
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-H-bonded self-assembled superstructures for ultrahigh-capacity and ultralong-life all-organic ammonium-ion batteries†
Pingxuan
Liu
a,
Ziyang
Song
 *a,
Qi
Huang
c,
Ling
Miao
a,
Yaokang
Lv
d,
Lihua
Gan
*ab and
Mingxian
Liu
*ab
*a,
Qi
Huang
c,
Ling
Miao
a,
Yaokang
Lv
d,
Lihua
Gan
*ab and
Mingxian
Liu
*ab
aShanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, 1239 Siping Rd., Shanghai, 200092, P. R. China. E-mail: 21310240@tongji.edu.cn; ganlh@tongji.edu.cn; liumx@tongji.edu.cn
bState Key Laboratory of Cardiovascular Diseases and Medical Innovation Center, Shanghai East Hospital, School of Medicine, Tongji University, 150 Jimo Rd., Shanghai 200120, P. R. China
cInstitute for Electric Light Sources, School of Information Science and Technology, Fudan University, 2005 Songhu Rd., Shanghai 200438, P. R. China
dCollege of Chemical Engineering, Zhejiang University of Technology, 18 Chaowang Rd., Hangzhou 310014, P. R. China
First published on 23rd April 2025
Abstract
All-organic ammonium-ion batteries (AOBs), utilizing lightweight organic electrodes and small, hydrated NH4+ charge carriers, are promising candidates for next-generation energy storage. However, the limited NH4+-accessible redox-active motifs in organics with high coordination barriers present significant challenges to the advancement of AOBs. Here, we designed multi-H-bonded donor–acceptor self-assembled ultralow-coordination-barrier organic superstructures (OSs) by integrating six-electron melem modules (H-bond donors) and three-electron cyanuric acid units (H-bond acceptors) via in-plane hydrogen bonding and out-of-plane π–π stacking. These OSs, featuring a low-energy-bandgap conjugated planar configuration and long-range π-electron delocalization pathways, enabled nearly complete utilization (99.3%) of intrinsic redox-active carbonyl/imine motifs with an ultralow activation energy (0.16 eV). Consequently, a high-kinetics and ultrastable 15-e− NH4+ coordination mechanism was activated within the OSs cathode, delivering an ultrahigh capacity (393 mA h g−1cathode) and exceptional cycling stability (60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles). Notably, the superior performance metrics of the OSs electrode enabled state-of-the-art AOBs with a record capacity (213 mA h g−1) and an unprecedented lifespan (100000 cycles). This work provides new insights into the structural engineering of multi-active, low-coordination-barrier OSs for advanced aqueous batteries.
000 cycles). Notably, the superior performance metrics of the OSs electrode enabled state-of-the-art AOBs with a record capacity (213 mA h g−1) and an unprecedented lifespan (100000 cycles). This work provides new insights into the structural engineering of multi-active, low-coordination-barrier OSs for advanced aqueous batteries.
Broader contextAqueous ammonium-ion batteries offer a highly safe, cost-effective, and environmentally friendly solution for next-generation energy storage. Non-metallic NH4+ charge carriers, with their small tetrahedral structures and light weights, demonstrate resource abundance, high migration rates, and strong H-bonding orientation, making them an ideal choice for overcoming the slow interfacial reactions of metal ions. A crucial task for NH4+ storage is the design of compatible electrode materials to fully unlock its potential. Electroactive organic materials stand out due to their molecular-level structural tunability and fast surface redox behavior, providing promising avenues for the development of all-organic ammonium-ion batteries (AOBs). However, the limited accessibility of NH4+ to redox-active motifs and the high coordination barriers in organic materials present significant challenges for advancing AOBs. Herein, we designed multi-H-bonded donor–acceptor self-assembled ultralow-coordination-barrier organic superstructures (OSs) by integrating melem modules (H-bond donors) and cyanuric acid units (H-bond acceptors) via hydrogen bonding and π–π interactions. The low-energy-bandgap conjugated plane and long-range π-electron delocalization of OSs electrode ensured full utilization of carbonyl/imine motifs via a high-kinetics and ultrastable two-step, 15 e−-NH4+ storage mechanism, enabling state-of-the-art symmetric AOBs with ultrahigh capacity and ultralong stability. Our work presents a promising approach for advancing the domain of multi-redox, low-energy-barrier superstructural organics for advanced energy storage. |
Introduction
Aqueous rechargeable batteries are gaining momentum as next-generation energy storage systems due to their cost-effectiveness, environmental sustainability, and intrinsic safety.1,2 Metallic ions (e.g., Zn2+, Mg2+, Ca2+, and Al3+) are typically employed as charge carriers in aqueous batteries, but their high charge density often leads to sluggish interfacial reaction kinetics.3–6 In contrast, non-metallic ions, such as H+ and NH4+, with their small hydrated structures and light weights, provide significant advantages, including resource abundance, high migration rates, and low reaction energy barriers. These features make them attractive shuttling carriers for overcoming the sluggish interfacial charge transfer with metallic ions in aqueous batteries.7,8 With the smallest size and lightest mass, H+ is considered an attractive charge carrier for enabling fast reaction kinetics, but it inevitably causes electrode corrosion, leading to degraded electrochemical stability.9 In contrast, NH4+ ions possess a unique tetrahedral geometry and strong preferential orientation, allowing flexible H-bonding interactions with host materials. This synergistically enhances both kinetic performance and structural stability during battery operation.10A crucial task for NH4+ storage is the design of compatible electrode materials to fully unlock its potential.11 To date, several inorganic materials, such as Prussian blue and Mn/V/Mo-based metal oxides, have been proposed for NH4+ storage, but they generally suffer from sluggish kinetics and poor cycling stability.12–15 An alternative approach has recently focused on π-conjugated organic materials due to their molecular-level structural tunability and rapid surface redox behavior, which enable high electrochemical activity and improved reaction kinetics for NH4+ storage.16 To date, various organic compounds, such as quinones,17 imines,18 azines,19 nitriles,20 and nitroaromatics,21 have been reported for fast NH4+ storage. However, they typically exhibit unsatisfactory capacities (<350 mA h g−1) due to a high proportion of redox-inactive motifs and substantial energy barriers for NH4+ diffusion within the active frameworks.22
Energy storage in organic electrode materials typically relies on coordination reactions between their redox-active groups and charge carriers.23 The lower the coordination barrier that redox-active groups must overcome to coordinate with NH4+ charge carriers, the more readily the coordination reaction can proceed, ultimately enhancing reaction kinetics and overall battery performance.24 Organic superstructures (OSs), derived from the epitaxial growth or self-assembly of low-dimensional building units via H-bonding interactions, exhibit ingenious hierarchical architectures and desirable functional properties.25 Generally, OSs can maximize the exposure of high-density redox-active sites inherited from their basic building units, and attain well-arranged consecutive geometries to afford fast NH4+-transfer paths, positioning them as promising candidates for large-scale energy systems.26 The structural and functional originality of OSs inspired us to consider whether multi-site-active H-bonding organic acceptors and donators as repeating units could be integrated to construct donator–acceptor-interlocking OSs with ultralow coordination barriers, thereby allowing for multielectron-redox NH4+ storage for revolutionizing battery metrics.27
Herein, we report multi-H-bonded donor–acceptor self-assembled OSs as highly redox-active, low-redox-barrier, and ultrastable electrode materials for activating superior NH4+ storage. Three-electron cyanuric acid (CA, H-bond acceptor) and six-electron 1,3,4,6,7,9,9b-heptaazaphenalen-2,5,8-triamine (HT, H-bond donor) were coupled to form an insoluble supramolecular organic superstructure through multiple H-bonds and π–π interactions. Featuring a low-energy-bandgap conjugated planar H-bonding configuration and long-range π-electron localization paths, the OSs achieved complete utilization of the built-in redox-active carbonyl/imine motifs with low-redox-energy barriers. Systematic studies revealed the reversible two-step 15 e− NH4+ coordination with carbonyl/imine sites of the OSs, which could facilitate achieving an ultrahigh capacity, rate performance, and long-term life for the constructed Zn||OSs battery. Besides, OS electrodes could be further extended to design superior symmetrical all-organic ammonium-ion batteries (AOBs), making such materials a competitive candidate for propelling advanced ammonium-ion batteries.
Results and discussion
Driven by their electron-withdrawing carbonyl and electron-donating amino groups, CA and HT can be integrated to form lock-and-key planar H-bonding supramolecular networks through their highly ordered 2D arrangement via multiple in-plane H-bonds (N–H⋯O and N–H⋯H, Fig. 1a and Fig. S1, ESI†). Then, the 2D supramolecular planes can self-assemble into 3D well-orchestrated superstructures driven by out-of-plane π–π stacking interactions (Fig. 1b). Fourier transform infrared (FT-IR) spectra demonstrated that the carbonyl vibration signals of CA were shifted towards higher frequency, while the melem vibrations of HT were moved to a lower frequency (Fig. S2, ESI†), providing firm evidence of the rich formation of H-bonds (N–H⋯O and N–H⋯H) between neighboring cyanuric acid and melem units. Furthermore, reduced density gradient (RDG) simulation28,29 displayed prominent green signals (−0.01 to 0.01 a.u.) and blue spikes (−0.04 to −0.02 a.u.) with the sign (λ2)ρ, which was further evidence of the H-bonding and π–π stacking interactions between the adjacent plane modules, consistent with the FT-IR spectra and X-ray diffraction (XRD) results (Fig. 1c). In particular, the sharp diffraction peak at 27.7° and the corresponding layer-spacing of 0.33 nm were due to the π–π stacking interactions of the 2D planar H-bonding supramolecular networks (Fig. S3, ESI†). | ||
| Fig. 1 Structural characterization of the OSs. (a) Two-dimensional supramolecular planar structure interconnected by multiple interlocking hydrogen bonds between CA and HT. (b) RDG map and corresponding isosurface. (c) XRD patterns. (d) SEM images recorded at different reaction times. (e) Elemental maps. (f) Electrical conductivities. (g) Eg values. | ||
The supramolecular self-assembly process took 60 min to get stable well-defined organic superstructures with multi-layered geometries (Fig. 1d), rich pore channels (Fig. S4, ESI†), high thermal stability (Fig. S5, ESI†) and a uniform C/N/O element distribution (Fig. 1e). In contrast with irregular CA and HT (Fig. S6, ESI†), the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images revealed the OSs comprised fibre-shaped superstructures built from sheet modules (Fig. S7, ESI†). The electronic conductivity of the OS cathode (obtained by mixing 60 wt% OSs, 30 wt% graphite and 10 wt% polytetrafluoroethylene) was determined as 50.46 S cm−1 using a four-probe method compared, which was higher than for the CA/HT cathodes (12.44/15.39 S cm−1), and which is favorable for highly efficient electron transfer for propelling the redox reactions (Fig. 1f). As is well-established, the optical energy gap (Eg) reflects the electron injection and conduction capabilities of organic materials, whereby a lower Eg helps to improve the electron-transfer efficiency to achieve fast charge-storage kinetics with low energy barriers.2,19 Thanks to their favourable multi-H-bonded donor–acceptor long-range superstructures, the OSs achieved a very low optical energy gap (Eg) of 2.16 eV, which was superior to that of CA (2.99 eV) and HT (2.65 eV), ensuring low-energy-barrier charge-storage kinetics (Fig. 1g). Owing to the longitudinal self-assembly of the π-conjugated aromatic planar sheets via rich H-bonds and π–π interactions (Fig. 1a–c and Fig. S2, S3, ESI†), the OSs possessed well-arranged fibre-shaped superstructure geometries (Fig. 1d and e), which provide long-range continuous charge migration paths with high conductivity (Fig. 1f) and a low energy gap (Fig. 1g), thereby contributing to efficient π-electron delocalization with low energy barriers. Overall, the OSs fused high-density redox-active sites, long-range low-barrier π-electron localization paths, and ultrastable superstructure skeletons, which could be expected to be highly competitive features for superior charge-storage metrics.
The electrochemical performances of the OSs as cathodes were studied in a Zn||OSs battery using Zn metal foil as the anode and 3 M NH4OTF/H2O solution as the electrolyte (OTF− = CF3SO3−, Fig. S8, ESI†). As depicted by the galvanostatic discharge/charge (GCD) curves (Fig. 2a), the OS cathode (derived from the reaction time of 60 min) delivered an ultrahigh capacity of 393 mA h g−1 at 0.2 A g−1, exceeding the performances of both Zn||CA (112 mA h g−1) and Zn||HT (165 mA h g−1) batteries (Fig. S9, ESI†). Upon increasing the current density to 50 A g−1 (i.e. 250-fold), the OSs still maintained a capacity of 157 mA h g−1 (Fig. 2b), highlighting their superior high-rate performance. Benefiting from their remarkable capacity together with an average discharge voltage of 0.8 V, the as-assembled battery was able to obtain a high energy output of 313 W h kg−1 at 170 W kg−1 (according to the mass loading of OSs in the cathode, i.e. ∼3 mg cm−2, Fig. S10 and Table S1, ESI†), which was higher than that with the 3 M Zn(OTF)2/H2O electrolyte (Fig. S11, ESI†), making it a strong contender compared to other recently reported organic cathode materials (Fig. 2c).30–44 Besides, the energy density of the Zn||OSs battery was calculated to be 271 W h kg−1 considering the mass loading of the OS cathode and the consumed NH4+ electrolyte.
 | ||
| Fig. 2 Electrochemical performances of the Zn||OSs cell. (a) GCD curves. (b) Rate capacities. (c) Ragone plots compared with other reported organic materials. (d) Cyclic lifespan (inset: SEM images before and after cycling, scale bar: 1 μm). (e) Lifetime comparison between OSs and other reported organic materials. (f) CV profiles at different scan rates. (g) Calculated b values. (h) Contribution proportions of diffusion and capacitive-controlled processes. | ||
Impressively, the Zn||OSs battery in 3 M NH4OTF/H2O electrolyte demonstrated a capacity retention of 85.8% and a Coulombic efficiency of 100% when the serve term was extended to 60000 cycles at 10 A g−1 (Fig. 2d and Table S1, ESI†), exceeding most other reported organic materials (Fig. 2e).30–33,38,39,41,43–49 Characterization of the OS cathode after prolonged cycling, including via SEM, and FT-IR and ultraviolet-visible (UV/vis) spectroscopies, indicated the outstanding stability of its structure, and regular functions, alongside its strong anti-dissolution ability (see the inset of Fig. 2d and Fig. S12, ESI†). Furthermore, the evolution of the Zn anodes through long-term cycling was examined by SEM images and XRD patterns. These revealed that during repeated Zn-plating/stripping processes, the pristine smooth Zn anode transformed into a rough and disordered surface structure without obvious dendrite formation (Fig. S13a–c, ESI†) or electrolyte penetration corrosion (Fig. S13d, ESI†). Considering the superior structural robustness of the OSs, the performance decline of the Zn||OSs cell could be attributed to structural degradation of the Zn electrode during the persistent operation (Fig. S14, ESI†).
The reaction kinetics of the Zn||OSs battery was evaluated using cyclic voltammetry (CV) measurements. The CV profiles of the OSs at different sweep rates displayed two pairs of stable redox signals (denoted as PR1, PR2, PO1, and PO2, Fig. 2f), indicating a two-step redox reaction. As the scan rate gradually increased, the peak shapes became almost the same, suggesting a high reversibility and low-ion-diffusion obstacles. Using the equation i = kvb (where i represents the current density, v is the scan rate, and k and b are constants), the b values calculated for four peaks (0.94–0.98) were found to approach to 1, revealing the rapid surface-controlled redox kinetics (Fig. 2g). In brief, the ultrafast and robust charge storage could be ascribed to the low-energy-bandgap sophisticated superstructures built with a conjugated planar configuration and long-range π-electron localization paths, which fully exposed the redox-active sites and maximized ion accessibility with ultralow coordination barriers, thereby promoting excellent electrochemical activity and durability.
To uncover the energy-storage mechanism of the OS cathode, various characterizations were employed to track its structural variation during charging/discharging. Two pairs of obvious platforms could be observed in the GCD curve (Fig. 3a), indicative of the two-step redox electrochemistry. The FT-IR spectra (Fig. 3b) displayed vibration signals belonging to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (1697/1778 cm−1) that sharply decreased to eventually disappear during discharging (state A → B), but were almost unchanged after further discharging (state B → C), alongside the appearance of C–O species (1163 cm−1). This result indicated the highly redox-active characteristics of the CO motifs in the OSs. Meanwhile, the generated peak at 2854 cm−1, belonging to the H-bond stretching mode (N–H⋯O),10 showed a strong uptrend during discharging and gradually lessened during charging, validating the H-bond generation/disappearance of the NH4+-coordinated carbonyl species. Moreover, a new signal (2923 cm−1) appeared that was assigned to N–H species of the NH4+ ions during discharging, proving NH4+ uptake. These results confirmed the coordination between the carbonyl motifs of the OS cathode and the NH4+ ions. Besides, the vibration intensity of CN at 1608 cm−1 showed a reversible weakening/strengthening trend (state B → C; state C → D) during discharging/charging, while the opposite variation was observed for the C–N bands at 1097 cm−1. Significantly, a new signal at 2956 cm−1 appeared upon discharging, confirming the redox coordination between the CN sites and NH4+ ions. Overall, the CO and CN motifs of the OS cathode initiated reversible electrochemical redox reactions with the NH4+ charge carriers.
O (1697/1778 cm−1) that sharply decreased to eventually disappear during discharging (state A → B), but were almost unchanged after further discharging (state B → C), alongside the appearance of C–O species (1163 cm−1). This result indicated the highly redox-active characteristics of the CO motifs in the OSs. Meanwhile, the generated peak at 2854 cm−1, belonging to the H-bond stretching mode (N–H⋯O),10 showed a strong uptrend during discharging and gradually lessened during charging, validating the H-bond generation/disappearance of the NH4+-coordinated carbonyl species. Moreover, a new signal (2923 cm−1) appeared that was assigned to N–H species of the NH4+ ions during discharging, proving NH4+ uptake. These results confirmed the coordination between the carbonyl motifs of the OS cathode and the NH4+ ions. Besides, the vibration intensity of CN at 1608 cm−1 showed a reversible weakening/strengthening trend (state B → C; state C → D) during discharging/charging, while the opposite variation was observed for the C–N bands at 1097 cm−1. Significantly, a new signal at 2956 cm−1 appeared upon discharging, confirming the redox coordination between the CN sites and NH4+ ions. Overall, the CO and CN motifs of the OS cathode initiated reversible electrochemical redox reactions with the NH4+ charge carriers.
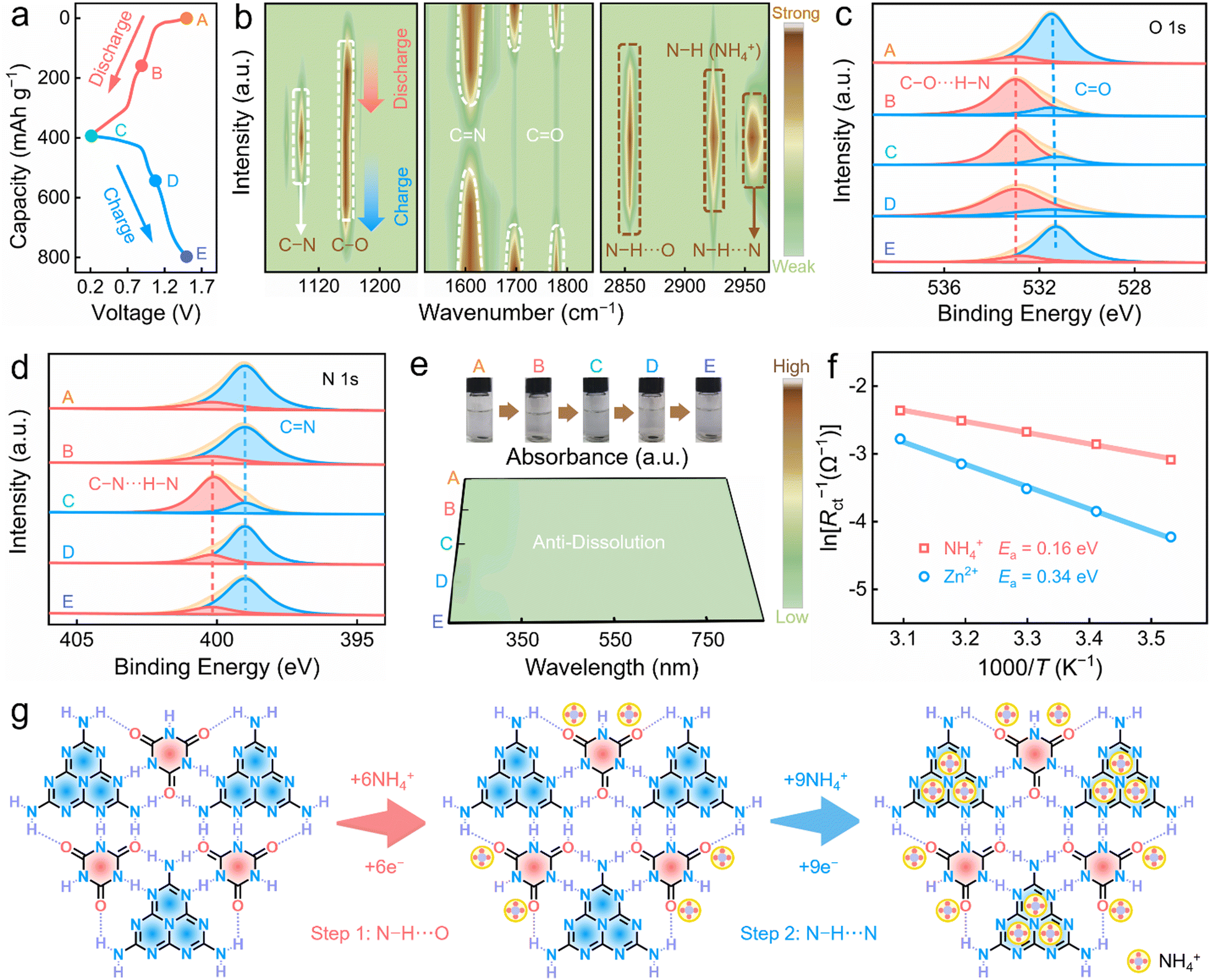 | ||
| Fig. 3 Structural evolution of the OSs cathode during (dis)charging. (a) Voltage-capacity curve measured at 0.2 A g−1. (b) Ex situ FT-IR spectra. (c) O 1s and (d) N 1s XPS spectra. (e) UV/vis spectra and images of the OSs cathodes in NH4OTF/H2O electrolytes at various electrochemical states. (f) Calculated Ea values. (g) H-bonding redox chemistry. | ||
Next, X-ray photoelectron spectroscopy (XPS) was performed to explore the H-bonding reaction mechanism between the NH4+ ions and CO/CN groups. In ex situ high-resolution O 1s XPS spectra of the OSs cathode (Fig. 3c), CO groups at 531.5 eV gradually weaken (state A → B) and are constant (state B → C) during discharging, along with the formation of C–O⋯H–N species at 533.0 eV, suggesting the redox interaction between NH4+ ions and carbonyl motifs. About ex situ high-resolution N 1s XPS spectra of the OSs cathode (Fig. 3d), the ratio of CN motifs at 399.0 eV remains almost unchanged (state A → B) and declines (state B → C). Meanwhile, a newly emerged signal (400.3 eV), resulting from the generation of C–N⋯H–N, could be observed. After charging, the signals of all the species reversibly returned to their initial levels through the decoordination of NH4+ from the redox-active sites. Besides, the ex situ XRD patterns and Raman spectra provided additional proof of a two-step NH4+ coordination with the CO and CN groups of the OS cathode (Fig. S15, ESI†). In conclusion, two findings were evident: (i) CO and CN can be recognized as redox-active sites for the cathodic electrochemical reactions; (ii) the OS electrode undergoes a two-step-continuous NH4+ storage process, with CO coordinating first and CN second.
UV/vis spectroscopy was next employed to study the dissolution behaviour of the OS cathode at five different voltage states in NH4OTF/H2O electrolyte. Here, no obvious absorption peaks were observed in the UV/vis spectra and the colourless nature of the NH4OTF/H2O electrolyte suggested the excellent anti-dissolution of the OSs (Fig. 3e). Furthermore, electrochemical impedance spectroscopy (EIS) was performed for the OS cathode at different temperatures (T) (Fig. S16, ESI†) to obtain the interfacial charge transfer resistance (Rct) for fitting the activation energy (Ea) according to the Arrhenius equation,50 where Ea reflects the interfacial coordination reaction process between the redox-active groups of organic electrodes and ionic carriers of electrolytes.23 Generally, a lower Ea indicates a lower reaction energy barrier, making it easier for ions to coordinate with the redox-active groups of organics, which helps to achieve high-kinetics redox reactions and efficient charge storage.24 NH4+ coordination had a Ea of 0.16 eV (Fig. 3f), which was lower than for the Zn2+ reaction (0.34 eV). This result implies that high-kinetics NH4+ uptake takes precedence over Zn2+ at the redox-sites of the OS cathode.
Considering the slight acidity of the NH4OTF/H2O electrolyte (pH 4.85), it was essential to probe H+ reactions during NH4+ electrochemistry. When replacing NH4OTF with HOTF/H2O electrolyte with the same pH, the capacity of the Zn||OSs battery was 6.5 mA h g−1 (Fig. S17, ESI†). This demonstrates the marginal role of H+ in the energy-storage process of the OS cathode, as also confirmed by the absence of Zn salts on the cathode surface (Fig. S18, ESI†). In addition, the element mapping images and XPS spectrum of the OS electrode at full discharge (Fig. S19, ESI†) revealed the absence of Zn signals, indicating that Zn2+ ions did not participate in NH4+ storage. Therefore, NH4+ serves as the exclusive effective shuttling carrier in NH4OTF/H2O electrolyte, which dominates the entire electrochemical redox reaction of the OS cathode. Besides, the experimental capacity of the OSs, excluding 3.3 mA h g−1 for the graphite conductive agent (Fig. S20, ESI†) and 6.5 mA h g−1 for H+ storage, was 383.2 mA h g−1, corresponding to a high utilization (99.3%) of CO/CN motifs and a 15 e− redox reaction. In general, OSs initiate a high-kinetics and stable two-step 15 e− NH4+ charge-storage process, involving six CO coordinating with 6 NH4+ ions and then nine CN couplings with 9 NH4+ ions (Fig. 3g).
Next, density functional theory (DFT) simulations were carried out to unravel the energy levels and redox activity of the OSs. Molecular electrostatic potential (MEP) simulation of an optimized OS skeleton (Fig. S21, ESI†) implied that the electronegative six carbonyl and nine triazine units were dual redox-active sites (red region) for NH4+ coordination (Fig. 4a). The OSs showed a highly π-conjugated superstructure, as revealed by the π-electron localization function (ELF-π), which can facilitate efficient electron delocalization through the aromatic backbone (Fig. 4b). Theoretically, a higher π-conjugation degree makes for a lower energy gap (ΔE) between the lowest unoccupied molecular orbital (LUMO) and the highest occupied molecular orbital (HOMO). The ΔE value of the OSs was 2.51 eV, exhibiting the favourable electronic configuration of the OSs to facilitate excellent electron-transfer efficiency with a low energy barrier (Fig. 4c). The anisotropy of the induced current density (ACID) calculation44 revealed that the clockwise diamagnetic continuous current flowed across the whole organic skeleton (Fig. 4d), reflecting the global π-aromaticity of the OSs.
 | ||
| Fig. 4 Theoretical study of the OSs redox reactions under different electrochemical conditions. (a) MEP distribution. (b) ELF-π color-filled map. (c) LUMO–HOMO energy levels. (d) ACID plots. (e) Optimized geometries and corresponding ΔG values after NH4+-coordination. (f) Differential charge isosurfaces at states I and II. | ||
The structural evolution of the OS cathode upon two-step NH4+ (de)coordination was further clarified. During the first discharge stage (step 1), six NH4+ ions coordinate with six carbonyl sites by delivering a negative Gibbs free energy (ΔG) of −6.76 eV (Fig. 4e). In the subsequent discharging process (step 2), nine NH4+ charge carriers couple with nine imine motifs of triazine units by a more negative ΔG value (−13.71 eV). Step 1 has a lower ΔG value compared to step 2, suggesting that step 1 and 2 can proceed sequentially. In accordance with the minimal energy theory, a robust two-step 15 e− redox process was triggered in the OS cathode, corresponding to two pairs of discharge platforms of the Zn||OSs cell (Fig. 2a and f). The differential charge isosurfaces were employed to investigate the binding nature of the NH4+-coordinated OSs (Fig. 4f). Based on the Bader charge analysis,51 the charge accumulation and depletion among NH4+ ions and CO/CN groups suggested their strong coordination to form robust ligand configurations involving notable charge shifts (2.67 e of state I; 6.21 e of state II). The rapid kinetics of NH4+ ions was capable of maximizing the utilization of the carbonyl/imine motifs in the ultralow-coordination-barrier OSs, thereby enhancing the electrochemical activity and durability.
As discussed above, the OSs could achieve full utilization of the high-density redox-sites, long-range low-barrier electron delocalization, and electrochemical ultrastability, and underwent two-step NH4+ storage in NH4OTF/H2O electrolyte (Fig. 3g), resulting in two redox couples at 0.69 V (6 e− transfer in the first step) and 1.01 V (9 e− transfer in the second step). Such structural and functional features make OSs promising to serve as both the cathode and anode for building advanced all-organic NH4+-ion batteries (AOBs, Fig. 5a), while avoiding excessive use and adverse structural degeneration of the Zn anodes. As a proof of concept, new-type symmetric AOBs were constructed using the original OSs as the cathode, the reduced product of OSs (OSs-R) as the anode, and 3 M NH4OTF/H2O solution as the electrolyte. The charge-storage process of the AOBs belonged to a rocking chair mechanism, which operated in the form of a NH4+ shuttle between the OS cathode and OSs-R anode. During the discharge process, the OSs cathode accepted NH4+ ions through the reduction of its CO groups, resulting in the formation of C–O bonds. Simultaneously, the OSs-R anode released NH4+ ions through the oxidation reaction, which could convert into CN bonds. The reverse redox reactions occurred during the charge process with the opposite structural evolution.
 | ||
| Fig. 5 Electrochemical evaluation of AOBs. (a) Schematic of the OSs||OSs-R cell. (b) GCD curves. (c) Rate metrics. (d) Long-term cycling performance. (e) Lifespan comparison with reported NH4+-ion full cells. | ||
Next, electrochemical measurements were conducted to evaluate the performances of the AOBs. The GCD curves of the OSs||OSs-R battery displayed a discharge capacity of 213 mA h g−1 at 0.2 A g−1 (Fig. 5b, based on the mass loading of OSs in the cathode). A high energy density of 80.9 W h kg−1 was obtained for the OSs||OSs-R battery by determination of the integral area of the GCD curve at 0.2 A g−1 (Fig. 5b, based on the mass loading of OSs in the cathode). With a 250-fold current density increase to 50 A g−1, the capacity still retained 96 mA h g−1, with high electrochemical reversibility within 90 cycles (Fig. 5c). The high capacity of the OSs||OSs-R cell is a key advantage over previously reported organic full batteries,4,9,52–59 making it a formidable choice for next-generation organic electrodes. Significantly, the OSs||OSs-R battery achieved an extraordinary cycling lifespan of 100000 cycles with 90.3% capacity retention at 5 A g−1 (Fig. 5d), which is the highest value among the NH4+-hosting full batteries reported to date (Fig. 5e).11,12,60–65 The comprehensive electrochemical performances of the OSs||OSs-R full cell, including its superior capacity, excellent rate performance and ultralong life, are far superior to those of the currently reported all-organic batteries (Table S2, ESI†).
Conclusion
In conclusion, multi-H-bonded donor–acceptor self-assembled OSs were designed as high-performance electrodes for AOBs. Three-electron CA (H-bond acceptor) and six-electron HT (H-bond donor) were integrated into lock-and-key supramolecular H-bonding networks via in-plane H-bonding and out-of-plane π–π stacking interactions. The OSs showed low-energy-barrier conjugated planar structures and long-range π-electron localization pathways, allowing for full accessibility (99.3%) of the carbonyl/imine redox-active sites with an ultralow activation energy (0.16 eV). Consequently, the OSs initiated a high-kinetics and ultrastable 15 e− NH4+ coordination process, delivering an ultrahigh capacity, large-current survivability, and long-lasting cycle life. More importantly, the fabricated OS electrode was further applied to construct superb symmetric AOBs with a record capacity (213 mA h g−1) and unprecedented stability (100000 cycles). This study proposes a new route for designing multi-redox-active and low-coordination-barrier organic materials for advanced NH4+-ion batteries.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 22272118, 22172111, and 22309134), the Science and Technology Commission of Shanghai Municipality, China (No. 22ZR1464100, 20ZR1460300, and 19DZ2271500), the China Postdoctoral Science Foundation (2022M712402), the Shanghai Rising-Star Program (23YF1449200), the Zhejiang Provincial Science and Technology Project (2022C01182), and the Fundamental Research Funds for the Central Universities (2023-3-YB-07).Notes and references
- P. Shi, J. Ma, M. Liu, S. Guo, Y. Huang, S. Wang, L. Zhang, L. Chen, K. Yang, X. Liu, Y. Li, X. An, D. Zhang, X. Cheng, Q. Li, W. Lv, G. Zhong, Y.-B. He and F. Kang, Nat. Nanotechnol., 2023, 18, 602–610 CrossRef CAS PubMed.
- Z. Ran, R. Wang, J. Fu, M. Yang, M. Li, J. Hu, J. He and Q. Li, Adv. Mater., 2023, 35, 2303849 CrossRef CAS PubMed.
- D. Zhang, Z. Song, L. Miao, Y. Lv, H. Duan, M. Li, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2025, 64, e202414116 CrossRef CAS PubMed.
- Z. Fu, H. Zhang, D. Geng, Z. Liu, Z. Zhang, X. Li and C. Yan, Adv. Energy Mater., 2024, 36, 2403934 Search PubMed.
- S. Zhang, Y. Zhu, D. Wang, C. Li, Y. Han, Z. Shi and S. Feng, Adv. Sci., 2022, 9, 2200397 CrossRef CAS PubMed.
- L.-W. Luo, C. Zhang, W. Ma, C. Han, X. Ai, Y. Chen, Y. Xu, X. Ji and J.-X. Jiang, Adv. Mater., 2024, 36, 2406106 CrossRef CAS PubMed.
- Y. Yuan, D. Cao, F. Cui, Y. Yang, C. Zhang, Y. Song, Y. Zheng, J. Cao, S. Chen, Y. Song, F. Wang and G. Zhu, Nat. Water, 2025, 3, 89–98 CrossRef CAS.
- M. Shi, P. Das, Z.-S. Wu, T.-G. Liu and X. Zhang, Adv. Mater., 2023, 35, 2302199 CrossRef CAS PubMed.
- Z. Tie, S. Deng, H. Cao, M. Yao, Z. Niu and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202115180 CrossRef CAS PubMed.
- Z. Tian, V. S. Kale, Y. Wang, S. Kandambeth, J. Czaban-Jóźwiak, O. Shekhah, M. Eddaoudi and H. N. Alshareef, J. Am. Chem. Soc., 2021, 143, 19178–19186 CrossRef CAS PubMed.
- H. Lu, J. Hu, K. Zhang, Y. Zhang, B. Jiang, M. Zhang, S. Deng, J. Zhao, H. Pang and B. Xu, Adv. Mater., 2024, 36, 2408396 CrossRef CAS PubMed.
- F. Ye, R. Pang, C. Lu, Q. Liu, Y. Wu, R. Ma and L. Hu, Angew. Chem., Int. Ed., 2023, 62, e202303480 CrossRef CAS PubMed.
- D. Yang, Y. Song, M.-Y. Zhang, Z. Qin, J. Liu and X.-X. Liu, Angew. Chem., Int. Ed., 2022, 61, e202207711 CrossRef CAS PubMed.
- G. Liang, Y. Wang, Z. Huang, F. Mo, X. Li, Q. Yang, D. Wang, H. Li, S. Chen and C. Zhi, Adv. Mater., 2020, 32, 1907802 CrossRef CAS PubMed.
- R. Zheng, Y. Li, H. Yu, X. Zhang, D. Yang, L. Yan, Y. Li, J. Shu and B.-L. Su, Angew. Chem., Int. Ed., 2023, 62, e202301629 CrossRef CAS PubMed.
- Y. Sun, B. Yin, J. Yang, Y. Ding, M. Li, H. Li, J. Li, B. Jia, S. Zhang and T. Ma, Energy Environ. Sci., 2023, 16, 5568–5604 RSC.
- Q.-Q. Sun, J.-Y. Du, T. Sun, Z.-B. Zhuang, Z.-L. Xie, H.-M. Xie, G. Huang and X.-B. Zhang, Adv. Mater., 2024, 36, 2313388 CrossRef CAS PubMed.
- F. Huang, W. Zhao, Y. Guo, Y. Mi, S. Gull, G. Long and P. Du, Adv. Funct. Mater., 2024, 34, 2407313 CrossRef CAS.
- J. Liu, K. Guo, W. Guo, J. Chang, Y. Li and F. Bao, Angew. Chem., Int. Ed., 2025, 64, e202424494 CrossRef CAS PubMed.
- P. Shao, Y. Liao, X. Feng, C. Yan, L. Ye and J. Yang, J. Colloid Interface Sci., 2023, 633, 199–206 CrossRef CAS PubMed.
- Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309446 CrossRef CAS PubMed.
- H. Zhang, Y. Tian, W. Wang, Z. Jian and W. Chen, Angew. Chem., Int. Ed., 2022, 61, e202204351 CrossRef CAS PubMed.
- Z. Lin, H.-Y. Shi, L. Lin, X. Yang, W. Wu and X. Sun, Nat. Commun., 2021, 12, 4424 CrossRef CAS PubMed.
- M. Liu, X. Li, M. Cui, F. Chen, J. Li, W. Shi, Y. Liu, X. Li, Y. Wang, W. Zhang, C. Shao and Y. Liu, Nat. Commun., 2024, 15, 10769 CrossRef PubMed.
- C. Ding, Y. Zhao, W. Yin, F. Kang, W. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202417988 CrossRef CAS PubMed.
- M.-P. Zhuo, J.-J. Wu, X.-D. Wang, Y.-C. Tao, Y. Yuan and L.-S. Liao, Nat. Commun., 2019, 10, 3839 CrossRef PubMed.
- B. Jiang, J. Zhang, K. Yu, Z. Jia, H. Long, N. He, Y. Zhang, Y. Zou, Z. Han, Y. Li and L. Ma, Adv. Mater., 2024, 36, 2404446 CrossRef CAS PubMed.
- T. Sun, W. Zhang, M. Shi, D. Li, Q. Sun, M. Cheng and Z. Tao, Angew. Chem., Int. Ed., 2025, 64, e202416845 CrossRef CAS PubMed.
- Z. Song, Q. Huang, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2025, 64, e202418237 CrossRef CAS PubMed.
- H. Zhang, L. Zhong, J. Xie, F. Yang, X. Liu and X. Lu, Adv. Mater., 2021, 33, 2101857 CrossRef CAS PubMed.
- Y. Zhang, Q. Huang, Z. Song, L. Miao, Y. Lv, L. Gan and M. Liu, Adv. Funct. Mater., 2025, 35, 2416415 CrossRef CAS.
- X. Wang, J. Tang and W. Tang, Adv. Funct. Mater., 2022, 32, 2200517 CrossRef CAS.
- S. Zheng, D. Shi, D. Yan, Q. Wang, T. Sun, T. Ma, L. Li, D. He, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202117511 CrossRef CAS PubMed.
- N. Patil, C. de la Cruz, D. Ciurduc, A. Mavrandonakis, J. Palma and R. Marcilla, Adv. Energy Mater., 2021, 11, 2100939 CrossRef CAS.
- X. Wang, J. Xiao and W. Tang, Adv. Funct. Mater., 2022, 32, 2108225 CrossRef CAS.
- Y. Yan, P. Li, Y. Wang, L. Bi, T. W. Lau, M. Miao, S. Yang, Q. Xiong, F. R. Lin, H.-L. Yip, J. Yin, C. Zhi and A. K. Y. Jen, Adv. Funct. Mater., 2025, 35, 2312332 Search PubMed.
- H. Cui, T. Wang, Z. Huang, G. Liang, Z. Chen, A. Chen, D. Wang, Q. Yang, H. Hong, J. Fan and C. Zhi, Angew. Chem., Int. Ed., 2022, 61, e202203453 CrossRef CAS PubMed.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116289 CrossRef CAS PubMed.
- W. Li, H. Xu, H. Zhang, F. Wei, L. Huang, S. Ke, J. Fu, C. Jing, J. Cheng and S. Liu, Nat. Commun., 2023, 14, 5235 CrossRef CAS PubMed.
- Q.-Q. Sun, T. Sun, J.-Y. Du, Z.-L. Xie, D.-Y. Yang, G. Huang, H.-M. Xie and X.-B. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202307365 CrossRef CAS PubMed.
- Y. Zhang, M. Li, Z. Li, Y. Lu, H. Li, J. Liang, X. Hu, L. Zhang, K. Ding, Q. Xu, H. Liu and Y. Wang, Angew. Chem., Int. Ed., 2024, 63, e202410342 CrossRef CAS PubMed.
- U. Mittal, F. Colasuonno, A. Rawal, M. Lessio and D. Kundu, Energy Storage Mater., 2022, 46, 129–137 CrossRef.
- L. Yan, Q. Zhu, Y. Qi, J. Xu, Y. Peng, J. Shu, J. Ma and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202211107 CrossRef CAS PubMed.
- Y. Zhang, Z. Song, Q. Huang, Y. Lv, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2025, 64, e202423936 CrossRef CAS PubMed.
- Z. Xu, M. Li, W. Sun, T. Tang, J. Lu and X. Wang, Adv. Mater., 2022, 34, 2200077 CrossRef CAS PubMed.
- H. Peng, S. Huang, V. Montes-García, D. Pakulski, H. Guo, F. Richard, X. Zhuang, P. Samorì and A. Ciesielski, Angew. Chem., Int. Ed., 2023, 62, e202216136 CrossRef CAS PubMed.
- T. Sun, W. Zhang, Z. Zha, M. Cheng, D. Li and Z. Tao, Energy Storage Mater., 2023, 59, 102778 CrossRef.
- Y. Wang, C. Wang, Z. Ni, Y. Gu, B. Wang, Z. Guo, Z. Wang, D. Bin, J. Ma and Y. Wang, Adv. Mater., 2020, 32, 2000338 CrossRef CAS PubMed.
- D. Du, J. Zhou, Z. Yin, G. Feng, W. Ji, H. Huang and S. Pang, Adv. Energy Mater., 2024, 14, 2400580 CrossRef CAS.
- N. Wang, R. Zhou, H. Li, Z. Zheng, W. Song, T. Xin, M. Hu and J. Liu, ACS Energy Lett., 2021, 6, 1141–1147 CrossRef CAS.
- D. Zhang, Y. Chen, X. Zheng, P. Liu, L. Miao, Y. Lv, Z. Song, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2025, 64, e202500380 Search PubMed.
- P. Yi, Z. Li, L. Ma, B. Feng, Z. Liu, Y. Liu, W. Lu, S. Cao, H. Fang, M. Ye and J. Shen, Adv. Mater., 2024, 36, 2414379 CrossRef CAS PubMed.
- J. Yang, H. Hua, H. Yang, P. Lai, M. Zhang, Z. Lv, Z. Wen, C. C. Li, J. Zhao and Y. Yang, Adv. Energy Mater., 2023, 13, 2204005 CrossRef CAS.
- L. Zhong, C. Liu, Y. Zhang, J. Li, F. Yang, Z. Zhang and D. Yu, Angew. Chem., Int. Ed., 2025, 64, e202413971 CrossRef CAS PubMed.
- M. Zhu, L. Zhao, Q. Ran, Y. Zhang, R. Peng, G. Lu, X. Jia, D. Chao and C. Wang, Adv. Sci., 2022, 9, 2103896 CrossRef CAS PubMed.
- X. Wang, J. Zhou and W. Tang, Energy Storage Mater., 2021, 36, 1–9 CrossRef.
- S. Wu, M. Taylor, H. Guo, S. Wang, C. Han, J. Vongsvivut, Q. Meyer, Q. Sun, J. Ho and C. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202412455 CrossRef CAS PubMed.
- X. Yan, F. Wang, X. Su, J. Ren, M. Qi, P. Bao, W. Chen, C. Peng and L. Chen, Adv. Mater., 2023, 35, 2305037 CrossRef CAS PubMed.
- R. Emanuelsson, M. Sterby, M. Strømme and M. Sjödin, J. Am. Chem. Soc., 2017, 139, 4828–4834 CrossRef CAS PubMed.
- H. Hong, J. Zhu, Y. Wang, Z. Wei, X. Guo, S. Yang, R. Zhang, H. Cui, Q. Li, D. Zhang and C. Zhi, Adv. Mater., 2024, 36, 2308210 CrossRef CAS PubMed.
- J. Han, M. Zarrabeitia, A. Mariani, M. Kuenzel, A. Mullaliu, A. Varzi and S. Passerini, Adv. Mater., 2022, 34, 2201877 CrossRef CAS PubMed.
- S. Zhang, K. Zhu, Y. Gao and D. Cao, ACS Energy Lett., 2023, 8, 889–897 CrossRef CAS.
- Q. Liu, F. Ye, K. Guan, Y. Yang, H. Dong, Y. Wu, Z. Tang and L. Hu, Adv. Energy Mater., 2023, 13, 2202908 CrossRef CAS.
- H. Li, J. Yang, J. Cheng, T. He and B. Wang, Nano Energy, 2020, 68, 104369 CrossRef CAS.
- L. Chen, W. Sun, K. Xu, Q. Dong, L. Zheng, J. Wang, D. Lu, Y. Shen, J. Zhang, F. Fu, H. Kong, J. Qin and H. Chen, ACS Energy Lett., 2022, 7, 1672–1678 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee00823a |
| This journal is © The Royal Society of Chemistry 2025 |