
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Designing multi-tentacle electrolytes to enable fast and deep cycling of aqueous Zn batteries at low temperatures†
Huimin
Wang‡
a,
Mingzi
Sun‡
 ab,
Yongqiang
Yang
a,
Junhua
Zhou
a,
Lingtao
Fang
c,
Qiyao
Huang
cd,
Bolong
Huang
*ab and
Zijian
Zheng
*acdefg
ab,
Yongqiang
Yang
a,
Junhua
Zhou
a,
Lingtao
Fang
c,
Qiyao
Huang
cd,
Bolong
Huang
*ab and
Zijian
Zheng
*acdefg
aDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong SAR, China. E-mail: zijian.zheng@polyu.edu.hk
bDepartment of Chemistry, City University of Hong Kong, Hong Kong SAR, China. E-mail: b.h@cityu.edu.hk
cSchool of Fashion and Textile, The Hong Kong Polytechnic University, Hong Kong SAR, China
dResearch Institute for Intelligent Wearable Systems (RI-IWEAR), The Hong Kong Polytechnic University, Hong Kong SAR, China
eResearch Institute for Smart Energy (RISE), The Hong Kong Polytechnic University, Hong Kong SAR, China
fThe Hong Kong Polytechnic University-Wenzhou Technology and Innovation Research Institute, Wenzhou, Zhejiang Province, China
gThe Hong Kong Polytechnic University-Daya Bay Technology and Innovation Research Institute, Huizhou, Guangdong Province, China
First published on 25th April 2025
Abstract
Rechargeable aqueous zinc batteries (AZBs) offer a safe and sustainable solution for large-scale energy storage, but the freezing of electrolytes prevents AZBs from working at low temperatures. Recent research has shown that the freezing point can be effectively lowered by using either concentrated salts or organic-rich electrolytes. However, these strategies result in either low oxidation stability or sluggish mass transport at low temperatures. Here, we report a multi-tentacle electrolyte (MTE) strategy that enables stable, fast and deep cycling of AZBs at −40 °C. The MTE leverages the abundant hydrogen-bonding sites of multi-tentacle salts and organics. Adding small amounts of multi-tentacle moieties not only effectively confines water molecules’ movement and prevents their icing even at −60 °C, but also maintains low viscosity and high ionic conductivity of the electrolyte at low temperatures. At −40 °C, Zn metal anodes could stably cycle for more than 1100 hours at a high current density of 2 mA cm−2 and a high capacity of 2 mA h cm−2; high-capacity AZBs (3.4 mA h cm−2) sustain 1000 stable cycling with 99.99% retention per cycle in the MTE. The MTE strategy is also versatile to high-voltage LiMn2O4 cathodes, which further enhances the energy density of AZBs to 154.4 W h kgLMO−1 at −40 °C.
Broader contextAqueous zinc batteries (AZBs) are promising for large-scale energy storage due to their safety, affordability, and environmental friendliness. However, their performance at low temperatures is severely limited by sluggish Zn2+ transport in both electrolytes and cathodes, leading to substantial energy and power losses. Traditional approaches to prevent electrolyte freezing—such as concentration salts or organic strategies—introduce new challenges including corrosion, reduced oxidation stability, and increased viscosity. To address these limitations, we report a novel multi-tentacle electrolyte (MTE) strategy. This approach utilizes multi-tentacle salts and organics that form extensive hydrogen bonds with water molecules, effectively inhibiting freezing while maintaining low viscosity and high ionic conductivity, attributed to minimal amounts of salts and organics used. Remarkably, the MTE concept extends beyond Zn2+ chemistry. By incorporating multi-tentacle Li salts, we demonstrate electrolytes with freezing points below −60 °C that enable swift Li+ transport in high-voltage LiMn2O4 cathodes. The resulting AZBs exhibit exceptional performance at −40 °C, including stable Zn plating/stripping at 2 mA cm−2 and extended cycle life in high-loading cells. This work provides a facile and feasible strategy for designing low-viscosity, temperature-tolerant electrolytes, suggesting an effective approach to improve the sluggish kinetics of AZBs at low temperatures. |
Introduction
Sustainable grid-scale energy storage solutions are important for integrating intermittent renewable energy sources, such as solar and wind, while reducing reliance on fossil fuels.1–4 Rechargeable aqueous zinc batteries (AZBs) are viewed as a promising option for grid-scale energy storage due to their low cost and high safety.5–8 Additionally, because zinc offers a high gravimetric capacity (820 mA h g−1) and volumetric capacity (5855 mA h cm−3), and a low electrode potential (−0.76 V versus the standard hydrogen electrode, SHE), its energy density is also highly competitive with other aqueous battery technologies. However, severe performance degradation of aqueous AZBs occurs at subzero temperatures as a result of the freezing of aqueous electrolytes. This seriously limits the use of AZBs in cold climates or high-latitude regions that are often abundant in renewable energy.9,10A general principle to mitigate the freezing challenge of AZBs is to interrupt the long-range ordered rearrangement of water molecules in the electrolyte during cooling. In this regard, three well-established strategies are reported to date, including the (i) concentrated salt strategy that dissolves large amounts of inorganic salts (e.g., chlorides11–13) in an aqueous electrolyte, (ii) organic-rich strategy that mixes large quantities of organic moieties (>50% by volume or mole fraction) with water,14–16 and (iii) anti-freezing hydrogel strategy that replaces a liquid electrolyte with a gel-like electrolyte.17–19 In the concentrated salt strategy, chloride salts are the most reported ones to offer high ionic conductivity at low temperatures. However, they are highly corrosive to the stainless-steel (SS) case of AZBs, and are easily oxidized with low oxidation stability.20 In the organic-rich strategy, the large amount of organic moieties will significantly increase the viscosity of the electrolyte at low temperatures,13,21–23 and hence the mass transport becomes very sluggish. A similar challenge also exists in the anti-freezing hydrogel strategy. Moreover, all these strategies inevitably reduce the amount of water in the electrolyte and concentrate Zn2+, which slows down the Zn2+ mass transport and Zn2+ desolvation process, and deteriorates the battery performance at low temperatures.24–27
Therefore, an ideal anti-freezing electrolyte suitable for high-performance AZBs should simultaneously possess the following characteristics at low temperatures: (i) high oxidation stability, (ii) high ionic conductivity, (iii) low viscosity, and (iv) fast desolvation kinetics. Confining a substantial proportion of water molecules using minimal amounts of non-corrosive salts and organics is the prerequisite. Several studies have aimed to achieve this, including efforts to reduce the organic proportion (e.g., 2m Zn(OTf)2 in trimethyl phosphate/water (40/60, vol.), yielding an organic-to-water molar ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :39)22 or decrease the salt concentration (e.g., 1m Zn(OAc)2 in formamide/water (50/50, vol.), resulting in a salt-to-solvent molar ratio of 1.5:30).28 However, maintaining low freezing points while minimizing the amounts of salts and organics remains challenging. Furthermore, the potential salts and organics for constructing such water-rich electrolytes as well as their low-temperature characteristics have not yet been systematically explored.
:39)22 or decrease the salt concentration (e.g., 1m Zn(OAc)2 in formamide/water (50/50, vol.), resulting in a salt-to-solvent molar ratio of 1.5:30).28 However, maintaining low freezing points while minimizing the amounts of salts and organics remains challenging. Furthermore, the potential salts and organics for constructing such water-rich electrolytes as well as their low-temperature characteristics have not yet been systematically explored.
