
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly dense atomic Fe–Ni dual metal sites for efficient CO2 to CO electrolyzers at industrial current densities†
Manman
Qi‡
 b,
Michael J.
Zachman‡
c,
Yingxin
Li‡
d,
Yachao
Zeng‡
b,
Sooyeon
Hwang
e,
Jiashun
Liang
a,
Mason
Lyons
f,
Qian
Zhao
g,
Yu
Mao
d,
Yuyan
Shao
g,
Zhenxing
Feng
f,
Ziyun
Wang
*d,
Yong
Zhao
*h and
Gang
Wu
*ab
b,
Michael J.
Zachman‡
c,
Yingxin
Li‡
d,
Yachao
Zeng‡
b,
Sooyeon
Hwang
e,
Jiashun
Liang
a,
Mason
Lyons
f,
Qian
Zhao
g,
Yu
Mao
d,
Yuyan
Shao
g,
Zhenxing
Feng
f,
Ziyun
Wang
*d,
Yong
Zhao
*h and
Gang
Wu
*ab
aDepartment of Energy, Environmental & Chemical Engineering, Washington University in St. Louis, St. Louis, MO 63130, USA. E-mail: gangw@wustl.edu
bDepartment of Chemical and Biological Engineering University at Buffalo, The State University of New York, Buffalo, NY 14260, USA
cCentre for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
dSchool of Chemical Sciences, The University of Auckland, Auckland 1010, New Zealand. E-mail: ziyun.wang@auckland.ac.nz
eCentre for Functional Nanomaterials Brookhaven National Laboratory Upton, Upton, NY 11973, USA
fSchool of Chemical, Biological, and Environmental Engineering Oregon State University, Corvallis, OR 97331, USA
gPacific Northwest National Laboratory, Richland, WA 99354, USA
hCommonwealth Scientific and Industrial Research Organization (CSIRO) Energy Centre, 10 Murray Dwyer Circuit, Mayfield West, NSW 2304, Australia. E-mail: y.zhao@csiro.au
First published on 5th May 2025
Abstract
Carbon-supported, atomically dispersed, nitrogen-coordinated metal sites (e.g., Fe and Ni) are arguably the most promising catalysts for the electrochemical reduction of CO2 to CO due to their unique catalytic properties and the use of earth-abundant elements. However, conventional single metal sites are constrained by their structural simplicity, causing either too weak or too strong absorption/desorption of multiple critical intermediates (e.g., *COOH and *CO). Current catalysts also suffer from ultra-low loadings (<1.0 wt%) of atomic metal active sites in catalysts, leading to inadequate performance for CO2-to-CO conversion. Here, we develop dual Ni/Fe metal site catalysts with significantly increased atomically dispersed metal loadings (up to 4.8 wt%). A gas-phase chemical vapor deposition (CVD) approach to introducing single Ni sites was integrated with Fe2O3/ZIF-8 precursors, followed by an optimal thermal activation. The optimized CVD-Ni/Fe–N–C catalyst exhibited remarkable electrocatalytic performance for the CO2 reduction to CO in a continuous membrane-electrode-assembly electrolyzer, achieving a maximum CO faradaic efficiency (FECO) of 96% at a current density of 700 mA cm−2 in a near-neutral electrolyte. Furthermore, a desirable but challenging acidic flow-cell electrolyzer was designed using this dual metal site catalyst to improve CO2 utilization, accomplishing a FECO of up to 95% at a CO partial current density close to 600 mA cm−2. Density functional theory (DFT) calculations suggest a synergetic effect between Fe–Ni pairs facilitating *COOH intermediate formation and *CO desorption simultaneously during CO2 to CO conversion. This is key to breaking the linear scaling relationship of conventional single-metal site catalysts during the CO2 reduction reaction.
Broader contextThe electrochemical reduction of carbon dioxide (CO2RR) powered by renewable energy offers a promising strategy for producing carbon-based fuels and chemicals. Among various CO2 reduction pathways, converting CO2 to CO is particularly practical due to its high selectivity (>90%) and energy efficiency. However, current electrocatalytic processes remain inefficient due to the stable chemical bonding strength of CO2 (806 kJ mol−1) and competition from the hydrogen evolution reaction. Carbon-supported, atomically dispersed, nitrogen-coordinated metal sites (e.g., Fe and Ni) are among the most promising catalysts for CO2-to-CO conversion due to their intrinsic catalytic activity, selectivity, and earth-abundance. Current catalysts, however, suffer from low metal loadings (<1.0 wt%) and limited performance. This work presents a high-density atomically dispersed dual-metal atomic Ni–Fe site catalyst with significantly increased metal loadings (4.8 vs. ∼1.0 wt%) accomplished by a chemical vapor deposition approach. The highly dense dual metal site catalyst achieved compelling CO2-to-CO conversion at industrial-level current densities in near-neutral membrane-electrode-assembly electrolyzers and challenging acidic flow-cell electrolyzers. Comprehensive experimental and theoretical studies reveal synergy between Fe–Ni pairs and catalytic mechanisms. This work would represent significant progress in developing highly efficient and low-cost catalysts and electrolyzers for industrial-scale electrochemical CO2 to CO conversion. |
Introduction
Electrochemical CO2 reduction reactions (CO2RRs) for producing valuable chemicals and fuels hold great promise to accomplish energy storage and conversion.1,2 CO is particularly noteworthy among the various CO2RR products due to its wide industrial applications and sufficiently high selectivity for easy subsequent separation from other products.3,4 Generally, the CO2RR performed at ampere-level current densities (≥300 mA cm−2) is highly desirable to mitigate capital costs and increase energy efficiency.5–7 Atomically dispersed metal–nitrogen co-doped carbons (M–N–C) have been demonstrated as the most promising catalysts for CO2-to-CO conversion due to their controllable metal coordination environment that allows the creation of favorable electronic structures to adsorb CO2 and intermediates.8–12Among explored single metal sites, atomic Fe and Ni catalysts are the most efficient CO2-to-CO electrocatalysts with high activity and selectivity.13–18 To synthesize these materials, the zinc-based zeolitic imidazolate framework (ZIF-8) is a desirable N–C precursor owing to its abundant micropores and the low boiling point (b.p.) of the Zn nodes.13–15,19,20 Conventional methods for introducing active metals to ZIF-8 are typically based on wet chemistry, followed by thermal activation.21 Preparing ZIF-8 precursors with Fe or Ni salts usually results in doped or spatially confined metal loading lower than 1.0 wt% after carbonization at high temperatures (>900 °C).14,15,19,22 However, increasing the metal precursor content during catalyst synthesis leads to inactive species like nanoclusters/particles after annealing rather than atomically dispersed active sites.23–25 Therefore, it remains a challenge to develop M–N–C catalysts with ultra-high metal site loading and increased site accessibility to realize efficient CO production at industrially desirable current densities (≥300 mA cm−2) and selectivities (≥95%).24,26–30
Generally, the electrochemical CO2-to-CO process involves multiple proton-coupled electron transfer steps and reaction intermediates (e.g., *COOH and *CO). These intermediates' optimal adsorption/desorption energies are required to facilitate the kinetics.3,10 Atomic Ni sites have a relatively weak binding of CO intermediates (*CO), which is desirable for *CO desorption and CO production.15,20 However, its sluggish kinetic at the first proton-coupled electron transfer hinders CO2 activation at low overpotentials.15,20 In contrast, atomic Fe active sites have high intrinsic activity for CO2 reduction at low overpotentials. Still, the strong binding strength of *CO to Fe active sites inhibits CO desorption and leads to insufficient current densities or large overpotentials.13,18,31–33 The inevitable limitations in linear scaling relationships between different adsorption and desorption species generally arise from the simplicity of single-metal site structures.34
In this regard, the dual-metal site catalysts could overcome the limitations of single isolated sites and yield a synergetic effect between the neighboring metal sites to regulate CO2 activation and intermediate formation/desorption.15,19,35–37 Currently, the limited synthesis methods remain significant challenges to simultaneously realize high-density active sites and well-defined dual-metal configuration for highly efficient CO2 to CO conversion.34,38
Notably, the reaction environment associated with electrolytes used for the CO2RR also plays a crucial role in catalytic activity and selectivity. Generally, alkaline or near-neutral electrolytes are applied to the CO2RR for a high Faraday efficiency (FE) due to the mitigated hydrogen evolution reaction (HER).17,39 Nonetheless, CO2 utilization is limited by the reaction of CO2 with bulk/local hydroxide to form carbonate or bicarbonate ions.39,40 The generated carbonates would further block the CO2 gas channels within the gas diffusion electrode (GDE) and cause the crossover of the (bi)carbonate anion, resulting in significant CO2 loss and low CO2 conversion efficiency.39–42 In contrast, CO2RR in acidic electrolytes can tackle the abovementioned problems by inhibiting carbonate generation. However, the competitive HER becomes dominant, resulting in low CO selectivity and energy efficiency. Studies have demonstrated that concentrating weakly hydrated cations in electrolytes, such as Cs+ and K+, can suppress the HER and enable efficient CO2RR in acids (pH ≤ 2) because these weakly hydrated cations decrease the proton concentration at the interfacial microenvironment of the electrode/electrolyte.42,43 Therefore, exploring highly selective catalysts for achieving promising CO2RR in acidic media is highly demanded.
