. Schadeck and M. Willert-Porada, Surf. Coat. Technol., 2017, 310, 51–58 CrossRef CAS.
- L. Zhu, H. Zhang, W. Li and H. Liu, J. Phys. Chem. Solids, 2009, 70, 45–54 CrossRef CAS.
- J. H. Park, C. Choi, J. B. Park, S. Yu and D.-W. Kim, Adv. Energy Mater., 2024, 14, 2302493 CrossRef CAS.
- X. Zhu, W. Zhang, Z. Peng, L. Pan, B. Li, Z. Zhang, J. Zhu, W. Meng, L. Dai, L. Wang and Z. He, Chem. Eng. J, 2024, 499, 156521 CrossRef CAS.
- K. Xia, L. Li, Y. Qiu, J. Weng, S. Shen, M. Chen, Y. Zhuang, Y. Wen, C. Yang, Z. Liu, M. Wu and Z. Zou, J. Mater. Chem. A, 2024, 12, 24175–24187 RSC.
- Q. Han, L. Cai, P. Huang, S. Liu, C. He, Z. Xu, H. Ying and W.-Q. Han, ACS Appl. Mater. Inter., 2023, 15, 48316–48325 CrossRef CAS PubMed.
- V.-C. Ho, H. Yen Nguyen Thi, T. Huong Pham, H.-G. Jung, J. Ho Kim, J. F. Kim and J. Mun, Chem. Eng. J., 2024, 496, 153845 CrossRef CAS.
- X. Sun, X. Lv, M. Zhang, K. Shi, Z. Li, X. Pan, T. Lian, R. Chen, F. Wu and L. Li, ACS Nano, 2024, 18, 8452–8462 CrossRef CAS PubMed.
- H. Yang, J. Wang, P. Zhang, X. Cheng, Q. Guan, J. Dong, B. Chen, L. Jia, J. Zhang, Y. Zhang, Y. Liu and H. Lin, J. Energy Chem., 2025, 100, 693–701 CrossRef CAS.
- M. Wei, K. Wang, Y. Zuo, L. Zhong, A. Züttel, Z. Chen, P. Zhang, H. Wang, S. Zhao and P. Pei, Adv. Funct. Mater., 2023, 33, 2302243 CrossRef CAS.
- Z. Li, L. Wu, S. Dong, T. Xu, S. Li, Y. An, J. Jiang and X. Zhang, Adv. Funct. Mater., 2021, 31, 2006495 CrossRef CAS.
- Q. Hu, J. Hou, Y. Liu, L. Li, Q. Ran, J. Mao, X. Liu, J. Zhao and H. Pang, Adv. Mater., 2023, 35, 2303336 CrossRef CAS PubMed.
- Y. Wu, Y. Zhang, Y. Ma, J. D. Howe, H. Yang, P. Chen, S. Aluri and N. Liu, Adv. Energy Mater., 2018, 8, 1802470 CrossRef.
- Q. Yang, Y. Guo, B. Yan, C. Wang, Z. Liu, Z. Huang, Y. Wang, Y. Li, H. Li, L. Song, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 2001755 CrossRef CAS PubMed.
- S. Kim, G. Hee Ryu and G. H. An, Appl. Surf. Sci., 2023, 635, 157634 CrossRef CAS.
- X. Xu, S. Li, H. Yan, J. Du, S. Yang and B. Li, Adv. Funct. Mater., 2024, 34, 2308661 CrossRef CAS.
- M. Liu, W. Yuan, G. Ma, K. Qiu, X. Nie, Y. Liu, S. Shen and N. Zhang, Angew. Chem., 2023, 135, e202304444 CrossRef.
- J. J. Conde, F. A. Paloma and A. M. Chaparro, Appl. Mater. Today, 2018, 13, 100–106 CrossRef.
- D. Han, S. Wu, S. Zhang, Y. Deng, C. Cui, L. Zhang, Y. Long, H. Li, Y. Tao, Z. Weng, Q.-H. Yang and F. Kang, Small, 2020, 16, 2001736 CrossRef CAS PubMed.
- L. Kang, M. Cui, F. Jiang, Y. Gao, H. Luo, J. Liu, W. Liang and C. Zhi, Adv. Energy Mater., 2018, 8, 1801090 CrossRef.
- H. J. Liu, C. Y. Yang, M. C. Han, C. Y. Yu, X. Li, Z. Z. Yu and J. Qu, Angew. Chem., 2023, 135, e202217458 CrossRef.
- S. Zhou, Y. Wang, H. Lu, Y. Zhang, C. Fu, I. Usman, Z. Liu, M. Feng, G. Fang, X. Cao, S. Liang and A. Pan, Adv. Funct. Mater., 2021, 31, 2104361 CrossRef CAS.
- S. Di, X. Nie, G. Ma, W. Yuan, Y. Wang, Y. Liu, S. Shen and N. Zhang, Energy Storage Mater., 2021, 43, 375–382 CrossRef.
- R. Wang, S. Xin, D. Chao, Z. Liu, J. Wan, P. Xiong, Q. Luo, K. Hua, J. Hao and C. Zhang, Adv. Funct. Mater., 2022, 32, 2207751 CrossRef CAS.
- H. Zhang, Y. Wu, J. Yu, T. Jiang and M. Wu, Adv. Funct. Mater., 2023, 34, 2301912 CrossRef.
- F. Ling, L. Wang, F. Liu, M. Ma, S. Zhang, X. Rui, Y. Shao, Y. Yang, S. He, H. Pan, X. Wu, Y. Yao and Y. Yu, Adv. Mater., 2023, 35, 2208764 CrossRef CAS PubMed.
- X. Zhou, B. Wen, Y. Cai, X. Chen, L. Li, Q. Zhao, S. Chou and F. Li, Angew. Chem., Int. Ed., 2024, 63, e202402342 CrossRef CAS PubMed.
- Y. Fang, P. Lei, H. Xing, K. Xu, M. Zhu, Z. Fan, K. Qi, Q. Wu and Y. Zhu, Energy Storage Mater., 2022, 53, 13–21 CrossRef.
- C. Deng, X. Xie, J. Han, Y. Tang, J. Gao, C. Liu, X. Shi, J. Zhou and S. Liang, Adv. Funct. Mater., 2020, 30, 2000599 CrossRef CAS.
- K. Wang, A. Paxson, T. I. Valdez, A. Erlat, P.-C. Lee, S. Yun, P. Khajanji, Z. Zhang, A. C. Kummel and P. Bandaru, ACS Appl. Mater. Inter., 2024, 16, 35043–35052 CrossRef CAS PubMed.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021 CrossRef CAS PubMed.
- R. Guo, X. Liu, F. Xia, Y. Jiang, H. Zhang, M. Huang, C. Niu, J. Wu, Y. Zhao, X. Wang, C. Han and L. Mai, Adv. Mater., 2022, 34, e2202188 CrossRef PubMed.
- P. Xiao, H. Li, J. Fu, C. Zeng, Y. Zhao, T. Zhai and H. Li, Energy Environ. Sci., 2022, 15, 1638–1646 RSC.
- K. Ouyang, D. Ma, N. Zhao, Y. Wang, M. Yang, H. Mi, L. Sun, C. He and P. Zhang, Adv. Funct. Mater., 2021, 32, e202416032 Search PubMed.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- M. Fu, H. Yu, S. Huang, Q. Li, B. Qu, L. Zhou, G.-C. Kuang, Y. Chen and L. Chen, Nano Lett., 2023, 23, 3573–3581 CrossRef CAS PubMed.
- D. Xie, Z. W. Wang, Z. Y. Gu, W. Y. Diao, F. Y. Tao, C. Liu, H. Z. Sun, X. L. Wu, J. W. Wang and J. P. Zhang, Adv. Funct. Mater., 2022, 32, 2204066 CrossRef CAS.
- M. Liu, W. Yuan, G. Ma, K. Qiu, X. Nie, Y. Liu, S. Shen and N. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202304444 CrossRef CAS PubMed.
- B. Lv, Q. Zong, Y. Yu, C. Liu, A. Pan, J. Wang, D. Tao, J. Zhang, Q. Zhang and G. Cao, Adv. Funct. Mater., 2024, 34, 2316535 CrossRef CAS.
- J. Yu, C. Chen, F. Shi, R. Li, F. Chen, J. Tang, K. C. Chan and Z.-L. Xu, Energy Storage Mater., 2023, 63, 102966 CrossRef.
- D. Xie, Y. Sang, D. H. Wang, W. Y. Diao, F. Y. Tao, C. Liu, J. W. Wang, H. Z. Sun, J. P. Zhang and X. L. Wu, Angew. Chem., Int. Ed., 2023, 62, e202216934 CrossRef CAS PubMed.
- M. Liu, W. Yuan, X. Qu, X. Ru, X. Li, T. Wang, X. Wang, Y. Wang, Y. Liu and N. Zhang, Energy Environ. Sci., 2024, 17(24), 9611–9622 RSC.
- A. Mitha, A. Z. Yazdi, M. Ahmed and P. Chen, ChemElectroChem, 2018, 5, 2409–2418 CrossRef CAS.
- B.-S. Lee, S. Cui, X. Xing, H. Liu, X. Yue, V. Petrova, H.-D. Lim, R. Chen and P. Liu, ACS Appl. Mater. Inter., 2018, 10, 38928–38935 CrossRef CAS PubMed.
- Y. Gao, Z. Yan, J. L. Gray, X. He, D. Wang, T. Chen, Q. Huang, Y. C. Li, H. Wang, S. H. Kim, T. E. Mallouk and D. Wang, Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed.
- Y. Huang, Z. Li, Z. Pei, Z. Liu, H. Li, M. Zhu, J. Fan, Q. Dai, M. Zhang and L. Dai, Adv. Energy Mater., 2018, 8, 1802288 CrossRef.
- Y. Da, F. Zhao, J. Shi and Z. Zhang, J. Electron. Mater., 2020, 49, 2479–2490 CrossRef CAS.
- J. Ren, J. Ma, J. Zhang, C. Fu and B. Sun, J. Alloys Compd., 2019, 808, 151708 CrossRef CAS.
- B. Li, K. Yang, J. Ma, P. Shi, L. Chen, C. Chen, X. Hong, X. Cheng, M.-C. Tang, Y.-B. He and F. Kang, Angew. Chem., Int. Ed., 2022, 134, e202212587 CrossRef.
- G. Lin, Y. Zhou, S. Zanna, A. Seyeux, P. Marcus and J. Swiatowska, J. Magnes. Alloy, 2024, 12, 3646–3660 CrossRef CAS.
- H. Tian, G. Feng, Q. Wang, Z. Li, W. Zhang, M. Lucero, Z. Feng, Z.-L. Wang, Y. Zhang, C. Zhen, M. Gu, X. Shan and Y. Yang, Nat. Commun., 2022, 13, 7922 CrossRef CAS PubMed.
- M. Fayette, H. J. Chang, X. Li and D. Reed, ACS Energy Lett., 2022, 7, 1888–1895 CrossRef CAS.
- Y. Zhao, S. Guo, M. Chen, B. Lu, X. Zhang, S. Liang and J. Zhou, Nat. Commun., 2023, 14, 7080 CrossRef CAS PubMed.
- S. B. Wang, Q. Ran, R. Q. Yao, H. Shi, Z. Wen, M. Zhao, X. Y. Lang and Q. Jiang, Nat. Commun., 2020, 11, 1634 CrossRef CAS PubMed.
- H. Meng, Q. Ran, T.-Y. Dai, H. Shi, S.-P. Zeng, Y.-F. Zhu, Z. Wen, W. Zhang, X.-Y. Lang and W.-T. Zheng, Nano-Micro Lett., 2022, 14, 128 CrossRef CAS PubMed.
- C. Hu, Z. Yang, Q. Zhang, M. Zhang, T. Wu, C. Xie, H. Wang, Y. Tang and H. Wang, Angew. Chem., Int. Ed., 2024, 63, e202409096 CrossRef CAS PubMed.
- Y. Nie, H. Chen, J. Wu, R. Nie, L. Yu, L. Liu and J. Xi, Chem. Eng. J., 2024, 498, 155615 CrossRef CAS.
- Y. Du, Y. Feng, R. Li, Z. Peng, X. Yao, S. Duan, S. Liu, S. C. Jun, J. Zhu, L. Dai, Q. Yang, L. Wang and Z. He, Small, 2024, 20, 2307848 CrossRef CAS PubMed.
- T. Wang, Y. Zhu, Y. Li, K. Yang, W. Lu, K. Peng and Z. Tian, Sustainable Energy Fuels, 2022, 7, 300–309 RSC.
- X. Liu, J. Xue, J. Cao, Z. Wang and X. Li, J. Energy Storage, 2024, 87, 111503 CrossRef.
- M. Deng, D. Höche, S. V. Lamaka, D. Snihirova and M. L. Zheludkevich, J. Power Sources, 2018, 396, 109–118 CrossRef CAS.
- S. K. Kaiser, Z. Chen, D. F. Akl, S. Mitchell and J. Perez-Ramirez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- Y. N. Jo, K. Prasanna, S. H. Kang, P. R. Ilango, H. S. Kim, S. W. Eom and C. W. Lee, J. Ind. Eng. Chem., 2017, 53, 247–252 CrossRef CAS.
- Y. Gouale, S. Khatbi and M. Essahli, Port. Electrochim. Act., 2018, 36, 89–99 CrossRef CAS.
- T. W. Cain, C. F. Glover and J. R. Scully, Electrochim. Acta, 2019, 297, 564–575 CrossRef CAS.
- M. Esmaily, J. E. Svensson, S. Fajardo, N. Birbilis, G. S. Frankel, S. Virtanen, R. Arrabal, S. Thomas and L. G. Johansson, Prog. Mater. Sci., 2017, 89, 92–193 CrossRef CAS.
- I.-J. Park, S.-R. Choi and J.-G. Kim, J. Power Sources, 2017, 357, 47–55 CrossRef CAS.
- L. Wang, R. Wang, Y. Feng, M. Deng and N. Wang, J. Electrochem. Soc., 2017, 164, A438–A446 CrossRef CAS.
- A. D. Südholz, N. T. Kirkland, R. G. Buchheit and N. Birbilis, Electrochem. Solid-State Lett., 2011, 14, C5–C7 CrossRef.
- X. Xia, J. Nie, C. Davies, W. Tang and N. Birbilis, Corrosion, 2015, 71, 1370–1386 CrossRef CAS PubMed.
- J. Ma, G. Wang, Y. Li, F. Ren and A. A. Volinsky, Ionics, 2018, 25, 2201–2209 CrossRef.
- R. L. Liu, J. R. Scully, G. Williams and N. Birbilis, Electrochim. Acta, 2018, 260, 184–195 CrossRef CAS.
- G. Williams, H. A.-L. Dafydd, H. N. McMurray and N. Birbilis, Electrochim. Acta, 2016, 219, 401–411 CrossRef CAS.
- B. Lenhart, M. Zuraw and W. Mustain, J. Electrochem. Soc., 2023, 170, 070501 CrossRef CAS.
- H. Zhang, L. Yang, H. Wang, B. Cui, J. Wang, X. Han and W. Hu, Adv. Funct. Mater., 2024, 34, 2312469 CrossRef CAS.
- Z. Yang, C. Hu, Q. Zhang, T. Wu, C. Xie, H. Wang, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2023, 135, e202308017 CrossRef.
- Y. N. Jo, H. S. Kim, K. Prasanna, P. R. Ilango, W. J. Lee, S. W. Eom and C. W. Lee, Ind. Eng. Chem. Res., 2014, 53, 17370–17375 CrossRef CAS.
- H. S. Kim, Y. N. Jo, W. J. Lee, K. J. Kim and C. W. Lee, Electroanal., 2015, 27, 517–523 CrossRef CAS.
- H. Moghanni-Bavil-Olyaei and J. Arjomandi, RSC Adv., 2015, 5, 91273–91279 RSC.
- H. Xiong, X. Yin, Y. Yan, Y. Dai, S. Fan, X. Qiao and K. Yu, J. Mater. Eng. Perform., 2016, 25, 3456–3464 CrossRef CAS.
- R. N. Mutlu and B. Yazıcı, J. Solid State Electrochem, 2018, 23, 529–541 CrossRef.
- J. Ma, G. Wang, Y. Li, C. Qin and F. Ren, J. Mater. Eng. Perform., 2019, 28, 2873–2880 CrossRef CAS.
- P. Wang, J. Li, Y. Guo, Z. Yang, F. Xia and J. Wang, Rare Met., 2011, 30, 639–643 CrossRef CAS.
- J. Xu, Q. Yang, C. Huang, M. S. Javed, M. K. Aslam and C. Chen, J. Appl. Electrochem., 2017, 47, 767–775 CrossRef CAS.
- M. A. Deyab, J. Power Sources, 2015, 280, 190–194 CrossRef CAS.
- N. Wang, X. Chen, H. Wan, B. Zhang, K. Guan, J. Yao, J. Ji, J. Li, Y. Gan, L. Lv, L. Tao, G. Ma, H. Wang, J. Zhang and H. Wang, Adv. Funct. Mater., 2023, 33, 2300795 CrossRef CAS.
- M. A. Deyab, J. Power Sources, 2015, 292, 66–71 CrossRef CAS.
- T. Wei, L. e Mo, Y. Ren, H. Zhang, M. Wang, Y. He, P. Tan, Z. Li, W. Chen and L. Hu, Energy Storage Mater., 2024, 70, 103525 CrossRef.
- X. Shen, W. Chen, H. Wang, L. Zhang, B. Hao, C. Zhu, X. Yang, M. Sun, J. Zhou, X. Liu, C. Yan and T. Qian, Chem. Sci., 2024, 15, 10182–10192 RSC.
- M. C. Han, J. H. Zhang, C. Y. Yu, J. C. Yu, Y. X. Wang, Z. G. Jiang, M. Yao, G. Xie, Z. Z. Yu and J. Qu, Angew. Chem., 2024, 136, e202403695 CrossRef.
- J. Huang, Y. Zhong, H. Fu, Y. Zhao, S. Li, Y. Xie, H. Zhang, B. Lu, L. Chen and S. Liang, Adv. Mater., 2024, 36, 2406257 CrossRef CAS PubMed.
- W. Xie, K. Zhu, W. Jiang, H. Yang, M. Ma, L. Zhao and W. Yang, Acs Nano, 2024, 18, 21184–21197 CrossRef CAS PubMed.
- Y. Qiang, S. Zhang, L. Guo, S. Xu, L. Feng, I. B. Obot and S. Chen, J. Cleaner Prod., 2017, 152, 17–25 CrossRef CAS.
- D. Gelman, H. Drezner, A. Kraytsberg, D. Starosvetsky and Y. Ein-Eli, J. Solid State Electrochem., 2018, 22, 2217–2226 CrossRef CAS.
- M. Abdallah, A. Y. El-Etre and M. F. Moustafa, Port. Electrochim. Act., 2009, 27, 615–630 CrossRef CAS.
- H. Zhou, Q. Huang, M. Liang, D. Lv, M. Xu, H. Li and W. Li, Mater. Chem. Phys., 2011, 128, 214–219 CrossRef CAS.
- Y. Li, Z. Hu and J. Kan, Metals, 2016, 6, 176 CrossRef.
- F. Yang, J. A. Yuwono, J. Hao, J. Long, L. Yuan, Y. Wang, S. Liu, Y. Fan, S. Zhao, K. Davey and Z. Guo, Adv. Mater., 2022, 34, e2206754 CrossRef PubMed.
- M. Kim, S. J. Shin, J. Lee, Y. Park, Y. Kim, H. Kim and J. W. Choi, Angew. Chem., Int. Ed., 2022, 134, e202211589 CrossRef.
- T. Xiao, J.-L. Yang, B. Zhang, J. Wu, J. Li, W. Mai and H. J. Fan, Angew. Chem., Int. Ed., 2024, 63, e202318470 CrossRef CAS PubMed.
- X. Wang, Y. Ying, S. Chen, Q. Meng, H. Huang and L. Ma, Nano Energy, 2024, 119, 109099 CrossRef CAS.
- Z. Shi, M. Li, X. Fu, Y. Zhang, S. Jiao and Y. Zhao, Adv. Funct. Mater., 2024, 34, 2316427 CrossRef CAS.
- D. Li, Y. Zhong, X. Xu, D. Zhou, Y. Tang, L. Wang, S. Liang, B. Lu, Y. Liu and J. Zhou, Energy Environ. Sci., 2024, 17, 8855–8865 RSC.
- T. Wu, C. Hu, Q. Zhang, Z. Yang, G. Jin, Y. Li, Y. Tang, H. Li and H. Wang, Adv. Funct. Mater., 2024, 2315716 CrossRef CAS.
- X. Feng, P. Li, J. Yin, Z. Gan, Y. Gao, M. Li, Y. Cheng, X. Xu, Y. Su and S. Ding, ACS Energy Lett., 2023, 8, 1192–1200 CrossRef CAS.
- Y. Nie, J. Gao, E. Wang, L. Jiang, L. An and X. Wang, Electrochim. Acta, 2017, 248, 478–485 CrossRef CAS.
- P. Ruan, X. Chen, L. Qin, Y. Tang, B. Lu, Z. Zeng, S. Liang and J. Zhou, Adv. Mater., 2023, 35, 2300577 CrossRef CAS PubMed.
- K. Liu, P. He, H. Bai, J. Chen, F. Dong, S. Wang, M. He and S. Yuan, Mater. Chem. Phys., 2017, 199, 73–78 CrossRef CAS.
- Z. Liu, Y. Zhao, G.-C. Han, J. Liu, Q. He and C. Chen, Ionics, 2016, 22, 2391–2397 CrossRef CAS.
- Y. Ein-Eli, M. Auinat and D. Starosvetsky, J. Power Sources, 2003, 114, 330–337 CrossRef CAS.
- M. Liang, H. Zhou, Q. Huang, S. Hu and W. Li, J. Appl. Electrochem., 2011, 41, 991–997 CrossRef CAS.
- M.-H. Lin, C.-J. Huang, P.-H. Cheng, J.-H. Cheng and C.-C. Wang, J. Mater. Chem. A, 2020, 8, 20637–20649 RSC.
- S. Hosseini, W. Lao-Atiman, S. J. Han, A. Arpornwichanop, T. Yonezawa and S. Kheawhom, Sci. Rep., 2018, 8, 14909 CrossRef PubMed.
- Y. Xiao, J. Shi, F. Zhao, Z. Zhang and W. He, J. Electrochem. Soc., 2018, 165, A47–A54 CrossRef CAS.
- R. Sun, D. Han, C. Cui, Z. Han, X. Guo, B. Zhang, Y. Guo, Y. Liu, Z. Weng and Q.-H. Yang, Angew. Chem., Int. Ed., 2023, 62, e202303557 CrossRef CAS PubMed.
- Y. Shang, V. Kundi, I. Pal, H. N. Kim, H. Zhong, P. Kumar and D. Kundu, Adv. Mater., 2024, 36, 2309212 CrossRef CAS PubMed.
- C. Yang, Z. Zhang, Z. Tian, K. Zhang, J. Li and Y. Lai, J. Electrochem. Soc., 2016, 163, A1836–A1840 CrossRef CAS.
- H. Liu, Z. Xin, B. Cao, Z. Xu, B. Xu, Q. Zhu, J.-L. Yang, B. Zhang and H. J. Fan, Adv. Funct. Mater., 2024, 34, 2309840 CrossRef CAS.
- F. Wang, W. Liang, X. Liu, T. Yin, Z. Chen, Z. Yan, F. Li, W. Liu, J. Lu, C. Yang and Q.-H. Yang, Adv. Energy Mater., 2024, 14, 2400110 CrossRef CAS.
- Z. Liang, C. Li, D. Zuo, L. Zeng, T. Ling, J. Han and J. Wan, Energy Storage Mater., 2023, 63, 102980 CrossRef.
- X. Gu, Y. Du, X. Ren, F. Ma, X. Zhang, M. Li, Q. Wang, L. Zhang, C. Lai and S. Zhang, Adv. Funct. Mater., 2024, 34, 2316541 CrossRef CAS.
- D.-Q. Cai, H. Cheng, J.-L. Yang, H. Liu, T. Xiao, X. Liu, M. Chen and H. J. Fan, Energy Environ. Sci., 2024, 17, 8349–8359 RSC.
- S. Mehta, S. Kaur, M. Singh, M. Kumar, K. Kumar, S. K. Meena and T. C. Nagaiah, Adv. Energy Mater., 2024, 14, 2401515 CrossRef CAS.
- X. Xiao, X. Ye, Z. Wu, X. Wu, J. Yu, L. Gu and S. Liu, Adv. Mater., 2024, 36, 2408706 CrossRef CAS PubMed.
- R. Chen, W. Zhang, Q. Huang, C. Guan, W. Zong, Y. Dai, Z. Du, Z. Zhang, J. Li, F. Guo, X. Gao, H. Dong, J. Zhu, X. Wang and G. He, Nano-Micro Lett., 2023, 15, 81 CrossRef CAS PubMed.
- M. Kim, S.-J. Shin, J. Lee, Y. Park, Y. Kim, H. Kim and J. W. Choi, Angew. Chem., Int. Ed., 2022, 61, e202211589 CrossRef CAS PubMed.
- Y. Lin, Z. Mai, H. Liang, Y. Li, G. Yang and C. Wang, Energy Environ. Sci., 2023, 16, 687–697 RSC.
- R. Han, T. Jiang, Z. Wang, R. Xue, X. Liu, Y. Tang, Z. Qi and Y. Ma, Adv. Funct. Mater., 2024, 2412255 Search PubMed.
- J. Zhang, Y. Liu, Y. Wang, Z. Zhu and Z. Yang, Adv. Funct. Mater., 2024, 34, 2401889 CrossRef CAS.
- C. Hou, S. Chen, Z. Wang, G. Wang and G. Dong, Mater. Corros., 2020, 71, 1480–1487 CrossRef CAS.
- M. A. Deyab, J. Power Sources, 2019, 412, 520–526 CrossRef CAS.
- M. A. Deyab, Electrochim. Acta, 2017, 244, 178–183 CrossRef CAS.
- J. Ma, W. Li, G. Wang, Y. Li, F. Ren and Y. Xiong, J. Electrochem. Soc., 2018, 165, A266–A272 CrossRef CAS.
- D. Gelman, I. Lasman, S. Elfimchev, D. Starosvetsky and Y. Ein-Eli, J. Power Sources, 2015, 285, 100–108 CrossRef CAS.
- E. Grishina, D. Gelman, S. Belopukhov, D. Starosvetsky, A. Groysman and Y. Ein-Eli, ChemSusChem, 2016, 9, 2103–2111 CrossRef CAS PubMed.
- Y. Liu, H. Zhang, Y. Liu, J. Li and W. Li, J. Power Sources, 2019, 434, 226723 CrossRef CAS.
- Q. X. Kang, Y. Wang and X. Y. Zhang, J. Alloys Compd., 2019, 774, 1069–1080 CrossRef CAS.
- C. Zhu, H. Yang, A. Wu, D. Zhang, L. Gao and T. Lin, J. Power Sources, 2019, 432, 55–64 CrossRef CAS.
- M. A. Deyab, J. Power Sources, 2016, 325, 98–103 CrossRef CAS.
- Y. Li, J. Ma, G. Wang, F. Ren, Y. Zhu and Y. Song, J. Electrochem. Soc., 2018, 165, A1713–A1717 CrossRef CAS.
- Y. Zhao, G. Huang, C. Zhang, C. Peng and F. Pan, Mater. Chem. Phys., 2018, 218, 256–261 CrossRef CAS.
- B. Liu, T. Lv, A. Zhou, X. Zhu, Z. Lin, T. Lin and L. Suo, Nat. Commun., 2024, 15, 2922 CrossRef CAS PubMed.
- S. Yang, Q. Wu, Y. Li, F. Luo, J. Zhang, K. Chen, Y. You, J. Huang, H. Xie and Y. Chen, Angew. Chem., Int. Ed., 2024, 136, e202409160 CrossRef.
- X. Wang, B. Wang, P. Lei, X. Wang, L. Zhou, J. Zhang, J. Zhang and J. Cheng, Energy Environ. Sci., 2024, 17, 6640–6655 RSC.
- Q. Fu, S. Hao, X. Zhang, H. Zhao, F. Xu and J. Yang, Energy Environ. Sci., 2023, 16, 1291–1311 RSC.
- Y. Zhao, Z. Chen, X. Gao, H. Dong, X. Zhao, G. He and H. Yang, Angew. Chem., Int. Ed., 2024, e202415251 Search PubMed.
- Y. Cui, W. Chen, W. Xin, H. Ling, Y. Hu, Z. Zhang, X. He, Y. Zhao, X.-Y. Kong, L. Wen and L. Jiang, Adv. Mater., 2024, 36, 2308639 CrossRef CAS PubMed.
- J.-L. Yang, T. Xiao, T. Xiao, J. Li, Z. Yu, K. Liu, P. Yang and H. J. Fan, Adv. Mater., 2024, 36, 2313610 CrossRef CAS PubMed.
- T. Qiu, T. Wang, W. Tang, Y. Li, Y. Li, X. Lang, Q. Jiang and H. Tan, Angew. Chem., 2023, 135, e202312020 CrossRef.
- L. Ma, S. Chen, D. Wang, Q. Yang, F. Mo, G. Liang, N. Li, H. Zhang, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9, 1803046 CrossRef.
- F. Mo, G. Liang, D. Wang, Z. Tang and C. Zhi, EcoMat, 2019, 1, e12008 CrossRef CAS.
- L. Han, H. L. Huang, J. F. Li, X. L. Zhang, Z. L. Yang, M. Xu and L. K. Pan, J. Mater. Chem. A, 2020, 8, 15042–15050 RSC.
- K. T. Leng, G. J. Li, J. J. Guo, X. Y. Zhang, A. X. Wang, X. J. Liu and J. Y. Luo, Adv. Funct. Mater., 2020, 30, 10 CrossRef.
- Z. Liu, D. Wang, Z. Tang, G. Liang, Q. Yang, H. Li, L. Ma, F. Mo and C. Zhi, Energy Storage Mater., 2019, 23, 636–645 CrossRef.
- H. Li, C. Han, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Wang, Z. Liu, Z. Tang, Y. Wang, F. Kang, B. Li and C. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC.
- Z. Shen, Y. Liu, Z. Li, Z. Tang, J. Pu, L. Luo, Y. Ji, J. Xie, Z. Shu and Y. Yao, Adv. Funct. Mater., 2024 DOI:10.1002/adfm.202406620.
- F. Mo, Z. Chen, G. Liang, D. Wang, Y. Zhao, H. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2020, 10, 2000035 CrossRef CAS.
- J. Zhao, K. K. Sonigara, J. Li, J. Zhang and L. Chen, Angew. Chem., Int. Ed., 2017, 56, 7979–7983 CrossRef.
- Q. Han, X. Chi, S. Zhang, Y. Liu, B. Zhou, J. Yang and Y. Liu, J. Mater. Chem. A, 2018, 6, 23046–23054 RSC.
- Z. Yang, Q. Zhang, T. Wu, Q. Li, J. Shi, J. Gan, S. Xiang, H. Wang, C. Hu and Y. Tang, Angew. Chem., Int. Ed., 2024, 63, e202317457 CrossRef CAS PubMed.
- S. Zhang, N. Yu, S. Zeng, S. Zhou, M. Chen, J. Di and Q. Li, J. Mater. Chem. A, 2018, 6, 12237–12243 RSC.
- S. Wu, S. Hu, Q. Zhang, D. Sun, P. Wu, Y. Tang and H. Wang, Energy Storage Mater., 2020, 31, 310–317 CrossRef.
- W. Xiong, T. K. A. Hoang, D. Yang, Y. Liu, M. Ahmed, J. Xu, X. Qiu and P. Chen, J. Energy Storage, 2019, 26, 100920 CrossRef.
- T. K. A. Hoang, M. Acton, H. T. H. Chen, Y. Huang, T. N. L. Doan and P. Chen, Mater. Today Energy, 2017, 4, 34–40 CrossRef.
- T. K. A. Hoang, T. N. L. Doan, J. H. Cho, J. Y. J. Su, C. Lee, C. Lu and P. Chen, ChemSusChem, 2017, 10, 2816–2822 CrossRef CAS PubMed.
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschamps and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- S. Liu, H. Lin, Q. Song, J. Zhu and C. Zhu, Energy Environ. Mater., 2023, 6, e12405 CrossRef CAS.
- X. Zhou, B. Wen, Y. Cai, X. Chen, L. Li, Q. Zhao, S.-L. Chou and F. Li, Angew. Chem., Int. Ed., 2024, 63, e202402342 CrossRef CAS PubMed.
- L. Fan, H. Lu, J. Leng, Z. Sun and C. Chen, J. Power Sources, 2015, 299, 66–69 CrossRef CAS.
- N. Wang, Y. Mu, W. Xiong, J. Zhang, Q. Li and Z. Shi, Corros. Sci., 2018, 144, 107–126 CrossRef CAS.
- B. J. Hopkins, Y. Shao-Horn and D. P. Hart, Science, 2018, 362, 658–661 CrossRef CAS PubMed.
- P. Xie, M. Z. Rong and M. Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1805–1809 CrossRef CAS PubMed.
- D. Raptis, A. K. Seferlis, V. Mylona, C. Politis and P. Lianos, Int. J. Hydrogen Energy, 2019, 44, 1359–1365 CrossRef CAS.
- C. Kim, J. Kim, S. Joo, Y. Yang, J. Shin, M. Liu, J. Cho and G. Kim, Angew. Chem., Int. Ed., 2019, 58, 9506–9511 CrossRef CAS PubMed.
- A. Z. Zhuk, A. E. Sheindlin, B. V. Kleymenov, E. I. Shkolnikov and M. Y. Lopatin, J. Power Sources, 2006, 157, 921–926 CrossRef CAS.
- T. Bellezze, G. Giuliani and G. Roventi, Corros. Sci., 2018, 130, 113–125 CrossRef CAS.
- S. Karimi, I. Taji, T. Hajilou, S. Palencsár, A. Dugstad, A. Barnoush, K. Verbeken, T. Depover and R. Johnsen, Corros. Sci., 2023, 214, 111031 CrossRef CAS.
- R. Meng, H. Li, Z. Lu, C. Zhang, Z. Wang, Y. Liu, W. Wang, G. Ling, F. Kang and Q. H. Yang, Adv. Mater., 2022, 34, e2200677 CrossRef PubMed.
- H. Tian, J. L. Yang, Y. Deng, W. Tang, R. Liu, C. Xu, P. Han and H. J. Fan, Adv. Energy Mater., 2023, 13, 2202603 CrossRef CAS.
- M. P. Gomes, I. Costa, N. Pébère, J. L. Rossi, B. Tribollet and V. Vivier, Electrochim. Acta, 2019, 306, 61–70 CrossRef CAS.
- G. L. de Gouveia, J. E. Spinelli and G. Y. Koga, Corros. Sci., 2024, 237, 112326 CrossRef CAS.
- R. De Motte, E. Basilico, R. Mingant, J. Kittel, F. Ropital, P. Combrade, S. Necib, V. Deydier, D. Crusset and S. Marcelin, Corros. Sci., 2020, 172, 108666 CrossRef CAS.
- M. Diaz-Ramos, V. Roche, R. Song, H. Fan, C. Bureau and J. C. Lepretre, Corros. Sci., 2023, 213, 110932 CrossRef CAS.
- J. M. Ferreira, Jr., M. Oliveira, G. F. Trindade, L. C. L. Santos, C. R. Tomachuk and M. A. Baker, Corros. Sci., 2018, 137, 13–32 CrossRef.
- S. M. Hoseinieh, A. M. Homborg, T. Shahrabi, J. M. C. Mol and B. Ramezanzadeh, Electrochim. Acta, 2016, 217, 226–241 CrossRef CAS.
- M. Sharifuzzaman, M. A. Zahed, M. S. Reza, M. Asaduzzaman, S. Jeong, H. Song, D. K. Kim, S. Zhang and J. Y. Park, Adv. Funct. Mater., 2023, 33, 2208894 CrossRef CAS.
- J. Chen, J. He and L. Li, Corros. Sci., 2022, 205, 110425 CrossRef CAS.
- J. H. Arellano-Perez, R. F. Escobar-Jimenez, D. Granados-Lieberma, J. F. Gomez-Aguilar, J. Uruchurtu-Chavarin and V. M. Alvarado-Martinez, J. Electroanal. Chem., 2019, 848, 113249 CrossRef CAS.
- C. Ma, S. Song, Z. Gao, J. Wang, W. Hu, Y. Behnamian and D.-H. Xia, Corros. Eng. Sci. Technol., 2017, 52, 432–440 CAS.
- I. B. Obot, I. B. Onyeachu, A. Zeino and S. A. Umoren, J. Adhes. Sci. Technol., 2019, 33, 1453–1496 CrossRef CAS.
- O. Ramos-Negrón, R. Escobar-Jiménez, J. Arellano-Pérez, J. Uruchurtu-Chavarín, J. Gómez-Aguilar and M. Lucio-García, J. Electroanal. Chem., 2019, 855, 113597 CrossRef.
- A. Legat and C. Zevnik, Corros. Sci., 1993, 35, 1661–1666 CrossRef CAS.
- C. Ma, Z. Wang, Y. Behnamian, Z. Gao, Z. Wu, Z. Qin and D.-H. Xia, Measurement, 2019, 138, 54–79 CrossRef.
- D. H. Xia, S. Z. Song and Y. Behnamian, Corros. Eng. Sci. Technol., 2016, 51, 527–544 CAS.
- A. Rana, K. Roy, J. N. Heil, J. H. Nguyen, C. Renault, B. M. Tackett and J. E. Dick, Adv. Energy Mater., 2024, 14, 2402521 CrossRef CAS.
- M. Peng, X. Tang, K. Xiao, T. Hu, K. Yuan and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202302701 CrossRef CAS PubMed.
- J. Fu, H. Wang, P. Xiao, C. Zeng, Q. Sun and H. Li, Energy Storage Mater., 2022, 48, 191–196 CrossRef.
- I. Serebrennikova, S. Lee and H. S. White, Faraday Discuss., 2002, 121, 199–210 RSC.
- H. Shi, Z. Tian, T. Hu, F. Liu, E.-H. Han, M. Taryba and S. V. Lamaka, Corros. Sci., 2014, 88, 178–186 CrossRef CAS.
- M. Li, Y. Wang, B. Blount, E. Gordon, J. A. Muñoz-Castañeda, Z. Ye and H. Ren, Nano Lett., 2022, 22, 6313–6319 CrossRef CAS PubMed.
- R. M. Souto, Y. González-García, J. Izquierdo and S. González, Corros. Sci., 2010, 52, 748–753 CrossRef CAS.
- N. Payne, L. Stephens and J. Mauzeroll, Corrosion, 2017, 73, 759–780 CrossRef CAS PubMed.
- C. S. Santos, A. Botz, A. S. Bandarenka, E. Ventosa and W. Schuhmann, Angew. Chem., Int. Ed., 2022, 61, e202202744 CrossRef CAS PubMed.
- A. Mishra, M. Zorigt, D. O. Kim and J. Rodriguez-Lopez, J. Am. Chem. Soc., 2024, 146, 8847–8851 CrossRef CAS PubMed.
- I. Traxler, T. D. Singewald, G. Schimo-Aichhorn, S. Hild and M. Valtiner, Corros. Rev., 2022, 40, 515–542 CrossRef CAS.
- U. M. Tefashe, M. E. Snowden, P. D. Ducharme, M. Danaie, G. A. Botton and J. Mauzeroll, J. Electroanal. Chem., 2014, 720, 121–127 CrossRef.
- P. Dauphin-Ducharme, R. Matthew Asmussen, U. M. Tefashe, M. Danaie, W. Jeffrey Binns, P. Jakupi, G. A. Botton, D. W. Shoesmith and J. Mauzeroll, J. Electrochem. Soc., 2014, 161, C557–C564 CrossRef.
- A. Asserghine, M. Medvidović-Kosanović, A. Stanković, L. Nagy, R. M. Souto and G. Nagy, Sens. Actuators, B, 2020, 321, 128610 CrossRef CAS.
- M. Zhang, H. Hua, P. Dai, Z. He, L. Han, P. Tang, J. Yang, P. Lin, Y. Zhang, D. Zhan, J. Chen, Y. Qiao, C. C. Li, J. Zhao and Y. Yang, Adv. Mater., 2023, 35, 2208630 CrossRef CAS PubMed.
- J. Izquierdo, L. Nagy, Á. Varga, J. J. Santana, G. Nagy and R. M. Souto, Electrochim. Acta, 2011, 56, 8846–8850 CrossRef CAS.
- I. Liberman, R. Ifraemov, R. Shimoni and I. Hod, Adv. Funct. Mater., 2022, 32, 2112517 CrossRef CAS.
- A. Mishra, M. Zorigt, D. O. Kim and J. Rodríguez-López, J. Am. Chem. Soc., 2024, 146, 8847–8851 CrossRef CAS PubMed.
- Y. Gan, B. Tan, Q. Hu, S. Zhang and W. Li, Corros. Sci., 2022, 208, 110619 CrossRef CAS.
- X. Li, P. Ye, A. Dou, Z. Jiang, A. Naveed, Y. Zhou, M. Su, P. Zhang and Y. Liu, J. Energy Storage, 2024, 76, 109874 CrossRef.
- Y. Yu, Y. X. Zuo, Z. H. Zhang, L. Wu, C. L. Ning and C. C. Zuo, Coatings, 2019, 9, 692 CrossRef CAS.
- X. Luo, R. Wang, L. Zhang, Z. Liu, H. Li, J. Mao, S. Zhang, J. Hao, T. Zhou and C. Zhang, ACS Nano, 2024, 18, 12981–12993 CrossRef CAS PubMed.
- X. Zhang, R. Wang, Z. Liu, Q. Ma, H. Li, Y. Liu, J. Hao, S. Zhang, J. Mao and C. Zhang, Adv. Energy Mater., 2024, 14, 2400314 CrossRef CAS.
- Y. Meng, L. Liu, D. Zhang, C. Dong, Y. Yan, A. A. Volinsky and L. N. Wang, Bioact. Mater., 2019, 4, 87–96 Search PubMed.
- J. A. Yuwono, N. Birbilis, R. Liu, Q. Ou, Q. Bao and N. V. Medhekar, J. Electrochem. Soc., 2017, 164, C918–C929 CrossRef CAS.
- X. Ni, J. Zhou, H. Ji, Y. Chen, H. Yu, Y. Zheng, T. Qian, M. Wang, L. Chen and C. Yan, Adv. Funct. Mater., 2023, 33, 2302293 CrossRef CAS.
- F. Ling, L. Wang, F. Liu, M. Ma, S. Zhang, X. Rui, Y. Shao, Y. Yang, S. He, H. Pan, X. Wu, Y. Yao and Y. Yu, Adv. Mater., 2023, 35, 2208764 CrossRef CAS PubMed.
- S.-J. Zhang, J. Hao, H. Li, P.-F. Zhang, Z.-W. Yin, Y.-Y. Li, B. Zhang, Z. Lin and S.-Z. Qiao, Adv. Mater., 2022, 34, 2201716 CrossRef CAS PubMed.
- Q. Shi, L. J. Rendek, W.-B. Cai and D. A. Scherson, Electrochem. Solid-State Lett., 2003, 6, E35 CrossRef CAS.
- C. Pan, L. Liu, Y. Li and F. Wang, Corros. Sci., 2013, 73, 32–43 CrossRef CAS.
- J. Cheng, J. Pan, T. Wang and X. Lu, Corros. Sci., 2018, 137, 184–193 CrossRef CAS.
- F. Zhang, J. Pan and C. Lin, Corros. Sci., 2009, 51, 2130–2138 CrossRef CAS.
- A. Almalla, O. Ozcan and J. Witt, Adv. Eng. Mater., 2022, 24, 2101342 CrossRef CAS.
- L. Wei and W. Qin, Corros. Sci., 2022, 206, 110525 CrossRef CAS.
- F. A. Martin, C. Bataillon and J. Cousty, Corros. Sci., 2008, 50, 84–92 CrossRef CAS.
- P. Yi, C. Dong, K. Xiao and X. Li, Corros. Sci., 2021, 181, 109244 CrossRef CAS.
- S. Montecinos and S. N. Simison, Appl. Surf. Sci., 2011, 257, 7732–7738 CrossRef CAS.
- X. Zhou, Q. Zhang, Z. Hao, Y. Ma, O. A. Drozhzhin and F. Li, ACS Appl. Mater. Inter., 2021, 13, 53227–53234 CrossRef CAS PubMed.
- A. Kreta, M. Gaberšček and I. Muševič, J. Mater. Res., 2021, 36, 79–93 CrossRef CAS.
- H. Wang, B. Brown, S. Nesic and A. Pailleret, NACE CORROSION, 2021, D091S033R003 Search PubMed.
- Y. Shi, L. Collins, N. Balke, P. K. Liaw and B. Yang, Appl. Surf. Sci., 2018, 439, 533–544 CrossRef CAS.
- S. D. Zhang, Z. W. Liu, Z. M. Wang and J. Q. Wang, Corros. Sci., 2014, 83, 111–123 CrossRef CAS.
- Y.-J. Cho, I.-J. Park, H.-J. Lee and J.-G. Kim, J. Power Sources, 2015, 277, 370–378 CrossRef CAS.
- Y. Zhong, Z. Cheng, H. Zhang, J. Li, D. Liu, Y. Liao, J. Meng, Y. Shen and Y. Huang, Nano Energy, 2022, 98, 107220 CrossRef CAS.
- E. Faegh, S. Shrestha, X. Zhao and W. E. Mustain, J. Appl. Electrochem., 2019, 49, 895–907 CrossRef CAS.
- Y. Huang, Q. Gu, Z. Guo, W. Liu, Z. Chang, Y. Liu, F. Kang, L. Dong and C. Xu, Energy Storage Mater., 2022, 46, 243–251 CrossRef.
- X. Wei, D. Desai, G. G. Yadav, D. E. Turney, A. Couzis and S. Banerjee, Electrochim. Acta, 2016, 212, 603–613 CrossRef CAS.
- A. Rana, A. Thakare, N. Kumar, B. Mukherjee, A. Torris, B. Das, S. Ogale and A. Banerjee, ACS Appl. Mater. Inter., 2023, 15, 23093–23103 CrossRef CAS PubMed.
- J. Scharf, L. Yin, C. Redquest, R. Liu, X. L. Quinn, J. Ortega, X. Wei, J. Wang, J.-M. Doux and Y. S. Meng, Adv. Energy Mater., 2021, 11, 2101327 CrossRef CAS.
- J. Wang, J. Peng, W. Huang, H. Liang, Y. Hao, J. Li, H. Chu, H. Wei, Y. Zhang and J. Liu, Adv. Funct. Mater., 2024, 34, 2316083 CrossRef CAS.
Footnote |
| † Xuejin Li and Pengyun Liu contribute equally to this review. |
| This journal is © The Royal Society of Chemistry 2025 |