
A 405 W h kg−1 Ah-level lithium–sulfur pouch battery stabilized over 200 cycles by an electron-triode-like GeS2–NiS2 heterostructure†
Xun
Jiao
a,
Li
Tan
a,
Xiaoxia
Tang
a,
Cheng
Tong
a,
Tao
Wang
*a,
Minhua
Shao
 bc,
Bin
Liu
d,
Cunpu
Li
*ae and
Zidong
Wei
ae
bc,
Bin
Liu
d,
Cunpu
Li
*ae and
Zidong
Wei
ae
aState Key Laboratory of Advanced Chemical Power Sources, School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China. E-mail: wangtaotao@cqu.edu.cn; lcp@cqu.edu.cn
bDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong
cGuangzhou Key Laboratory of Electrochemical Energy Storage Technologies, Fok Ying Tung Research Institute, The Hong Kong University of Science and Technology, Guangzhou 511458, China
dDepartment of Materials Science and Engineering, City University of Hong Kong, Kowloon, Hong Kong
eSuining Lithium Battery Research Institute of Chongqing University (SLiBaC), Suining 629000, China
First published on 18th March 2025
Abstract
Lithium–sulfur batteries (LSBs) form soluble polysulfides (LiPSs) during discharge, leading to decline in cycling performance, especially the failure of pouch batteries. The failure may be due to the fact that conventional sulfur hosts can only adsorb LiPSs and cannot rapidly inject and transfer electrons in electrochemical reactions. The sluggish electrochemical interconversion of LiPSs leads to continuous loss of active sulfur materials, which is a barrier to long-life commercial LSBs. Herein, an electron-triode-like GeS2–NiS2 heterostructure is successfully designed and synthesized to serve as a catalytic sulfur host. An Ohmic contact rather than a Schottky contact is formed between GeS2 and NiS2, which is proven using the ultraviolet photoelectron spectra and X-ray absorption fine structure spectra. Therefore, the LiPSs can be interconverted with an electron-triode-like model: NiS2 acts as the emitter and injects a batch of electrons into the LiPSs (the collector) collectively through the GeS2 base electrode, with a maximum reaction current amplification factor (βR) of 105.87. In situ XRD and ex situ AFM indicate that the collective injection of electrons can achieve an earlier deposition of Li2S as early as ∼80% of SOC. Ultimately, the S@GeS2–NiS2/rGO battery achieves a high specific capacity of 1007.8 mA h g−1 at 0.5C. The 1.2 Ah pouch battery can achieve a high energy density of 405 W h kg−1 and work stably for 200 cycles, highlighting its great potential for practical applications.
Broader contextThe decarbonization of transportation and grid infrastructure urgently demands electrochemical energy storage systems transcending the 350 W h kg−1 threshold. In addition, in order to reduce the dependence on heavy metals such as Co, a sulfur material, which is abundant in resources, has become a widely researched cathode. While lithium–sulfur batteries (LSBs) theoretically satisfy this demand through sulfur's ultrahigh capacity (1675 mA h g−1) and global abundance (≈5% of terrestrial crust), their commercialization is hindered by two interlinked sustainability challenges, such as shuttling losses of lithium polysulfides (LiPSs) and excessive electrolyte usage (E/S ratio > 5 μL mg−1), contradicting green chemistry principles. Addressing these challenges, we propose a GeS2–NiS2 heterostructured sulfur host mimicking solid-state triode operation: the Ohmic contact creates an electron-rich region at the interface of the heterostructure, to rapidly inject batch of electrons into soluble LiPSs. The electron injection effect enables a LiPS conversion reaction current amplification factor (βR) of 105.87. Therefore, earlier Li2S nucleation from 20% to 80% of SOC via the electron-triode-like GeS2–NiS2 heterostructure. The developed 1.2 Ah pouch batteries deliver a 405 W h kg−1 energy density, which is one of the available methods to achieve these targets, and the state-of-the-art 200 cycles with 81.2% capacity retention under lean electrolyte conditions. |
1. Introduction
The quest for low-cost and high-energy-density devices has prompted extensive research into high-quality rechargeable batteries beyond traditional lithium-ion batteries.1–3 Among the prospective candidates, lithium–sulfur batteries (LSBs) have the advantages of high theoretical specific capacity (1675 mA h g−1), abundant sulfur source, and inexpensive cost.4–7 Nevertheless, the application of LSBs is still hindered by poor cycling stability due to their intrinsic defects.8–10 The inherently low electronic conductivity of sulfur and its product Li2S results in slow redox kinetics and low rate capacity. The intermediate products of the reaction, soluble lithium polysulfides (LiPSs), are prone to migrate from the cathode to the anode in the liquid electrolyte, thereby corroding lithium metal and leading to an irreversible loss of active materials.11–14 Consequently, LSBs suffer from insufficient reaction kinetics, poor cycle life, and rapid capacity decay during cycling.To tackle these issues, various catalysts have been developed as sulfur hosts.15–19 These studies have successfully strengthened the ability to adsorb LiPSs, thereby extending the life-span of LSBs.20–24 However, in a practical high-load lithium–sulfur pouch battery, the electrons will not transfer/distribute evenly in the electrodes: the electrons will concentrate at the edge regions and the tab-near regions (Scheme 1(a)).25 The uneven electron distribution should be responsible for the failure of the pouch battery. Taking discharge as an example, S8 will undergo multiple soluble LiPSs, including Li2S8, Li2S6, and Li2S4, and ultimately form the final solid product Li2S. Therefore, LiPSs will generate unevenly, and will dissolve in the electrolyte and migrate to the anode side, eventually leading to rapid capacity degradation and long-life cycle failure of the pouch battery.26–28
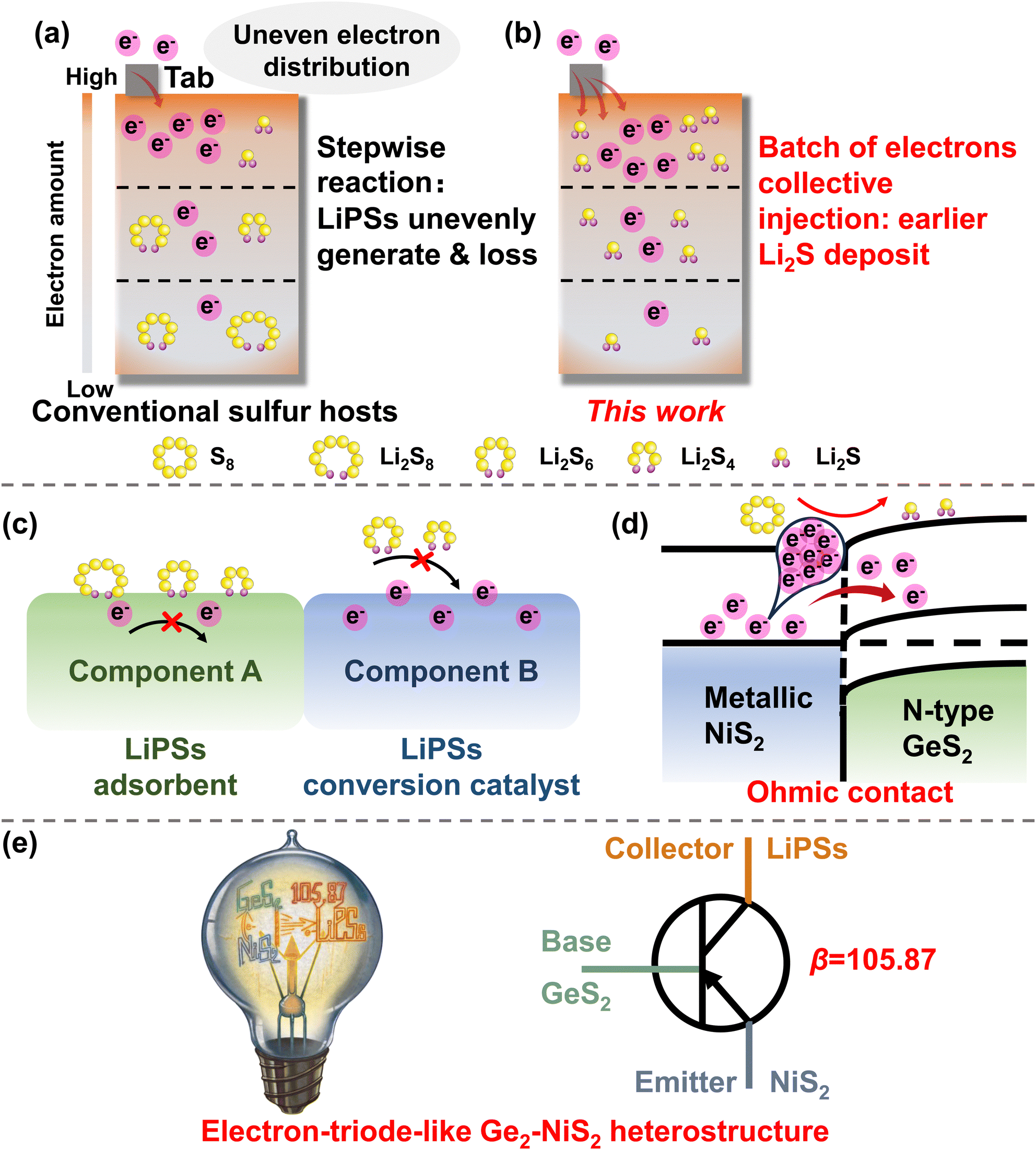 | ||
| Scheme 1 (a) and (b) Schematic diagram of the lithium–sulfur pouch battery, and the current will result in an uneven electron distribution manner because of the continuous reaction of the transferred electrons. (a) LiPSs are generated unevenly due to the uneven distribution of electrons. (b) This work develops a method to inject a batch of electrons from the electrode to LiPSs, to accelerate the multi-electron experience through soluble LiPSs, achieving earlier Li2S deposition. (c) Conventional heterostructures work separately: one component is used as the LiPS adsorbent and the other is used as the LiPS conversion catalyst. (d) In this work, an electron-triode-like GeS2–NiS2 heterostructure with an Ohmic contact forms an electron-rich region at the heterointerfaces to collectively inject into S8 to obtain Li2S without the accumulation of soluble LiPSs. (e) Electron-triode-like GeS2–NiS2 heterostructure achieves 105.87 times current amplification (βR = 105.87) during cycling, greatly retarding the accumulation of soluble LiPSs. | ||
As the unevenly distributed electrons are inevitable, it is important to develop methods to inject a batch of electrons from the electrode to the LiPSs, to accelerate the multi-electron experience through the soluble LiPSs, and realize an earlier Li2S deposition (Scheme 1(b)). In this regard, we designed and synthesized an electron-triode-like GeS2–NiS2 heterostructure to realize the collective injection of electrons by amplifying the reaction current. Unlike conventional heterostructures, where one component works as the LiPS adsorbent and the other works as the LiPS conversion catalyst (Scheme 1(c)), the GeS2–NiS2 heterostructure possesses an “Ohmic contact” behavior. In the theory of semiconductor physics, the contacts for the two components in a heterostructure include Schottky contacts and Ohmic contacts.29,30 For Schottky contacts, the interface forms a potential barrier with very low conductivity to electrons, called the electron barrier layer. For Ohmic contacts, the interface has a very low resistance and is a highly conductive region for electrons, called the anti-blocking layer. In the designed electron-triode-like GeS2–NiS2 heterostructure, the “Ohmic contact” between the metallic component (NiS2) and the semiconductor (GeS2), as shown in Scheme 1(d), will form a highly conductive heterointerface. In this electron-rich region, a large number of free-moving electrons are gathered.31 When LiPSs are close to the heterointerface, abundant S–S and Li–S bonds formed can effectively adsorb LiPSs through the orbital effect. The electrons accumulated at the interface can be rapidly transferred into the LiPSs, and the conversion of LiPSs is facilitated by the successive sequential action of electrons. During the electrochemical process, the base GeS2 can effectively adsorb LiPSs, the emitter NiS2 can rapidly inject a batch of electrons, and the collector LiPSs can receive these electrons to amplify the reaction current (βR = 105.87) (Scheme 1(e)). In addition, reduced graphene oxide (rGO) is incorporated into the GeS2–NiS2 heterostructure. rGO has been reported to serve as an excellent electron conductor. Although it possesses a lower work function, its low polarity nature ensures that it does not interfere with the designed Ohmic contact between GeS2 and NiS2, which originates from the orbital interaction between GeS2 and NiS2.32–34
Because of the above advantages, the GeS2–NiS2 heterostructure can support a long-term cycling and achieve robust LBSs. As expected, the S@GeS2–NiS2/rGO cathode battery exhibits a high specific capacity of 878.5 mA h g−1 at a high current density of 2C, and an extremely low-capacity decay of 0.046% per cycle after 1000 long-term cycles. The 1.2 Ah pouch battery constructed with GeS2–NiS2/rGO can achieve a high energy density of 405 W h kg−1 and provide a stable cycling capacity of 200 cycles.
2. Results and discussion
2.1. Structural characterization and analysis
GeS2–NiS2/rGO was synthesized by high-temperature calcination followed by a multiple-step solvothermal method with GeO2 as a Ge source, anhydrous NiCl2 as a Ni source, and anhydrous ethanol as a solvent. The reference samples GeS2/rGO and NiS2/rGO were also synthesized using similar methods with GeO2 as the Ge source and anhydrous NiCl2 as the Ni source, respectively (For details, see the Experimental section in the ESI†). The material ratios were optimized based on the cyclic voltammetry (CV) of symmetric batteries and cycle capacity tests of the GeS2–NiS2/rGO heterostructure (Fig. S1, ESI†). Coupled plasma-optical emission spectroscopy (ICP-OES) was used to determine the ratios of GeS2 and NiS2 in the heterostructure (Table S1, ESI†). The heterostructure with three different ratios (GeS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NiS2 = 1:1, 1:2 and 1:3) were prepared in this work. As shown in Fig. S1a (ESI†), when GeS2:NiS2 = 1:2, the GeS2–NiS2/rGO heterostructure exhibits the best catalytic performance for Li2S6. In addition, the S@GeS2–NiS2/rGO battery has the highest discharge capacity and the best capacity retention when GeS2:NiS2 = 1:2 (Fig. S1b, ESI†). In the heterostructure, electrons are transferred from NiS2 to GeS2, forming an electron-rich region at the interface. NiS2 has high electrical conductivity and can effectively enhance the electron transfer efficiency. However, a 1:1 ratio has an insufficient NiS2 content, and a 1:3 ratio may result in too little GeS2, which affects electron acceptance and structural stability. Therefore, a heterostructure with a 1:2 ratio of GeS2 to NiS2 was chosen as a sulfur host for the study, achieving improved catalytic performance and excellent electrochemical cycling performance.
:NiS2 = 1:1, 1:2 and 1:3) were prepared in this work. As shown in Fig. S1a (ESI†), when GeS2:NiS2 = 1:2, the GeS2–NiS2/rGO heterostructure exhibits the best catalytic performance for Li2S6. In addition, the S@GeS2–NiS2/rGO battery has the highest discharge capacity and the best capacity retention when GeS2:NiS2 = 1:2 (Fig. S1b, ESI†). In the heterostructure, electrons are transferred from NiS2 to GeS2, forming an electron-rich region at the interface. NiS2 has high electrical conductivity and can effectively enhance the electron transfer efficiency. However, a 1:1 ratio has an insufficient NiS2 content, and a 1:3 ratio may result in too little GeS2, which affects electron acceptance and structural stability. Therefore, a heterostructure with a 1:2 ratio of GeS2 to NiS2 was chosen as a sulfur host for the study, achieving improved catalytic performance and excellent electrochemical cycling performance.
The morphology and structure of GeS2–NiS2/rGO are characterized to understand the microstructure and fine heterointerfaces. From Fig. 1(a), we can confirm that the GeS2–NiS2/rGO exists as uniformly dispersed nanoparticles from scanning electron microscopy (SEM). The transmission electron microscopy (TEM) image in Fig. 1(b) further indicates that GeS2–NiS2 nanoparticles (70–90 nm) are firmly attached onto the rGO sheets. The corresponding energy dispersive X-ray (EDX) elemental mappings in Fig. 1(c) show that the elements S, Ni and Ge are distributed evenly. The high-resolution TEM (HRTEM) image (Fig. 1(d)) and corresponding fast Fourier transform (FFT) patterns (Fig. 1(e)) exhibit the crystal structure of the GeS2–NiS2/rGO material. The lattice distances of 0.58 and 0.28 nm can be attributed to the (111) plane of orthorhombic GeS2 (Fdd2) and the (200) plane of cubic NiS2 (Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), respectively.
), respectively.
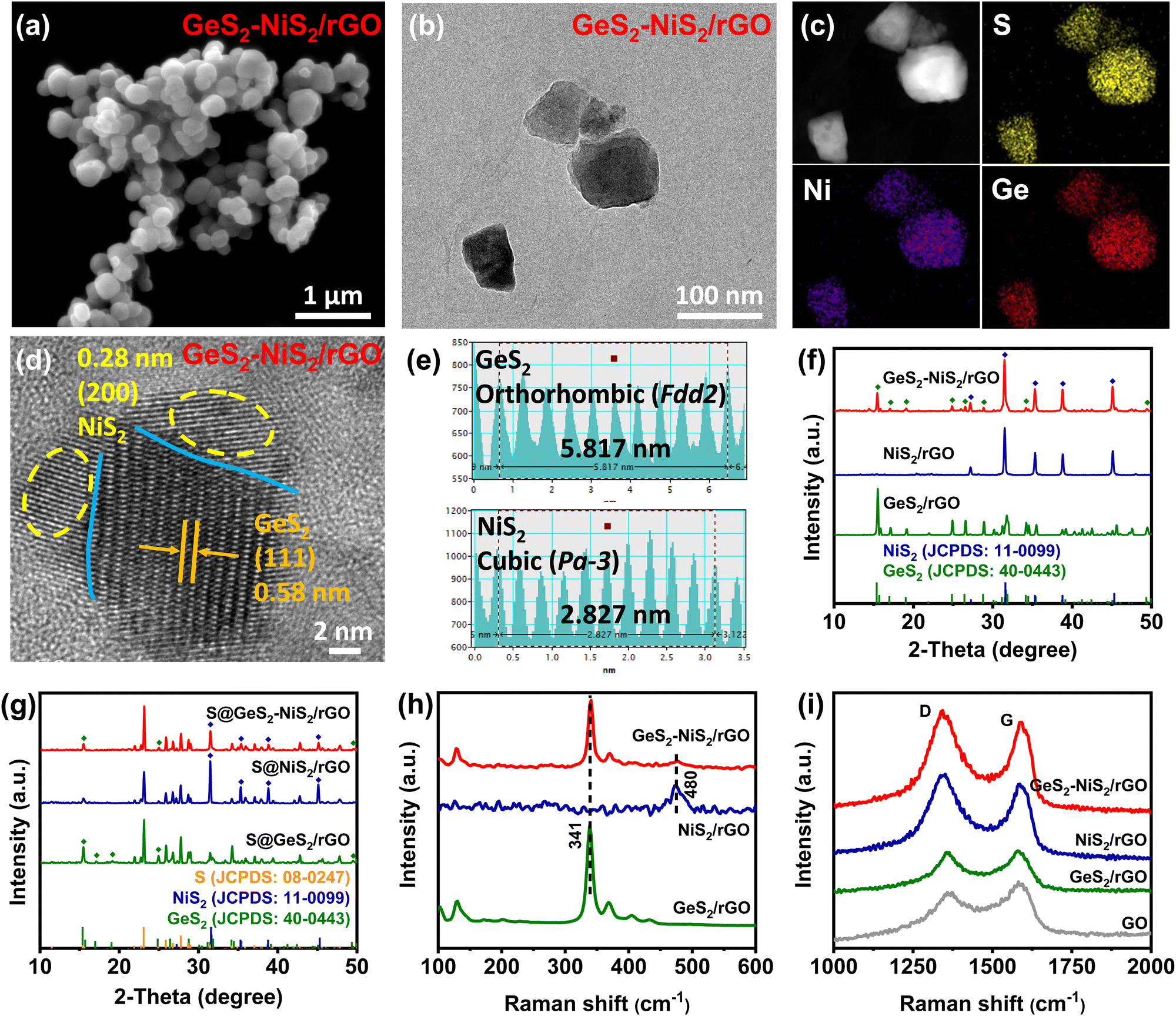 | ||
| Fig. 1 (a) SEM image of GeS2–NiS2/rGO. (b) TEM image and (c) corresponding EDX elemental mappings of GeS2–NiS2/rGO. (d) HRTEM image of GeS2–NiS2/rGO and (e) corresponding reduced FFT patterns. (f) XRD patterns of GeS2–NiS2/rGO, NiS2/rGO and GeS2/rGO. (g) XRD patterns of S@GeS2–NiS2/rGO, S@NiS2/rGO and S@GeS2/rGO. (h) Raman spectra of GeS2–NiS2/rGO, NiS2/rGO and GeS2/rGO. (i) The D-band and G-band of GeS2–NiS2/rGO, NiS2/rGO, GeS2/rGO and GO. | ||
Powder X-ray diffraction (XRD) spectra provide further evidence of the successful synthesis of GeS2–NiS2/rGO. Fig. 1(f) clearly shows the coexistence of GeS2 (JCPDS: 40-0443) and NiS2 (JCPDS: 11-0099) in GeS2–NiS2/rGO.35,36 Moreover, the Rietveld refinement result of the GeS2–NiS2/rGO heterostructure is given in Fig. S2 (ESI†). The calculated refinement reliability parameters of Rwp = 8.76% and Rp = 7.85% indicate that the refinement results are reliable, and the crystal structure is determined to be NiS2 and GeS2 phases. According to the Rietveld refinement results, the content of GeS2 and NiS2 in the heterostructure can be calculated to be 55.1% and 26.4%, respectively, which is consistent with the ICP results. The corresponding diffraction peaks of GeS2 and NiS2 are also exhibited in the XRD pattern after sulfur loading (Fig. 1(g)), which further indicates the successful synthesis of the S@GeS2–NiS2 material.
GeS2–NiS2/rGO, NiS2/rGO and GeS2/rGO are used as sulfur hosts by a melt impregnation method and tested with a thermogravimetric (TG) analyzer (Fig. S3, ESI†). The test results show that the sulfur content of three materials is 72.9%, 70.2% and 73.2%, respectively. The composition and molecular structure of GeS2–NiS2/rGO are further characterized by Raman spectroscopy (Fig. 1(h and i)). Fig. 1(h) shows two typical Raman peaks at 341 and 480 cm−1, corresponding to GeS2 and NiS2, respectively.37,38 Notably, in GeS2–NiS2/rGO, NiS2/rGO and GeS2/rGO, the intensity ratios of the D-band to G-band are all higher than that of GO, which confirms the reduction of GO (Fig. 1(i)).39 The above results suggest the successful formation of the GeS2–NiS2/rGO material.
2.2. Heterointerface states of GeS2–NiS2
To understand the correlation of changes in the electronic structure, density functional theory (DFT) calculations were performed. Density of states (DOS) results are illustrated for GeS2–NiS2 (Fig. 2(a)), NiS2 (Fig. 2(b)) and GeS2 (Fig. 2(c)), respectively. The calculation results indicate that NiS2 has no band gap and exhibits metallic properties, and GeS2 has a significant band gap and is a semiconductor. The GeS2–NiS2 heterostructure exhibits a combination of GeS2 and NiS2, where a more contribution from Ni can be observed at the region near the Fermi level (Ef). The differences in DOS are evident throughout the Ef, where GeS2 shows poor electron transfer ability due to the wide band gap, while NiS2 and GeS2–NiS2 provide high electron conductivity without a band gap across the Ef. Thus, the calculations of DOS indicate that the formation of heterointerfaces in GeS2–NiS2 improves electrical conductivity during sulfur redox reactions.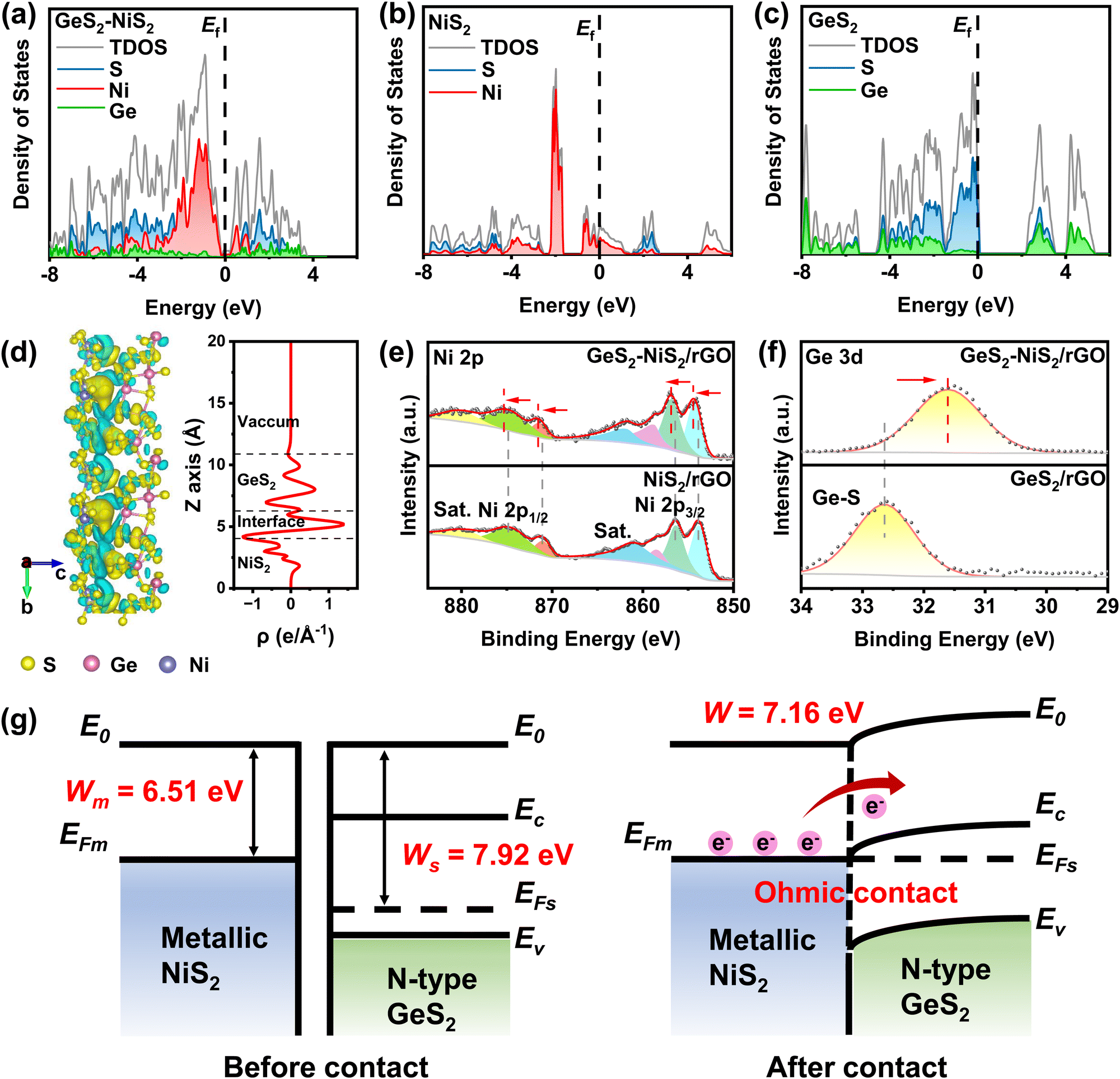 | ||
| Fig. 2 (a)–(c) Calculated pDOS near the Fermi level of GeS2–NiS2 (a), NiS2 (b) and GeS2 (c). The combination of the n-type semiconductor GeS2 with high conductivity NiS2 results in a highly conductive region with a very low contact resistance at the heterointerfaces. (d) Differential charge density and planar average density in the Z-direction of GeS2–NiS2 (yellow: electron accumulation; cyan: electron depletion). The strong bonding interactions of the GeS2–NiS2 heterostructure form rich catalytic heterointerfaces. (e) High-resolution Ni 2p XPS spectra of GeS2–NiS2/rGO and NiS2/rGO. (f) High-resolution Ge 3d XPS spectra of GeS2–NiS2/rGO and GeS2/rGO. The electron transfer from NiS2 to GeS2 in the GeS2–NiS2 heterostructure. (g) Schematic diagram of band structures of NiS2 and GeS2 before and after contacts (Eo: vacuum energy level, EFm: Fermi level, EFs: band gap, Ec: conduction band, and Ev: valence band). The Ohmic contact between GeS2 and NiS2 forms an electron-rich region at the GeS2–NiS2 heterointerfaces. | ||
Based on experimental characterization, GeS2 (111) and NiS2 (200) models were conducted to further clarify the catalytic effect of heterointerfaces at the atomic level by DFT calculations. Fig. 2(d) shows the differential charge density and planar average density in the Z-direction of GeS2–NiS2. Both the interfacial charge gain/loss region and the plane-averaged density amplitude are significant, indicating the electron transfer between GeS2 and NiS2. In the GeS2–NiS2 heterostructure, electron transfer at the interface leads to a change in the center of charge distribution. Bader charge analysis calculated the charge redistribution at the heterointerfaces and quantified an average gain of 0.57 electrons for GeS2 from NiS2. An electron exchange region of approximately 10 Å was identified in the GeS2–NiS2 heterostructure. This finding indicates that the S–S bonds effectively bridge the GeS2 and NiS2 phases through orbital interactions, thereby facilitating the formation of the Ohmic contact.
The charge transfer was also confirmed by comparing the independent X-ray photoelectron spectroscopy (XPS) spectra of NiS2, GeS2, and GeS2–NiS2 (Fig. 2(e, f) and Fig. S4, ESI†). The band corresponding to adventitious C–C at 284.8 eV is used as a reference. The peaks located at 284.8, 286.4 and 288.9 eV are attributed to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O and O–CC, respectively (Fig. S4a, c and e, ESI†).40 For the S 2p high resolution spectrum, the peaks located at 163.0 and 164.3 eV are assigned to the 2p3/2 and 2p1/2 of S2− and S22−, respectively (Fig. S4b, d and f, ESI†).41,42 In the Ni 2p spectrum of GeS2–NiS2/rGO, the fitted XPS peaks at 854.5 and 856.9 eV are Ni 2p3/2, and the peaks at 871.6 and 875.4 eV are Ni 2p1/2, accompanied by three shake-up satellite peaks centered at 858.9, 862.1 and 88.0.6 eV (Fig. 2(e)).43,44 Similar peaks are also observed in the NiS2/rGO material. As expected, compare with NiS2/rGO, significant positive shifts of Ni 2p are observed in the GeS2–NiS2/rGO material. In addition, the Ge 3d spectrum in Fig. 2(f) shows that the peak at 31.6 eV is typical of the bonding of the Ge4+ ion in GeS2.45 Similarly, the Ge 3d spectrum of GeS2–NiS2/rGO shows an obvious negative shift compared to that of GeS2/rGO. These shifts indicate the electron loss of NiS2 when in contact with GeS2 at the heterointerfaces, suggesting the electron gain of GeS2 and the charge transfer at the heterointerfaces, which agrees with the DFT results.