In this work, we report a new electrolyte design strategy, namely multi-tentacle electrolyte (MTE), which can construct a series of high-performance low-temperature electrolytes without the need for high-concentration organic moieties or inorganic salts. The key idea of the multi-tentacle strategy is to introduce small amounts of multi-tentacle salts and organics, which can form abundant hydrogen bonds (H-bonds) with water molecules, into the electrolyte. As such, without increasing the viscosity or sacrificing the mass transport properties significantly, the multi-tentacle moieties can effectively confine water molecules’ movement, prevent their icing, and preserve high ionic conductivity at low temperatures.
As a proof of concept, we demonstrate a series of MTEs that are primarily composed of multi-tentacle Zn salts (x), multi-tentacle organics (y), and water, at a molar ratio of 1.5x:4y![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) :24H2O. Such MTEs exhibit anti-freezing properties under −60 °C, even with a minimal salt-to-solvent ratio of 1.5:28 and an organic-to-water ratio of 4:24. This MTE possesses a low viscosity of 142.8 mPa s and a high ionic conductivity of 4.7 mS cm−1 at −40 °C, which enables the fast and deep cycling of AZBs at low temperatures. Specifically, Zn anodes in MTEs sustain 1100-h high-current (2 mA cm−2) and high-capacity (2 mA h cm−2) cycling even at −40 °C, achieving an average coulombic efficiency of 98.6%. Additionally, Zn batteries using high-loading NaV3O8·1.5H2O (NVO) cathodes (∼17 mg cm−2) reach an areal capacity of ∼3.4 mA h cm−2 even at −40 °C and achieve 1000 stable cycling with 99.99% retention per cycle in the MTE. Importantly, this strategy can be extended from multi-tentacle Zn salts to multi-tentacle Li salts, so that high-voltage cathodes such as LiMn2O4 (LMO) can be used to pair with Zn anodes. We demonstrate smooth cycling of Zn||LMO in MTEs at −40 °C, with outstanding capacity retention (75% of its practical capacity at room temperature) and a high output voltage (1.72 V). Our findings pave the way for the development of high-performance Zn batteries with enhanced low-temperature capabilities.
:24H2O. Such MTEs exhibit anti-freezing properties under −60 °C, even with a minimal salt-to-solvent ratio of 1.5:28 and an organic-to-water ratio of 4:24. This MTE possesses a low viscosity of 142.8 mPa s and a high ionic conductivity of 4.7 mS cm−1 at −40 °C, which enables the fast and deep cycling of AZBs at low temperatures. Specifically, Zn anodes in MTEs sustain 1100-h high-current (2 mA cm−2) and high-capacity (2 mA h cm−2) cycling even at −40 °C, achieving an average coulombic efficiency of 98.6%. Additionally, Zn batteries using high-loading NaV3O8·1.5H2O (NVO) cathodes (∼17 mg cm−2) reach an areal capacity of ∼3.4 mA h cm−2 even at −40 °C and achieve 1000 stable cycling with 99.99% retention per cycle in the MTE. Importantly, this strategy can be extended from multi-tentacle Zn salts to multi-tentacle Li salts, so that high-voltage cathodes such as LiMn2O4 (LMO) can be used to pair with Zn anodes. We demonstrate smooth cycling of Zn||LMO in MTEs at −40 °C, with outstanding capacity retention (75% of its practical capacity at room temperature) and a high output voltage (1.72 V). Our findings pave the way for the development of high-performance Zn batteries with enhanced low-temperature capabilities.
Results and discussion
Design principle of MTEs
Concentrated salts, e.g., chlorides, disrupt the intermolecular network in water clusters, preventing their contraction and crystallization at low temperatures (Fig. 1a). However, corrosive chlorides can damage SS cell cases, and higher salt concentrations increase ion attraction.11,29 Alternatively, concentrated organic strategies that create organic-in-water regimes can also disrupt the ordered structure of water clusters (Fig. 1b). Yet, this method may raise electrolyte viscosity due to the high proportion of organic content, hindering rapid mass transport under cold conditions.14,23,30 Both conventional concentrated salt and organic strategies require substantial amounts of salts or organics, resulting in high salt-to-solvent or organic-to-water molar ratios, to suppress the rearrangement of water molecules at low temperatures. Reducing these proportions inevitably leads to electrolyte freezing at higher temperatures.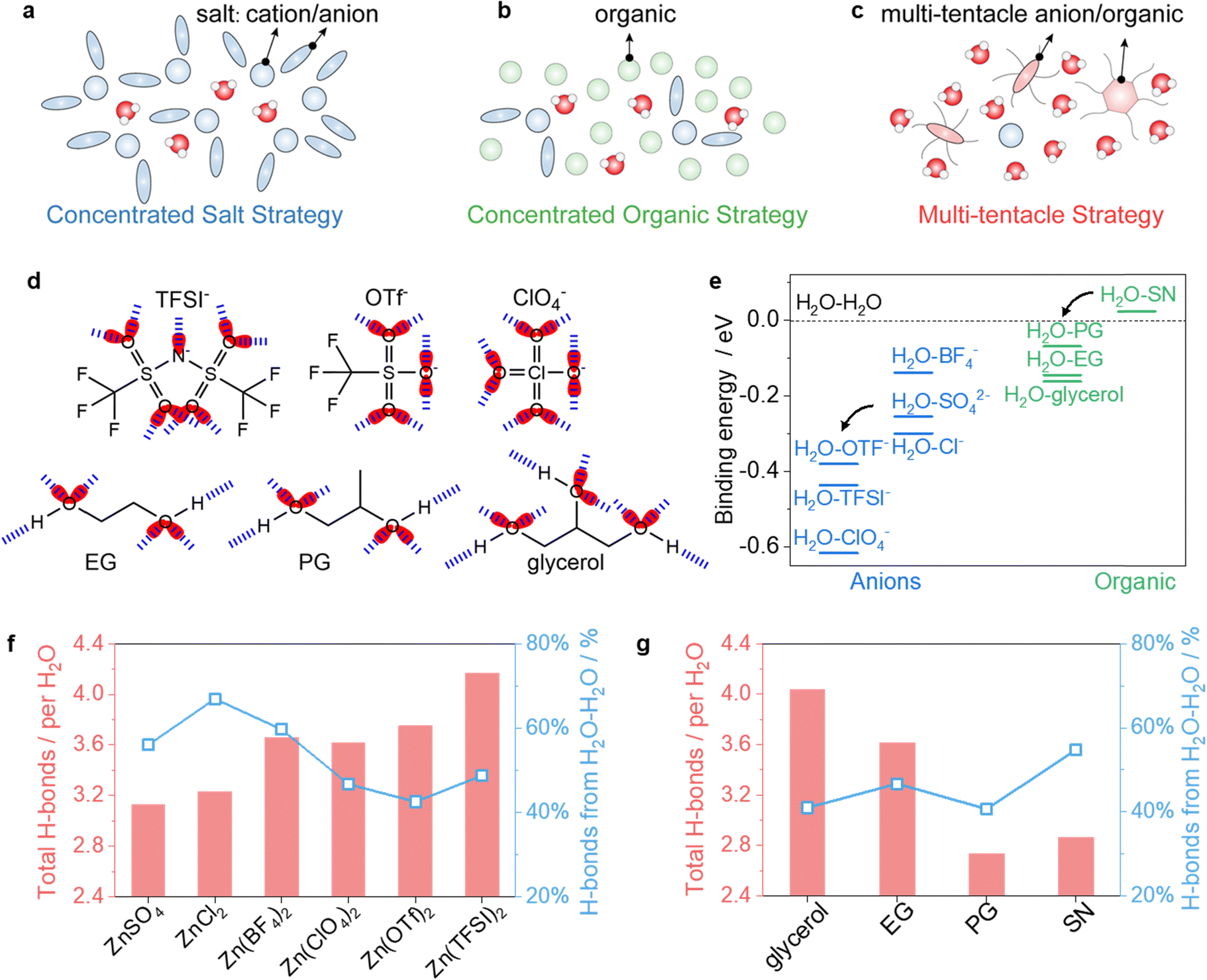 | ||
| Fig. 1 Electrolyte design strategy. (a)–(c) Illustration of low-temperature electrolyte strategies, including the concentrated salt strategy (a), concentrated organic strategy (b), and multi-tentacle strategy (c). (d) Potential H-bonding sites in multi-tentacle anions and organics. Red ellipses indicate electron lone pairs that can act as H-bond acceptors, while the H atom in the –OH group of organics serves as an H-bond donor. Blue dashed lines represent possible H-bonds. (e) Binding energy between water and various species relative to the binding energy between water molecules. (f) and (g) H-bond analysis for electrolytes with the composition of 1.5x:4EG:24H2O with x varying from ZnSO4, ZnCl2, Zn(BF4)2, Zn(ClO4)2, Zn(OTf)2 and Zn(TFSI)2 (f) and 1.5Zn(ClO4)2:4y:24H2O with y varying from glycerol, EG, PG and SN (g). | ||
In contrast, MTEs effectively confine substantial amounts of water even at −60 °C by adding only small amounts of multi-tentacle salts (x) and organics (y), at a molar ratio of 1.5x:4y:24H2O (Fig. 1c). Fig. 1d shows the multi-tentacle anions such as TFSI−, OTf−, and ClO4−, and multi-tentacle organics such as ethylene glycol (EG), propylene glycol (PG), and glycerol, which contain multiple sites acting as H-bond acceptors or donors (Fig. S1, ESI†). Theoretical calculations show that multi-tentacle anions exhibit less negative electrostatic potential (ESP) values compared to other anions like SO42− (Fig. S2, ESI†). Less negative ESP values of anions correspond to weaker binding with Zn2+, suggesting that multi-tentacle anions are more involved in H-bonding with water rather than in coordinating Zn2+, when compared to anions like SO42−, Cl−, and BF4−(Fig. S3, ESI†).