Herein, we developed an effective chemical vapor deposition (CVD) method to prepare a high-density atomically dispersed Fe/Ni dual metal site electrocatalyst for CO2 to CO conversion. Fe species in the form of Fe2O3 nanoparticles were pre-confined in ZIF-8 precursors, while tunable Ni species were introduced from nickel(II) acetylacetonate (Ni(acac)2) through the CVD process. The optimal CVD-Ni/Fe–N–C catalyst contains a significantly increased loading of atomic metal sites up to 4.8 wt%, nearly five times higher than traditional M–N–C catalysts directly from ZIF-8 precursors.14,20 The dual metal site catalyst exhibited an industry-relevant current density of 700 mA cm−2 with a FECO of 96% in a neutral-electrolyte-based membrane-electrode-assembly (MEA) electrolyzer. More importantly, in a challenging acidic flow cell system, the dual metal catalyst also demonstrated compelling FECO at 95% at a CO partial current density (JCO) of ∼570 mA cm−2, capable of retaining promising stability up to 20 hours at 300 mA cm−2 without apparent performance loss. Density functional theory (DFT) calculations further elucidated that, due to the regulated electronic structures, a synergy between Fe–Ni pairs could break the linear scaling relationship and simultaneously facilitate the formation of *COOH intermediate and promote *CO desorption during the CO2 to CO conversion, with simultaneously enhanced activity and selectivity in a wide potential range.
Results and discussion
Dual metal site catalyst synthesis
The high-density atomically dispersed dual site Ni/Fe–N–C catalyst was synthesized through a CVD approach, as illustrated in Fig. 1a. The catalysts with different Fe-to-Ni ratios (mass ratio of Fe and Ni metals in the precursors) and a single metal site Ni–N–C catalyst were prepared using a similar procedure. Typically, organic compound Ni(acac)2 (b.p. ∼232 °C) was used as a gaseous Ni precursor and placed upstream of a tube furnace with Ar as carrier gas. An Fe2O3@ZIF-8 composite precursor was prepared by encapsulating nano-sized Fe2O3 (∼5 nm) into ZIF-8 during the synthesis and located downstream. Due to the unique cavity structure of the ZIF-8 precursors,44 the guest molecule Ni(acac)2 can be trapped or deposited at the surface of the Fe2O3@ZIF-8 precursor during the CVD at mild temperatures around 240 °C. | ||
| Fig. 1 (a) Schematic of the CVD process to prepare high-density atomic dispersed Ni/Fe dual metal site catalysts. (b) SEM and (c) HAADF-STEM images of carbon shell and Ni core (C@Ni) nanoparticles deposited on the exterior surface of the Fe2O3@ZIF-8 precursors after the CVD at 400 °C. (d) and (e) HAADF-STEM image of a single C@Ni particle (d) and corresponding EELS line-scan profile across the center of the particle (e). HAADF-STEM image (f) of the intermediate precursor after the CVD showing the co-existence of C@Ni and Fe2O3 nanoparticles and corresponding EELS maps of C (red), Ni (green), Fe (yellow), N (blue), and the overlapped EELS map including C, Ni, and Fe elements. (g) The metal ratios of the Fe–N–C, the CVD-Ni/Fe–N–C, and the CVD-Ni–N–C catalysts were determined by using ICP-OES. | ||
We found that the gaseous Ni(acac)2 species deposited on the Fe2O3@ZIF-8 and converted into Ni nanoparticles coated with thin carbon layers, as shown in Fig. 1b and c. Electron energy-loss spectroscopy (EELS) line profiles verified the typical Ni-core and C-shell structure covering the Fe2O3/ZIF-8 particles. Integrated Ni L- and C K-edges quantitatively showed that the carbon shell has a thickness of around 3 nm (Fig. 1d and e). The projected EELS maps of C (red), Ni (green), Fe (yellow), and N (blue) on the Fe2O3@ZIF-8 particles verify the carbon shell and Ni core (Ni@C) structures (Fig. 1f). The uniform carbon shell might be derived from the gaseous Ni(acac)2 (Fig. S1, ESI†). The possible mechanism is that the Ni core was firstly formed after the decomposition of Ni(acac)2. It then catalyzes the growth of carbon shell from hydrocarbon species likely from the thermal decomposition of acac or/and ZIF-8.45,46 EELS also indicates the co-existence of Fe and Ni species on the intermediate precursor after the CVD (Fig. S2, ESI†). This is a critical factor in generating Fe–Ni pairs in the subsequent high-temperature pyrolysis processes (900 °C), which ZIF-8 nanocrystals began to completely carbonize and convert to nitrogen-doped carbon. Meanwhile, the introduced Ni species and pre-doped Fe2O3 nanoparticles simultaneously transformed into atomically dispersed metal sites coordinated with N ligands embedded in the ZIF-8-derived carbon to obtain the final catalysts.
With the increase in Ni(acac)2 during the CVD, the Zn content in catalysts gradually reduces. These opposite trends suggest that Ni sites could replace the original Zn sites and coordinate with N in ZIF-8 precursors (Fig. 1g). Similar to the process verified by Jia et al.,47 the Zn sites in ZIF-8 could serve as the active site templates. Therefore, at high temperatures, the pre-confined Fe and externally introduced Ni compete with Zn to coordinate with N in ZIF-8s, which also helps the removal of Zn. The vapor deposition of Ni(acac)2 over Fe2O3@ZIF-8 yields dense Ni/Fe metal sites. As evidenced by the ICP-OES results, when the mass of Ni(acac)2 was increased upstream during the CVD, the Ni content in dual-atom catalysts rose accordingly (Fig. S3 and Tables S1, S2, ESI†) and eventually reached saturation. The highest Ni content in dual-site CVD-Ni/Fe–N–C catalysts was up to 4.1 wt%, with an atomic Fe content of 0.7 wt%.
As a comparison, the individual CVD-Ni–N–C catalyst was prepared by replacing the Fe2O3@ZIF-8 with Fe-free ZIF-8 precursors in the downstream zone with the optimal Ni(acac)2 weight in the upstream. The Ni content in the obtained CVD-Ni–N–C catalyst is 3.8 wt%. Notably, we also reversed the order to introduce different metals during the catalyst synthesis. For example, Fe(acac)3 was utilized during the CVD, making Fe(acac)3 uniformly deposited onto Ni-doped ZIF-8 precursors. Unfortunately, the total metal loading, especially Ni content, is relatively low due to the challenges of directly doping Ni into ZIF-8. Also, although the boiling point of Fe(acac)3 is approximately 110 °C. However, it's also noted that it decomposes at this temperature, which is unsuitable for CVD. Therefore, the exchange of incorporation order is possible but produces relatively low metal loading, which is ineffective for improving mass activity of studied catalysts. Hence, the optimal synthetic process is to gaseously deposit sufficient Ni(acac)2 onto the Fe2O3@ZIF-8 precursor to yield highly dense dual metal site catalysts with exceptional activity and selectivity to be discussed below.
Catalytic properties of CO2 to CO conversion
We first performed electrochemical CO2 reduction in an H-type cell with 0.5 M potassium bicarbonate (KHCO3) as the electrolyte to investigate intrinsic catalytic activities and selectivities of various catalysts for the CO2 to CO conversion. We tuned the Ni content in the dual metal site catalysts to determine the interplay between Fe and Ni sites for the CO2RR. The CO2RR activity and selectivity were improved simultaneously as the Ni content in catalysts increased, as shown in Fig. S4 (ESI†). Specifically, FECO and CO partial current density (JCO) of the CVD-Ni11/Fe15–N–C catalysts were 67%, −18 mA cm−2 at −0.7 VRHE. The CVD-Ni82/Fe14–N–C catalyst achieved 94% and −75 mA cm−2, respectively. The improved CO2RR performance could be attributed to the increased Ni contents in the catalysts. With further increasing the mass of Ni(acac)2 upstream, the Ni content was saturated with a mass ratio of 3.8 wt% (0.3 wt% lower than CVD-Ni82/Fe14–N–C, probably due to the measurement error), with reduced FECO (91%) and JCO (−70 mA cm−2). The excessively induced Ni is likely to cause the formation of Ni/Fe nanoclusters/particles (which favors the HER process) rather than atomically dispersed active sites.29,30,48 It should be noted that the control Zn–N–C sample derived from Ni/Fe-free ZIF-8 precursors with identical annealing exhibited negligible catalytic performance towards CO2RR, which agrees with our previous works.13,14,49Generally, a larger charge capacity in the single metal sites will lower the electrochemical barrier towards the CO2RR with enhanced activity.50 Hence, the electrochemical active surface areas (ECSAs) of the dual-metal sites CVD-Ni/Fe–N–C and single-metal sites catalysts were evaluated by recording their double-layer capacitances (Cdl) at different CV scanning rates, as shown in Fig. S5 (ESI†).51Cdl or ECSAs tend to decrease when the Ni content in the catalysts increases because excess Ni(acac)2 catalyzes the formation of carbon shells, reducing the micropore surface areas within catalysts and compromising catalytic performance.
Thermal activation often significantly impacts the intrinsic activity and selectivity of single metal site catalysts regarding their adsorption energies of CO2 molecules and intermediates.13,25 We also investigated the effects of thermal activation temperature and duration on catalyst structures and electrocatalytic properties for these CVD-Ni/Fe–N–C catalysts. We have controlled different temperatures (T = 800, 900, 1000, and 1100 °C) during thermal activation with the same amount of Ni in the CVD, as shown in Fig. S6 (ESI†). An initial increase in temperatures from 800 to 900 °C leads to improvement in both activity and selectivity due to the formation of active metals sites with optimal coordination and bond length between metal and nitrogen ligands.13,14 However, further rising temperatures from 900 to 1100 °C maintained the CO selectivity of the CO2RR (FECO: ∼97%) but decreased the reaction rates, evidenced by the drop of JCO from −51 (900 °C) to −32 mA cm−2 (1100 °C) at −0.7 VRHE. The loss of N at higher temperatures and a reduced active site density may cause the activity loss.13,25 Prolonging the thermal treatment duration at 900 °C from 1 to 3 hours. The measured JCO was increased remarkably without compromising the CO selectivity due to the continuing evaporation of the unfavorable Zn elements from ZIF-8 precursors (Fig. S7 and S8, ESI†). These results highlight the importance of preparation parameters on dual-site CVD-Ni/Fe–N–C catalysts to significantly promote CO2RR performance via creating a favorable atomically dispersed coordinate environment for high-density metal sites. To be concise and clear, we labeled the dual metal site with optimal Ni content, carbonization temperature (900 °C), and duration (3 h) as the CVD-Ni/Fe–N–C in the rest of this work.
These two single-metal-site catalysts (i.e., CVD-Ni–N–C and Fe–N–C) and the optimal dual-metal-site CVD-Ni/Fe–N–C catalyst for CO production were compared by potential-dependent CO2RR operation (Fig. 2a and b). The onset potential observed with the CVD-Ni/Fe–N–C catalyst was more positive than the CVD-Ni–N–C catalyst, implying the benefits of combining Fe and Ni sites for enhancing the CO2RR kinetics.15,19 At a relatively low potential range from −0.5 to −0.7 V, the CVD-Ni/Fe–N–C catalyst exhibited remarkably higher JCO and FECO than those of Fe–N–C and the CVD-Ni–N–C catalysts (Fig. 2c). For example, the FECO and JCO can reach 98% and −55 mA cm−2, respectively, at −0.6 VRHE, outperforming the CVD-Ni–N–C (94% and −21 mA cm−2) and the Fe–N–C (90% and −26 mA cm−2). As the CVD-Ni/Fe–N–C and CVD-Ni–N–C have similar high metal loadings (4.1 wt% for CVD-Ni/Fe–N–C, 3.8 wt% for CVD-Ni–N–C), the superior activity and selectivity of the CVD-Ni/Fe–N–C could be due to the likely synergistic effect between Fe and Ni sites, which will be discussed in the theory part. The newly achieved CO2 to CO conversion performance is among the best-reported M–N–C catalysts in H-cell tests (Table S3, ESI†), probably benefiting from the high-density, surface-exposed active sites and possible synergy between Fe and Ni sites. The turnover frequency (ToF) values toward CO2 to CO conversion were further calculated to determine the enhanced intrinsic activity of dual metal sites over single ones. The metal site densities in studied catalysts were quantified by using surface-sensitive XPS. The ToFs at −0.7 VRHE are ranked from high to low: CVD-Ni/Fe–N–C (6.3 × 103 h−1) > CVD-Ni–N–C (5.9 × 103 h−1) > Fe–N–C (5.6 × 103 h−1). The ECSA-normalized current density of the CVD-Ni/Fe–N–C catalyst was around three times higher than the Fe–N–C and the CVD-Ni–N–C counterparts (Fig. 2e and Fig. S5, ESI†), further verifying the higher intrinsic activity of the dual-metal-site CVD-Ni/Fe–N–C catalyst. The promising stability of the CVD-Ni/Fe–N–C catalysts was demonstrated in an H-cell at −0.7 VRHE with FECO above 90% for up to 20 hours. (Fig. 2f).
 | ||
| Fig. 2 Electrocatalytic CO2 reduction performance in H-cells with CO2-saturated 0.5 M KHCO3 electrolyte. (a) LSV curves of the Fe–N–C, the CVD-Ni/Fe–N–C, and the CVD-Ni–N–C catalysts were recorded at a scan rate of 5 mV s−1 and 90% iR compensation. (b) FEs of CO for the Fe–N–C, the CVD-Ni/Fe–N–C, and the CVD-Ni–N–C catalysts. (c) JCO for the Fe–N–C, the CVD-Ni/Fe–N–C, and the CVD-Ni–N–C catalysts at different potentials. (d) and (e) calculated TOFs (d) and ECSA-normalized JCO (e) for various catalysts. (f) A short-term stability test for the CVD-Ni/Fe–N–C at −0.7 V vs. RHE in H-cells. | ||
Catalyst CO2RR performance in MEAs and flow cells
To explore the viability of the CVD-Ni/Fe–N–C catalyst for practical applications, we performed CO2 reduction in a zero-gap membrane-electrode-assembly (MEA) electrolyzer with 0.1 M KHCO3 as the electrolyte. The catalyst was spray-coated onto a gas diffusion layer (GDL) to construct the triple-phase boundary (TPB) and guarantee the CO2 reaction took place in a gaseous phase, overcoming the mass transport limitation of CO2 in the H-type device.26,27 Fig. S9 (ESI†) displays the schematic illustration of a zero-gap MEA. The CVD-Ni–N–C cathode showed a maximum JCO current density of 500 mA cm−2 with the FECO of 91% at 3.9 V (Fig. 3a). Furthermore, the CVD-Ni/Fe–N–C cathode exhibited better performance for the CO2 to CO conversion under a wide range of industrially desirable operating current densities: FECO > 90% at 300 to 700 mA cm−2 with a cell voltage from 3.0 to 3.8 V (Fig. 3b). A maximum JCO as obtained at 675 mA cm−2 with the highest FECO of 96% at 3.8 V, outperforming most of the previously reported CO2 to CO catalysts tested in MEAs and flow cells (Table S4, ESI†). | ||
| Fig. 3 (a) and (b) Faradaic efficiency and cell voltages of the CVD-Ni–N–C (a) and the CVD-Ni/Fe–N–C (b) cathodes with catalyst loading of 1 mg cm−2 in MEAs with 0.1 M KHCO3 as the electrolyte. (c) and (d) The faradaic efficiency and cathodic potentials vs. Ag/AgCl at different current densities for the CVD-Ni–N–C (c) and the CVD-Ni/Fe–N–C (d) cathodes in acidic flow cells with 0.025 M H2SO4 and 3 M KCl as the catholyte and 0.5 M H2SO4 as the anolyte. (e) A short-term stability test for the CVD-Ni/Fe–N–C operated at 300 mA cm−2 in an acidic flow cell. The triangle points correspond to FE for CO (red) and H2 (grey), and the line corresponds to the potential. Both the MEA and flow cell tests were conducted without iR compensation. | ||
To further explore the potential of the developed CVD-Ni/Fe–N–C catalyst in the challenging acidic media, we performed CO2 reduction in an electrolyte of 0.025 M H2SO4 and 3 M KCl in a slim-flow cell. We adopted a 100 nm-thick Ag sputtering-coated ePTFE film (Ag loading: ∼0.1 mg cm−2) as the substrate to improve gas diffusion and support catalysts.42 Carbon black serving as the catalyst on the identical substrate was tested to exclude the side effect of the Ag layer on the ePTFE support. As shown in Fig. S10 (ESI†), the highest FECO of the carbon-coated ePTFE-Ag substrate reached 54.5% of FECO at 100 mA cm−2 at −2.3 V vs. Ag/AgCl. The FECO decreased to 19.6% at 400 mA cm−2 with voltage at −4.2 V vs. Ag/AgCl. In contrast, when using the CVD-Ni/Fe–N–C and CVD-Ni–N–C catalysts as the cathode, the FECO and current density have been significantly promoted (Fig. 3c and d). For instance, the CVD-Ni–N–C cathode exhibited a maximum FECO of 90% with current dentistry at 500 mA cm−2 at −3.8 V vs. Ag/AgCl (Fig. 3c). Furthermore, at −3.8 V, current dentistry of 500 mA cm−2 was achieved on the CVD-Ni/Fe–N–C cathode with an FECO of 93%, showing improvements in the activity and selectivity. Notably, the CVD-Ni/Fe–N–C exhibited a maximum FECO of 95% at 600 mA cm−2 at −4.5 V vs. Ag/AgCl (Fig. 3d). The FECO of the CVD-Ni/Fe–N–C cathode maintained >90% FECO during 20-hour electrolysis at an industrial required current density of 300 mA cm−2 (Fig. 3e), further evidencing the encouraging stability of the CVD-Ni/Fe–N–C catalyst under actual CO2RR conditions. The CVD-Ni/Fe–N–C and CVD-Ni–N–C catalysts outperformed most reported M–N–C catalysts in acidic CO2 electrolysis (Table S5, ESI†). A more detailed reaction mechanism in acidic media for the CO2 to CO conversion was discussed in the theoretical part.
Catalyst morphologies and atomic structures
First, the morphology and structure of studied catalysts were characterized using scanning electron microscopy (SEM) and aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The Fe–N–C catalyst presented typical rhombic dodecahedral structures and uniform nanoparticles around 100 nm in diameter (Fig. 4a and Fig. S11a, b, ESI†).52,53 In contrast, the CVD-Ni/Fe–N–C catalyst presents a slight collapse in catalyst particles and concave surfaces. Interestingly, numerous carbon nanotubes are formed at the surface of the CVD-Ni/Fe–N–C catalyst (Fig. 4b and Fig. S11c, d, ESI†), similar to the CVD-Ni–N–C catalyst (Fig. 4c and Fig. S11e, f, ESI†). A control sample was prepared by replacing the Ni(acac)2 with Mg(acac)2 at the upstream to exclude the side effects of acetylacetonate (acac) ligand. The obtained CVD-Mg/Fe–N–C catalyst (Fig. S11g and h, ESI†) showed a similar structure to Fe–N–C without nanotubes, suggesting that Ni played a crucial role in generating branched nanotubes at the surface of the CVD-Ni/Fe–N–C and the CVD-Ni–N–C catalysts. No metallic nanoparticles were observed in STEM images of all studied catalysts, indicating that active Fe and Ni species were dispersed uniformly at the atomic level (Fig. S12, ESI†). In addition, we found that Ni has a unique catalytic effect on the graphite planes on the carbon support compared with Fe (Fig. S12c, g, h, k and l, ESI†). Powder X-ray diffraction (XRD) analysis displayed two broad peaks centered around 28° and 44°, which were assigned to the (002) and (101) facets of graphitic carbon (Fig. S13, ESI†).19 The calculated interplanar spacing of (002) and (101) facets were around 1.64 and 1.07 Å, respectively.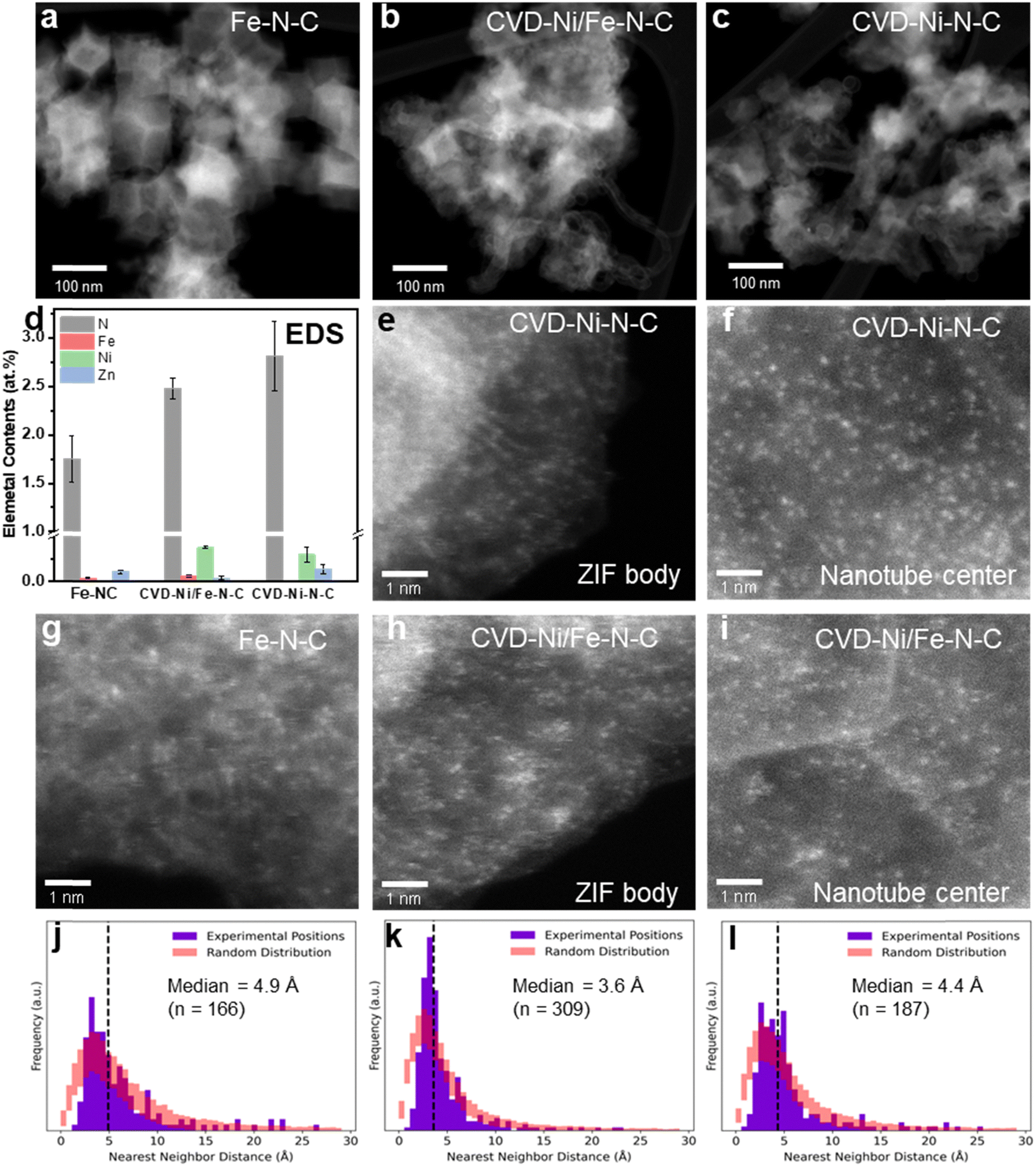 | ||
| Fig. 4 Morphologies and atomic dispersion of metal sites in studied M–N–C catalysts. (a)–(c) HAADF-STEM images of the Fe–N–C (a), the CVD-Ni/Fe–N–C (b), and the CVD-Ni–N–C (c). (d) Elemental Fe, Ni, Zn, and N compositions from EDS analysis. (e) and (f) HAADF-STEM images showing the atomic dispersion of the CVD-Ni–N–C in the ZIF-8-derived carbon particle body (e) and nanotube center (f). (g)–(i) HAADF-STEM images of the Fe–N–C (g) and the CVD-Ni/Fe–N–C carbon particle body (h) and nanotube center (i). (j)–(l) The corresponding nearest neighbor distance statistics for the Fe–N–C (j), and the carbon particle body (k) and nanotube center (l) of the CVD-Ni/Fe–N–C catalysts. The red bars represent the range of results (one standard deviation above and below the mean) from one hundred sets of random atom positions generated using the same number of atoms and field of view as each associated experimental image. | ||
EELS point spectra taken directly on individual metal sites in the CVD-Ni/Fe–N–C catalyst confirmed the co-existence of Fe and Ni atoms near one another and near nitrogen (Fig. S14, ESI†), implying that Ni and Fe sites were likely coordinated with nitrogen in carbon.15,20 Similarly, according to the EELS point spectra of the CVD-Ni–N–C catalyst, single Ni sites were coordinated with nitrogen in carbon (Fig. S15, ESI†). Energy-dispersive X-ray spectroscopic (EDS) mapping of the CVD-Ni/Fe–N–C and CVD-Ni–N–C catalysts demonstrated a homogeneous distribution of metal sites on the nitrogen-doped carbon support (Fig. S16 and S17, ESI†). Fig. 4d and Table S6 (ESI†) show EDS quantifications of the catalysts’ Fe, Ni, Zn, and N contents. The atomic ratio of Fe in the Fe–N–C catalyst was 0.04 at%, Ni in CVD-Ni–N–C catalyst was 0.30 at%, and Fe and Ni in the CVD-Ni/Fe–N–C catalyst were 0.06 and 0.38 at%, respectively. Moreover, the nitrogen content in the CVD-Ni/Fe–N–C (2.5 at%) and the CVD-Ni–N–C (2.8 at%) was increased compared to that of the Fe–N–C (1.8 at%), implying the potentially enhanced capability of hosting high-density atomic metal sites.24,25 Highly dense metal sites (bright spots, i.e., atoms heavier than carbon) were apparent in the CVD-Ni–N–C catalyst's ZIF-8-derived carbon particles (Fig. 4e) and nanotubes (Fig. 4f and Fig. S18, ESI†). Fig. 4g shows the well-defined atomic dispersion of the Fe–N–C catalysts. Furthermore, the CVD-Ni/Fe–N–C catalyst exhibits a markedly enhanced atom density compared with the Fe–N–C (Fig. 4h, i and Fig. S19, ESI†). Combining the EDS (Fig. 4d) and ICP-OES (Fig. 1g) results, the metal loading in the CVD-Ni/Fe–N–C was significantly higher than the Fe–N–C catalyst, signifying enhanced atomic dispersion. High-temperature treatments often lead to a direct atomization of small nanoparticles into atomic sites.53 Hence, we believe that the pre-confined Fe2O3 nanoparticles and induced carbon-encapsulated Ni nanoparticles (Ni@C) atomized into atomically dispersed sites to enrich active site density on the ZIF-8-derived carbon. Carbon-shell and Ni-core structures self-catalyzed to form extended nanotubes with dense atomic sites in the nanotube center and edge, further increasing the active sites density from the CVD approach.
To explore whether the metal sites tended to be more closely associated with one another, rather than the case of a random distribution, we compared the nearest-neighbor distributions of metal sites in both Fe–N–C and CVD-Ni/Fe–N–C against those for randoms sets of atomic positions, using the same number of atoms and field of view as the images (Fig. 4j–l). The range of nearest neighbor values (one standard deviation above and below the mean) returned for the sets of random atom positions are shown on each plot in red. The results indicate a greater tendency for nearest neighbors to be situated approximately 3–5 Å apart compared to a random distribution. Notably, the CVD-Ni/Fe–N–C particle deviates most significantly from the random distribution, exhibiting a taller and sharper peak in this range than the Fe–N–C ZIF body or the CVD-Ni/Fe–N–C nanotube. These statistical results indicate that the metal sites in the CVD-Ni/Fe–N–C catalyst tended to be more associated with each other over distances of a few Å than random metal site positions. The above analysis validated the successful preparation of high-density atomically dispersed pair metal sites in the CVD-Ni/Fe–N–C catalysts using the CVD process. Notably, the CVD-Ni/Fe–N–C catalyst with extended nanotubes and well-defined morphology can facilitate electron transfer and create CO2 transport highways within catalyst layers.
X-ray photoelectron spectroscopy (XPS) was acquired to investigate chemical compositions and nitrogen dopants at the surface of studied catalysts. The quantified surface elemental contents are listed in Table S7 and Fig. S20a (ESI†). The N content in the CVD-Ni/Fe–N–C (8.0 at%) was 1.6 times higher than in the Fe–N–C (5.0 at%). The Fe content in the CVD-Ni/Fe–N–C (0.80 at%) catalyst increased notably compared with the Fe–N–C (0.08 at%). Ni contents were similar between the CVD-Ni/Fe–N–C (0.81 at%) and CVD-Ni–N–C (0.90 at%) catalysts, which were higher than our previous Ni–N–C catalysts derived from wet-chemistry from ZIF-8 precursors.14 The N1s spectrum demonstrated that catalysts likely have five types of nitrogen, which are pyridinic-N (398.58 eV), metal–N (399.60 eV), pyrrolic-N (400.99 eV), graphitic-N (402.49 eV), and oxidic-N (405.00 eV) as shown in Fig. S20b (ESI†).15 The metal–N percentage of the Fe–N–C catalyst was 0.8 at%, which was increased to 1.7 at% in the CVD-Ni/Fe–N–C, probably due to the extra induced Ni improved metal loading. Additionally, pyridinic-N and pyrrolic-N were the two dominant N-types in the prepared catalysts, of which pyridinic-N was in higher content (Fig. S20c and Table S8, ESI†). The increased surface nitrogen content can potentially increase the formation probability of atomically dispersed metal sites. The results verified that the effective CVD approach simultaneously increases the metal loading and nitrogen content. Notably, the surface deposition using gaseous metal precursors can ensure the active sites are exposed at the surface, further contributing to the metal site utilization and mass transport.
The high-resolution C1s spectra exhibited dominant peaks assigned to sp2-hybridized graphitic carbon (Fig. S20d, ESI†), indicating a nearly identical carbon structure in all studied Fe–N–C, CVD-Ni/Fe–N–C, and CVD-Ni–N–C catalysts. Raman spectroscopy was further employed to study the defective features of nitrogen-doped carbon in catalysts. The intensity ratio between the disordered carbon (D band, ∼1353 cm−1) and graphitic carbon (G band, ∼1580 cm−1) revealed a similar graphitization degree regardless of the metal loading and the Fe to Ni ratios (Fig. S21, ESI†), suggesting nearly identical carbon structures in studied catalysts. Sufficient surface areas are essential for high-density and uniform dispersion of metal sites in catalysts.23,54 The measured Brunauer–Emmett–Teller (BET) specific surface area of the CVD-Ni/Fe–N–C catalysts is 541.7 m2 g−1, which was close to the Fe–N–C (516.1 m2 g−1) and higher than the CVD-Ni–N–C (469.9 m2 g−1) (Fig. S22 and Table S9, ESI†). Porosity analyses indicate that the volume of mesoporosity of both CVD-Ni/Fe–N–C(0.14 cm3 mg−1) and CVD-Ni–N–C (0.12 cm3 mg−1) catalysts are higher than that of Fe–N–C (0.09 cm3 mg−1), likely due to the concaved surfaces and extended nanotubes in the catalysts. Such unique catalyst structures can promote the construction of 3D-interconnected porosity in the catalyst layer, facilitating the mass transport of CO2 and CO.55
The dual metal CVD-Ni/Fe–N–C and single metal site control samples were further characterized using X-ray absorption spectroscopy (XAS).56,57 The X-ray absorption near-edge structure (XANES) spectra for Fe K-edge of the CVD-Ni/Fe–N–C (∼7113 eV) and Fe–N–C (∼7115 eV) positioned in the middle of FePc and Fe foil references, suggesting the valence of Fe is positive but lower than +2 (Fig. 5a). Moreover, the absorption edge energy of Fe K-edge of the CVD-Ni/Fe–N–C presented lower energy than the single metal site-based Fe–N–C, manifesting a decreased valence of Fe in the CVD-Ni/Fe–N–C. The XANES spectra of Ni K-edge of CVD-Ni/Fe–N–C (∼8339 eV) and CVD-Ni–N–C (8338 eV) between those of Ni foil and phthalocyanine nickel (NiPc) reveal the valence of Ni as being between 0 and +2 valence as well (Fig. 5b).15 Importantly, the valence of Ni in the CVD-Ni/Fe–N–C was slightly higher than in the CVD-Ni–N–C. Hence, the measured energy shifts of Fe and Ni in XANES spectra confirmed the possible synergistic effect between Fe–Ni pairs and electron transfer from Ni to Fe in the dual-site CVD-Ni/Fe–N–C catalyst. These different characteristic features among single-metal sites, dual-metal sites, and their counter samples suggested they have different local coordination environments. The second rectangles (black solid) (Fig. 5a and b) corresponded to the dipole-allowed 1s–4pz transition in the disordered D4h configuration. The intensities of the Fe–N–C, the CVD-Ni–N–C, and the CVD-Ni/Fe–N–C catalysts were significantly weaker than those of standard FePc and NiPc references. These results imply that the local coordination structure of Fe and Ni sites in the CVD-Ni/Fe–N–C was highly disordered and defective, which was probably caused by the asymmetric and unsaturated M–N coordination.16
 | ||
| Fig. 5 (a) and (b) Fe K-edge (a) and Ni K-edge (b) XANES spectra for these M–N–C catalysts and references. (c) and (d) FT-EXAFS spectra Fe K-edge (c) and Ni K-edge (d) for various M–N–C catalysts and references. (e) The R-space EXAFS-fitting curves of the CVD-Ni/Fe–N–C at Fe K-edge, insert image is the fitting model, and (f) Ni K-edge. The brown, light grey, grey, and dark brown balls represent C, N, Ni, and Fe atoms, respectively. | ||
The extended X-ray absorption fine structure (EXAFS) characterization was further employed to analyze the coordination environment of metal sites. The Fourier transform k3-weighted χ(k) function of spectra (FT-EXAFS) for Fe sites in the CVD-Ni/Fe–N–C and the Fe–N–C catalysts and Ni sites in the CVD-Ni/Fe–N–C and CVD-Ni–N–C catalysts show similar prominent peaks around 1.3–1.5 Å, which are likely belonging to the first shell coordination Fe–N and Ni–N scattering paths (Fig. 5c and d).13,31 The absence of second shell coordination of Fe–Fe (∼2.20 Å) and Ni–Ni (∼2.17 Å) strong metal–metal scattering signals suggests that the Fe and Ni sites in the CVD-Ni/Fe–N–C are atomically dispersed.15,25,31
Since bridged structures are common in dual metal site M–N–C catalysts,20 we proposed three possible bimetal–nitrogen fitting models: double-bridged (2-N), single-bridged (1-N), and non-bridged (0-N), based on the EXAFS experimental spectra. The EXAFS fitted results are listed in Fig. 5e, f and Fig. S23–S25 (ESI†). The double-bridged (2-N) model converged to the most likely structures of the CVD-Ni/Fe–N–C. The coordination numbers derived from Fe EXAFS analysis are marginally higher than theoretically predicted; however, the fitted amplitude reduction factor (S02) remains within a physically reasonable range (0.7 < S02 < 1), supporting the validity of the structural model. In contrast, fitting the single N-bridged and non-bridged models resulted in larger contributions from second shell scattering to account for the lack of the 2.46 Å Fe–Ni bond. Meanwhile, these models were constrained to yield physically realistic results, and the fitting statistics were undesirable, as shown by the R-factor > 2% (Fig. S26, ESI†). The detailed fitting parameters with the double-bridged (2-N) model involving the metal–N and metal–metal coordination numbers are summarized in Table S10 (ESI†), and a model structure with the coordinated atoms labeled (Fig. S27, ESI†). Specifically, the coordination numbers of Fe–N and Ni–N sites were 2.5 and 4.0, respectively. The notable signals at approximately 2.5 to 2.6 Å observed for both FePc and NiPc are primarily attributed to multiple scattering contributions from their coordinated ligands (Fig. S28, ESI†). In contrast, as for the CVD-Ni/Fe–N–C catalyst, both the Fe and Ni sites were embedded in the defective carbon support, significantly suppressing scattering contribution from the second coordination layer due to the disorder effects.16,20 Thus, based on the experimental and fitted FT-EXAFS spectra in R space, we could confirm that the synergistic effect between Fe and Ni pairs and the unsaturated Fe–N and Ni–N coordination environment of the CVD-Ni/Fe–N–C with a possible structure illustrated in Fig. 5e, where Fe and Ni share two bridging N atoms, forming a dual-metal center.
Elucidate the possible dual metal site synergy
DFT calculations were performed to elucidate the intermediate geometry, reaction kinetics, and charge transfer during the CO2 to CO conversion on the Fe–N–C, CVD-Ni–N–C, and CVD-Ni/Fe–N–C catalysts. According to EXAFS analysis (Fig. 5e) and previous literature,11 the possible active site structures of Fe–N–C, CVD-Ni–N–C, and CVD-Ni/Fe–N–C catalysts are shown in Fig. S29 (ESI†). Before the desorption of CO(g) product, CO2RR undergoes two elementary steps generating *COOH and *CO intermediates. The CO2RR mechanism of the dual-metal-site CVD-Ni/Fe–N–C is shown in Fig. 6a. As previously seen in Fig. 2a, the onset potential of the CVD-Ni/Fe–N–C is more positive than the CVD-Ni–N–C catalysts, closed to the Fe–N–C. However, the FECO from Fig. 2b indicated that the potential for optimal CO(g) selectivity is at −0.6 to −0.7 V vs. RHE. Thus, the applied potential for the Gibbs free energy calculations was set based on −0.7 V vs. RHE for DFT calculations using the computational hydrogen electrode method.58 Fig. S30–S32 (ESI†) illustrates the different geometric adsorption of *CO, *COOH, and *H on the three catalysts. Free energy diagrams of CO2RR steps for these catalysts are shown in Fig. 6b. The free energy of these three studied catalysts for the initial *COOH step correlates well with the LSV results (Fig. 2a) concerning their catalytic activity. Compared to the Fe–N–C, the CVD-Ni/Fe–N–C shows further reduced free energy (0.10 eV) to form the critical *COOH, which undergoes an exothermic reaction from CO2(g). In contrast, the CVD-Ni–N–C catalyst showed the highest free energy (0.52 eV) at the *COOH step. Higher free energy at the *COOH step signifies that the CVD-Ni–N–C catalyst is less susceptible to *COOH attachment than the Fe–N–C and the CVD-Ni/Fe–N–C catalysts. Concerning the formation of *CO, all three catalysts undergo an exothermic reaction pathway to produce *CO. As for the third step to desorb *CO to produce CO(g), the CVD-Ni/Fe–N–C catalyst shows lower free energy by 0.07 eV, and both Ni-containing catalysts exhibit exothermic reaction pathways to desorb CO. On the contrary, the Fe–N–C catalyst shows the lowest free energy and undergoes an uphill step to produce CO(g), indicating an unfavorable step. This is related to the much stronger adsorption of *CO on the Fe–N–C than on the CVD-Ni/Fe–N–C and the Ni–N–C catalysts.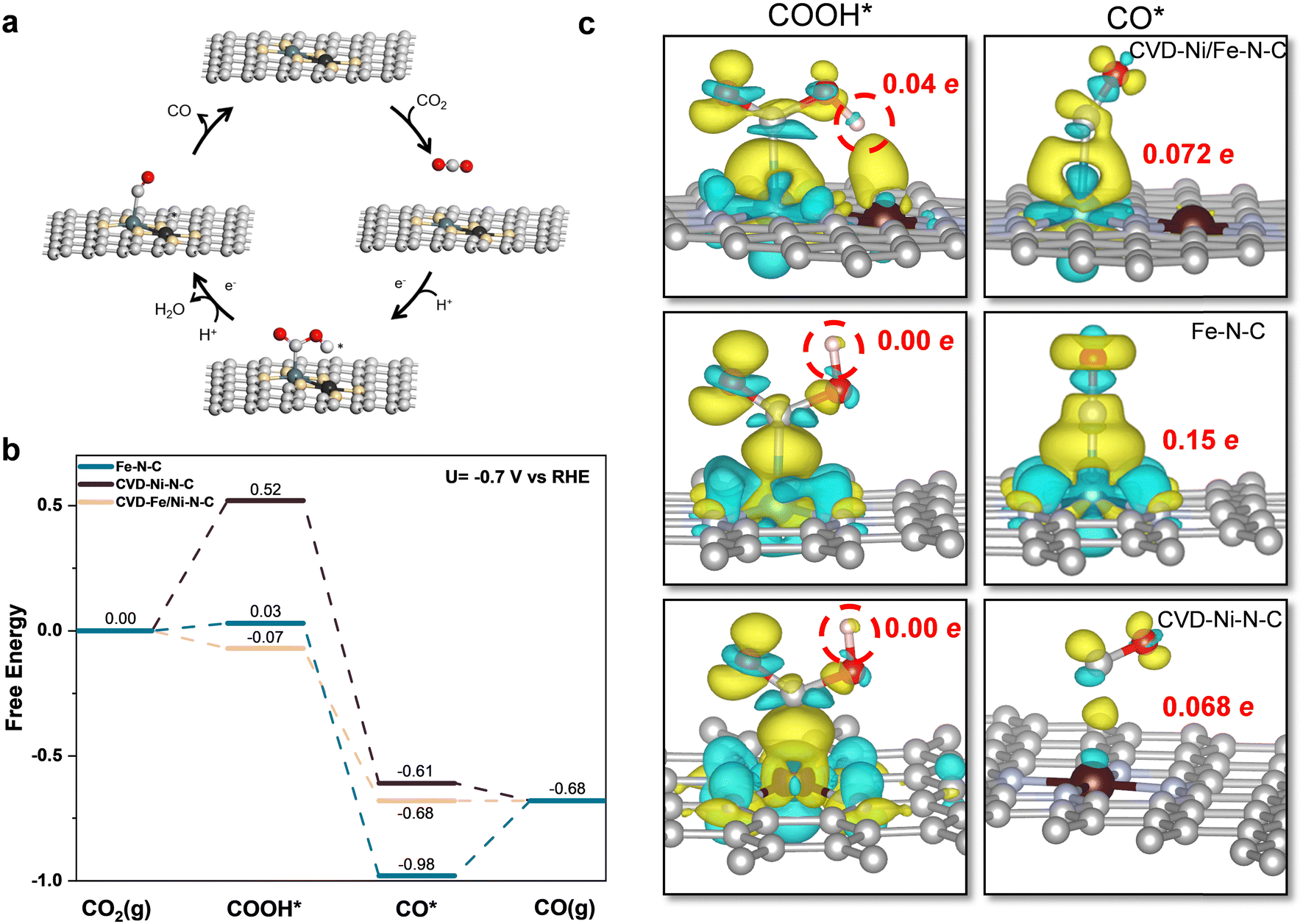 | ||
| Fig. 6 (a) Schematic of CO2 to CO conversion on Ni/Fe dual metal sites in the CVD-Ni/Fe–N–C catalyst, where O atoms in red and H atoms in white. (b) Free energy profile of CO2 to CO for the Fe–N–C, the CVD-Ni–N–C, and the CVD-Ni/Fe–N–C catalysts at −0.7 V vs. RHE. (c) Charge density difference before and after CO and COOH adsorption upon the dual Ni/Fe, Fe, and Ni sites. Red numbers mean the electrons transfer to H (in *COOH) and *CO. Yellow represents electron accumulation, and cyan represents electron depletion; the value of iso-surface is 0.002. | ||
The above theoretical results pinpoint that the presence of Fe sites is more favorable regarding the *COOH step, and the presence of Ni sites holds more advantages regarding the *CO step. Thus, the combination of Fe and Ni sites generates a synergy effect, in which the advantages of both metal sites are leveraged. As a result, the CVD-Ni/Fe–N–C catalyst exhibits a stronger *COOH adsorption and a more susceptible dissociation of *CO, making the CO2RR more spontaneous and maximizing CO(g) selectivity.
Furthermore, the thermodynamic driving force for CO2RR is directly influenced by electrode potential and the local pH values due to proton-coupled electron transfer (PCET) steps. We further calculate the Gibbs free energies at different pH values and present in Fig. S33 (ESI†) along with a brief discussion. Likewise, the Ni/Fe dual sites yield similar synergy to promote CO2 to CO conversion in wide pH values from 0 to 14.
The charge density difference of *COOH and *CO was then investigated and shown in Fig. 6c. The *COOH adsorption configuration in the Fe–Ni dual metal system orients the hydrogen atom downwards, as opposed to the upward orientation observed in Ni or Fe systems. This could be attributed to the Fe–Ni dual metal site; when *COOH is located on the Fe site, the downward H could form a hydrogen bond with the neighboring Ni site, thus giving it additional adsorption strength. This hydrogen bond can be captured by the electron transfer between Ni and H (as shown in Fig. 6c, a clear yellow region between Ni and H and 0.04 e electron transfer). In the case of *CO, the greater the electron gains by *CO, the stronger its adsorption. Fig. 6c shows that CO gains the most electrons (0.15 e) on the Fe site, leading to the strongest *CO adsorption (unfavorable for CO desorption). Within the Fe–Ni dual metal site, *CO adsorption configuration on the Fe site is not as vertical as in a single Fe site system, potentially leading to compromised electron transfer (0.072 e) and weaker *CO adsorption.
The HER is the primary competitive reaction in the CO2RR. Free energy diagrams of the HER process for the Fe–N–C, Ni–N–C, and CVD-Ni/Fe–N–C catalysts were presented in Fig. S34 (ESI†). The Gibbs free energy change of the rate-determining step for the HER (* → *H) was compared to the Gibbs free energy changes of CO2 → *COOH and *COOH → *CO. The calculated theoretical overpotentials of the HER (free energy of H* adsorption) on the Fe–N–C, Ni–N–C, and CVD-Ni/Fe–N–C are compared in Table S11 (ESI†). For Fe–N–C and CVD-Ni/Fe–N–C, the ΔG of the HER is greater than that of *COOH, thus signifying that the CO2RR suppresses the HER during CO2RR.
Conclusions
In summary, an effective approach to integrating a CVD to deposit gaseous Ni species onto Fe-containing ZIF-8 precursors was developed to successfully construct atomically dispersed Ni/Fe dual-metal-site catalysts with high metal loadings and more accessible active sites. Due to the unique gaseous phase deposition, various Ni loadings anchored in a porous nitrogen-doped carbon derived from ZIF-8 were tunable to regulate catalyst structures and morphologies for maximum catalytic performance. The optimal Fe/Ni dual metal site catalyst achieved exceptional CO2 to CO conversion at nearly 100% faradaic efficiency and industrial current densities. In particular, a FECO of 96% with maximum JCO at ∼700 mA cm−2 was demonstrated in MEA tests when using 0.1 M KHCO3 as the electrolyte. More importantly, the CVD-Ni/Fe–N–C catalyst exhibited FECO of 95% with maximum JCO at ∼600 mA cm−2 in challenging acidic electrolytes.Extensive electron microscopy and spectroscopy characterization suggest a high probability of double-bridged (2-N) Ni–Fe dual metal sites in the optimal catalyst. Furthermore, theoretical investigations reveal that the Fe and Ni sites in catalysts synergistically facilitate CO2 to CO conversion under a wide pH range, capable of reducing the formation energy barrier of *COOH and favoring *CO desorption simultaneously. Hence, the unique Ni–Fe pair sites lead to compelling CO2 to CO conversion activity and selectivity. This work provides an effective strategy for synthesizing atomically dispersed dual-metal-site catalysts with significantly increased atomic metal loading, holding great potential for viable CO2 to CO conversion at industry-level current densities.
Author contributions
G. W. and Y. Zeng conceived the concept and methods. M. Q., J. L., and Y. Zeng conducted catalyst synthesis and electrochemical measurements. M. J. Z and S. H. performed electron microscopy analysis. Y. L., Y. M., and Z. W. carried out DFT calculations. M. L. and Z. F. conducted XAS analysis. Q. Z. and Y.S. conducted XPS analysis. Y. Zhao performed electrolyzer tests. All authors wrote the manuscript. G. Wu supervised the execution of the overall project.Data availability
The data supporting this article have been included in the ESI.† The datasets generated and analyzed during the current study are also available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. Wu thanks the National Science Foundation (CBET-1804326) support and the start-up fund at Washington University in St. Louis. The atomic-scale HAADF-STEM and associated statistical analysis of atom positions, single-atom point EELS analyses along with overall EDS quantification portions of this research were supported by the Centre for Nanophase Materials Sciences (CNMS), which is a US Department of Energy, Office of Science User Facility at Oak Ridge National Laboratory. Other electron microscopy research was conducted at the Center for Functional Nanomaterials at Brookhaven National Laboratory. XPS analysis was performed at the Pacific Northwest National Laboratory. Y. Zhao acknowledges the Australian Research Council Discovery Early Career Researcher Award (DE250101462) funded by the Australian Government.Notes and references
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, 2102886 CrossRef CAS PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- H. Tabassum, W. Chen, B. Ma, L. Feng, X. Yang, Y. Li, M. Lucero, M. Lyons, Z. Feng, S. Hwang, X. Zhang, X. Hai, G. Wu and R. Zou, Appl. Catal., B, 2025, 365, 124922 CrossRef CAS.
- H. Tabassum, X. Yang, R. Zou and G. Wu, Chem. Catal., 2022, 2, 1561–1593 CrossRef CAS.
- F. Pan, L. Fang, B. Li, X. Yang, T. O’Carroll, H. Li, T. Li, G. Wang, K.-J. Chen and G. Wu, J. Am. Chem. Soc., 2024, 146, 1423–1434 CrossRef CAS PubMed.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X. M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef CAS PubMed.
- Y. Li, H. Wang, X. Yang, T. O'Carroll and G. Wu, Angew. Chem., Int. Ed., 2024, 63, e202317884 CrossRef CAS PubMed.
- N. Mohd Adli, W. Shan, S. Hwang, W. Samarakoon, S. Karakalos, Y. Li, D. A. Cullen, D. Su, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 1022–1032 CrossRef CAS PubMed.
- Y. Li, N. M. Adli, W. Shan, M. Wang, M. J. Zachman, S. Hwang, H. Tabassum, S. Karakalos, Z. Feng, G. Wang, Y. C. Li and G. Wu, Energy Environ. Sci., 2022, 15, 2108–2119 RSC.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- Y. Zhou, Q. Zhou, H. Liu, W. Xu, Z. Wang, S. Qiao, H. Ding, D. Chen, J. Zhu, Z. Qi, X. Wu, Q. He and L. Song, Nat. Commun., 2023, 14, 3776 CrossRef PubMed.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–692 CrossRef CAS.
- Q. Hao, H.-X. Zhong, J.-Z. Wang, K.-H. Liu, J.-M. Yan, Z.-H. Ren, N. Zhou, X. Zhao, H. Zhang, D.-X. Liu, X. Liu, L.-W. Chen, J. Luo and X.-B. Zhang, Nat. Synth., 2022, 1, 719–728 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- Y. Li, W. Shan, M. J. Zachman, M. Wang, S. Hwang, H. Tabassum, J. Yang, X. Yang, S. Karakalos, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2022, 61, e202205632 CrossRef CAS PubMed.
- G. Wu and P. Zelenay, Nat. Rev. Mater., 2024, 9, 643–656 CrossRef CAS.
- C. Yan, H. Li, Y. Ye, H. Wu, F. Cai, R. Si, J. Xiao, S. Miao, S. Xie, F. Yang, Y. Li, G. Wang and X. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- Y. Li, Z. He, F. Wu, S. Wang, Y. Cheng and S. Jiang, Mater. Rep. Energy, 2023, 3, 100197 CAS.
- L. Zhao, Y. Zhang, L.-B. Huang, X.-Z. Liu, Q.-H. Zhang, C. He, Z.-Y. Wu, L.-J. Zhang, J. Wu, W. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
- L. Han, H. Cheng, W. Liu, H. Li, P. Ou, R. Lin, H.-T. Wang, C.-W. Pao, A. R. Head, C.-H. Wang, X. Tong, C.-J. Sun, W.-F. Pong, J. Luo, J.-C. Zheng and H. L. Xin, Nat. Mater., 2022, 21, 681–688 CrossRef CAS PubMed.
- K. Liu, W. A. Smith and T. Burdyny, ACS Energy Lett., 2019, 4, 639–643 CrossRef CAS PubMed.
- X. Sheng, W. Ge, H. Jiang and C. Li, Adv. Mater., 2022, 34, 2201295 CrossRef CAS PubMed.
- T. N. Nguyen and C.-T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. T. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed.
- X. Hai, S. Xi, S. Mitchell, K. Harrath, H. Xu, D. F. Akl, D. Kong, J. Li, Z. Li, T. Sun, H. Yang, Y. Cui, C. Su, X. Zhao, J. Li, J. Pérez-Ramírez and J. Lu, Nat. Nanotechnol., 2022, 17, 174–181 CrossRef CAS PubMed.
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K.-J. Noh, K.-S. Lee and J. W. Han, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS.
- F. Pan, B. Li, E. Sarnello, S. Hwang, Y. Gang, X. Feng, X. Xiang, N. M. Adli, T. Li, D. Su, G. Wu, G. Wang and Y. Li, Nano Energy, 2020, 68, 104384 CrossRef CAS.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef CAS PubMed.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schafer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- D. Yao, C. Tang, X. Zhi, B. Johannessen, A. Slattery, S. Chern and S. Z. Qiao, Adv. Mater., 2023, 35, e2209386 CrossRef PubMed.
- Y. N. Gong, C. Y. Cao, W. J. Shi, J. H. Zhang, J. H. Deng, T. B. Lu and D. C. Zhong, Angew. Chem., Int. Ed., 2022, 61, e202215187 CrossRef CAS PubMed.
- X. Yang, C. Priest, Y. Hou and G. Wu, SusMat, 2022, 2, 569–590 CrossRef CAS.
- X. Yang, M. Wang, M. J. Zachman, H. Zhou, Y. He, S. Liu, H.-Y. Zang, Z. Feng and G. Wu, Small Sci., 2021, 1, 2100046 CrossRef CAS PubMed.
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O’Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CAS.
- H. Li, H. Li, P. Wei, Y. Wang, Y. Zang, D. Gao, G. Wang and X. Bao, Energy Environ. Sci., 2023, 16, 1502–1510 RSC.
- J. Y. T. Kim, P. Zhu, F.-Y. Chen, Z.-Y. Wu, D. A. Cullen and H. Wang, Nat. Catal., 2022, 5, 288–299 CrossRef CAS.
- B. Pan, J. Fan, J. Zhang, Y. Luo, C. Shen, C. Wang, Y. Wang and Y. Li, ACS Energy Lett., 2022, 7, 4224–4231 CrossRef CAS.
- Z. Jiang, Z. Zhang, H. Li, Y. Tang, Y. Yuan, J. Zao, H. Zheng and Y. Liang, Adv. Energy Mater., 2022, 13, 202203603 Search PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O’Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- J. B. James and Y. Lin, J. Phys. Chem. C, 2016, 120, 14015–14026 CrossRef CAS.
- X. Yan, M. Gu, Y. Wang, L. Xu, Y. Tang and R. Wu, Nano Res., 2020, 13, 975–982 CrossRef CAS.
- L. Jiao, J. Li, L. L. Richard, Q. Sun, T. Stracensky, E. Liu, M. T. Sougrati, Z. Zhao, F. Yang, S. Zhong, H. Xu, S. Mukerjee, Y. Huang, D. A. Cullen, J. H. Park, M. Ferrandon, D. J. Myers, F. Jaouen and Q. Jia, Nat. Mater., 2021, 20, 1385–1391 CrossRef CAS PubMed.
- Z. Li, D. He, X. Yan, S. Dai, S. Younan, Z. Ke, X. Pan, X. Xiao, H. Wu and J. Gu, Angew. Chem., Int. Ed., 2020, 59, 18572–18577 CrossRef CAS PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- S. Fu, B. Izelaar, M. Li, Q. An, M. Li, W. de Jong and R. Kortlever, Nano Energy, 2025, 133, 110461 CrossRef CAS.
- Y. Zeng, C. Li, B. Li, J. Liang, M. J. Zachman, D. A. Cullen, R. P. Hermann, E. E. Alp, B. Lavina, S. Karakalos, M. Lucero, B. Zhang, M. Wang, Z. Feng, G. Wang, J. Xie, D. J. Myers, J.-P. Dodelet and G. Wu, Nat. Catal., 2023, 6, 1215–1227 CrossRef CAS.
- S. Liu, C. Li, M. J. Zachman, Y. Zeng, H. Yu, B. Li, M. Wang, J. Braaten, J. Liu, H. M. Meyer, M. Lucero, A. J. Kropf, E. E. Alp, Q. Gong, Q. Shi, Z. Feng, H. Xu, G. Wang, D. J. Myers, J. Xie, D. A. Cullen, S. Litster and G. Wu, Nat. Energy, 2022, 7, 652–663 CrossRef CAS.
- H. Jin, R. Zhao, P. Cui, X. Liu, J. Yan, X. Yu, D. Ma, W. Song and C. Cao, J. Am. Chem. Soc., 2023, 145, 12023–12032 CrossRef CAS PubMed.
- H. Wang, Y. Li, M. Wang, S. Chen, M. Yao, J. Chen, X. Liao, Y. Zhang, X. Lu, E. Matios, J. Luo, W. Zhang, Z. Feng, J. Dong, Y. Liu and W. Li, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219043120 CrossRef CAS PubMed.
- J. Wang, B. Li, Y. Li, X. Fan, F. Zhang, G. Zhang and W. Peng, Adv. Sci., 2021, 8, e2101824 CrossRef PubMed.
- M. Wang, L. Árnadóttir, Z. J. Xu and Z. Feng, Nano-Micro Lett., 2019, 11, 47 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01081k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |