
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-conversion-efficiency and stable six-electron Zn–I2 batteries enabled by organic iodide/thiazole-linked covalent organic frameworks†
Wenyan
Du
a,
Qi
Huang
b,
Xunwen
Zheng
a,
Yaokang
Lv
 d,
Ling
Miao
a,
Ziyang
Song
a,
Lihua
Gan
ac and
Mingxian
Liu
*ac
d,
Ling
Miao
a,
Ziyang
Song
a,
Lihua
Gan
ac and
Mingxian
Liu
*ac
aShanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai, 1239 Siping Rd., 200092, P. R. China. E-mail: liumx@tongji.edu.cn
bInstitute for Electric Light Sources, School of Information Science and Technology, Fudan University, 2005 Songhu Rd., Shanghai 200438, P. R. China
cState Key Laboratory of Cardiovascular Diseases and Medical Innovation Center, Shanghai East Hospital, School of Medicine, Tongji University, 150 Jimo Rd., Shanghai 200120, P. R. China
dCollege of Chemical Engineering, Zhejiang University of Technology, 18 Chaowang Rd, Hangzhou 310014, P. R. China
First published on 9th April 2025
Abstract
Six-electron I−/I5+ redox chemistry gives a promising platform to achieve high-capacity Zn–I2 batteries, but faces limited conversion efficiency and instability of IO3− species. Here, we design a thiazole-linked covalent organic framework (TZ-COF) hosted organic trimethylsulfonium iodide (C3H9IS/TZ-COFs) electrode in a 1-methyl-3-propylimidazolium bromide (MPIBr)-containing electrolyte to stimulate I−/I0/I+/I5+ iodine conversion chemistry with better electrochemical efficiency and stability. Compared with inorganic symmetric I2 molecules, the more easily exposed I− center of polar C3H9IS combines with the oxygen in H2O to form HIO3, which initiates 6e− I−/IO3− conversion through I+ activation of MPIBr, thus reducing the oxidation/reduction potential gap to achieve 97% iodine conversion efficiency. Meanwhile, thiazole units of TZ-COFs enable strong chemical adsorption with IO3− species to improve redox stability with high reversibility due to reduced energy barriers (−5.1 vs. −3.5 eV in activated carbon (AC) host) and upgraded conversion kinetics (activation energy: 0.21 vs. 0.38 eV in AC). Such a stable and high-efficiency 6e− iodine conversion gives C3H9IS/TZ-COFs electrodes record high capacity (1296 mA h g−1) and energy density (1464 W h kg−1), and superior cycling stability (1200 cycles). These findings constitute a major advance in the design of iodine redox chemistry towards state-of-the-art Zn–I2 batteries.
Broader contextRecently, 6e− iodine conversion (I−/IO3−) chemistry has been achieved via hetero-halogen chemistry activation, which boosts iodine conversion kinetics and reversibility to achieve Zn–I2 batteries with high capacity and energy density. Nevertheless, the electrochemical formation of hypervalent iodine (I5+) is impeded by low conversion efficiency and unstable intermediates, limiting further exploration of multi-electron iodine electrochemistry in the realm of energy storage. In this work, we design a thiazole-linked covalent organic framework (TZ-COF) hosted organic trimethylsulfonium iodide (C3H9IS/TZ-COFs) electrode in a 1-methyl-3-propylimidazolium bromide (MPIBr)-containing electrolyte to stimulate I−/I0/I+/I5+ iodine conversion chemistry with better electrochemical efficiency and stability. The high iodine conversion efficiency (97%) and stable 6e− iodine conversion bring the capacity and energy density of Zn–I2 batteries to a new level. This work broadens the horizons of multielectron redox chemistry for building advanced Zn–I2 batteries. |
Introduction
Rechargeable zinc–iodine batteries have garnered significant attention due to environmental friendliness, high abundance, and diversity in the valence of iodine.1–6 However, a traditional iodine electrode operates as a one-electron transfer I−/I2 redox couple, exhibiting a relatively low theoretical capacity (211 mA h g−1), a low discharge plateau (0.54 V vs. standard hydrogen potential), and consequently insufficient energy density.7–13 To overcome these limitations, activating the 2 e− I−/I+ redox couple at high discharge voltage via the halogen reaction between I− species and halides (e.g., Br−, Cl−) has been widely reported for achieving better Zn–I2 batteries.14–16 Recently, the Liang group reported a 6e− iodine conversion (I−/IO3−) enabled by interhalogen chemistry between I− and Br− (I− + Br− → I+Br− + 2e−), which offers an electrochemical–chemical loop that boosts the kinetics and reversibility of the iodine conversion reaction, facilitating a high capacity of 1200 mA h g−1 (80.9% iodine conversion efficiency when deducting the capacity contribution of the activated carbon (AC) host) and an energy density of 1357 W h kg−1.17 The Li group activated the 6e− I−/IO3− redox process using highly concentrated hetero-halogen (I− and Br−) electrolytes to achieve a high capacity of 840 A h Lcatholyte−1 (945 mA h g−1, 74.6% iodine conversion efficiency) and energy density of 1200 W h Lcatholyte−1 (1350 W h kg−1).18 Nevertheless, the electrochemical formation of hypervalent iodine (I5+) is impeded by low conversion efficiency and unstable intermediates, limiting further exploration of multielectron iodine electrochemistry in the realm of energy storage. Highly soluble IO3− species are difficult to be stabilized by a common carbon host due to weak physical adsorption.19–22 More importantly, a high energy barrier (6.01 eV), caused by a huge potential gap of 0.66 V between the I−/I0 couple (0.54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) V) and I0/I5+ couple (1.20V), hinders the reversible electrochemical redox reaction from I− to IO3−, leading to slow reaction kinetics and limited conversion efficiency.23–30 These two issues bring challenges in establishing efficient iodine redox chemistry with more electron transfer for building better aqueous Zn–I2 batteries.31–38
V) and I0/I5+ couple (1.20V), hinders the reversible electrochemical redox reaction from I− to IO3−, leading to slow reaction kinetics and limited conversion efficiency.23–30 These two issues bring challenges in establishing efficient iodine redox chemistry with more electron transfer for building better aqueous Zn–I2 batteries.31–38
In this work, we report high-conversion-efficiency and stable 6e− Zn–I2 batteries enabled by organic iodide/thiazole-linked covalent organic frameworks (C3H9IS/TZ-COFs). The C3H9IS/TZ-COFs electrode in a 1-methyl-3-propylimidazolium bromide (MPIBr)-containing electrolyte stimulates I−/I0/I+/I5+ iodine conversion with superior electrochemical efficiency and stability. Compared with conventional I2, C3H9IS with a polar iodide center is more likely to combine with the oxygen in H2O to form IO3− species, initiating 6e− I−/I0/I+/I5+ iodine conversion chemistry through I+ activation of MPIBr, thereby reducing the potential gap between oxidation and reduction and improving the electrochemical efficiency. Moreover, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/C–S motifs of thiazole units in TZ-COFs coordinate with IO3− solving the solubility and stability issues, and simultaneously catalyze reversible iodine conversion from I− to IO3− due to the reduced energy barriers and the boosted redox reaction kinetics. The high iodine conversion efficiency (97%) and stable 6e− iodine conversion bring the capacity and energy density of C3H9IS/TZ-COFs electrodes to a new level. This work broadens the horizons of multielectron redox chemistry for building advanced Zn–I2 batteries.
N/C–S motifs of thiazole units in TZ-COFs coordinate with IO3− solving the solubility and stability issues, and simultaneously catalyze reversible iodine conversion from I− to IO3− due to the reduced energy barriers and the boosted redox reaction kinetics. The high iodine conversion efficiency (97%) and stable 6e− iodine conversion bring the capacity and energy density of C3H9IS/TZ-COFs electrodes to a new level. This work broadens the horizons of multielectron redox chemistry for building advanced Zn–I2 batteries.
Results and discussion
Naphthalene-2,6-diamine (ND), benzene-1,3,5-tricarbaldehyde (TA) and sulfur can undergo an oxidative annulation reaction to form highly crystalline TZ-COFs under solvothermal conditions (Fig. 1a).39 Powder X-ray diffraction (PXRD) patterns of TZ-COFs exhibit typical diffraction peaks at 3.41° and 6.85° belonging to the (110) and (200) planes (Fig. 1b), confirming a highly crystalline structure. Fourier transformed infrared (FT−IR) spectra show new peaks of CN groups at 1609 cm−1 and C−S groups at 673 cm−1 (Fig. 1c), indicating the formation of TZ-COFs. TZ-COFs are highly stable in boiling H2O, 12 M HCl or KOH solution for 24 h, suggesting excellent acid–alkali resistance and thermal stability (Fig. 1b, c and Fig. S1, ESI†). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images reveal the rod-like geometry of TZ-COFs with a lattice space of 0.3 nm (Fig. 1d and e). CN/C−S motifs of thiazole units in TZ-COFs show negative molecular electrostatic potential (MEP),40 and are the desirable active sites (blue area) to anchor iodine species (Fig. 1f and Fig. S2, ESI†), while the remaining domains (red area) around the aromatic ring maintain electron localization. Besides, TZ-COFs have a low optical energy gap (Eg) of 2.01 eV (Fig. 1g), which affords desirable electron conductivity and swift charge transport for stimulating high-kinetics redox reactions. The localized orbital locator-π (LOL-π) color-filled map41 unravels the connected isosurfaces of TZ-COFs, validating the highly conjugated organic structure and the electron delocalization effect (Fig. 1h).
 | ||
| Fig. 1 Structural characterization of TZ-COFs. (a) Synthesis process of TZ-COFs. (b) XRD patterns. (c) FT-IR spectra. (d) SEM and (e) TEM images. (b) Optimized molecular structures and energy levels. (f) ESP simulation. (e) Eg values. (h) LOL-π map. | ||
Due to the exposed polar iodide center and high stability, C3H9IS is elaborately selected as the iodine source, which includes the C3H9S+ chain and the I− anion connected by a weak ionic bond (Fig. S3, ESI†). To solve the problems of inevitable dissolution in aqueous electrolytes and low conductivity, C3H9IS was encapsulated in porous TZ-COFs (Fig. S4, ESI†) via the fusion-diffusion method to obtain the C3H9IS/TZ-COFs electrode. The electrochemical performances of the C3H9IS/TZ-COFs electrode were studied using a three-electrode Swagelok cell with a Ti mesh as the counter electrode and Hg/HgCl2 as the reference electrode in an aqueous Zn(OTF)2-MPIBr electrolyte (Fig. S5, ESI†).
Galvanostatic charge–discharge (GCD) curves of the C3H9IS/TZ-COFs electrode show an ultrahigh capacity of 1296 mA h g−1 (Fig. 2a), which exceeds those of C3H9IS/N-COFs (1202 mA h g−1), C3H9IS/activated carbon (AC) (1023 mA h g−1), and TZ-COFs (34 mA h g−1) electrodes (Fig. 2b and Fig. S6, S7, ESI†). A small voltage polarization of 0.28 V enabling 97% iodine conversion efficiency can be observed in the GCD curves of the C3H9IS/TZ-COFs electrode, indicating a fast reduction/oxidation kinetics process. With the increase of current density from 1 to 5 A g−1, the C3H9IS/TZ-COFs electrode still holds a high capacity of 762 mA h g−1, suggesting the inhibition of the iodine species shuttle and improved conversion kinetics during (dis)charging. The high capacity and average discharge voltage endow the C3H9IS/TZ-COFs electrode with a record high energy density of 1464 W h kg−1 (based on the mass loading of C3H9IS in the electrode, Fig. 2c),7,8,14,15,17,18,26,42–48 rendering it the state-of-the-art Zn–I2 battery electrode (Table S1, ESI†). Significantly, the C3H9IS/TZ-COFs electrode delivers a high-capacity retention of 83.6% over 1200 cycles (Fig. 2d), outperforming C3H9IS/N-COFs (30% over 540 cycles) and C3H9IS/AC electrodes (10% over 420 cycles). Compared with soluble C3H9IS/N-COFs and C3H9IS/AC electrodes, the C3H9IS/TZ-COFs electrode prevents the iodine shuttle and thus promotes iodine conversion for activating better capacity storage. Furthermore, the electrochemical impedance spectra after cycling also display a small diffusion resistance (Rct) for the C3H9IS/TZ-COFs electrode (Fig. S8, ESI†), implying rapid redox kinetics of iodine species in TZ-COFs.
 | ||
| Fig. 2 Electrochemical metrics of Zn–I2 batteries. (a) GCD curves. (b) Rate capacities. (c) Contour plots of energy density compared with reported Zn–I2 batteries. (d) Cycling stability. (e) CV curves at various scan rates. (f) Capacitive and diffusion-controlled contribution of the C3H9IS/TZ-COFs electrode at various scan rates. | ||
The charge storage kinetics of the C3H9IS/TZ-COFs electrode was further studied by Dunn's method.49–53 Cyclic voltammetry (CV) profiles distinctly exhibit three pairs of redox signals (denoted as PR1, PR2, PR3, PO1, PO2 and PO3, Fig. 2e), indicative of a multi-step electrochemistry. The peak shape is almost the same with the increase of the scan rate from 0.1 to 0.5 mV s−1, substantiating the superb electrochemical reversibility and ion diffusion kinetics. The relationship between current (i) and scan rate (v) can be expressed as i = kvb, where k is constant.54–56 Plotting log(i) against log(v) yields high power-exponent b values of 0.92–0.84 for the eight redox peaks (Fig. S10, ESI†), signifying the fast surface-controlled charge-storage kinetics. Almost 95% of the total stored charge is contributed by the surface redox reaction at 0.5 mV s−1 (Fig. S11, ESI†), along with the slight diffusion-limited process (5%). With incremental scan rate, the capacitive contribution dominates the diffusion-limited contribution, and gradually increases from 83 to 95% (Fig. 2f). This contributes to the surface-dominated capacitive charge storage for fast charge carrier transport in the TZ-COFs/C3H9IS electrode, which well explains its superior high-rate capability as reflected in the GCD curves (Fig. 2a).
To verify the redox mechanism of the C3H9IS/TZ-COFs electrode in the Zn(OTF)2-MPIBr electrolyte, ex situ Raman spectra were performed to reveal its structural evolution at different voltage states (Fig. 3a and b).18,57,58 At the initial state, a signal at 110 cm−1, the vibrational peak of C3H9S−I, corresponds to I−, which confirms the presence of C3H9IS in TZ-COFs. Upon increasing the voltage to 0.54 V, a new peak is identified at 182 cm−1 ascribed to I2,1,18,46 indicating the progress of the first redox stage for I−/I2 (corresponding to 0.54/0.41 V in CV profiles). When I− ions are oxidized into iodine at the electrode, it spontaneously reacts with I− forming I3− species (2I− − 2e− → I2, I2 + I− → I3−). Given the fact that I3− cannot be effectively distinguished by the Raman spectrum because it is roughly at the same position as the I− peak, we further confirm the generated I− through the UV-vis spectra (325 nm for I3−, Fig. 3c). As the voltage increases to 0.93 V, a distinctive peak appears at 214 cm−1, which is associated with the formation of IBr species, suggesting the I2/IBr redox reaction.17 During charging to 1.20 V, two new signals emerge at 310/320 cm−1 and 780 cm−1 ascribed to IO3− species, confirming the I+ to IO3− conversion.18,59,60 Overall, a multi-step multielectron redox reaction can be clarified for the C3H9IS/TZ-COFs electrode, involving I−/I2 (0.54/0.41V), I2/I+ (0.93/0.80V) and I+/IO3− (1.20/1.03 V) conversions. During subsequent discharging, all peaks return to their initial levels, suggesting the highly reversible I−/I0/I+/I5+ conversion redox chemistry. In the ex situ UV-vis spectra of the C3H9IS/TZ-COFs electrode at different states (Fig. 3c), a prominent peak at 225 nm at the initial state is observed, which corresponds to I− from C3H9IS. As the voltage increases to 0.54 V, new peaks at 287/445 nm appear, indicating the presence of I2 species.1 As the voltage increases to 0.93 V, new peaks at 254 and 205 nm appear, indicating the presence of IBr species and the formation of IO3− species, respectively.17,18 Furthermore, the charging of the cell gives rise to the formation of Br2 which is ascribed to the oxidation of IBr interhalogen. Upon charging to 1.20 V, the signal peak of IO3− at 205 nm becomes stronger, while the characteristic peak of I+ disappears, indicating I+ to IO3− conversion.
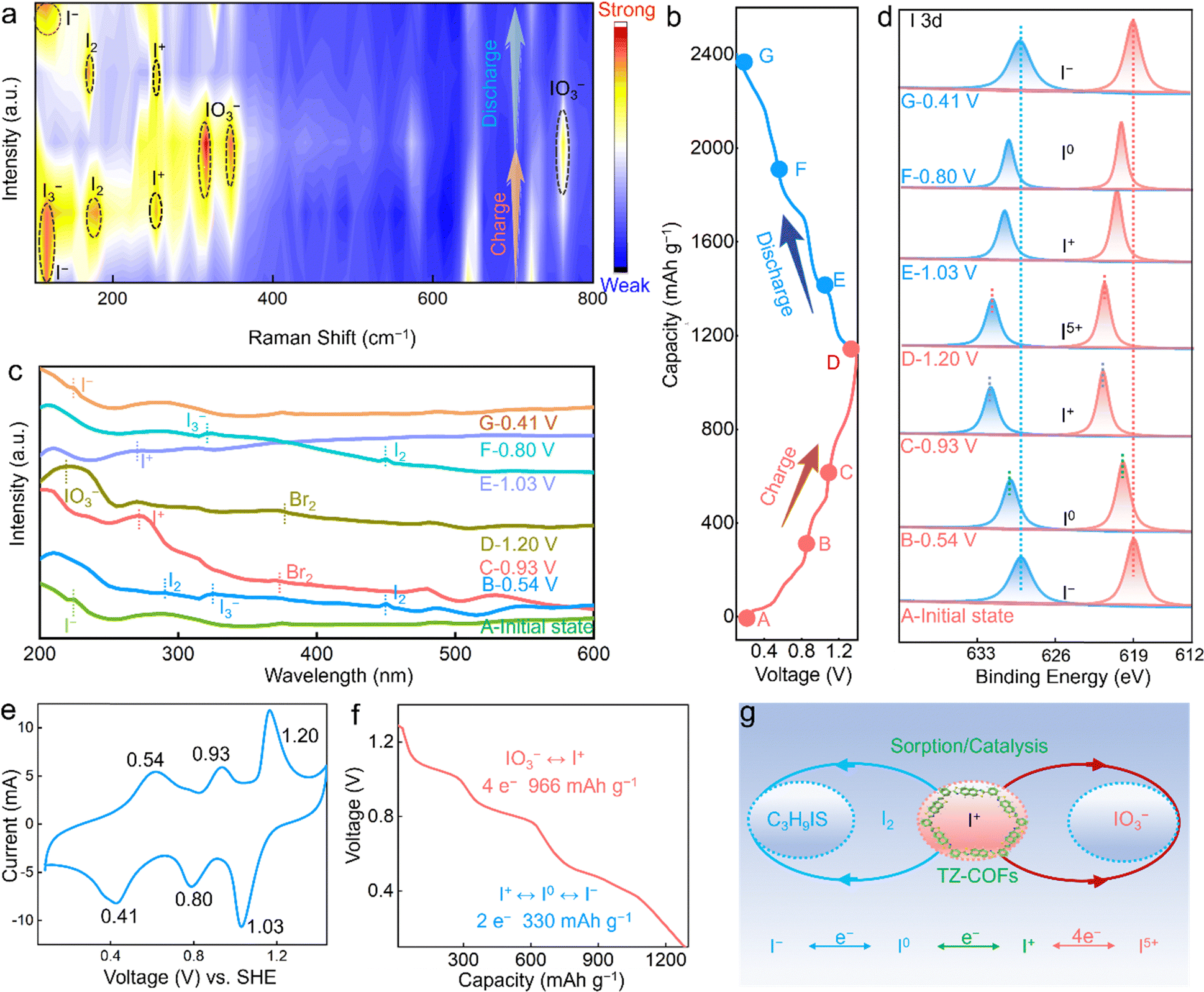 | ||
| Fig. 3 Iodine redox conversion mechanism. (a) Raman spectra. (b) GCD curves. (c) UV-vis spectra. (d) High-resolution I 3d XPS spectra of the C3H9IS electrode at various charge/discharged states. (e) CV curve of the C3H9IS/TZ-COFs electrode. (f) GCD curves of the C3H9IS/TZ-COFs electrode at 1 A g−1. (g) Iodine redox chemistry during the operation of Zn–I2 batteries. | ||
The valence states of iodine species in the C3H9IS/TZ-COFs electrode were further evidenced by X-ray photoelectron spectroscopy (XPS) at different voltages (Fig. 3d). Two signals appearing at high binding energies of 628.4/619.6 eV at 0.54 V correspond to I2 species. With the deepening of the charging to 0.93 V, higher binding energy peaks at 630.1/622.4 eV can be observed, which originate from the I+ ion. Upon further charging to 1.20 V, the binding energy peaks of iodine species continuously shift, accompanied by suggesting the formation of I5+ ions. These results indicate a highly reversible I−/I2/I+/I5+ redox reaction, and are consistent with Raman spectral results. The changes on the surface of the C3H9IS/TZ-COFs electrode during charging–discharging process were also inspected using ex situ SEM images (Fig. S12, ESI†). During initial charging to 0.54 V, bulk I2 particles can be observed on the electrode. Formed oil droplets upon further charging indicate the presence of IO3−. Upon further charging to 1.20 V, most IO3− droplets are converted into mist matter that covers the electrode surface. Upon discharge, the misty matter gradually disappears, and the bulk I2 species are regenerated. Ex situ Raman spectra (Fig. 3a) and XPS spectra (Fig. 3d) confirm the generation of I+ species, implying the formation of IBr intermediates (I− + Br− → I+Br− + 2e−) during multielectron I−/IO3− iodine conversion.
The CV curve of the C3H9IS/TZ-COFs electrode based on the Zn(OTF)2-MPIBr electrolyte exhibits three pairs of redox peaks at 0.54/0.41, 0.93/0.80, and 1.20/1.03 V corresponding to the I−/I2, I2/I+, and I+/IO3− couples (Fig. 3e and Fig. S13, ESI†), respectively. Specifically, the C3H9IS/TZ-COFs electrode initiates 2 e− conversion from I+ to I− to deliver a capacity of 330 mA h g−1 and 4 e− IO3−/I+ conversion to deliver an ultrahigh capacity of 966 mA h g−1 (Fig. 3f) with a discharge potential of 1.13 V of the Zn–I2 system, exhibiting 97% iodine conversion efficiency. The self-discharge behavior of the Zn–I2 battery is observed under the fully charged condition of 1.3 V, which shows a high-capacity retention of 93% after a rest step of 3 days, indicating good anti-dissolution and slight shuttle behavior (Fig. S14, ESI†). By contrast, three redox signals for I− to IO3−conversion in the C3H9IS/AC electrode were observed in the first curve, but this conversion is irreversible in the second cycle, highlighting the key role of TZ-COFs in catalyzing reversible 6e− conversion from I− to IO3− (Fig. S15–S17, ESI†). The CV curve of the Zn‖I2/TZ-COFs battery shows only a pair of redox peaks at 0.54/0.42 V (Fig. S18, ESI†), corresponding to a typical I−/I2 conversion reaction, which contributes a capacity of 154 mA h g−1 at 1 A g−1.[1e, 2c, 7a] The excellent performance of the C3H9IS/TZ-COFs electrode highlights the key role of the C3H9IS/TZ-COFs electrode in the MPIBr-containing electrolyte to stimulate I−/I0/I+/I5+ iodine conversion chemistry with high electrochemical efficiency and stability (Fig. 3g and Fig. S19–S21, ESI†).
The binding energies of I2 and C3H9IS with H2O were calculated (Fig. S22, ESI†). C3H9IS/H2O interaction (−0.81 eV) is stronger than that of I2/H2O (−0.42 eV). Compared with I2, C3H9IS in TZ-COFs can start multielectron iodine conversion with much lower binding energy due to the more easily exposed I− center to combine with the oxygen in H2O to form HIO3.61,62 Therefore, the difference in reaction energy barriers between I2 and C3H9IS is partly responsible for different redox voltages of the C3H9IS/TZ-COFs electrode. However, the reasons for this large difference in iodine conversion voltages are complicated and still need a further extensive and in-depth study. Overall, a highly reversible I−/I2/I+/I5+ redox reaction is triggered in the C3H9IS/TZ-COFs electrode (Fig. 3g), contributing to record high capacity and energy density. The whole conversion of iodine species can be expressed as follows:
| I− − e− ↔ I0 0.54 V | (step 1) |
| I0 + Br− − e− ↔ I+Br− 0.93 V | (step 2) |
| I+ + 3H2O − 4 e− ↔ IO3− + 6H+ 1.20 V | (step 3) |
Generally, the dissolution and shuttling issues of soluble IO3− species from electrodes hinder the cycling durability of Zn–I2 batteries.63 In this regard, CN/C−S motifs of thiazole units in TZ-COFs are favorable for binding iodine species to relieve their loss during battery operation (Fig. S23, ESI†). TZ-COFs coordinate with IO3− through CN/C−S motifs of thiazole units to ensure the redox reversibility, and catalyze a new reversible 6e− conversion of I− to IO3− to further boost capacity (Fig. S24 and S25, ESI†). Density functional theory (DFT) calculations were performed to understand in-depth the binding affinity between TZ-COFs and iodine species (Fig. 4a and b). The adsorption energies of TZ-COFs with I−, I2, I3−, I+ and IO3− (from −5.59 to −0.012 eV) are lower than those of N-COFs (from −5.22 to −0.011 eV), suggesting the strong interaction between TZ-COFs and iodine species. Of note, TZ-COFs show a more negative adsorption energy with IO3− than N-COFs, highlighting the significant effect of thiazole units of TZ-COFs in catalyzing reversible I−/IO3− conversion.
 | ||
| Fig. 4 Theoretical simulation of the iodine conversion process at different electrochemical states. (a) and (b) The optimized adsorption configurations of TZ-COFs and COFs. (c) HOMO and LUMO of TZ-COFs and N-COFs. (d) Interaction between the iodine species and TZ-COFs monomers. (e) The optimized charge-density-difference isosurfaces of IO3− adsorption on TZ-COFs. (f) Calculated cohesive energies of iodine species during TZ-COFs and AC. (g) Calculated Ea values of the C3H9IS/TZ-COFs electrode and the C3H9IS/AC electrode. | ||
Fig. 4c shows the energy levels of the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO) for TZ-COFs and N-COFs.40,64–66 N-COFs (only containing CN groups) with structures similar to TZ-COFs involving CN/C−S groups were designed as an example for comparison to study the role of catalytic sites. The energy gap (ΔE) of TZ-COFs is calculated to be 3.14 eV (Fig. 4c), which is lower than that of N-COFs (3.31 eV). It indicates that synergistic CN/C−S sites are more favorable for boosting iodine conversion with low energy barriers. Consequently, the C3H9IS/TZ-COFs electrode delivers higher conversion efficiency/cycling stability (97%/1200 cycles, Fig. 2d) than C3H9IS/N-COFs (94%/540 cycles). Furthermore, MEP simulation and charge density difference isosurfaces suggest a strong chemical interaction between TZ-COF and IO3− (Fig. 4d and e), accompanied by electron accumulation (green area) and depletion (yellow area) to form a stable configuration for triggering improved electrochemical activity and durability.
The role of TZ-COFs in activating and stabilizing the 6e− transfer redox reaction from I− to IO3− can be further understood from DFT calculations.67,68 The Gibbs free energy (ΔG) values of iodine species in TZ-COFs at various electrochemical states are negative than that of AC (Fig. 4f), suggesting an energy-favorable 6e− I−/I2/I+/IO3− redox conversion with low energy barriers. Of note, TZ-COFs catalyze a reversible 6e− conversion from I− to IO3− due to reduced energy barriers (−5.1 vs. −3.5 eV in the AC host). Besides, the activation energy (Ea) is 0.21 eV for the C3H9IS/TZ-COFs electrode (Fig. 4g and Fig. S26, S27, ESI†) based on the Arrhenius equation, which is much lower than that of the C3H9IS/AC electrode (0.38 eV). TZ-COFs activate high-kinetics interfacial charge mobility and redox reactions to catalyze a highly reversible I−/I2/I+/IO3− conversion reaction.
Overall, the C3H9IS/TZ-COFs electrode in the Zn(OTF)2-MPIBr electrolyte exhibits high-efficiency and stable 6e− iodine conversion chemistry. Compared with inorganic symmetric I2 molecules, the more easily exposed I− center of polar C3H9IS combines with the oxygen in H2O to form HIO3, due to the lower binding energy of C3H9IS/H2O (−0.81 vs. −0.42 eV for I2/H2O), which initiates I−/IO3− conversion. Meanwhile, thiazole units of TZ-COFs enable strong chemical adsorption with IO3− species to improve redox stability with high reversibility due to reduced energy barriers (−5.1 vs. −3.5 eV in the AC host) and upgraded conversion kinetics (activation energy: 0.21 vs. 0.38 eV in AC). Owing to the halogen reaction between I− species and Br− halides, MPIBr can effectively activate I+ to reduce the oxidation/reduction potential gap (0.39 V), which constitutes an important step to propel I−/I0/I+/I5+ conversion with a lower energy barrier (0.21 eV). The synergy of C3H9IS, TZ-COFs and MPIBr endows the Zn–I2 batteries with ultrahigh capacity and energy density, and superior cycling stability, constituting a major advance in the design of better Zn–I2 batteries.
Conclusion
In summary, a high conversion-efficiency and stable 6e− I−/I2/I+/IO3− iodine conversion chemistry are proposed, which are enabled by the TZ-COFs/C3H9IS electrode in the Zn(OTF)2-MPIBr electrolyte for high-performance Zn–I2 batteries. Compared with inorganic I2 molecules, owing to a more easily exposed I− center to combine with the oxygen in H2O to form HIO3, C3H9IS with a polar iodine center through I+ activation of MPIBr initiates 6e− I−/IO3− conversion to reduce the oxidation/reduction potential gap to show 97% iodine conversion efficiency. Meanwhile, CN/C−S groups of thiazole units in TZ-COFs enable strong chemical adsorption with IO3− species to improve redox stability. Besides, thiazole units of TZ-COFs catalyze a reversible 6e− conversion from I− to IO3− due to reduced energy barriers and boosted conversion kinetics. Consequently, the TZ-COFs/C3H9IS electrode delivers a record high capacity and energy density, and long life, becoming the state-of-the-art Zn–I2 battery electrode in comprehensive performances. This work provides a new avenue to develop efficient and stable multielectron iodine conversion chemistry for building better aqueous Zn–I2 batteries.
Data availability
The data that support the findings of this study are available upon reasonable request from the corresponding author.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22272118, 22172111, and 22309134), the Science and Technology Commission of Shanghai Municipality, China (No. 22ZR1464100, 20ZR1460300, and 19DZ2271500), China Postdoctoral Science Foundation (2022M712402), Shanghai Rising-Star Program (23YF1449200), Zhejiang Provincial Science and Technology Project (2022C01182), and the Fundamental Research Funds for the Central Universities (2023-3-YB-07).References
- F. Zhu, Z. Li, Z. Wang, Y. Fu and W. Guo, J. Am. Chem. Soc., 2024, 146, 11193–11201 CAS.
- S. Bi, H. Wang, Y. Zhang, M. Yang, Q. Li, J. Tian and Z. Niu, Angew. Chem., Int. Ed., 2023, 62, e202312982 CrossRef CAS PubMed.
- J. Hao, L. Yuan, Y. Zhu, X. Bai, C. Ye, Y. Jiao and S. Z. Qiao, Angew. Chem., Int. Ed., 2023, 62, e202310284 CrossRef CAS PubMed.
- P. Li, Y. Wang, Q. Xiong, Y. Hou, S. Yang, H. Cui, J. Zhu, X. Li, Y. Wang, R. Zhang, S. Zhang, X. Wang, X. Jin, S. Bai and C. Zhi, Angew. Chem., Int. Ed., 2023, 62, e202303292 CrossRef CAS PubMed.
- N. Li, Z. Yang, Y. Li, D. Yu, T. Pan, Y. Chen, W. Li, H. Xu, X. Guo and H. Pang, Adv. Energy Mater., 2024, 14, 2402846 CrossRef CAS.
- C. Wang, G. Gao, Y. Su, J. Xie, D. He, X. Wang, Y. Wang and Y. Wang, Nat. Commun., 2024, 15, 6234 CrossRef CAS PubMed.
- C. Wang, X. Ji, J. Liang, S. Zhao, X. Zhang, G. Qu, W. Shao, C. Li, G. Zhao, X. Xu and H. Li, Angew. Chem., Int. Ed., 2024, 63, e202403187 CrossRef CAS PubMed.
- M. Wang, Y. Meng, M. Sajid, Z. Xie, P. Tong, Z. Ma, K. Zhang, D. Shen, R. Luo, L. Song, L. Wu, X. Zheng, X. Li and W. Chen, Angew. Chem., Int. Ed., 2024, 63, e202404784 CrossRef CAS PubMed.
- J. He, Y. Mu, B. Wu, F. Wu, R. Liao, H. Li, T. Zhao and L. Zeng, Energy Environ. Sci., 2024, 17, 323–331 RSC.
- L. Xiang, S. Yuan, F. Wang, Z. Xu, X. Li, F. Tian, L. Wu, W. Yu and Y. Mai, J. Am. Chem. Soc., 2022, 144, 15497–15508 CrossRef CAS PubMed.
- Q. Chen, S. Chen, J. Ma, S. Ding and J. Zhang, Nano Energy, 2023, 117, 108897 CrossRef CAS.
- J.-L. Yang, H.-H. Liu, X.-X. Zhao, X.-Y. Zhang, K.-Y. Zhang, M.-Y. Ma, Z.-Y. Gu, J.-M. Cao and X.-L. Wu, J. Am. Chem. Soc., 2024, 146, 6628–6637 CrossRef CAS PubMed.
- S. J. Zhang, J. Hao, H. Li, P. F. Zhang, Z. W. Yin, Y. Y. Li, B. Zhang, Z. Lin and S. Z. Qiao, Adv. Mater., 2022, 34, 2201716 CrossRef CAS PubMed.
- Y. Zou, T. Liu, Q. Du, Y. Li, H. Yi, X. Zhou, Z. Li, L. Gao, L. Zhang and X. Liang, Nat. Commun., 2021, 12, 70 CrossRef PubMed.
- S. Lv, T. Fang, Z. Ding, Y. Wang, H. Jiang, C. Wei, D. Zhou, X. Tang and X. Liu, ACS Nano, 2022, 16, 20389–20399 CrossRef CAS PubMed.
- X. Jin, L. Song, C. Dai, Y. Xiao, Y. Han, X. Li, Y. Wang, J. Zhang, Y. Zhao, Z. Zhang, N. Chen, L. Jiang and L. Qu, Adv. Mater., 2022, 34, 2109450 CrossRef CAS PubMed.
- W. Ma, T. Liu, C. Xu, C. Lei, P. Jiang, X. He and X. Liang, Nat. Commun., 2023, 14, 5508 CrossRef CAS PubMed.
- C. Xie, C. Wang, Y. Xu, T. Li, Q. Fu and X. Li, Nat. Energy, 2024, 9, 714–724 CrossRef CAS.
- X. Li, W. Xu and C. Zhi, Nat. Rev. Chem., 2024, 8, 359–375 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, M. Yu, Y. Jiao and Y. Huang, Nat. Commun., 2022, 13, 6489 CrossRef CAS PubMed.
- T. Liu, C. Lei, H. Wang, C. Xu, W. Ma, X. He and X. Liang, Sci. Bull., 2024, 69, 1674–1685 CrossRef CAS PubMed.
- X. Li, S. Wang, D. Zhang, P. Li, Z. Chen, A. Chen, Z. Huang, G. Liang, A. L. Rogach and C. Zhi, Adv. Mater., 2023, 36, 2304557 CrossRef PubMed.
- M. Liu, Q. Chen, X. Cao, D. Tan, J. Ma and J. Zhang, J. Am. Chem. Soc., 2022, 144, 21683–21691 CrossRef CAS PubMed.
- F. Wei, T. Zhang, H. Xu, Y. Peng, H. Guo, Y. Wang, S. Guan, J. Fu, C. Jing, J. Cheng and S. Liu, Adv. Funct. Mater., 2024, 34, 2310693 CrossRef CAS.
- Z. Chen, F. Wang, R. Ma, W. Jiao, D. Li, A. Du, Z. Yan, T. Yin, X. Yin, Q. Li, X. Zhang, N. Yang, Z. Zhou, Q.-H. Yang and C. Yang, ACS Energy Lett., 2024, 9, 2858–2866 CrossRef CAS.
- C. Guo, Y. Cao, Y. Gao, C. Zhi, Y. X. Wang, Y. Luo, X. J. Yang and X. Luo, Adv. Funct. Mater., 2024, 34, 2314189 CrossRef CAS.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- L. Huang, W. Li, F. Wei, S. Ke, H. Chen, C. Jing, J. Cheng and S. Liu, Chem, 2024, 10, 3103 Search PubMed.
- Z. Wei, Z. Huang, G. Liang, Y. Wang, S. Wang, Y. Yang, T. Hu and C. Zhi, Nat. Commun., 2024, 15, 3841 CrossRef CAS PubMed.
- Y. Wang, C. Lei, W. Guan, W. Shi, R. Shen, S. X.-A. Zhang and G. Yu, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2401060121 CrossRef CAS PubMed.
- X. Li, M. Li, Z. Huang, G. Liang, Z. Chen, Q. Yang, Q. Huang and C. Zhi, Energy Environ. Sci., 2021, 14, 407–413 RSC.
- P. Li, S. Yang, J. Zhu, S. Wang, Y. Hou, H. Cui, Z. Chen, R. Zhang, Z. Wu, Y. Wang, Z. Wei, X. Liu, S. Zhang, X. Li and C. Zhi, Matter, 2024, 7, 1867–1878 CrossRef CAS.
- C. Dai, L. Hu, H. Chen, X. Jin, Y. Han, Y. Wang, X. Li, X. Zhang, L. Song, M. Xu, H. Cheng, Y. Zhao, Z. Zhang, F. Liu and L. Qu, Nat. Commun., 2022, 13, 1863 CrossRef CAS PubMed.
- P. Li, X. Li, Y. Guo, C. Li, Y. Hou, H. Cui, R. Zhang, Z. Huang, Y. Zhao, Q. Li, B. Dong and C. Zhi, Adv. Energy Mater., 2022, 12, 2103648 CrossRef CAS.
- S. Wang, Y. Wang, Z. Wei, J. Zhu, Z. Chen, H. Hong, Q. Xiong, D. Zhang, S. Li, S. Wang, Y. Huang and C. Zhi, Adv. Mater., 2024, 36, 2401924 CrossRef CAS PubMed.
- C. Xu, C. Lei, P. Jiang, W. Yang, W. Ma, X. He and X. Liang, Joule, 2024, 8, 461–481 CrossRef CAS.
- L. Zhou, L. Zhao, X. Gao, C. Chi, Z. Qiu, Y. Song, T. Cai, P. Liu, X. Li, Z. Fan, Q. Xue, Z. Yan, Y. Cui and W. Xing, ACS Energy Lett., 2023, 8, 5152–5160 CrossRef CAS.
- R. Yang, W. Yao, L. Zhou, F. Zhang, Y. Zheng, C. S. Lee and Y. Tang, Adv. Mater., 2024, 36, 2314247 CrossRef CAS PubMed.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS PubMed.
- Z. Song, L. Miao, H. Duan, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2024, 63, e202401049 CrossRef CAS PubMed.
- Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309446 CrossRef CAS PubMed.
- Y. Wang, X. Jin, J. Xiong, Q. Zhu, Q. Li, R. Wang, J. Li, Y. Fan, Y. Zhao and X. Sun, Adv. Mater., 2024, 36, 2404093 CrossRef CAS PubMed.
- W. Li, L. Huang, H. Zhang, Y. Wu, F. Wei, T. Zhang, J. Fu, C. Jing, J. Cheng and S. Liu, Matter, 2023, 6, 2312–2323 CrossRef CAS.
- S. Wang, Z. Huang, B. Tang, X. Li, X. Zhao, Z. Chen, C. Zhi and A. L. Rogach, Adv. Energy Mater., 2023, 13, 2300922 CrossRef CAS.
- T. Liu, C. Lei, H. Wang, J. Li, P. Jiang, X. He and X. Liang, Adv. Mater., 2024, 36, 2405473 CrossRef CAS PubMed.
- J. Hu, Z. Zhang, T. Deng, F. C. Cui, X. Shi, Y. Tian and G. Zhu, Adv. Mater., 2024, 36, 2401091 CrossRef CAS PubMed.
- Z. Zhang, W. Ling, N. Ma, J. Wang, X. Chen, J. Fan, M. Yu and Y. Huang, Adv. Funct. Mater., 2023, 34, 2310294 CrossRef.
- W. Li, H. Xu, H. Zhang, F. Wei, T. Zhang, Y. Wu, L. Huang, J. Fu, C. Jing, J. Cheng and S. Liu, Energy Environ. Sci., 2023, 16, 4502–4510 RSC.
- Y. Zhang, P. Chen, Q. Wang, Q. Wang, K. Zhu, K. Ye, G. Wang, D. Cao, J. Yan and Q. Zhang, Adv. Energy Mater., 2021, 11, 2101712 CrossRef CAS.
- Q. Liu, F. Ye, K. Guan, Y. Yang, H. Dong, Y. Wu, Z. Tang and L. Hu, Adv. Energy Mater., 2022, 13, 2202908 CrossRef.
- G. Li, L. Sun, S. Zhang, C. Zhang, H. Jin, K. Davey, G. Liang, S. Liu, J. Mao and Z. Guo, Adv. Funct. Mater., 2023, 34, 2301291 CrossRef.
- Y. Zhang, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2024, 63, e202316835 CrossRef CAS PubMed.
- L.-P. Hou, X.-Q. Zhang, N. Yao, X. Chen, B.-Q. Li, P. Shi, C.-B. Jin, J.-Q. Huang and Q. Zhang, Chem, 2022, 8, 1083–1098 CAS.
- P. Jia, J. Wang, T. Zheng, C. Tao, G. Yila, L. Wang, Y. Wang and T. Liu, Angew. Chem., Int. Ed., 2024, 63, e202401055 CrossRef CAS PubMed.
- X. Zheng, Z. Song, D. Zhang, W. Du, L. Miao, Y. Lv, L. Xie, L. Gan and M. Liu, J. Mater. Chem. A, 2024, 12, 15352–15360 RSC.
- Y. Chen, L. Miao, Z. Song, H. Duan, Y. Lv, L. Gan and M. Liu, Adv. Funct. Mater., 2024, 34, 2409428 CrossRef CAS.
- Y. An, Y. Zhong, T. Sun, H. Wang, Z. Hu, H. Liu, S. Liu, Y. Kong and J. Xu, Dalton Trans., 2019, 48, 13074–13080 RSC.
- K.-P. Shing, B. Cao, Y. Liu, H. K. Lee, M.-D. Li, D. L. Phillips, X.-Y. Chang and C.-M. Che, J. Am. Chem. Soc., 2018, 140, 7032–7042 CrossRef CAS PubMed.
- W. Zong, J. Li, C. Zhang, Y. Dai, Y. Ouyang, L. Zhang, J. Li, W. Zhang, R. Chen, H. Dong, X. Gao, J. Zhu, I. P. Parkin, P. R. Shearing, F. Lai, K. Amine, T. Liu and G. He, J. Am. Chem. Soc., 2024, 146, 21377–21388 CrossRef PubMed.
- A. Y. Ya, Russ. Chem. Rev., 1982, 51, 566 CrossRef.
- H. Pan, B. Li, J. Yang, W. Liu, W. Luo and B. Chen, J. Hazard. Mater., 2023, 460, 132423 CrossRef CAS PubMed.
- X. Guo, H. Xu, Y. Tang, Z. Yang, F. Dou, W. Li, Q. Li and H. Pang, Adv. Mater., 2024, 36, 2408317 CrossRef CAS PubMed.
- K. Wang, H. Li, Z. Xu, Y. Liu, M. Ge, H. Wang, H. Zhang, Y. Lu, J. Liu, Y. Zhang, Y. Tang and S. Chen, Adv. Energy Mater., 2024, 14, 2304110 CrossRef CAS.
- Y. Zhang, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Adv. Funct. Mater., 2024, 34, 2405710 CrossRef CAS.
- Q. Huang, L. Huang, Y. Jin, Y. Sun, Z. Song and F. Xie, Chem. Eng. J., 2024, 482, 148912 CrossRef CAS.
- Q. Huang, Y. Jin, L. Huang, Y. Cong and Z. Xu, J. Mater. Chem. A, 2023, 11, 12297–12307 RSC.
- Q. An, L. Wang, G. Zhao, L. Duan, Y. Sun, Q. Liu, Z. Mei, Y. Yang, C. Zhang and H. Guo, Adv. Mater., 2023, 36, 2305818 CrossRef PubMed.
- H. Duan, K. Li, M. Xie, J.-M. Chen, H.-G. Zhou, X. Wu, G.-H. Ning, A. I. Cooper and D. Li, J. Am. Chem. Soc., 2021, 143, 19446–19453 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00365b |
| This journal is © The Royal Society of Chemistry 2025 |