Besides, we also monitored the binding energy changes of water–anions and water–organics with density functional theory (DFT) calculations. As shown in Fig. 1e, the combination of water and multi-tentacle anions (TFSI−, OTf−, and ClO4−) releases more energy than combinations with weaker H-bond anions (e.g., SO42−, BF4−, and Cl−). A similar trend is observed with organics: the combination of water and multi-tentacle organics (PG, EG, and glycerol) releases more energy than that with succinonitrile (SN). These results demonstrate the superior H-bonding tendencies of multi-tentacle anions and organics with water.
Furthermore, we monitored the overall H-bond numbers arising from the H2O–H2O, H2O–anion, and H2O–organic interactions, and the percentage of H2O–H2O H-bonds via molecular dynamics (MD) simulations. As shown in Fig. 1f, MTEs containing Zn(TFSI)2, Zn(OTF)2, or Zn(ClO4)2 exhibit much higher overall H-bond numbers than electrolytes using conventional salts such as ZnCl2 and ZnSO4. Moreover, the percentage of H2O–H2O H-bonds in MTEs is less than 50%, which is significantly lower than those using ZnCl2, ZnSO4, and Zn(BF4)2 (55%–70%). This suggests a more disrupted intermolecular network within water clusters, stronger confinement of water molecules, and consequently, a reduced freezing tendency in multi-tentacle salt-based electrolytes. A similar trend is observed in MTEs containing glycerol, EG, or PG, when compared to the electrolyte containing SN. Computational studies on intermolecular interactions and mean square displacement (MSD) of ions (Fig. S4 and S5, ESI†) further demonstrate a lower freezing tendency in electrolytes using multi-tentacle salts and organics. These results indicate that water molecules preferably form H-bonds with multi-tentacle salts and organics, so that the formation of long-range H-bond networks among water molecules is disrupted.
To quantify the theory, we then experimentally assessed the water-holding ability of different salts and organics using 1H nuclear magnetic resonance (1H-NMR) tests. Salts interact with water molecules via coordination (cation–water) or the H-bond (anions–water) effects, while organics hold water only through the H-bond effect. Both interactions affect the electron density around the H atom of water molecules, shifting the 1H signal upfield or downfield. We first recorded the chemical shifts (σ) of 1H of water in different electrolytes and then calculated the change of chemical shift (|Δσ|) to obtain |Δσ1| and |Δσ2|, where |Δσ1| represents the shift due to the coordination effect21 and |Δσ2| represents the shift due to the H-bond effect.31,32 Therefore, log(|Δσ1| + |Δσ2|) represents the water-holding ability of different salts or organics in the electrolytes.
Specifically, introducing ZnSO4 into water causes a downfield shift of the 1H signal of water molecules (Fig. 2a). This downshift is ascribed to |Δσ1| because of the strong coordination between Zn2+ and water molecules, and the negligible H-bond effect between SO42− and water. An upfield shift occurs when replacing SO42− with Cl−, BF4−, ClO4−, OTf−, and TFSI−. This upshift is ascribed to |Δσ2| because of the H-bonds between these anions with water molecules. As shown in Fig. 2b, the water-holding ability of different salts, expressed as log(|Δσ1| + |Δσ2|), suggests a decreasing water-holding ability in the order: TFSI− > OTf− > ClO4− > BF4− > Cl−> SO42−.
 | ||
| Fig. 2 Water-holding ability. (a)–(f) Evolution of 1H NMR spectra from H2O (a), (c) and (e) and assessment of the water-holding ability via log(|Δσ1| + |Δσ2|) (b), (d) and (f) in mixtures of 1.5x:24H2O (a) and (b), 4y:24H2O (c) and (d) and 1.5x:4EG:24H2O (e) and (f). x: salts. y: organics. (g) DSC curves of electrolytes with the composition of 1.5x:4y:24H2O during heating from −65 to 20 °C. | ||
We also assessed the water-holding ability of various organics in organic–water mixtures. As shown in Fig. 2c, adding organics to water only results in an upfield shift of |Δσ2| due to the H-bond effects (|Δσ1| equals 0). The log(|Δσ1| + |Δσ2|) indicates a decreasing water-holding ability in the order: glycerol > EG > PG> diglyme > glyme > N-methylacetamide (NMA) > SN (Fig. 2d). Diglyme, glyme, and NMA exhibit a small |Δσ2|, due to fewer H-bond sites (Fig. S6, ESI†), while SN shows incompatibility with water, resulting in immiscible phases.
In electrolytes of 1.5x:4EG:24H2O (Fig. 2e and f), the log(|Δσ1| + |Δσ2|) follows an decreasing order of Zn(TFSI)2 > Zn(OTf)2 > Zn(ClO4)2 > Zn(BF4)2 > ZnCl2 > ZnSO4. This trend aligns with the above discussion and indicates that more water molecules are confined via coordination and H-bond effects when using multi-tentacle salts and EG.
Differential scanning calorimetry (DSC) tests were further employed to check the water-holding ability in a wide temperature window of −65 to 20 °C. MTEs using multi-tentacle salts and multi-tentacle organics remain liquid even at temperatures as low as −60 °C (Fig. 1g). In contrast, electrolytes using organics that have weaker water-holding abilities (e.g., SN, glyme, or diglyme) or Zn salts containing weak H-bonding anions (e.g., ZnSO4, ZnCl2 or Zn(BF4)2), lead to solid-to-liquid phase transitions during DSC tests, indicating electrolyte freezing. This highlights the critical role of multi-tentacle moieties in forming anti-freezing MTEs. Additionally, even in the electrolytes containing multi-tentacle moieties, reducing x from 1.5 to 1 in the formula 1x:4y:24H2O or reducing y from 4 to 3.5 in the formula 1.5x:3.5y:24H2O results in electrolyte freezing (Fig. S7, ESI†). It occurs due to decreased amounts of multi-tentacle moieties and thereby weakened water-holding effects. Therefore, maintaining adequate concentrations of both multi-tentacle salts and organic components is essential for preventing freezing in these electrolyte systems. 1.5 multi-tentacle salt and 4 multi-tentacle organics are used in our formula.
Physical and chemical properties of MTEs
We compare the salt-to-solvent and organic-to-water ratios of MTEs with established strategies in Fig. 3a. Our approach achieves competitively low salt-to-solvent (1.5:28) and organic-to-water (4:24) ratios. The ionic conductivity of MTEs is summarized in Fig. 3b, showing comparable or superior conductivity to those based on concentrated salt or organic strategies. Specifically, the electrolyte 1.5Zn(ClO4)2:4EG:24H2O (denoted as MTE-EG-Zn(ClO4)2) exhibits ionic conductivity comparable to the state-of-the-art concentrated salt electrolyte, 7.5m ZnCl2,29 across a wide temperature range of −40 to 25 °C, and is approximately 50 times greater (at −40 °C) than that of the state-of-the-art concentrated organic-based electrolyte, 4m hydrated Zn(BF4)2 in EG.14 Thus, MTE-EG-Zn(ClO4)2 was selected for further studies.
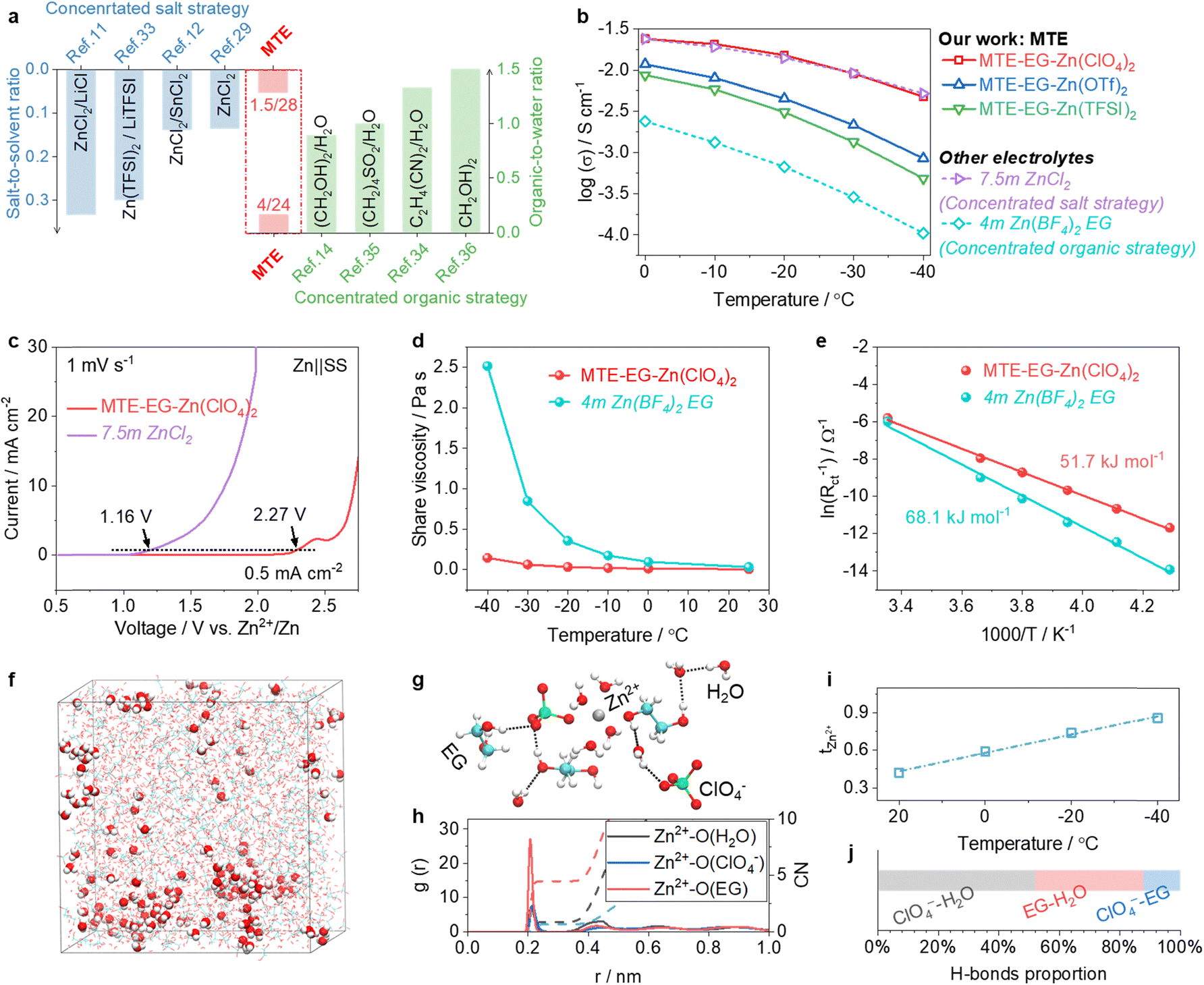 | ||
| Fig. 3 Physical and chemical properties of MTEs. (a) Comparison of salt-to-solvent and organic-to-water molar ratios between MTEs and other reported low-temperature electrolyte strategies.11,12,14,15,29,33–36 (b) Comparison of ionic conductivities between MTEs and electrolytes using concentrated salt (7.5m ZnCl2) and concentrated organic (4m hydrated Zn(BF4)2 EG) strategies. (c) Comparison of the chemical/electrochemical stability of MTE-EG-Zn(ClO4)2 and 7.5m ZnCl2 in Zn||SS cells. (d) Evolution of shear viscosities of MTE-EG-Zn(ClO4)2 and 4m hydrated Zn(BF4)2 EG as a function of temperature. (e) Comparison of desolvation energy of Zn2+ in MTE-EG-Zn(ClO4)2 and 4m hydrated Zn(BF4)2 EG. (f)–(h) Snapshot of a typical MD simulated cell (f) and representative Zn2+-solvation structure (g), and the radial distribution function (RDF) and average coordination number (CN) of Zn2+ (h) in MTE-EG-Zn(ClO4)2. (i) and (j) Evolution of the transference number of Zn2+ with lowering temperature (i) and proportion of H-bonds (j) in MTE-EG-Zn(ClO4)2. | ||
MTEs demonstrate high chemical and electrochemical stabilities, far outperforming concentrated salt electrolytes. As a proof of concept, we assessed the side-reaction currents during the anodic scan in different electrolytes using Zn||SS cells, in which the SS was used as the working electrode (Fig. 3c). In the reference electrolyte of 7.5m ZnCl2, SS corrosion occurs at 1.16 V (vs. Zn2+/Zn). The corrosion current is 0.5 mA cm−2. It then sharply rises to 30 mA cm−2 at ∼1.9 V, indicating severe decomposition of the electrolyte. In contrast, MTE-EG-Zn(ClO4)2 shows excellent compatibility with SS and enhanced oxidation stability. The onset corrosion potential of MTE-EG-Zn(ClO4)2 is 2.27 V. Electrolyte decomposition is not observed until the potential exceeds 2.5 V (vs. Zn2+/Zn). This stability allows for the use of low-cost SS cell cases and high-voltage cathodes in low-temperature environments.
Meanwhile, MTEs outperform the concentrated organic electrolyte in their low viscosity and rapid desolvation process of Zn2+ at low temperatures. As a proof of concept, we first investigate the evolution of viscosities of MTE-EG-Zn(ClO4)2 and reference electrolyte 4m Zn(BF4)2 in EG as a function of decreasing temperatures (Fig. 3d). At 25 °C, the viscosities of these electrolytes are similar. When the temperature decreases to −40 °C, the viscosity of 4m Zn(BF4)2 in EG increases sharply to 2516 mPa s, while that of MTE-EG-Zn(ClO4)2 is only 142.8 mPa s. The low viscosity of MTE-EG-Zn(ClO4)2 ensures the swift Zn2+ transport, as represented by its high conductivity at −40 °C (Fig. 3b). As shown in Fig. 2e, the low viscosity also benefits a swifter desolvation process: the desolvation energy of MTE-EG-Zn(ClO4)2 is 51.7 kJ mol−1, much lower than that of 4m Zn(BF4)2 EG (68.1 kJ mol−1).
The low viscosity and facile desolvation process at low temperatures is attributed to the high proportion of spatial-confined water molecules in MTE-EG-Zn(ClO4)2. The 3D snapshot (Fig. 3f) and representative solvation structures (Fig. 3g and Fig. S8, ESI†) of MTE-EG-Zn(ClO4)2 in the MD simulation demonstrate that most water molecules in the electrolyte are confined through coordination and H-bond effects. Only 5.4% of water molecules are neither involved in coordination nor H-bonding with anions and organics, which are shown as ball-and-stick structures in the 3D snapshot (Fig. 3f and Fig. S9, ESI†). Instead of a fully hydrated solvation shell Zn2+(H2O)6, radial distribution functions (RDFs) show that anions and EG molecules will replace partial water molecules in the solvation sheath in MTE-EG-Zn(ClO4)2 (Fig. 3h). This feature not only favors high mass transport of charge carriers in MTEs but also reduces water decomposition tendency at high and low potentials.20,37 Additionally, the transference number of Zn2+ (tZn2+) in MTE-EG-Zn(ClO4)2 increases as the temperature decreases, reaching 0.86 at −40 °C (Fig. 3i and Fig. S10, ESI†). This suggests a reduced migration trend of multi-tentacle anions under the applied electric field at low temperatures. As shown in Fig. 3j, the proportion of H-bonds from ClO4−-water in MTE-EG-Zn(ClO4)2 dominates, accounting for over 50% (Fig. S11, ESI†). Therefore, unlike at room temperature, where anions migrate freely, low temperatures pronounce the water-binding effect of anions to suppress the orderly rearrangement of water molecules. This leads to reduced anion migration and higher transference numbers of Zn2+ at low temperatures.
In summary, MTEs confine a substantial fraction of water molecules through minimal amounts of non-corrosive multi-tentacle salts and organics, achieving competitively low salt-to-solvent and organic-to-water ratios. This strategy, distinct from other well-established electrolyte strategies,14,29,38 enables MTEs to simultaneously deliver high ionic conductivities down to −40 °C, high oxidation stability and low viscosities. These properties ensure stable battery performance under extreme conditions, including low temperatures and high areal current densities/capacities. Additionally, MTEs exhibit exceptional compatibility with stainless-steel casings, enabling the use of low-cost cell housings while eliminating corrosion-related maintenance costs.
Electrochemical performance of batteries using MTE-EG-Zn(ClO4)2
The plating and stripping of Zn in MTE-EG-Zn(ClO4)2 exhibit high stability and reversibility with moderate overpotential, even at high current density and low temperatures. At −40 °C, Zn||Cu cells using MTE-EG-Zn(ClO4)2 sustain over 1400 hours at 1 mA cm−2/1 mA h cm−2 and 1100 hours at 2 mA cm−2/2 mA h cm−2, achieving impressive average coulombic efficiencies of 98.3% and 98.6%, respectively (Fig. 4a). This performance is accompanied by small overpotentials of 560 mV (at 1 mA cm−2) and 620 mV (at 2 mA cm−2) (Fig. 4b). The Zn||Cu cells at 2 mA cm−2/2 mA h cm−2 show overlapping voltage profiles across cycles (Fig. S12, ESI†), demonstrating stable Zn plating/stripping in MTE-EG-Zn(ClO4)2 even under −40 °C and high current density/capacity. In sharp contrast, the voltage profiles of Zn plating under the same conditions quickly drop below −3.0 V (vs. Zn2+/Zn) when using 4m hydrated Zn(BF4)2 EG (Fig. 4b), attributed to the slow Zn2+ transport and desolvation process in the concentrated organic-based electrolyte. Additionally, symmetric Zn||Zn cells using MTE-EG-Zn(ClO4)2 also show high stability. It sustains operation for over 550 hours at −40 °C when evaluated at a high current density of 2 mA cm−2 (Fig. 4c), while Zn||Zn cells using 4m hydrated Zn(BF4)2 EG under the same current density cannot cycle. Fig. 4d compares the applicable current densities in the MTE with literature reports, showing the outstanding fast kinetics advantage of the MTE even at −40 °C. | ||
| Fig. 4 Electrochemical performance of batteries using MTE-EG-Zn(ClO4)2. (a) and (b) Coulombic efficiency (a) and voltage profiles (b) of Zn plating/stripping in Zn||Cu asymmetrical cells evaluated at 1 mA cm−2/1 mA h cm−2 and 2 mA cm−2/2 mA h cm−2 in MTE-EG-Zn(ClO4)2 and 4m hydrated Zn(BF4)2 EG at −40 °C. (c) Long-term stability of Zn||Zn symmetric cells evaluated at 2 mA cm−2/2 mA h cm−2 in MTE-EG-Zn(ClO4)2 and 4m hydrated Zn(BF4)2 EG at −40 °C. (d) Comparison of current densities employed at low temperatures between our work and other reported low-temperature electrolytes.14–16,21–23,28,34,35,39 (e) and (f) Top (e) and cross-sectional (f) views of Zn deposits in MTE-EG-Zn(ClO4)2 after 1 hour of plating at 10 mA cm−2. (g) Operando observation of Zn deposits in MTE-EG-Zn(ClO4)2 at 10 mA cm−2 after 0, 10, 20, and 30 min of plating. (h) and (i) Long-term stability (h) and voltage profiles (i) of Zn||NVO (∼17 mg cm−2) cells using MTE-EG-Zn(ClO4)2 and 4m hydrated Zn(BF4)2 EG evaluated at 0.1, 0.2, 0.5 and 1C at −40 °C. (j) Voltage profiles of Zn||NVO pouch cells at 0.2C and varying temperatures. | ||
MTEs also facilitate a dense growth of Zn metal with the suppressed hydrogen evolution reaction (HER). Top and cross-sectional SEM images of Zn deposits in MTE-EG-Zn(ClO4)2 reveal a highly compact Zn layer, indicating dense and dendrite-free growth (Fig. 4e, f and Fig. S13, ESI†). We attribute this dendrite-free growth to the high Zn2+ transference number in MTE-Zn(ClO4)2 (Fig. 3i), which promotes uniform Zn2+ flux—especially at low-temperatures—preventing localized Zn2+ accumulation and dendritic growth. In contrast, Zn deposits in a commonly used electrolyte, e.g., 2m ZnSO4 in water, are highly porous with the obvious formation of Zn4SO4(OH)6·4H2O by-products (Fig. S14, ESI†). Operando optical study shows marked bubbling of Zn plating in 2m ZnSO4 (Fig. S15, ESI†) as a result of the hydrogen evolution reaction, while no bubble is observed when using MTE-EG-Zn(ClO4)2 (Fig. 4g). This result is consistent with the LSV analysis: the HER current in 2m ZnSO4 can be readily observed at −83 mV (versus Zn2+/Zn), while it is delayed to −147 mV in MTE-EG-Zn(ClO4)2 (Fig. S16, ESI†). Besides, Tafel plots of Zn in MTE-EG-Zn(ClO4)2 show a positive-shift of potentials and reduced currents are achieved, manifesting a lower corrosion rate and suppressed HER in MTE-EG-Zn(ClO4)2 (Fig. S17, ESI†). The electrochemical stability window of MTE-EG-Zn(ClO4)2 is extended to 2.53 V, compared to 2.33 V in the conventional 2m ZnSO4 aqueous electrolyte (Fig. S18, ESI†). This enhancement is attributed to the suppressed activity of water molecules, resulting from their confinement by anions and organic components via H-bond effects.
Zn||NVO full cells using MTEs show high available capacities and rate capability at low temperatures. In the high-loading mode with a NVO areal mass of ∼17 mg cm−2, the available capacities of Zn||NVO cells cycled under 0.1, 0.2, 0.5 and 1C (1C = 350 mA g−1) at −40 °C are 185.61, 141.35, 106.08, and 36.75 mA h g−1, respectively (Fig. 4i). In sharp contrast, Zn||NVO cells using 4m hydrated Zn(BF4)2 EG show significantly lower capacities of only 9.3 mA h g−1 at 0.1C and 0.85 mA h g−1 at 0.2C (Fig. 4i).
The long-term stability of Zn||NVO at −40 °C is further assessed (Fig. 4h). High-loading NVO cathodes in MTE-EG-Zn(ClO4)2 show an initial areal capacity of ∼3.4 mA h cm−2 (equivalent to 185.6 mA h g−1) at 0.1C, which corresponds to 53% of the practical capacity (350 mA h g−1) at 25 °C (Fig. 4h). After extensive cycling, the cells maintain areal capacities of 2.45 mA h cm−2 at 0.2C, 1.72 mA h cm−2 at 0.5C, and 0.71 mA h cm−2 at 1C after 300, 200, and 1000 cycles, respectively (Fig. 4h). In marked contrast, NVO cathodes using 4m hydrated Zn(BF4)2 EG show a negligible capacity, approaching 0 mA h cm−2. High-loading Zn||NVO cells using MTE-EG-Zn(ClO4)2 also exhibit impressive available capacities when evaluated at 0.2 and 1C across a temperature range of 0 to −40 °C (Fig. S19, ESI†). To assess the robustness of the MTE-Zn(ClO4)2 electrolyte under temperature variations, Zn||Zn symmetric cells and Zn||NVO full cells were cycled under repeated cooling (−40 °C) and heating (25 °C). As shown in Fig. S20 (ESI†), the Zn||Zn cells display stable voltage profiles, while the Zn||NVO cells retain consistent discharge capacities—confirming the electrolyte's ability to support reversible Zn plating/stripping and cathode operation across a wide temperature range.
Even when scaled up to a pouch cell measuring 3 × 4 cm2, the high-loading NVO cathode retains capacities of 298.64 (0 °C), 237.21 (−10 °C), 197.14 (−20 °C), 138.52 (−30 °C), and 92.44 (−40 °C) mA h g−1 at 0.2C (Fig. 4j). Additionally, the Zn||NVO pouch cell exhibits stable cycling stability, reaching 80% capacity retention after 400 cycles at 0.5C and −40 °C (Fig. S21, ESI†).
Universality with low-charged multi-tentacle Li salts
The multi-tentacle strategy is not limited to Zn2+. As a proof of concept, we show here that partially replacing Zn salts with low-charged multi-tentacle Li salts preserves the low freezing point of lower than −60 °C. As shown in Fig. 5a, with a composition of 1LiTFSI and 0.5Zn(OTf)2, the resulting dual-cation electrolytes (1LiTFSI + 0.5Zn(OTf)2):4EG:24H2O, denoted as MTE-EG-LiZn, maintain the liquidity at −60 °C. It also works when replacing EG with other multi-tentacle organics, including PG (MTE-PG-LiZn) and glycerol (MTE-glycerol-LiZn). Notably, substituting the multi-tentacle LiTFSI and Zn(OTf)2 salts in MTE-EG-LiZn with Li2SO4 and ZnSO4 salts results in endothermic peaks at −34 °C in the DSC curves (Fig. 5a). This highlights the critical role of multi-tentacle moieties in constructing MTEs.
 | ||
| Fig. 5 Universality with low-charged multi-tentacle Li salts. (a) DSC curves of MTE-EG-LiZn, MTE-PG-LiZn, MTE-glycerol-LiZn and counterpart electrolytes containing Li2SO4 and ZnSO4. (b) and (c) Temperature-dependent conductivity (b) and evolution of DSC curves (c) with increasing water doses n in the formula (1LiTFSI + 0.5Zn(OTf)2):4EG:nH2O. (d) Evolution of diffusion coefficients of Li+ in the LMO cathode host with decreasing temperatures. (e) Average output voltage, discharge capacity (based on the mass of LMO) and energy efficiency of LMO||Zn evaluated at 0.2C and different low temperatures. (f) Long-term stability of Zn||LMO cells (LMO loading: ∼18 mg cm−2) at 0 °C/1C, −20 °C/0.5C and −30 °C/0.2C. (g) Voltage profiles of the Zn||LMO pouch cell (LMO loading: ∼18 mg cm−2) evaluated at 0, −10, −20, and −30 °C. (h) Comparison of the output energy densities of Zn batteries with MTEs and other reported Zn batteries.14,15,21,34,35,39 The energy density is based on the mass of active materials on cathodes. | ||
We also investigate the upper limit of water molecules that multi-tentacle moieties can hold in the composition of (1LiTFSI + 0.5Zn(OTf)2):4EG:nH2O. As shown in Fig. 5b, the ionic conductivities increase with water doses n, peaking at n = 24. Further increasing water dose (n = 32) results in a conductivity plunge at −40 °C. This drop in conductivity is due to the icing of this electrolyte. As shown in Fig. 5c, the endothermic peak can only be observed in the electrolyte of n = 32, showing a freezing point of ∼−30 °C. Therefore, multi-tentacle moieties in the composition can hold up to 24 water molecules (MTE-EG-LiZn), without icing down to −60 °C. MTE-EG-LiZn was selected for the subsequent study due to its high ionic conductivity (Fig. 5b). This electrolyte also enables a highly stable and reversible Zn anode in a full temperature window of −40 to 25 °C as discussed in Fig. S22 (ESI†).
MTE-EG-LiZn enables the rapid Li intercalation/de-intercalation in high-voltage LMO cathodes, promoting the high-capacity recovery and output voltage of LMO cathodes at low temperatures. The Li+ diffusion coefficients in LMO at low temperatures were evaluated with a galvanostatic intermittent titration technique (GITT) (Fig. 5d and Fig. S23, ESI†). The diffusion coefficients of Li+ in LMO cathodes exhibit a mild decrease as the temperature falls and a high Li+-insertion state of 85% at −40 °C can be achieved (Fig. 5d), indicating the high utilization of LMO capacity at low temperatures.