C, C–O and O–CC, respectively (Fig. S4a, c and e, ESI†).40 For the S 2p high resolution spectrum, the peaks located at 163.0 and 164.3 eV are assigned to the 2p3/2 and 2p1/2 of S2− and S22−, respectively (Fig. S4b, d and f, ESI†).41,42 In the Ni 2p spectrum of GeS2–NiS2/rGO, the fitted XPS peaks at 854.5 and 856.9 eV are Ni 2p3/2, and the peaks at 871.6 and 875.4 eV are Ni 2p1/2, accompanied by three shake-up satellite peaks centered at 858.9, 862.1 and 88.0.6 eV (Fig. 2(e)).43,44 Similar peaks are also observed in the NiS2/rGO material. As expected, compare with NiS2/rGO, significant positive shifts of Ni 2p are observed in the GeS2–NiS2/rGO material. In addition, the Ge 3d spectrum in Fig. 2(f) shows that the peak at 31.6 eV is typical of the bonding of the Ge4+ ion in GeS2.45 Similarly, the Ge 3d spectrum of GeS2–NiS2/rGO shows an obvious negative shift compared to that of GeS2/rGO. These shifts indicate the electron loss of NiS2 when in contact with GeS2 at the heterointerfaces, suggesting the electron gain of GeS2 and the charge transfer at the heterointerfaces, which agrees with the DFT results.
Ultraviolet photoelectron spectra (UPS) were used to further characterize the surface electron transfer of GeS2–NiS2/rGO (Fig. 2(g) and Fig. S5, ESI†). According to the UPS results shown in Fig. S5 (ESI†), the work function (EΦ) of NiS2/rGO (6.51 eV) is lower than that of GeS2/rGO (7.92 eV). The Mott–Schottky plot was measured in the dark from an alternating current frequency of 1 KHz to determine the semiconductor type of GeS2 (Fig. S6, ESI†). As shown in Fig. S6 (ESI†), a positive slope of the plot around 0 V can be observed, indicating that GeS2 is an n-type semiconductor.46 According to the theory of semiconductor physics, based on the combination of the n-type semiconductor GeS2 and higher conductivity NiS2, there exists an Ohmic contact between GeS2 and NiS2 without a Schottky barrier.47 The contact resistance of the heterointerfaces is very small, creating an electron-rich region, which reduces the power dissipation of GeS2–NiS2 and increases the electron mobility.48
According to the DFT calculation of adsorption energy (Fig. S7–S10, ESI†), all the sulfur species (Li2S, Li2S2, Li2S4, Li2S6, Li2S8, and S8) have the strongest adsorption capacities (Eb) when adsorbed onto GeS2–NiS2, compared to those when adsorbed onto NiS2 and GeS2, which suggests that the heterointerfaces of GeS2–NiS2 are effective cooperative sites for LiPSs. In the GeS2–NiS2 heterostructure, the electron-rich region is formed at the interfaces by the orbital effect of S–S bonds, which can accumulate a large number of free-moving electrons. In addition, when LiPSs are close to the GeS2–NiS2 heterostructure, the orbital effect of S and Li in LiPSs can form abundant S–S and Li–S bonds with S in the heterostructure (Fig. S7, ESI†). Therefore, the abundant S–S and Li–S bonds at the heterointerface can effectively adsorb LiPSs through the orbital effect. As a result, the electrons accumulated at the heterointerface can be rapidly transferred to LiPSs to promote their catalytic conversion and achieve the rapid generation of Li2S. Therefore, compared to NiS2 and GeS2, the GeS2–NiS2 heterostructure exhibits enhanced adsorption ability for LiPSs (Figure S10, ESI†).
X-ray absorption fine structure (XAFS) spectroscopy is used to further demonstrate the electron transfer behavior in the electron-triode-like GeS2–NiS2 heterostructure. Fig. 3(a) exhibits the X-ray absorption near-edge structure (XANES) spectra of the Ge K-edge in GeO2, Ge foil, GeS2/rGO and GeS2–NiS2/rGO, respectively. Compared with GeS2/rGO, the K-edge of Ge in GeS2–NiS2/rGO shifts towards a lower energy value, suggesting that Ge is reduced to a lower oxidation state, which is consistent with the XPS results for Ge 3d (Fig. 2f).49 In the meantime, as shown in Fig. 3(b), after the formation of the GeS2–NiS2/rGO heterostructure, the Ni K-edge shifts to a higher energy value compared to NiS2/rGO, indicating that Ni loses electrons and is oxidized to a higher state, corresponding to the XPS results of Ni 2p (Fig. 2(e)).50 These results demonstrate that electrons transfer from NiS2 to GeS2 in the heterostructure, forming highly conductive heterointerfaces with free-moving electrons.
 | ||
| Fig. 3 (a) Ge K-edge XANES spectra of GeO2, Ge foil, GeS2/rGO and GeS2–NiS2/rGO. (b) Ni K-edge XANES spectra of NiO, Ni foil, NiS2/rGO and GeS2–NiS2/rGO. Electron transfer from NiS2 to GeS2 in the GeS2–NiS2 heterostructure. (c) Ge R-space EXAFS analysis of GeO2, Ge foil, GeS2/rGO and GeS2–NiS2/rGO. (d) Ni R-space EXAFS analysis of NiO, Ni foil, NiS2/rGO and GeS2–NiS2/rGO. (e) WT contour plots of Ge for Ge foil, GeS2/rGO and GeS2–NiS2/rGO at R-space. (f) WT contour plots of Ni for Ni foil, NiS2/rGO and GeS2–NiS2/rGO at R-space. The successfully formed electron-triode-like GeS2–NiS2/rGO heterostructure forms an electron-rich region, which will facilitate the collective injection of electrons during reaction processes. | ||
According to the R-space of the extended X-ray absorption fine structure (EXAFS) for Ge in Fig. 3(c), the main peak appears near 2.0 Å for GeS2/rGO and GeS2–NiS2/rGO, corresponding to the Ge–S bond.51 The Ge–S peak intensity of GeS2–NiS2/rGO is lower than that of GeS2/rGO, which can be attributed to the formation of S–Ni and Ge–Ni bonds in the GeS2–NiS2/rGO heterostructure. For the R-space EXAFS spectrum of Ni (Fig. 3(d)), the main peak near 2.0 Å corresponds to the Ni–S bond in NiS2/rGO and GeS2–NiS2/rGO.52 As the peak intensity change in Ge–S, the intensity of the Ni–S bond in GeS2–NiS2/rGO is also weaker than that in NiS2/rGO, because of the formation of S–Ge and Ni–Ge bonds in the heterostructure interface. These results indicate the abundant formation of the GeS2–NiS2/rGO heterostructure and the existence of new coordination structures.
The wavelet transform (WT) further proves the spatial structural information in the GeS2–NiS2/rGO heterostructure.53,54 As shown in the WT contours of Ge (Fig. 3(e)), the intensity maximum of the Ge–Ge bond in Ge foil can be observed at k = 9.1 Å−1. In the GeS2/rGO and GeS2–NiS2/rGO samples, the intensity maximum coordination structure can be observed at k = 7.7 Å−1, which can be assigned to the Ge–S bond. Moreover, the GeS2–NiS2/rGO sample presents a new coordination structure at k = 13.8 Å−1, referred to as the formation Ge–Ni bond. Interestingly, compared to the Ge–S bond, the intensity of the newly formed Ge–Ni bond is not significant, indicating that the scattering signal of Ge–Ni is relatively weak, which is due to the electron transfer from NiS2 to GeS2 in the heterostructure. The WT contours of Ni show a maximum intensity at k = 5.3 Å−1 (Fig. 3(f)), corresponding to Ni–S in NiS2/rGO and GeS2–NiS2/rGO. In addition, a new coordination structure, referred to as the Ni–Ge bond, is formed at k = 11.7 Å−1 in the GeS2–NiS2/rGO heterostructure. Unlike the newly formed Ge–Ni bond peak of Ge (Fig. 3(e)) for GeS2–NiS2/rGO, the intensity of the Ni–Ge bond is significant, even comparable to the existing Ni–S bond. This is identical to our electron-triode-like material design, where the GeS2 obtains electrons from NiS2 in the heterostructure; therefore, the scattering ability of Ge in GeS2 is relatively high to produce a higher Ni–Ge signal intensity. These results show that in the electron-triode-like GeS2–NiS2/rGO heterostructure, electrons are transferred from NiS2 to GeS2, and electron-rich heterointerfaces are formed, which will facilitate the collective injection of electrons during electrochemical reactions.
2.3. Electrocatalysis for polysulfide redox
Rapid injection of a batch of electrons can amplify the reaction current during the electrochemical processes, thereby achieving rapid catalytic conversion of LiPSs. CV tests with symmetric batteries were performed using 0.5 M Li2S6 electrolyte within a voltage window of −1.0 to 1.0 V, to analyze the redox kinetics of LiPSs in different sulfur hosts (Fig. 4(a)). The GeS2–NiS2/rGO material shows the highest peak currents and the lowest polarization voltage compared with the NiS2/rGO and GeS2/rGO materials, which indicates that GeS2–NiS2/rGO can significantly promote the catalytic conversion of LiPSs. | ||
| Fig. 4 (a) CV tests for the symmetrical batteries based on GeS2–NiS2/rGO, NiS2/rGO and GeS2/rGO materials with a scan rate of 18 mV s−1. The GeS2–NiS2/rGO material exhibits the highest peak currents and the lowest polarization voltages, promoting the catalytic conversion of LiPSs. (b) Amplified current plot of GeS2–NiS2/rGO versus NiS2/rGO from the CV results of symmetrical batteries. The electron-triode-like GeS2–NiS2/rGO realizes the collective injection of electrons and amplifies the reaction current (βR = 105.87 and βO = 30.24). (c) UV-vis absorption spectra of the adsorbed solutions. (d) Li2S precipitation experiments with the Li2S8 cathode catholyte at 2.05 V of GeS2–NiS2/rGO, NiS2/rGO and GeS2/rGO. (e) Li2S dissolution experiments of three materials at 2.40 V. (f) CV profiles of S@GeS2–NiS2/rGO, S@NiS2/rGO and S@GeS2/rGO cathode batteries in the range of 1.7–2.8 V at a scan rate of 0.1 mV s−1. (g) NTR values of three batteries at different current scanning rates. (h) Galvanostatic discharge/charge profiles of three batteries at a current rate of 0.1C and the corresponding (i) Rct values of the different sample's electrodes. | ||
As the electron injection process from GeS2–NiS2/rGO to LiPSs can be treated like an electron triode, current amplification factors (β) for LiPS reduction and oxidation can be quantitatively calculated with eqn (1) and (2), respectively:
 | (1) |
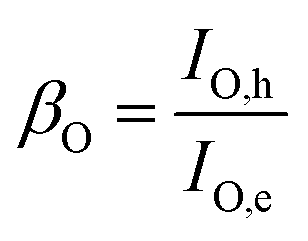 | (2) |
The ultraviolet-visible (UV-vis) absorption spectra of adsorbed solutions can reflect the adsorption capacity of polysulfides.55 As can be seen in Fig. 4(c), the Li2S6 absorption peak intensity of GeS2–NiS2/rGO is the weakest among different materials (GeS2–NiS2/rGO < NiS2/rGO < GeS2/rGO < rGO). Fig. S11 (ESI†) exhibits the optical images of the adsorption ability for the above materials after 1 and 8 h, respectively. The absorption wavelengths of S62− species range from 300 to 500 nm (around 315, 355, and 475 nm).56 In the Li2S6 solution of rGO, the color and the absorbance associated with Li2S6 remain almost unchanged. In the Li2S6 solutions of NiS2/rGO and GeS2/rGO, the adsorption peaks decrease and the solutions become transparent after the adsorption for 8 h. Compared with other samples, GeS2–NiS2/rGO has the lightest color after adsorption, accompany with the weakest adsorption peak from the UV-vis absorption spectra, indicating the strongest adsorption ability for LiPSs.
Except for the adsorption of soluble LiPSs, the ability to convert soluble LiPSs to insoluble Li2S in LSBs is a more important factor for a sulfur host. To study the advantage of the GeS2–NiS2/rGO material on the liquid–solid conversion of LiPSs, the potentiostatic discharge curves at 2.05 V were recorded. The curves were fitted to obtain the deposition of Li2S (Fig. 4(d)) for GeS2–NiS2/rGO, GeS2/rGO, and NiS2/rGO, respectively.57 The peak current of Li2S deposition for GeS2–NiS2/rGO is much higher and appears earlier (0.65 mA at 3151 s) compared with GeS2/rGO (0.18 mA at 5022 s) and NiS2/rGO (0.14 mA at 5145 s). Also, the GeS2–NiS2/rGO material shows the highest Li2S deposition capacity (198.21 mA h g−1), compared with the GeS2/rGO (167.58 mA h g−1) and NiS2/rGO (135.12 mA h g−1) materials, indicating rapid LiPS conversion and effective sulfur utilization by GeS2–NiS2/rGO. In our previous work, it was confirmed that due to the partial lattice matching nature between Fdd2 GeS2 and Fmm Li2S, it can guide the multi-site nucleation and rapid deposition of Li2S.35 Therefore, the Li2S deposition and dissolution capacity of GeS2/rGO is higher than that of NiS2/rGO. Furthermore, the Li2S dissociation is another critical indicator to demonstrate the advantages of GeS2–NiS2/rGO in promoting the reverse conversion of Li2S to S8. As shown in Fig. 4(e), the Li2S dissolution capacity of GeS2–NiS2/rGO (535.92 mA h g−1) is much higher than that of GeS2/rGO (366.17 mA h g−1) and NiS2/rGO (286.44 mA h g−1). In general, we can confirm that GeS2–NiS2/rGO exhibits excellent catalytic activity and fast redox kinetics for both charge and discharge processes.
The S@GeS2–NiS2/rGO, S@NiS2/rGO and S@GeS2/rGO cathodes were prepared by a simple melt-diffusion method. Fig. 4(f) exhibits the CV profiles of S@GeS2–NiS2/rGO, S@NiS2/rGO and S@GeS2/rGO cathode coin batteries with lithium metal as the anode, to demonstrate the redox reaction process in a LSB. The two distinct cathodic peaks at about 2.30 and 2.05 V are attributed to the reduction of solid S8 to long-chain soluble LiPSs and the subsequent conversion to insoluble Li2S2/Li2S, respectively. The anodic peaks are the oxidation of Li2S2/Li2S to LiPSs and then to S8.58 The CV profile of the S@GeS2–NiS2/rGO battery shows the highest peak currents and the lowest overpotentials than those of the S@NiS2/rGO and S@GeS2/rGO batteries, suggesting accelerated redox kinetics of LiPS conversion.
The CV profiles of different batteries at various scan rates were carried out to quantitatively evaluate the reaction kinetics of LiPSs and Li2S according to the nucleation transformation ratio (NTR) (Fig. 4(g) and Fig. S12, ESI†).59,60
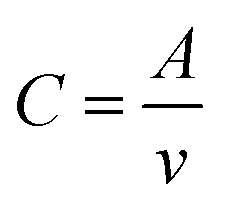 | (3) |
 | (4) |
Fig. 4(h) displays the galvanostatic discharge and charge curves of S@GeS2–NiS2/rGO, S@NiS2/rGO and S@GeS2/rGO batteries at 0.1C (1C = 1675 mA g−1). The initial discharge capacity of S@GeS2–NiS2/rGO is 1325.4 mA h g−1, which is higher than those of S@NiS2/rGO (1135.8 mA h g−1) and S@GeS2/rGO (1013.6 mA h g−1). The polarization voltage between the discharge and charge curves of the battery (ΔE, taken at 50% depth of discharge) is reported in Fig. 4(h) and Fig. S13, ESI.†61 A smaller ΔE value corresponds to a faster redox kinetics and a smaller Ohmic polarization of a battery. C1 and C2 are the capacities of the two discharge platforms, respectively. The ratio of C2 to C1 is used as another key indicator to analyze the catalytic activity of different sulfur hosts.62 As displayed in Fig. S13 (ESI†), the S@GeS2–NiS2/rGO battery exhibits the lowest ΔE and the highest C2/C1 compared to those of the S@GeS2/rGO and S@NiS2/rGO batteries.
The electron conduction of the materials and electrodes was tested using direct current (DC) resistance and electrochemical impedance spectroscopy (EIS) tests to demonstrate the enhanced conductivity of the GeS2–NiS2/rGO heterostructure (Fig. 4(i) and Fig. S14, ESI†). Electronic conductivity of different samples was investigated using a CHI1140C workstation with a constant voltage of 1.0 V over 1500 s (Fig. S14a and Table S2, ESI†). Fig. S14a and Table S2 (ESI†) show that GeS2–NiS2/rGO has the largest σ value, demonstrating the enhanced electrical conductivity of the heterostructure. The electronic conductivity of different electrodes, with or without sulfur loading, was tested using EIS. The GeS2–NiS2/rGO, NiS2/rGO, GeS2/rGO, S@GeS2–NiS2/rGO, S@NiS2/rGO, and S@GeS2/rGO electrodes were assembled into symmetric batteries (Fig. S14b and c, ESI†). Fig. 4(i) summarizes the charge transfer resistance (Rct) values for the different electrodes. Among GeS2–NiS2/rGO, NiS2/rGO, and GeS2/rGO electrodes, the GeS2–NiS2/rGO electrode has the smallest Rct (10.4 Ω), indicating improved electron conduction. What's more, the S@GeS2–NiS2/rGO electrode (20.1 Ω) has a smaller Rct compared to the S@NiS2/rGO (33.2 Ω) and S@GeS2/rGO (57.6 Ω) electrodes. These results demonstrate that the GeS2–NiS2/rGO heterostructure has excellent electrical conductivity with the best LiPS catalytic and reaction kinetics.
To devote into the understanding of electron injection functions in advancing Li2S deposition by the electron-triode-like GeS2–NiS2/rGO, in situ XRD spectra were recorded during the first discharge/charge processes of the S@GeS2–NiS2/rGO (Fig. 5(a)), S@GeS2/rGO (Fig. 5(b)) and S@NiS2/rGO (Fig. 5(c)) coin batteries. The major peaks detected at 22.7°, 25.4°, 26.3°, and 27.33° are indexed to be α-S8.63 The intensity of the characteristic peaks of α-S8 diffraction decreases gradually as the discharge process proceeds for all the batteries. In the meantime, a distinct broad peak appears at 24–25.5°, corresponding to the phase transition from α-S8 to LiPSs.64,65 Then, the characteristic peak of Li2S appeared at 26.5–27°, indicating the conversion of LiPSs into the final discharge product Li2S. The abundant S–S and Li–S bonds formed between LiPSs and GeS2–NiS2/rGO realized the collective electron injection. As expected, the Li2S characteristic peak appears earliest (prior ∼ 80% SOC) in S@GeS2–NiS2/rGO among the three batteries (∼40% SOC for S@GeS2/rGO and ∼20% SOC for S@NiS2/rGO). The earlier Li2S deposition as well as the extremely high current amplification factors (βR and βO) verified that the GeS2–NiS2/rGO heterostructure can boost the LiPS conversion to Li2S via the directional movement of electrons. All these results indicate that the electron-triode-like GeS2–NiS2 heterostructure can greatly accelerate electron injection/extraction and promote the catalytic conversion of LiPSs.
 | ||
| Fig. 5 (a)–(c) The first discharge/charge curves and corresponding in situ XRD contour plots of S@GeS2–NiS2/rGO (a), S@GeS2/rGO (b) and S@NiS2/rGO (c) batteries. The electron-triode-like GeS2–NiS2 heterostructure can inject a batch of electrons into LiPSs and achieve earlier Li2S deposition (∼80% SOC). (d)–(f) Ex situ AFM images of S@GeS2–NiS2/rGO (d), S@GeS2/rGO (e) and S@NiS2/rGO (f) electrodes cycled to different voltages. The S@GeS2–NiS2/rGO electrode consistently maintains a flat and homogeneous surface, indicating non-accumulation and rapid conversion of LiPSs. | ||
The catalytic conversion of LiPSs by the electron-triode-like GeS2–NiS2 can greatly maintain the stability of the electrode. We utilize ex situ atomic force microscopy (AFM) to understand the interface evolution of different electrodes in the z-direction during the discharge and charge processes (Fig. 5(d and e) and Fig. S15–S17, ESI†). AFM is a powerful tool for monitoring the surface changes in an electrode, especially for a surface with material conversion and volume changes.66 We have introduced the ex situ AFM to monitor the surface evolution for the cathode of a coin LSB for the first time, to understand the GeS2–NiS2 function on morphological stability. For a sulfur cathode, when S8 is first converted to LiPSs, which are soluble to the electrolyte, soluble LiPSs can travel on the electrode surface or shuttle to the electrolyte. Also, the S8 to 8Li2S material conversion will experience a ∼79% volume expansion, resulting in a dimension change, especially in the z-direction.67 An ideal collective electron injection can realize an earlier Li2S deposition rapidly and smoothly, by making a dent in the detainment of soluble LiPSs, thereby obtaining a uniform electrode surface during the discharge/charge process. As shown in Fig. 5(d and e), the fresh electrode surfaces of all three materials are uniform and flat, with no obvious particles. During the discharge process, there is no significant change on the surface of the S@GeS2–NiS2/rGO electrode (Fig. 5(d)). After discharge to 1.7 V, the surface of the S@GeS2–NiS2/rGO electrode remains dense and homogeneous. This is due to the rapid injection of free-moving electrons in the electron-triode-like GeS2–NiS2 heterostructure, which results in rapid conversion of S8 to LiPSs, and finally to the uniformly deposited Li2S on the surface of the S@GeS2–NiS2/rGO electrode. By comparison, the surfaces of S@GeS2/rGO (Fig. 5(e)) and S@NiS2/rGO (Fig. 5(f)) electrodes appear as large areas of fluctuation in the z-direction at 2.1 and 2.4 V, respectively, indicating the inhomogeneous S8 and LiPS conversion on the electrode surfaces. After discharge to 1.7 V, both the S@GeS2/rGO and S@NiS2/rGO electrodes form rough surfaces with large peaks and valleys in the z-direction. These results imply that the LiPSs are not evenly generated and converted to Li2S, resulting in rough electrode surfaces. Among them, the S@NiS2/rGO electrode exhibits the most aggregated structure, suggesting its maximum coverage of LiPSs, which corresponds to the results of in situ XRD. Similar results are observed during the subsequent charge process (Fig. S17, ESI†), where the S@GeS2–NiS2/rGO electrode maintains a relatively flat and homogeneous surface until fully charge to 2.8 V. The surface of the S@GeS2/rGO and S@NiS2/rGO electrodes is consistently covered with a large number of inhomogeneous particles, demonstrating the loss and accumulation of LiPSs during cycling. These results indicate that a batch of electrons in the electron-triode-like GeS2–NiS2 heterostructure can rapidly inject into soluble LiPSs without accumulation, achieving rapid conversion of LiPSs and earlier deposition of Li2S in the S@GeS2–NiS2/rGO battery.
2.4. Electrochemical performances of LSBs
To study in-depth the charge-carrier kinetics of the different hosts, ex situ EIS measurements were carried out during various states for the S@GeS2–NiS2/rGO, S@NiS2/rGO and S@GeS2/rGO batteries after 20 cycles (Fig. 6(a, b) and Fig. S18, ESI†). Fig. 6(a) presents the galvanostatic discharge/charge profile with the specified points at 0.1C. The corresponding ex situ EIS curves of S@GeS2–NiS2/rGO are given in Fig. 6(b). The fitting EIS results for the S@GeS2–NiS2/rGO, S@NiS2/rGO and S@GeS2/rGO batteries are summarized in Table S3 (ESI†). During the discharge process, the Rct value of the S@GeS2–NiS2/rGO battery first increases slightly from 14.5 Ω (A point) to 23.6 Ω (C point), then decreases significantly and finally falls to 5.6 Ω (F point). After full charging, the Rct value of the S@GeS2–NiS2/rGO battery decreases to 3.1 Ω. These small Rct values correspond to the relatively complete transformation of sulfur species during cycling. In contrast, the S@NiS2/rGO and S@GeS2/rGO batteries show higher Rct values (For details, see Table S3, ESI†) and change drastically throughout the discharge/charge processes, suggesting continuous accumulation and severe migration of LiPSs.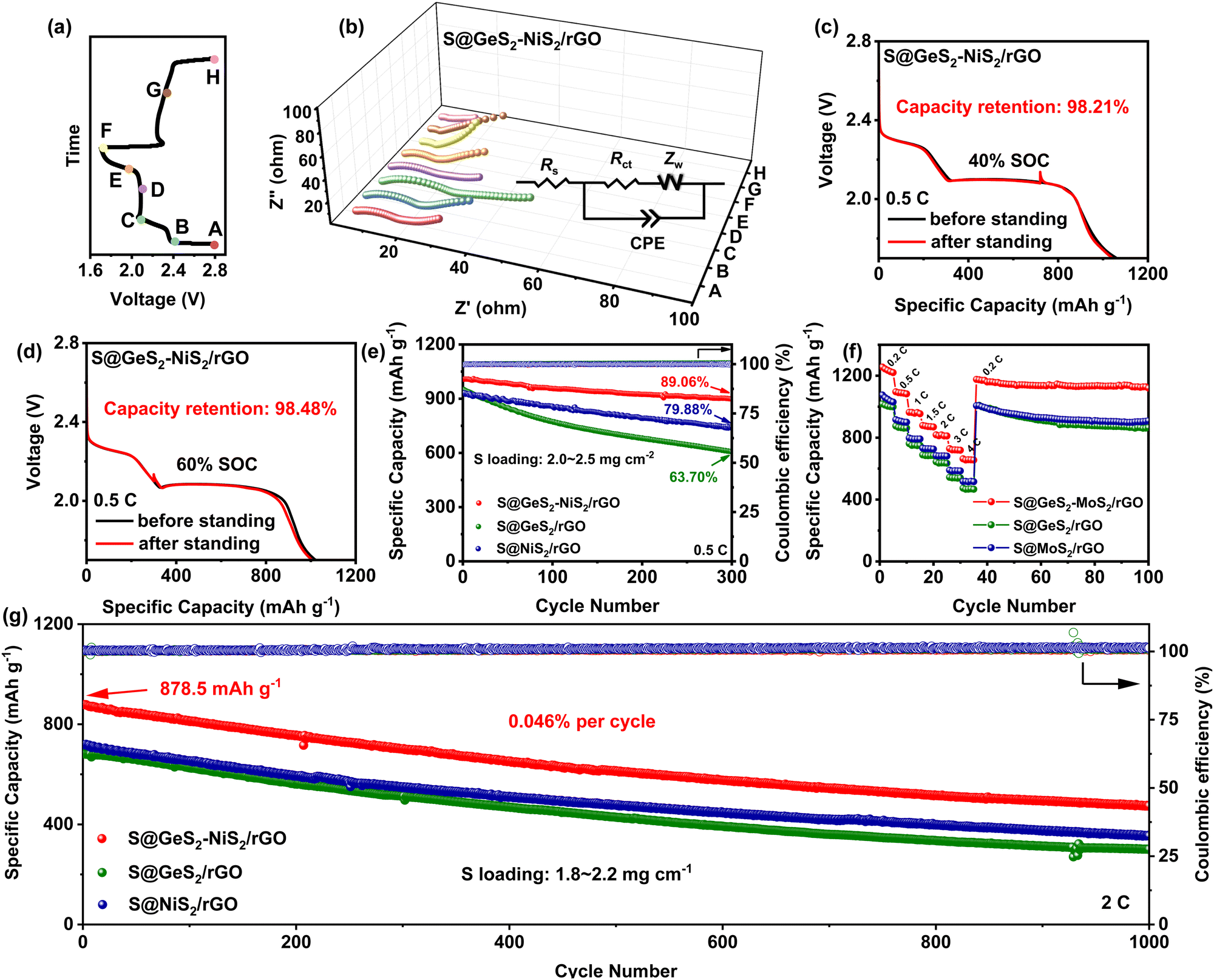 | ||
| Fig. 6 (a) The galvanostatic discharge/charge profile of S@GeS2–NiS2/rGO with the specified points for ex situ EIS tests. (b) Ex situ EIS profiles of the S@GeS2–NiS2/rGO battery. (c) 40% and (d) 60% SOC profiles of the galvanostatic discharge of the S@GeS2–NiS2/rGO battery at 0.5C. (e) Cycling stability of S@GeS2–NiS2/rGO, S@GeS2/rGO and S@NiS2/rGO batteries at 0.5C for 300 cycles. (f) Rate performances of different batteries at current densities from 0.2 to 4C. (g) Long-term cycling performances of different batteries at 2C over 1000 cycles. Among the three batteries, S@GeS2–NiS2/rGO has the highest discharge capacity and the most stable cycling performance, suggesting that LiPS loss during cycling is effectively suppressed. | ||
Due to the inevitable production of soluble LiPSs within different states of charge (SOC) ranges, evaluating the self-discharge performances of batteries is essential (Fig. 6(c, d) and Fig. S19, ESI†). In LSBs, about 60% of SOC corresponds to the conversion of Li2S6 to Li2S4, and about 40% of SOC corresponds to the reaction of Li2S4 to Li2S2/Li2S.68 Batteries are discharged to 60% or 40% of SOC for 48 h of standing, after 10 normal cycles. The capacity retention of S@GeS2–NiS2/rGO is 98.21% and 98.48% at 40% of SOC (Fig. 6(c)) and 60% of SOC (Fig. 6(d)), respectively, which is higher than that of S@NiS2/rGO (96.22% at 40% of SOC and 96.64% at 60% of SOC) and S@GeS2/rGO (94.65% at 40% of SOC and 95.94% at 60% of SOC). These results indicate that the GeS2–NiS2/rGO material can effectively reduce the loss of LiPSs and avoid the self-discharge of LSBs.
The effect of the GeS2–NiS2 heterostructure on the capacity and cycling performance of LSBs was further evaluated. The initial discharge capacity of S@GeS2–NiS2/rGO is 1007.8 mA h g−1 at 0.5C, which is superior to that of S@GeS2/rGO (951 mA h g−1) and S@NiS2/rGO (932.5 mA h g−1) (Fig. 6(e)). The S@GeS2–NiS2/rGO battery also maintains a high-capacity retention rate of 89.06% after 300 cycles. The rate capabilities of different cathode batteries were tested at current densities ranging from 0.2 to 4C (Fig. 6(f) and Fig. S20, ESI†). The S@GeS2–NiS2/rGO battery displays the best rate capacities of 1235, 1089.1, 957.4, 871.4, 817.6, 720.8 and 657.1 mA h g−1 at 0.2, 0.5, 1, 1.5, 2, 3 and 4 C, respectively. What's more, an impressive discharge capacity of 1170.4 mA h g−1 can be achieved in the S@GeS2–NiS2/rGO battery when the current density returns to 0.2C. Even after 100 consecutive cycles, the capacity of S@GeS2–NiS2/rGO can remain at 1120.8 mA h g−1, achieving an ultra-low-capacity decay. Compared with S@GeS2/rGO and S@NiS2/rGO, the S@GeS2–NiS2/rGO battery exhibits excellent rate performance. The galvanostatic discharge/charge voltage curves of S@GeS2–NiS2/rGO, S@GeS2/rGO and S@NiS2/rGO batteries at various current densities are further investigated (Fig. S20, ESI†). Two typical discharge plateaus of S@GeS2–NiS2/rGO are observed even at 4C, indicating the superior redox catalytic ability for LiPS conversion. The discharge/charge voltage curves of the S@GeS2–NiS2/rGO battery exhibits the smallest polarization potential compared to those of S@GeS2/rGO and S@NiS2/rGO batteries, indicating boosted redox kinetics of the GeS2–NiS2/rGO material.
The long-term cycling capabilities were measured at a high current rate of 2C to study the cycling stability of the S@GeS2–NiS2/rGO battery. As illustrated in Fig. 6(g), the S@GeS2–NiS2/rGO battery can maintain a high specific capacity of 474.6 mA h g−1 over 1000 cycles, with a low-capacity decay rate of 0.046% per cycle and coulombic efficiency over 99.5%. In comparison, the S@GeS2/rGO and S@NiS2/rGO batteries can only deliver capacities of 300.8 and 354.8 mA h g−1, respectively. These results demonstrate that the designed GeS2–NiS2/rGO material can realize a long-cycle LSB via the electron-triode-like approach.
To evaluate the practical feasibility of the GeS2–NiS2/rGO material, a lithium–sulfur pouch battery was fabricated using S@GeS2–NiS2/rGO as the cathode and a lithium–copper strip (80 m lithium) as the anode. In this lithium–sulfur pouch battery, the sulfur loading is 6.2 mg cm−2, the electrolyte/sulfur ratio (E/S) is 4 L mg−1, and the negative/positive ratio (N/P) is 1.6:1. Detailed information of the pouch cell is provided in the “Pouch battery assembly and Measurements” section of the ESI† and Table S4. The cycling performance (Fig. 7(a)) and the corresponding discharge/charge curves (Fig. 7(b)) are exhibited. The pouch battery attains a high specific capacity and maintains good cycling stability at 0.1C after 200 cycles. In addition, the 1.2 Ah pouch battery with an energy density of up to 405 W h kg−1 can power an electric fun (Fig. 7(c)). These above results indicate that the electron-triode-like GeS2–NiS2 heterostructure can successfully increase the life-span of a practical pouch LSB, under the uneven electron distribution nature of from the tab to the electrode. Overall, the GeS2–NiS2 heterostructure is highly competitive compared to previously reported pouch batteries (Fig. 7(d)).69–73
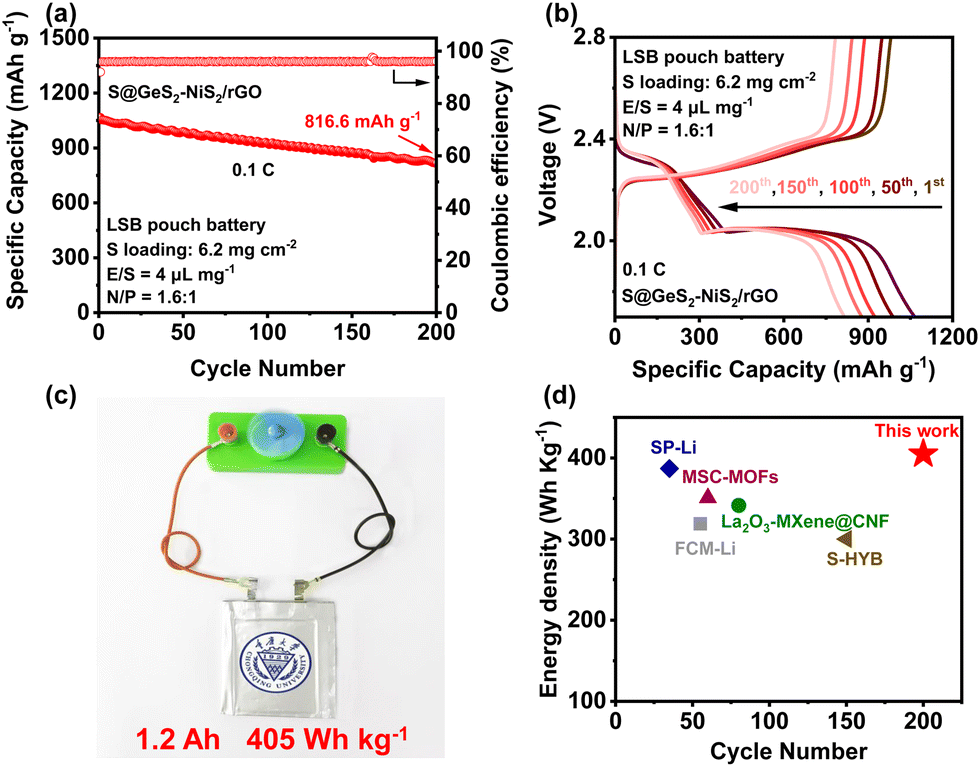 | ||
| Fig. 7 (a) Cycling performance of the S@GeS2–NiS2/rGO pouch battery at 0.1C and (b) corresponding galvanostatic discharge/charge curves. (c) A electric fan is powered by the S@GeS2–NiS2/rGO pouch battery. (d) Comparison of the energy density of different lithium–sulfur pouch batteries. The S@GeS2–NiS2/rGO battery maintains a high capacity after 200 cycles, indicating its potential electrochemical performance in practical applications.69–73 | ||
3. Conclusions
In this work, an electron-triode-like GeS2–NiS2/rGO heterostructure is successfully designed as a sulfur host to improve the electrochemical performance of LSBs. Theoretical calculations and experimental results show that the n-type semiconductor GeS2 forms an Ohmic contact with the metallic component NiS2, creating an electron-rich region at the heterointerfaces, which is verified by UPS and XAFS. The GeS2–NiS2 heterostructure can inject a batch of electrons into the LiPSs, accelerating the multi-electron experience through the LiPSs without accumulation of soluble LiPSs. Thus, the GeS2–NiS2 heterostructure can amplify the reaction current (up to βR = 105.87) of LSBs during cycling and enable earlier deposition of Li2S (from ∼20% to ∼80% SOC), which is verified by in situ XRD and ex situ AFM. Because of these outstanding advantages, the specific capacity of the S@GeS2–NiS2/rGO battery reaches 878.5 mA h g−1 at a high current density of 2C, and the capacity decay rate is as low as 0.046% after 1000 long-term cycles. Moreover, a 405 W h kg−1 1.2 Ah pouch battery under high sulfur loading (6.2 mg cm−2), lean-electrolyte (E/S = 4 L mg−1) and low N/P ratio (1.6:1) conditions is realized. This study elucidates the effectiveness of electron-triode-like GeS2–NiS2 in promoting catalytic conversion of LiPSs, providing inspiration for the design and development of heterostructure catalysts for high-performance lithium–sulfur pouch batteries.
Author contributions
All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors thank the National Natural Science Foundation of China (Grant No. 92372202, 22478043 and 22075033) and the Guangzhou Science and Technology Bureau (2024A03J0609).Notes and references
- C. D. Quilty, D. Wu, W. Li, D. C. Bock, L. Wang, L. M. Housel, A. Abraham, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Chem. Rev., 2023, 123, 1327–1363 CrossRef CAS PubMed.
- F. Degen, M. Winter, D. Bendig and J. Tübke, Nat. Energy, 2023, 8, 1284–1295 CrossRef CAS.
- Y. Che, X. Hu, X. Lin, J. Guo and R. Teodorescu, Energy Environ. Sci., 2023, 16, 338–371 Search PubMed.
- M. Zhao, H.-J. Peng, B.-Q. Li and J.-Q. Huang, Acc. Chem. Res., 2024, 57, 545–557 CAS.
- H. Li, Y. Li and L. Zhang, SusMat, 2022, 2, 34–64 CrossRef CAS.
- R. Chu, T. T. Nguyen, Y. Bai, N. H. Kim and J. H. Lee, Adv. Energy Mater., 2022, 12, 2102805 CrossRef CAS.
- K. Liu, H. Yuan, X. Wang, P. Ye, B. Lu, J. Zhang, W. Lu, F. Jiang, S. Gu, J. Chen, C. Yan, Y. Li, Z. Xu and Z. Lu, ACS Appl. Mater. Interfaces, 2023, 15, 31478–31490 CrossRef CAS PubMed.
- F. Zhao, J. Xue, W. Shao, H. Yu, W. Huang and J. Xiao, J. Energy Chem., 2023, 80, 625–657 CrossRef CAS.
- Z.-X. Chen, Q. Cheng, X.-Y. Li, Z. Li, Y.-W. Song, F. Sun, M. Zhao, X.-Q. Zhang, B.-Q. Li and J.-Q. Huang, J. Am. Chem. Soc., 2023, 145, 16449–16457 CrossRef CAS PubMed.
- R. Fang, K. Chen, Z. Sun, G. Hu, D.-W. Wang and F. Li, Interdiscip. Mater., 2023, 2, 761–770 Search PubMed.
- Z. Li, L.-P. Hou, N. Yao, X.-Y. Li, Z.-X. Chen, X. Chen, X.-Q. Zhang, B.-Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202309968 CrossRef.
- R. Deng, M. Wang, H. Yu, S. Luo, J. Li, F. Chu, B. Liu and F. Wu, Energy Environ. Mater., 2022, 5, 777–799 CrossRef CAS.
- X. Chen, H. Ji, Z. Rao, L. Yuan, Y. Shen, H. Xu, Z. Li and Y. Huang, Adv. Energy Mater., 2022, 12, 2102774 CrossRef CAS.
- Y. Liu, C. Cui, Y. Liu, W. Liu and J. Wei, RSC Adv., 2020, 10, 7384–7395 RSC.
- Y. Li, Y. Deng, J.-L. Yang, W. Tang, B. Ge and R. Liu, Adv. Funct. Mater., 2023, 33, 2302267 CrossRef CAS.
- L.-P. Hou, Y. Li, Z. Li, Q.-K. Zhang, B.-Q. Li, C.-X. Bi, Z.-X. Chen, L.-L. Su, J.-Q. Huang, R. Wen, X.-Q. Zhang and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202305466 Search PubMed.
- T. Wang, J. He, Z. Zhu, X.-B. Cheng, J. Zhu, B. Lu and Y. Wu, Adv. Mater., 2023, 35, 2303520 Search PubMed.
- Y. Kong, L. Wang, M. Mamoor, B. Wang, G. Qu, Z. Jing, Y. Pang, F. Wang, X. Yang, D. Wang and L. Xu, Adv. Mater., 2024, 36, 2310143 CrossRef CAS PubMed.
- A. Eftekhari and D.-W. Kim, J. Mater. Chem. A, 2017, 5, 17734–17776 RSC.
- A. I. Kamisan, T. I. T. Kudin, A. S. Kamisan, A. F. C. Omar, M. F. M. Taib, O. H. Hassan, A. M. M. Ali and M. Z. A. Yahya, Int. J. Hydrogen Energy, 2022, 47, 8630–8657 CrossRef CAS.
- B. Zhang, C. Luo, Y. Deng, Z. Huang, G. Zhou, W. Lv, Y.-B. He, Y. Wan, F. Kang and Q.-H. Yang, Adv. Energy Mater., 2020, 10, 2000091 Search PubMed.
- W. Yao, W. Zheng, J. Xu, C. Tian, K. Han, W. Sun and S. Xiao, ACS Nano, 2021, 15, 7114–7130 CrossRef CAS PubMed.
- S. Wang, S. Feng, J. Liang, Q. Su, F. Zhao, H. Song, M. Zheng, Q. Sun, Z. Song, X. Jia, J. Yang, Y. Li, J. Liao, R. Li and X. Sun, Adv. Energy Mater., 2021, 11, 2003314 CrossRef CAS.
- J. Liu, C. Lin, Q. Xie, D.-L. Peng and R.-J. Xie, Chem. Eng. J., 2022, 430, 133099 CrossRef CAS.
- C. Zhao, H. Jeong, I. Hwang, T. Li, Y. Wang, J. Bai, L. Li, S. Zhou, C. C. Su, W. Xu, Z. Yang, M. Almazrouei, C.-J. Sun, L. Cheng, G.-L. Xu and K. Amine, Joule, 2024, 8, 3397–3411 CrossRef CAS.
- G. Chen, L. Yan, H. Luo and S. Guo, Adv. Mater., 2016, 28, 7580–7602 CrossRef CAS PubMed.
- X. Long, Z.-H. Luo, W.-H. Zhou, S.-K. Zhu, Y. Song, H. Li, C.-N. Geng, B. Shi, Z.-Y. Han, G.-M. Zhou, W. Lv and J.-J. Shao, Energy Storage Mater., 2022, 52, 120–129 CrossRef.
- Y. Li, J. Zhang, Q. Chen, X. Xia and M. Chen, Adv. Mater., 2021, 33, 2100855 CrossRef CAS PubMed.
- Y. Zheng, J. Gao, C. Han and W. Chen, Cell Rep. Phys. Sci., 2021, 2, 1–27 Search PubMed.
- G. Jiang, X. Hu and L. Sun, ACS Appl. Electron. Mater., 2023, 5, 3071–3077 CrossRef CAS.
- H. Chen, Y. Qiu, Z. Cai, W. Liang, L. Liu, M. Li, X. Hou, F. Chen, X. Zhou, T. Cheng, L. He, J. Wang, X. Zhang, S. Dou and L. Li, Angew. Chem., Int. Ed., 2025, e202423357 CAS.
- Q. Su, W. Wang, J. Chen, J. Ji, W. Wang, W. Ren, L. Zhang, J. Xie and Q. An, Adv. Funct. Mater., 2024, 2419594 CrossRef.
- W.-G. Lim, S. Kim, C. Jo and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 18746–18757 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- X. Jiao, X. Tang, J. Li, Y. Xiang, C. Li, C. Tong, M. Shao and Z. Wei, Chem. Sci., 2024, 15, 7949–7964 RSC.
- Y. Jin, Y. Liu, L. Song, J. Yu, K. Li, M. Zhang and J. Wang, Chem. Eng. J., 2022, 430, 133029 CrossRef CAS.
- D. Feng, Q. Liu, Z. Li and T. Zeng, ACS Appl. Energy Mater., 2021, 4, 6096–6105 Search PubMed.
- X. Lou, P. Yan, B. Jiao, Q. Li, P. Xu, L. Wang, L. Zhang, M. Cao, G. Wang, Z. Chen, Q. Zhang and J. Chen, Nat. Commun., 2024, 15, 2730 Search PubMed.
- X. Ou, Z. Xiao, J.-F. Zhang, C. Wang, D. Wang, B. Zhang and Y. Wu, ACS Nano, 2020, 14, 13952–13963 Search PubMed.
- S. Luo, J. Shang, Y. Xu, H. Cheng, L. Zhang and Y. Tang, Adv. Funct. Mater., 2024, 34, 2403166 Search PubMed.
- Z. Xiao, C. Wang, B. Luo, L. Cao, R. Huang, J. Zhang, B. Zhang and X. Ou, Appl. Surf. Sci., 2021, 554, 149596 CrossRef CAS.
- K. Liang, K. Marcus, S. Zhang, L. Zhou, Y. Li, S. T. De Oliveira, N. Orlovskaya, Y.-H. Sohn and Y. Yang, Adv. Energy Mater., 2017, 7, 1701309 CrossRef.
- S.-A. He, Z. Cui, Q. Liu, G. He, D. J. Brett, W. Luo, R. Zou and M. Zhu, Small, 2021, 17, 2104186 CrossRef CAS PubMed.
- Y. Lin, Z. Qiu, D. Li, S. Ullah, Y. Hai, H. Xin, W. Liao, B. Yang, H. Fan, J. Xu and C. Zhu, Energy Storage Mater., 2018, 11, 67–74 CrossRef.
- H. Chen, C. Keiser, S. Du, H.-J. Gao, P. Sutter and E. Sutter, Phys. Chem. Chem. Phys., 2017, 19, 32473–32480 RSC.
- W. Xiong, J. Lin, H. Wang, S. Li, J. Wang, Y. Mao, X. Zhan, D.-Y. Wu and L. Zhang, J. Energy Chem., 2023, 81, 492–501 CrossRef CAS.
- W. Song, K. C. Chong, G. Qi, Y. Xiao, G. Chen, B. Li, Y. Tang, X. Zhang, Y. Yao, Z. Lin, Z. Zou and B. Liu, J. Am. Chem. Soc., 2024, 146, 3303–3314 CrossRef CAS PubMed.
- N. Zhang, Y. Hu, L. An, Q. Li, J. Yin, J. Li, R. Yang, M. Lu, S. Zhang, P. Xi and C. Yan, Angew. Chem., Int. Ed., 2022, 61, e202207217 CrossRef CAS PubMed.
- Q. Huang, G.-J. Xia, B. Huang, D. Xie, J. Wang, D. Wen, D. Lin, C. Xu, L. Gao and Z. Wu, et al. , Energy Environ. Sci., 2024, 17, 5260–5272 RSC.
- B. Zhang, J. Wang, G. Liu, C. M. Weiss, D. Liu, Y. Chen, L. Xia, P. Zhou, M. Gao and Y. Liu, et al., Nature, Catalysis, 2024, 7, 441–451 CAS.
- I. Pethes, V. Nazabal, R. Chahal, B. Bureau, I. Kaban, S. Belin and P. Jóvári, J. Alloys Compd., 2016, 673, 149–157 CrossRef CAS.
- J. Yin, J. Jin, H. Zhang, M. Lu, Y. Peng, B. Huang, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2019, 131, 18849–18855 Search PubMed.
- H. Niu, H. Liu, L. Yang, T. Kang, T. Shen, B. Jiang, W.-H. Huang, C.-C. Chang, Y. Pei and G. Cao, et al. , Nat. Commun., 2024, 15, 9421 Search PubMed.
- P. Wang, X. Bai, H. Jin, X. Gao, K. Davey, Y. Zheng, Y. Jiao and S.-Z. Qiao, Adv. Funct. Mater., 2023, 33, 2300687 CrossRef CAS.
- Y. Cui, X. Zhou, X. Huang, L. Xu and S. Tang, ACS Appl. Mater. Interfaces, 2023, 15, 49223–49232 Search PubMed.
- J. Häcker, D. H. Nguyen, T. Rommel, Z. Zhao-Karger, N. Wagner and K. A. Friedrich, ACS Energy Lett., 2021, 7, 1–9 Search PubMed.
- S. Tian, G. Liu, S. Xu, C. Han, K. Tao, J. Huang and S. Peng, Adv. Funct. Mater., 2024, 34, 2309437 CrossRef CAS.
- S. Haldar, M. Wang, P. Bhauriyal, A. Hazra, A. H. Khan, V. Bon, M. A. Isaacs, A. De, L. Shupletsov, T. Boenke, J. Grothe, T. Heine, E. Brunner, X. Feng, R. Dong, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2022, 144, 9101–9112 CrossRef CAS PubMed.
- R. Xu, H. Tang, Y. Zhou, F. Wang, H. Wang, M. Shao, C. Li and Z. Wei, Chem. Sci., 2022, 13, 6224–6232 Search PubMed.
- T. Wang, Q. Dong, C. Li and Z. Wei, Acta Phys.-Chim. Sin., 2024, 40, 2303061 CrossRef.
- C. Zhang, R. Du, J. J. Biendicho, M. Yi, K. Xiao, D. Yang, T. Zhang, X. Wang, J. Arbiol, J. Llorca, Y. Zhou, J. R. Morante and A. Cabot, Adv. Energy Mater., 2021, 11, 2100432 Search PubMed.
- Y. Wu, D. Li, J. Pan, Y. Sun, W. Huang, M. Wu, B. Zhang, F. Pan, K. Shi and Q. Liu, J. Mater. Chem. A, 2022, 10, 16309–16318 RSC.
- Y. Luo, Z. Fang, S. Duan, H. Wu, H. Liu, Y. Zhao, K. Wang, Q. Li, S. Fan, Z. Zheng, W. Duan, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215802 Search PubMed.
- X. Wang, X. Zhang, Y. Zhao, D. Luo, L. Shui, Y. Li, G. Ma, Y. Zhu, Y. Zhang, G. Zhou, A. Yu and Z. Chen, Angew. Chem., Int. Ed., 2023, 135, e202306901 CrossRef.
- B. Li, P. Wang, B. Xi, N. Song, X. An, W. Chen, J. Feng and S. Xiong, Nano Res., 2022, 15, 8972–8982 Search PubMed.
- S.-Y. Lang, R.-J. Xiao, L. Gu, Y.-G. Guo, R. Wen and L.-J. Wan, J. Am. Chem. Soc., 2018, 140, 8147–8155 Search PubMed.
- X. Jiao, X. Tang, J. Li, C. Li, Q. Liu and Z. Wei, Small, 2023, 19, 2304780 Search PubMed.
- H. Zhang, M. Zhang, R. Liu, T. He, L. Xiang, X. Wu, Z. Piao, Y. Jia, C. Zhang, H. Li, G. Xu, F. Zhou and Y. Mai, Nat. Commun., 2024, 15, 5451 Search PubMed.
- Z. Huang, Y. Zhu, Y. Kong, Z. Wang, K. He, J. Qin, Q. Zhang, C. Su, Y. L. Zhong and H. Chen, Adv. Funct. Mater., 2023, 33, 2303422 Search PubMed.
- H. Lu, Q. Zeng, L. Xu, Y. Xiao, L. Xie, J. Yang, J. Rong, J. Weng, C. Zheng, Q. Zhang and S. Huang, Angew. Chem., Int. Ed., 2024, 136, e202318859 Search PubMed.
- W. Chen, Y. Hu, Y. Liu, S. Wang, A. Hu, T. Lei, Y. Li, P. Li, D. Chen and L. Xia, et al. , Adv. Mater., 2024, 2312880 CrossRef CAS PubMed.
- L.-L. Su, N. Yao, Z. Li, C.-X. Bi, Z.-X. Chen, X. Chen, B.-Q. Li, X.-Q. Zhang and J.-Q. Huang, Angew. Chem., Int. Ed., 2024, 136, e202318785 Search PubMed.
- M. Liao, Y. Xu, M. M. Rahman, S. Tan, D. Wang, K. Wang, N. K. Dandu, Q. Lu, G. Li and L. Le, et al. , Nat. Sustainability, 2024, 1–10 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00615e |
| This journal is © The Royal Society of Chemistry 2025 |