Zn||LMO full cells in MTE-EG-LiZn, assessed at 0.2C and 1C across a range of low temperatures, demonstrate high available capacities (Fig. S24, ESI†). The key parameters, including average output voltage, specific capacity, and energy efficiency at 0.2C, are summarized in Fig. 5e. Notably, approximately 75% of the practical capacity (120 mA h g−1) can still be retained even at −40 °C, corresponding to an average output voltage of 1.72 V and a specific energy of 154.4 W h kgLMO−1 (Fig. 5e). Energy efficiency, defined as the ratio of the energy output to the energy input, is a key metric for energy storage systems operating at low temperatures. It can be significantly affected by additional voltage drops in both electrolytes and cathodes due to reduced charge-carrier mobility at lower temperatures. In MTE-EG-LiZn, the energy efficiency of Zn||LMO at −40 °C still exceeds 90%, indicating the rapid Li+ diffusion in both the electrolyte and MTE-EG-LiZn (Fig. 5e).
Furthermore, when increasing the mass loading of LMO to ∼18 mg cm−2, LMO cathodes retain over 99% of their initial capacities after 100 cycles, corresponding to 1.86 (0 °C/1C), 1.38 (−20 °C/0.5C), and 1.34 (−30 °C/0.2C) mA h cm−2, respectively (Fig. 5f and Fig. S25, ESI†). Additionally, a 27.4 mA h Zn||LMO pouch cell with an N/P ratio of 2.87 recovers 93.5%, 90.9%, 81.7%, and 79.1% of the practical capacity when evaluated at 0, −10, −20, and −30 °C, respectively, corresponding to energy densities of 138.2, 132.6, 117.2, and 113.8 W h kg−1 based on the total mass of LMO and Zn (Fig. 5g). Such high-capacity recovery is attributed to the fast mass transport of charge carriers in both the electrolyte and cathodes.
Fig. 5h and Table S1 (ESI†) summarize the mass loading, areal capacity, areal current, testing temperature, and specific energy between our work and other reported low-temperature Zn batteries. The use of MTEs facilitates competitive discharge capacity and output voltage in Zn batteries with NVO and LMO cathodes, even under high cathode loading conditions. Specifically, the high-loading Zn||NVO full cell delivers an energy density of 110.4 W h kgNVO−1 at 0.2C and −40 °C. This energy density can be further increased to 154.4 W h kgLMO−1 when employing LMO cathodes (0.2C and −40 °C). Consequently, MTEs enable competitive specific energies at low temperatures, outperforming other reported literature in this domain.
Conclusions
In summary, we have reported a new type of low-temperature-tolerant aqueous electrolyte, namely MTE, to enable high-performance AZBs at extremely low temperatures. The key attribute of MTE is the ability to confine a large portion of water molecules by using just small amounts of multi-tentacle species that form strong coordination and H-bonds with water. These multi-tentacle species include, but are not limited to, salts such as Zn(ClO4)2, Zn(OTf)2, Zn(TFSI)2, and LiTFSI, and organic moieties such as EG, PG and glycerol. In our examples, we show impressively low salt-to-solvent and organic-to-water ratios of 1.5:28 and 4:24 in MTEs, respectively. Under these low concentration conditions, the MTEs not only maintain liquid states even at −60 °C, but also outperform conventional low-temperature electrolytes utilizing either concentrated salt or concentrated organic strategies, in terms of chemical/electrochemical stability, viscosity, and ionic transportation kinetics. As a result, even at −40 °C, Zn metal anodes could stably cycle for more than 1100 hours at a high current density of 2 mA cm−2 and a high capacity of 2 mA h cm−2, with an average coulombic efficiency close to 99%. Furthermore, AZBs using VNO and LMO cathodes exhibit high discharge capacity and long-term stability at low temperatures. Specifically, Zn batteries using high-loading NVO cathodes (∼17 mg cm−2) reach an areal capacity of ∼3.4 mA h cm−2 even at −40 °C, and achieve 1000 stable cycles with 99.99% retention per cycle in MTE. Zn||LMO batteries reach an outstanding capacity retention (75% of their practical capacity at room temperature) and a high output voltage (1.72 V) at −40 °C. The swift mass transport in both electrolytes and cathodes leads to competitive specific energy compared to other reported work. The high-loading Zn||NVO full cell delivers an energy density of 110.4 W h kgNVO−1 even at 0.2C and −40 °C. This energy density can be further increased to 154.4 W h kgLMO−1 when employing LMO cathodes. The MTE strategy offers a new insight to prevent water molecules from icing, and therefore, contributes to the design of a low-viscosity, temperature-tolerant electrolyte that fundamentally addresses the sluggish kinetics of aqueous electrolytes at low temperatures. Currently, multi-tentacle salts such as Zn(TFSI)2, Zn(OTf)2, Zn(ClO4)2, and LiTFSI serve as proofs-of-concept to validate the MTE strategy, with Zn(ClO4)2 achieving a 95% cost reduction compared to Zn(TFSI)2 (Alfa Aesar, 2025). Future work should focus on developing additional cost-effective multi-tentacle salts for large-scale applications.
Author contributions
H. W. and M. S. H. W.: conceptualization, methodology, and writing. M. S.: computational study. Y. Y., J. Z., L. F. and Q. H.: methodology. B. H.: computational study and review. Z. Z.: conceptualization, funding acquisition, writing, review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the NSFC/RGC Collaborative Research Scheme (CRS_PolyU504/22), the RGC Research Impact Fund (R5019-22), the NSFC's Young Scientist Fund (52203318), the RGC General Research Fund (15304023 and 15304724), and the RGC Young Collaborative Research Grant (C1003-23Y).References
- J. J. Xu, J. X. Zhang, T. P. Pollard, Q. D. Li, S. Tan, S. Y. Hou, H. L. Wan, F. Chen, H. X. He, E. Y. Hu, K. Xu, X. Q. Yang, O. Borodin and C. S. Wang, Nature, 2023, 614, 694–700 CrossRef CAS PubMed.
- C. Grey and J. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed.
- Y. F. Meng, D. Zhou, R. L. Liu, Y. Tian, Y. F. Gao, Y. Wang, B. Sun, F. Y. Kang, M. Armand, B. H. Li, G. X. Wang and D. Aurbach, Nat. Energy, 2023, 11, DOI:10.1038/s41560-023-01339.
- F. Ai, Z. Y. Wang, N. C. Lai, Q. L. Zou, Z. J. Liang and Y. C. Lu, Nat. Energy, 2022, 7, 417–426 CrossRef CAS.
- H. Jiang, L. T. Tang, Y. K. Fu, S. T. Wang, S. K. Sandstrom, A. M. Scida, G. X. Li, D. Hoang, J. Hong, N. C. Chiu, K. C. Stylianou, W. F. Stickle, D. H. Wang, J. Li, P. A. Greaney, C. Fang and X. L. Ji, Nat. Sustainability, 2023, 6, 806–815 CrossRef.
- L. S. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Y. Yang, L. Chen, J. Vatamanu, E. Y. Hu, M. J. Hourwitz, L. Ma, M. Ding, Q. Li, S. Y. Hou, K. Gaskell, J. T. Fourkas, X. Q. Yang, K. Xu, O. Borodin and C. S. Wang, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
- L. Ma, M. A. Schroeder, O. Borodin, T. P. Pollard, M. S. Ding, C. S. Wang and K. Xu, Nat. Energy, 2020, 5, 743–749 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. L. Fan, W. Sun, F. D. Han, A. Faraone, J. A. Dura, K. Xu and C. S. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- S. L. Liu, R. Z. Zhang, J. F. Mao, Y. L. Zhao, Q. Cai and Z. P. Guo, Sci. Adv., 2022, 8, 24 Search PubMed.
- Z. Khan, D. Kumar and X. Crispin, Adv. Mater., 2023, 35, 31 Search PubMed.
- C. Y. Yang, J. L. Xia, C. Y. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan, E. Thimsen, J. J. Xu, W. T. Song, E. Y. Hu, X. Ji, S. Y. Hou, X. Y. Zhang, M. S. Ding, S. Hwang, D. Su, Y. Ren, X. Q. Yang, H. Wang, O. Borodin and C. S. Wang, Nat. Sustainability, 2023, 6, 325–335 CrossRef.
- L. S. Cao, D. Li, F. A. Soto, V. Ponce, B. Zhang, L. Ma, T. Deng, J. M. Seminario, E. Y. Hu, X. Q. Yang, P. B. Balbuena and C. S. Wang, Angew. Chem., Int. Ed., 2021, 60, 18845–18851 CrossRef CAS PubMed.
- S. C. Liu, H. Zhu, B. H. Zhang, G. Li, H. K. Zhu, Y. Ren, H. B. Geng, Y. Yang, Q. Liu and C. C. Li, Adv. Mater., 2020, 32, 10 Search PubMed.
- D. Han, C. Cui, K. Zhang, Z. Wang, J. Gao, Y. Guo, Z. Zhang, S. Wu, L. Yin, Z. Weng, F. Kang and Q.-H. Yang, Nat. Sustainability, 2022, 5, 205–213 CrossRef.
- Q. Y. Ma, R. Gao, Y. Z. Liu, H. Z. Dou, Y. Zheng, T. Or, L. X. Yang, Q. Y. Li, Q. Cu, R. F. Feng, Z. Zhang, Y. H. Nie, B. H. Ren, D. Luo, X. Wang, A. P. Yu and Z. W. Chen, Adv. Mater., 2022, 34, 2207344 CrossRef CAS PubMed.
- J. N. Hao, L. B. Yuan, C. Ye, D. L. Chao, K. Davey, Z. P. Guo and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- Y. Shi, R. Wang, S. S. Bi, M. Yang, L. L. Liu and Z. Q. Niu, Adv. Funct. Mater., 2023, 33, 10 Search PubMed.
- S. Y. Zhao, Y. Y. Zuo, T. Liu, S. Zhai, Y. W. Dai, Z. J. Guo, Y. Wang, Q. J. He, L. C. Xia, C. Y. Zhi, J. Bae, K. L. Wang and M. Ni, Adv. Energy Mater., 2021, 11, 29 Search PubMed.
- Y. Yan, S. Duan, B. Liu, S. Wu, Y. Alsaid, B. Yao, S. Nandi, Y. Du, T. W. Wang and Y. Li, Adv. Mater., 2023, 35, 2211673 CrossRef CAS PubMed.
- D. Dong, T. Wang, Y. Sun, J. Fan and Y.-C. Lu, Nat. Sustainability, 2023, 6, 1474–1484 CrossRef.
- N. N. Chang, T. Y. Li, R. Li, S. N. Wang, Y. B. Yin, H. M. Zhang and X. F. Li, Energy Environ. Sci., 2020, 13, 3527–3535 RSC.
- W. Wang, S. Chen, X. L. Liao, R. Huang, F. M. Wang, J. L. Chen, Y. X. Wang, F. Wang and H. Wang, Nat. Commun., 2023, 14, 12 CrossRef PubMed.
- X. Bai, M. Sun, J. Yang, B. Deng, K. Yang, B. Huang, W. Hu and X. Pu, Energy Environ. Sci., 2024, 17, 7330–7341 RSC.
- Q. Zhang, K. X. Xia, Y. L. Ma, Y. Lu, L. Li, J. Liang, S. L. Chou and J. Chen, ACS Energy Lett., 2021, 6, 2704–2712 CrossRef CAS.
- N. Wang, X. L. Dong, B. L. Wang, Z. W. Guo, Z. Wang, R. H. Wang, X. Qiu and Y. G. Wang, Angew. Chem., Int. Ed., 2020, 59, 14577–14583 CrossRef CAS PubMed.
- Y. H. Li, X. F. Dong, Z. W. Xu, M. L. Wang, R. F. Wang, J. Xie, Y. J. Ding, P. C. Su, C. Y. Jiang, X. M. Zhang, L. Y. Wei, J. F. Li, Z. Q. Chu, J. Y. Sun and C. Huang, Adv. Mater., 2023, 35, 15 Search PubMed.
- D. G. Vazquez, T. P. Pollard, J. Mars, J. M. Yoo, H.-G. Steinrück, S. E. Bone, O. V. Safonova, M. F. Toney, O. Borodin and M. R. Lukatskaya, Energy Environ. Sci., 2023, 16, 1982–1991 RSC.
- C. You, R. Wu, X. Yuan, L. Liu, J. Ye, L. Fu, P. Han, Y. J. E. Wu and E. Science, Energy Environ. Sci., 2023, 16, 5096–5107 RSC.
- Q. Zhang, Y. Ma, Y. Lu, L. Li, F. Wan, K. Zhang and J. Chen, Nat. Commun., 2020, 11, 4463 CrossRef CAS PubMed.
- W. Deng, Z. Deng, Y. Chen, R. Feng and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316499 CrossRef CAS PubMed.
- Y. Yang, W. Yang, R. Zhu, G. Wu, Y.-K. Choe, K. Sho, H. Yang and E. Yoo, Energy Storage Mater., 2024, 72, 103691 CrossRef.
- N. Dubouis, P. Lemaire, B. Mirvaux, E. Salager, M. Deschamps and A. Grimaud, Energy Environ. Sci., 2018, 11, 3491–3499 RSC.
- Y. W. Zhao, Y. Lu, H. P. Li, Y. B. Zhu, Y. Meng, N. Li, D. H. Wang, F. Jiang, F. N. Mo, C. B. Long, Y. Guo, X. L. Li, Z. D. Huang, Q. Li, J. C. Ho, J. Fan, M. L. Sui, F. R. Chen, W. G. Zhu, W. S. Liu and C. Y. Zhi, Nat. Commun., 2022, 13, 12 CrossRef PubMed.
- W. H. Yang, X. F. Du, J. W. Zhao, Z. Chen, J. J. Li, J. Xie, Y. J. Zhang, Z. L. Cui, Q. Y. Kong, Z. M. Zhao, C. G. Wang, Q. C. Zhang and G. L. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- X. D. Lin, G. D. Zhou, M. J. Robson, J. Yu, S. C. T. Kwok and F. Ciucci, Adv. Funct. Mater., 2022, 32, 7 Search PubMed.
- L. Geng, J. Meng, X. Wang, C. Han, K. Han, Z. Xiao, M. Huang, P. Xu, L. Zhang and L. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202206717 CrossRef CAS PubMed.
- K. Zhu, J. Luo, D. Zhang, N. Wang, S. Pan, S. Zhou, Z. Zhang, G. Guo, P. Yang and Y. Fan, Adv. Mater., 2024, 36, 2311082 CrossRef CAS PubMed.
- Y. Zheng, W. Zhang, F. Liu, Q. Liang, W. Li, X. Zhou, N. Yue and W. Zheng, Energy Storage Mater., 2024, 67, 103326 CrossRef.
- H. Y. Qiu, X. F. Du, J. W. Zhao, Y. T. Wang, J. W. Ju, Z. Chen, Z. L. Hu, D. P. Yan, X. H. Zhou and G. L. Cui, Nat. Commun., 2019, 10, 12 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee01316j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |