
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A semi-crystalline polymer binder with enhanced electrical conductivity and strong underwater adhesion in aqueous sodium–air batteries†
Jeonguk
Hwang
 a,
Min Hoon
Myung
a,
Jee Ho
Ha
a,
Seungwoo
Choi
a,
Soon-Jae
Jung
a,
Seunghyun
Lee
a,
Jinwoo
Park
a,
Young-Ryul
Kim
a,
Hyo
Jin
a,
Nyung Joo
Kong
a,
Youngsik
Kim
a,
Hyun-Wook
Lee
a,
Hyunhyub
Ko
a,
Tae Joo
Shin
b,
Seok Ju
Kang
a,
Myung-Jin
Baek
*a and
Dong Woog
Lee
*a
a,
Min Hoon
Myung
a,
Jee Ho
Ha
a,
Seungwoo
Choi
a,
Soon-Jae
Jung
a,
Seunghyun
Lee
a,
Jinwoo
Park
a,
Young-Ryul
Kim
a,
Hyo
Jin
a,
Nyung Joo
Kong
a,
Youngsik
Kim
a,
Hyun-Wook
Lee
a,
Hyunhyub
Ko
a,
Tae Joo
Shin
b,
Seok Ju
Kang
a,
Myung-Jin
Baek
*a and
Dong Woog
Lee
*a
aSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan, 44919, Republic of Korea. E-mail: bagmj100@unist.ac.kr; dongwoog.lee@unist.ac.kr
bGraduate School of Semiconductor Materials and Devices Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan, 44919, Republic of Korea
First published on 31st March 2025
Abstract
Common polymer binders are insulators, which significantly diminish the battery performance owing to their low electron mobility. For aqueous sodium–air batteries (SABs) to exhibit reliable performance as energy storage systems, polymer binders should possess high electrolyte wettability, strong underwater adhesion, high crystallinity, and conductivity to efficiently transport electrons to current collectors without degradation or dissolution over time. In this study, the electrochemical performance of SABs was significantly improved using a newly developed binder containing poly(ethylene glycol), catechol, and anthracene (At) functional groups. Versatile analysis of the polymer, including two-dimensional grazing incidence-wide-angle X-ray diffraction and polarized optical microscopy, showed that the enhanced SAB performance can be attributed to the At groups which have high crystallinity due to π–π stacking, thereby lowering the resistance and increasing the electrical conductivity. Because of the catechol and PEG groups, the binder also exhibited reliable underwater adhesion, and electrolyte wettability, which are essential for aqueous SAB binders. Moreover, the binder effectively prevented carbon corrosion of the carbon current collector in the air electrode. We believe that the synthesized semi-crystalline polymer binder can be applied to various batteries to improve their electrochemical performance and stability.
Broader contextSodium–air batteries (SABs) are gaining attention as a cost-effective and eco-friendly next-generation energy storage system due to the abundance of sodium resources for cathodes. However, SABs generally exhibit high overpotentials, which degrade electrochemical performance and corrode carbon current collectors due to undesired carbon oxidation reactions. One solution is to introduce suitable electrocatalysts that can lower the overpotential. To accomplish this, a polymer binder that can stably attach the electrocatalyst to the current collector is required. Commercial binders such as polyvinylidene fluoride (PVDF) and Nafion have been commonly used; however, these binders have several limitations including (i) poor adhesion leading to catalyst desorption, (ii) low electrical conductivity, (iii) low electrolyte wettability, and (iv) their classification as per- and polyfluoroalkyl substances (PFAS). In this study, we synthesized a multifunctional semi-crystalline binder that is fluorine-free and exhibits strong adhesion, hydrophilicity, and electrical conductivity. SABs utilizing this binder demonstrated improvements in electrochemical performance, including higher energy efficiency, power density, lower overpotential and resistance, and enhanced long-term cycling stability. The mechanism has shown that the binder's high electrical conductivity and strong catalyst immobilization onto the current collectors suppress carbon current collector corrosion and maintain stable electrochemical reactions during extended operation time. |
Introduction
Aqueous sodium–air batteries (SABs) are a class of metal–air batteries that are relatively low-cost and eco-friendly compared to lithium-ion batteries (LIBs) due to their abundance of sodium sources (e.g., NaCl and NaOH) for cathodes.1–3 Moreover, SABs have received significant attention as next-generation energy storage systems owing to their high theoretical energy densities.4,5 Seawater batteries are a class of sodium–air batteries that use abundant natural seawater as a cathode active material.6–8 Using abundant seawater as the sodium source for SABs eliminates the need for additional processing and significantly reduces the manufacturing costs of energy storage and energy conversion devices.9However, despite these advantages, SABs typically exhibit high overpotential owing to the oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR), which reduce charge/discharge and energy efficiency.10–12 and increase side reactions such as the chlorine evolution reaction13 and carbon corrosion.7,14 Consequently, lowering the overpotential has been one of the main focuses of SABs, and the use of electrocatalysts is considered to be an effective method.15–18
In order to attach the electrocatalyst to the cathode, glue, known as a polymer binder, should be utilized. Conventionally, polymer binders such as polyvinylidene fluoride (PVDF), Nafion, and carboxymethyl cellulose (CMC) are used to ensure the structural integrity of air electrodes for SABs.19–21 However, hydrophobic binders such as PVDF and Nafion show limitations in performance improvement due to catalyst desorption and poor aqueous electrolyte wettability, while hydrophilic binders (CMC, polyacrylic acid, polyethylene oxide, etc.) have been reported to suffer from hydration-induced softening and dissolution.22–24 The PVDF binder, as an insulating polymer with low electrical conductivity (<3.41 × 10−13 S cm−1),25 weakens the performance of the SABs. Additionally, in 2023, the European Union issued a report on the restriction of perfluorinated compounds, including perfluorooctanoic acid, perfluorooctanoate (PFOA) salts, and PFOA-related substances used in the synthesis of PVDF, as part of their defluorination policy.26 Consequently, the development of a polymer binder that exhibits the following characteristics is of great interest: (i) cost-effectiveness, (ii) eco-friendliness, (iii) good wettability without dissolution, (iv) reliable underwater adhesion, and (v) suitability as an alternative to per- and polyfluoroalkyl substance (PFAS).27
In this study, we developed a semi-crystalline polymer binder that simultaneously satisfies the aforementioned characteristics by incorporating and optimizing the ratios of three different monomers (Fig. 1): dopamine-m-acrylamide (DMA), poly(ethylene glycol) (PEG) acrylate, and butyl anthracene acrylate. The catechol functional group in dopamine-m-acrylamide can provide strong underwater adhesion with various substrates,28–31 and the PEG group is known to exhibit inherent hydrophilicity32 with high ion conductivity.33 The anthracene (At) moiety, which has been widely utilized in various applications such as secondary battery binders,34 organic thin-film transistors,35 organic light-emitting diodes,36 and organic solar cells,37,38 facilitates efficient electron transport via hopping conduction through π–π interactions. Moreover, the At moiety contributes to the crystallinity of the polymer through π–π stacking, enabling a balance between crystalline and amorphous characteristics. When semi-crystalline polymers are utilized as battery binders, they enable efficient coating on current collectors while simultaneously enhancing electron transport, thereby improving the overall battery performance.39,40
 | ||
| Fig. 1 Aqueous sodium–air batteries (Scheme). (a) air electrode assembled with a polymer binder, Pt catalyst, and carbon current collector in aqueous media, and (b) molecular design of a multi-functional binder with underwater adhesion and electrical conductivity. | ||
A sodium–air battery (SAB) assembled with the developed polymer binder exhibited significantly better electrochemical performance than the SAB with a bare carbon current collector, conventional binders such as PVDF, and the previously developed catechol-based polymer binder.41 Additionally, the antioxidant properties of catechol groups effectively prevented the corrosion of the carbon current collector. The superior properties of the developed binder were examined using versatile techniques and confirmed to be due to (i) the high crystallinity of the At group in water, resulting in high electrical conductivity, (ii) strong underwater adhesion of catechol groups, and (iii) the excellent hydrophilic properties of PEG.
Results and discussion
Characterization of the synthesized binder
The multi-functional polymer binder was designed to contain catechol, PEG, and At groups to improve the adhesive strength, wettability, and electrical conductivity, respectively, in aqueous environments (Fig. 1b and Scheme S1, Fig. S1–S4, Table S1; details of the synthesis procedures are provided in the ESI†). The 1H-NMR analysis of DPTA631, containing 10% At, revealed that its spectrum (Fig. S4b, ESI†) confirmed acrylate peaks from the At monomer (6.0–6.4 ppm) in trace amounts. This indicates that the residual trace amount of monomer in the DPTA631 polymer does not affect its semi-crystallinity or conductivity. To further explore whether the At content impacts crystallinity and conductivity, a binder (DPTA622) with an additional 10% At content was synthesized and analyzed. Additionally, to focus on the role of At, a previously reported DPA binder (Scheme S1, ESI†) was used as a control which exhibited strong adhesion and excellent wettability in water.41 The glass transition temperature (Tg) of DPA64 was measured and found to be −31.2 °C, while Tg and the melting temperature (Tm) of DPTA631 were measured and found to be −58.4 °C, and 113.1 °C, respectively (Fig. S5, ESI†). DPTA631 exhibited only Tg and Tm transitions, similar to other semi-crystalline polymers.42–44 Furthermore, the melting enthalpy of DPTA631 was determined to be 0.22 J g−1. The synthesized DPTA631 binder had a 10 mol% PEG group reduction compared to DPA64; however, it was expected to ensure sufficient aqueous electrolyte wettability for efficient electrochemical reactions. To verify the hydrophilicity of the synthesized DPTA631 polymer, the water contact angles of the DPTA631 and DPA64 coated glass surfaces were measured. Both the DPTA631 and DPA64 coated surfaces showed sufficiently low water contact angles of 16.3 ± 1.2° and 13.2 ± 1.4°, respectively, owing to the incorporated PEG groups, while DPTA631 showed a slightly higher (∼3°) contact angle due to the reduction in the PEG content and incorporation of the At group. Furthermore, DPTA622 showed an elevated contact angle of 23.8 ± 0.9° due to the increased presence of the hydrophobic At moiety (Fig. S6, ESI†). The increased At content above 10 mol% decreased hydrophilicity due to the smaller PEG group composition, thereby diminishing the coating property and electrolyte wettability, and electrochemical performance (Fig. S6–S8, ESI†). Therefore, we optimized the DPTA631 binder, which contains 10 mol% of At monomer.Electrochemical performance of SABs using synthesized polymer binders
To elucidate the effect of the conductive binder on the cathode performance of the SABs, the electrochemical performance was tested by assembling the SABs with cathodes containing the synthesized binders and Pt/C catalyst (see the preparation of electrodes in the Methods section). The theoretical value of the charge/discharge voltage of the SABs (seawater used in this study) is Vt_c = 3.48 and Vt = 2.90 V, respectively (1st subscript indicates theoretical, t, or measured, m, the 2nd subscript indicates charge, c, or discharge, d).45,46The electrochemical properties of the bare carbon current collector without a binder and catalyst are shown in Fig. S9 (ESI†). The bare carbon current collector exhibited a high voltage gap and overpotential at 100th charge/discharge cycles owing to carbon corrosion caused by the high overpotential, resulting in poor battery performance.14Fig. 2a and Fig. S10 (ESI†) show the galvanostatic charge/discharge profiles of the SABs using each binder for 100 cycles. It was confirmed that the voltage gap (ΔVm = Vm_c − Vm_d, Fig. 2a) and the overpotential (ΔVm − ΔVt, Fig. 2b) of the PVDF increased with the number of charge/discharge cycles and reached relatively high values of 1.5 V and 0.90 V, respectively, at the 100th cycle. Meanwhile, both DPA64 and DPTA631 incorporated SABs were relatively stable for up to 100 cycles without a noticeable increase in overpotential. Nevertheless, the DPTA631 containing SAB exhibited a 48% smaller overpotential (0.30 V) compared to those of DPA64 (0.58 V) and PVDF (0.90 V) at the 100th cycle. Moreover, DPTA631 exhibited outstanding cycle life and stability during long-term galvanostatic charge/discharge measurements conducted over 1200 h (300 cycles), as shown in Fig. S11 (ESI†).47–49 It is known that with an increasing number of charge/discharge cycles, the carbon electrode surface is oxidized, and the overpotential gradually increases, causing side reactions such as carbon corrosion.7,14 These phenomena degrade the performance of SABs; however, the synthesized polymer binders with electrocatalysts seemed to increase the rate of OER/ORR, preventing overpotential build-up and suppressing carbon oxidation and corrosion, thereby exhibiting excellent electrochemical performance. The significantly more stable and lower overpotential of SABs with catechol-containing binders should be due to the strong underwater adhesion of catechol groups, which inhibits the desorption of the electrocatalyst. The average overpotential values for all cycles are shown in Table S2 (ESI†). In addition, the lower overpotential of DPTA631 compared to that of DPA64 is due to the higher electrical conductivity of the At group (which will be thoroughly discussed in the latter sections).
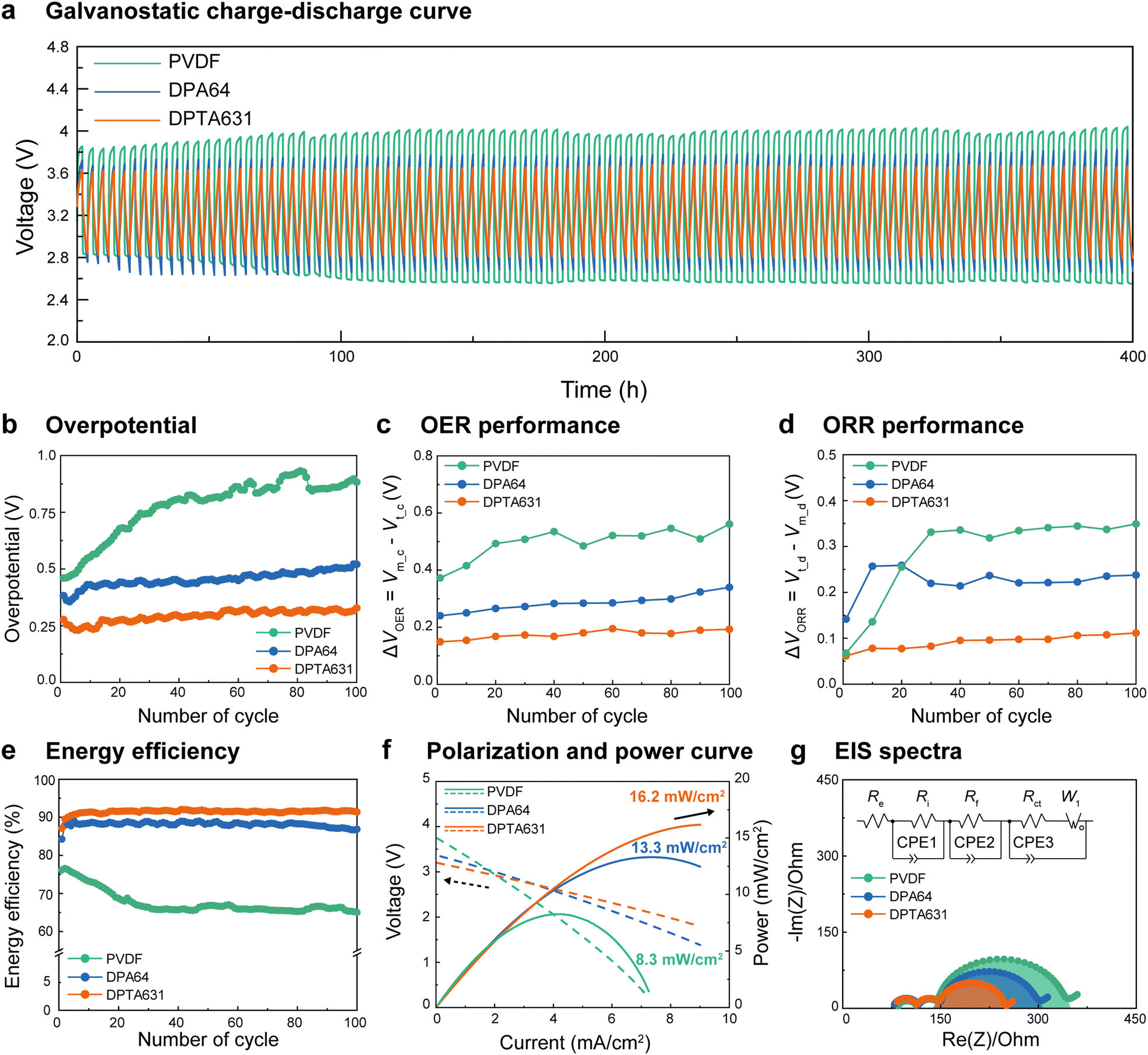 | ||
| Fig. 2 Electrochemical performance of SABs. (a) Galvanostatic charge/discharge profiles, (b) overpotential, (c) OER and (d) ORR performance, (e) energy efficiency, (f) polarization curves and power densities, and (g) electrochemical impedance spectroscopy (EIS) spectra and equivalent circuit of SABs assembled with PVDF, DPA64 and DPTA631. | ||
Fig. 2c and d show the OER and ORR performance (ΔVOER = Vm_c − Vt_c and ΔVORR = Vt_d − Vm_d, respectively; smaller values indicate higher performance) of the assembled SABs. The PVDF exhibited significantly poor OER and ORR performance due to the low adhesion and catalyst detachment. In contrast, the DPTA631 showed high OER (∼50% smaller ΔVOER) and ORR performances (∼46% smaller ΔVORR) compared to DPA64 after 100 cycles. Additionally, the average OER and ORR with variations are presented in Fig. S12 (ESI†). The significant enhancement of the OER and ORR performance of DPTA631 should be due to the better electrical conductivity triggered by At (which will be evaluated in the latter sections).
Fig. 2e shows the energy efficiencies (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Vm_d/Vm_c) of the SABs. The energy efficiency of PVDF continued to decrease from the initial charge/discharge, reaching approximately 65% in the 100th cycle. The initial energy efficiency of the DPA64 binder (at the 6th cycle) was ∼88%, which was stable up to 80 cycles, followed by a slight decrease (∼85% at the 100th cycle). Meanwhile, the DPTA631 binder showed a higher and more stable energy efficiency throughout 100 cycles (92% from the 6th cycle to the 100th cycle) compared to DPA64. Thus, at the 100th cycle, DPTA631 exhibited ∼7% and ∼27% higher energy efficiency compared to DPA64 and PVDF, respectively, which represents a huge improvement in energy storage systems.
Vm_d/Vm_c) of the SABs. The energy efficiency of PVDF continued to decrease from the initial charge/discharge, reaching approximately 65% in the 100th cycle. The initial energy efficiency of the DPA64 binder (at the 6th cycle) was ∼88%, which was stable up to 80 cycles, followed by a slight decrease (∼85% at the 100th cycle). Meanwhile, the DPTA631 binder showed a higher and more stable energy efficiency throughout 100 cycles (92% from the 6th cycle to the 100th cycle) compared to DPA64. Thus, at the 100th cycle, DPTA631 exhibited ∼7% and ∼27% higher energy efficiency compared to DPA64 and PVDF, respectively, which represents a huge improvement in energy storage systems.
Fig. 2f shows the polarization and power curves (after three charge/discharge cycles) of the SABs. The maximum power of PVDF is 8.3 mW cm−2 at 4.2 mA cm−2, owing to the slow electrochemical reaction (OER and ORR). The SAB containing DPTA631 shows significantly improved performance (∼22%), showing the maximum power of 16.2 mW cm−2 at 9 mA cm−2, compared to that of SAB containing DPA64 (13.2 mW cm−2 at 7.8 mA cm−2). In order to test the stability of the SABs, polarization and power curves were also evaluated after the 30th charge/discharge cycle, and we confirmed that the decreased power was smaller for DPTA631 (from 16.2 to 15.6 mW cm−2, ∼3.8% decrease) compared to that of DPA64 (from 13.2 to 12.0 mW cm−2, ∼9.9% decrease) (Fig. S13, ESI†). Specifically, PVDF was measured at 6.3 mW cm−2, with a 25% drop in the 30th charge/discharge cycle. This is likely due to the decrease in battery performance caused by catalyst detachment and carbon corrosion starting around the 25th cycle (Fig. 2b–e).
To analyze the effects of the polymer binders on the resistance of the battery system and its components, electrochemical impedance spectroscopy (EIS) was performed (Fig. 2g). Each resistance is described as a semicircle fitted into an equivalent circuit.50,51 In the battery system, the resistances of NASICON and the liquid electrolyte (Re, Ri, and Rf) were measured and found to be similar regardless of the binder type. The charge transfer resistance (Rct) is indicative of the resistance of the carbon current collector and the electrode; a smaller Rct of the electrode suggests faster electron, ion, and charge transfer. This allows for a direct assessment of the impact of the binder and catalyst on the SAB performance. The semicircle diameter corresponding to Rct was smaller for DPTA631 (56.3 Ω) and DPA64 (129.0 Ω) compared to PVDF (197.4 Ω) (Table S3, ESI†). Notably, DPTA631 exhibited reduced resistance and prevented carbon corrosion due to the crystallinity and conductivity of the At group. To evaluate the impact of the binder on the stability of SAB electrodes, EIS was conducted on SAB electrodes subjected to 30 cycles of electrochemical reactions. The electrodes containing DPA64 and DPTA631 showed a slight increase in Rct compared to the initial SAB electrodes, whereas the electrodes with bare carbon and PVDF exhibited a significant enhancement in Rct by more than 20% (Fig. S14 and Table S3, ESI†). The increase in the resistance of the PVDF-based electrode is likely due to catalyst detachment. In comparison, that of the bare carbon electrode is attributed to the carbon corrosion of the current collector.
Crystallinity of the polymer binder
It is previously known that when an aromatic group with conductivity undergoes π–π stacking, the crystallinity of the polymer increases,52–55 possibly enhancing the electron transfer.56–58 In addition, polymer binders with aromatic groups or crystallinity are known to improve battery performance.52,55,56 To investigate the crystallinity of the synthesized polymers, two-dimensional grazing incidence wide-angle X-ray diffraction (GI-WAXD) patterns, polarized optical microscopy (POM), X-ray diffraction (XRD), and transmission electron microscopy (TEM) were obtained (Fig. 3 and Fig. S15–S17, ESI†). Fig. 3a, c and d show that DPTA631 has strong random semi-crystallinity, and the very narrow peak signals indicate that the side chains of DPTA631 form large crystalline domains. The 2D GI-WAXD patterns of the DPTA631 polymer (Fig. 3c and d) show diffraction patterns with a ring shape along the Debye ring, indicating random orientation. Such a random orientation in the DPTA631 polymer is expected to provide efficient three-dimensional pathways for electron transport, leading to improved electrochemical performances. Using the Scherrer equation, the domain size of the DPTA631 polymer (0.75 Å−1 peak in Fig. 3c) was calculated to be 35.7 nm, which is within a similar range as the polymer crystal size.59,60 XRD analysis also revealed that the At-acrylate monomer exhibited strong crystallinity due to the π–π stacking of anthracene moieties. In contrast, the randomly structured DPTA631 polymer displayed multiple weak semi-crystalline peaks, differing from the XRD pattern of the At-acrylate monomer (001, 011, and 002) (Fig. S16, ESI†).61 Moreover, Fig. S17 (ESI†) shows the presence of random semi-crystallinity with varying domains and spacings in DPTA631. These findings collectively suggest that in an aqueous environment, the hydrophobic attraction between the At moieties should enhance the crystallinity of DPTA631 (Fig. 3e). | ||
| Fig. 3 Crystallinity of electron-conductive binders. GI-WAXD patterns of (a) DPTA631 and (b) DPA64. GI-WAXD line-cut profiles of the (c) horizontal and (d) vertical directions. (e) TEM images of DPTA631 in solution, forming crystals via π–π stacking of the anthracene moiety. (f) Photograph and POM images of DPTA631. All crystallinity analyses were conducted at room temperature. | ||
The DPA64 polymer was found to be amorphous in all measurements (Fig. 3b–d and Fig. S15, S16, ESI†). Typical acrylate polymers such as DPA64 generally act as insulators and inhibit effective electron transfer. This results in a high overpotential and increased side reactions in the batteries, leading to carbon corrosion. In the case of DPTA631, a semi-crystalline structure with large crystalline domains induced by π–π stacking of At moieties and intra-and inter-molecular interactions of the catechol and PEG groups should enhance the electron transport properties, thereby improving the cycling stability and power density of SABs (Fig. 2).62,63
Evaluation of electrical conductivity
As seen in Fig. 4a, the indicative peaks for the catechol and At groups were between 250–300 and 330–400 nm, respectively.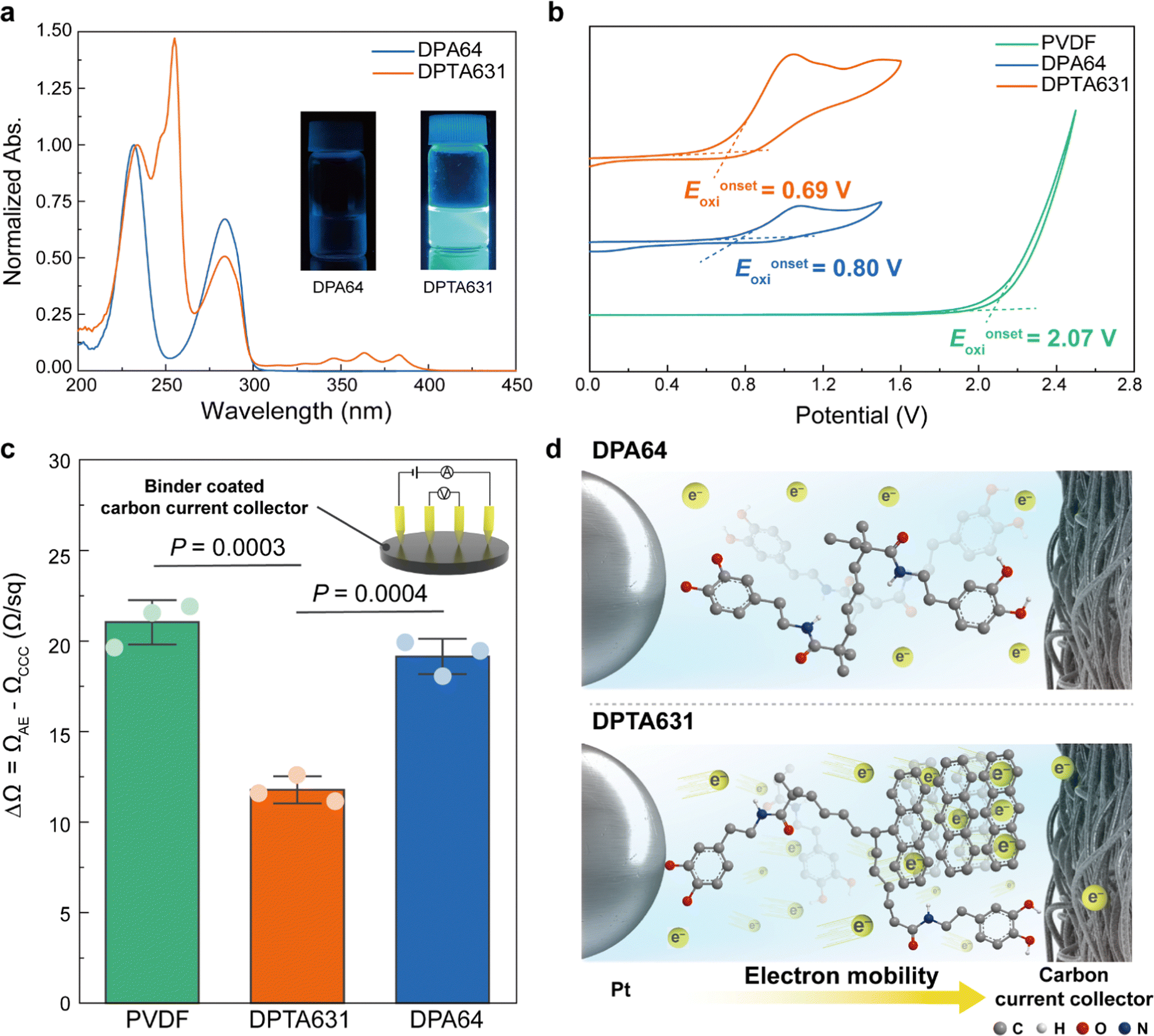 | ||
| Fig. 4 Electrochemical performance improvement mechanism of the synthesized binders. (a) UV-vis spectra of polymers (inset showed fluorescence images of each polymer solution at 365 nm) (b) cyclic voltammetry of polymers, (c) surface resistance of the carbon current collector and polymer-coated carbon current collector measured by the four-point probe method. Values indicate mean ± s.d. (n = 3 independent samples). Statistical significance and P values were evaluated using a two-sided t-test. (d) Electron-transfer mechanism with polymer binders in the SAB cathode. | ||
The DPTA631 polymer exhibited the absorption spectrum of the At group at a wavelength of 300–400 nm and fluorescence when exposed to UV at 365 nm. In Fig. 4b, PVDF exhibits a high oxidation potential (Eonsetoxi = 2.07 V) attributed to the high electronegativity of fluorine atoms.64 The high oxidation potential of the PVDF binder obstructs electron transport at the air electrode, thereby reducing the SAB performance (see Fig. 2). It has been previously reported that aromatic groups can induce a reduction in oxidation potential.57,65 The oxidation potential of DPTA631 (Eonsetoxi = 0.69 V) was reduced by 0.11 V compared to that of DPA64 (Eonsetoxi = 0.80 V) under the influence of the At group, facilitating electron transfer (Fig. 4b). The sheet resistance of the polymer-coated carbon current collectors was measured using the four-point probe method. High-resolution transmission electron microscopy (HR-TEM) analysis was performed to determine the coating thicknesses of DPTA631 and DPA64, which were measured and found to be 609 nm and 342 nm, respectively (Fig. S18, ESI†). The Fig. 4c shows the sheet resistance increment (ΔΩ) of the polymer-coated carbon current collector (ΩAE) compared to that of the bare carbon current collector (ΩCCC = 35.6 ± 1.8 Ω sq−1). According to the results, PVDF exhibited a ΔΩ value of 21.0 Ω sq−1, whereas DPA64 showed a slight reduction in ΔΩ (approximately 19.1 Ω sq−1). DPTA631 demonstrated the lowest electrical resistance (around 11.8 Ω sq−1), indicating enhanced electrical conductivity due to the At moiety. Moreover, the differences in the surface resistance of DPTA631 were significant compared to PVDF (P = 0.0003) and DPA64 (P = 0.0004), respectively (Fig. 4c). Overall, higher electron mobility (Fig. 4d) was achieved because of the lower oxidation potential (Fig. 4b) and lower resistance (Fig. 4c) of DPTA631.
Carbon corrosion analysis
Previous studies have reported that carbon corrosion of carbon current collectors degrades the performance of SABs.13,41 Differential electrochemical mass spectrometry (DEMS) is a highly useful analytical technique for real-time monitoring of gases generated during electrochemical reactions, enabling the identification of reaction mechanisms and side reactions.66–68 In Fig. 5, the carbon corrosion of carbon current collectors and its causes were analyzed using DEMS and scanning electron microscopy (SEM). Fig. 5a shows the amount of CO2 generated from the constant current charge/discharge results performed for 120 minutes at a charging current density of 0.25 mA cm−2. The CO2 evolution is caused by carbon oxidation, also known as carbon corrosion. It was observed that the carbon current collectors with DPA64 or DPTA631 binders produced considerably less CO2 following charge/discharge cycles than those using bare carbon. The PVDF binder exhibited poor wettability with seawater, which made the DEMS analysis inadequate.67 The average CO2 evolution for bare carbon was measured and found to be 0.75 ± 0.26 nmol min−1, while DPA64 showed a value of 0.15 ± 0.11 nmol min−1 of CO2. Furthermore, DPTA631 outperformed DPA64 CO2 evolution by 0.12 ± 0.07 nmol min−1 (Fig. S19, ESI†). Previous studies have reported that the catechol group has an oxidation inhibition effect.41,69 The results of reduced CO2 in the DEMS analysis with DPA64 and DPTA631 binders were attributed to the catechol group inhibiting the oxidation and corrosion of the carbon current collectors. Moreover, the electron mobility enhancement by the At group in DPTA631 was thought to further suppress the carbon corrosion compared to DPA64.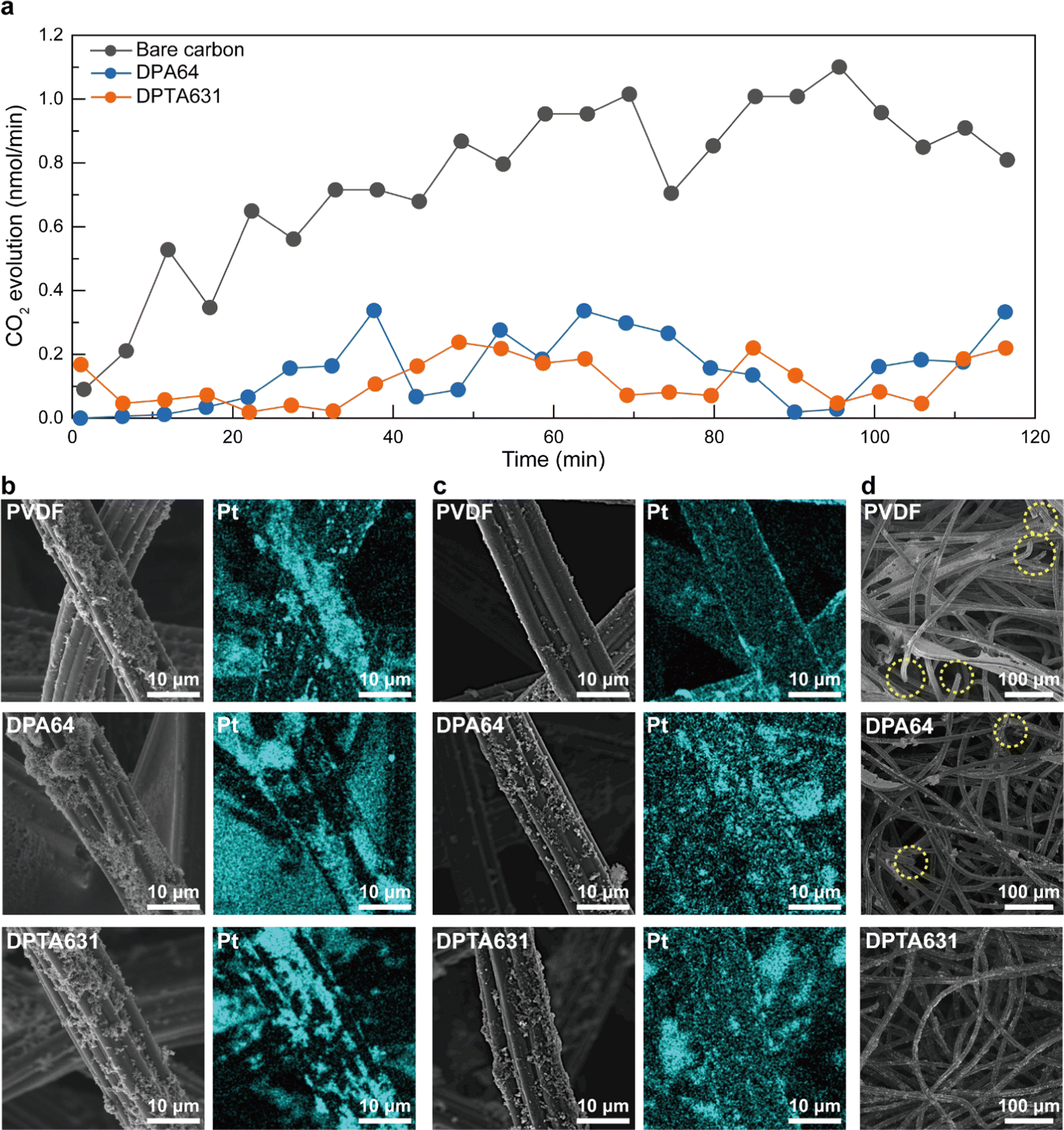 | ||
| Fig. 5 Evaluation of carbon corrosion and catalyst detachment at the air electrode. (a) In situ DEMS analysis of the CO2 evolution quantification result. SEM and elemental mapping images (Pt) of air electrodes (b) before (c) after 100 charge/discharge cycles, and (d) SEM images showing carbon corrosion at the air electrode. | ||
The SEM analysis of carbon current collectors before and after 100 charge/discharge cycles with each binder is shown in Fig. 5b and c. Regardless of the type of binder, the catalyst was successfully coated on the carbon current collectors before the cycle test, though its distribution was not entirely uniform. Compared to the PVDF binder, DPA64 and DPTA631 exhibited a slightly more uniform distribution of the Pt catalyst (Fig. 5b). However, after 100 charge/discharge cycles, the carbon current collector with the PVDF binder showed the catalyst detachment and severe carbon corrosion concerns (Fig. 5d). In contrast, DPA64 and DPTA631 binders demonstrated that the Pt/C catalyst remained intact even after the cycle tests, with significantly reduced carbon corrosion compared to PVDF (Fig. 5b–d). This confirms that the synthesized DPTA631 binder exhibits superior performance in preventing the carbon corrosion of current collectors due to its hydrophilicity, high underwater adhesion, and electrical conductivity.
Adhesive performance of synthesized binders
The bonding mechanism between the catechol group of the synthesized binder and the Pt catalyst has been reported to involve strong metal coordination and bidentate hydrogen bonding.29,70,71 To confirm the actual interaction between the catechol groups and Pt/C, the FT-IR spectra of the binder-Pt/C mixture were compared with those of the bare binder (DPTA631 or DPA64). The difference between the spectra of DPTA631 and DPA64 exhibited a decrease in the intensity of the C–O peak (1100 cm−1), representing the PEG group, and an increase in the signal of the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak (1650 cm−1) in the aromatic groups. Furthermore, the FT-IR spectra of mixtures showed a decrease in hydroxyl groups upon an increase in Pt/C content (Fig. 6c), indicating metal coordination between the catechol groups of the binder and Pt/C. In order to measure the adhesive properties quantitatively, the adhesion force between the polymer binders and Pt was evaluated using both microscale (surface forces apparatus (SFA), Fig. 6a) and macroscale analysis (lap shear test, Fig. 6b) (Fig. 6d and e). The PVDF measured no adhesive force with the Pt surface at all scales. DPA64 exhibited higher adhesion force (98.88 ± 5.82 mN) to the Pt surface compared to that of DPTA631 (64.78 ± 7.29 mN). All measurements showed cohesive failure, indicating that the measured values were cohesive forces and that adhesive forces should be higher than the measured values. The superior adhesion between the synthesized binders and Pt surface immediately formed even after a short contact time (5 s). The lower cohesive force of DPTA631 is probably due to the stronger bidentate hydrogen bonding compared to the π–π stacking of At groups. Furthermore, the differences in the macroscopic lap shear strength of DPA64 (9.11 ± 2.04 kPa) and DPTA631 (7.36 ± 1.73 kPa) statistically were non-significant (P = 0.406, Fig. 6e). The adhesion and cohesion properties of the synthesized binder seem to be strong enough to immobilize Pt/C onto carbon current collectors for a significant operation time.35 The immobilization of Pt/C on the carbon current collector was analyzed using SEM (Fig. 5b and c). After 100 charge/discharge cycles in an aqueous environment, the polymer binder and Pt catalyst (Pt peak) remained intact. These results, together with the electrochemical performance (Fig. 2 and 4), indicate that the slightly lower adhesion performance of DPTA631 is negligible and completely overcompensated by its better electrical conductivity compared to that of DPA64.
C peak (1650 cm−1) in the aromatic groups. Furthermore, the FT-IR spectra of mixtures showed a decrease in hydroxyl groups upon an increase in Pt/C content (Fig. 6c), indicating metal coordination between the catechol groups of the binder and Pt/C. In order to measure the adhesive properties quantitatively, the adhesion force between the polymer binders and Pt was evaluated using both microscale (surface forces apparatus (SFA), Fig. 6a) and macroscale analysis (lap shear test, Fig. 6b) (Fig. 6d and e). The PVDF measured no adhesive force with the Pt surface at all scales. DPA64 exhibited higher adhesion force (98.88 ± 5.82 mN) to the Pt surface compared to that of DPTA631 (64.78 ± 7.29 mN). All measurements showed cohesive failure, indicating that the measured values were cohesive forces and that adhesive forces should be higher than the measured values. The superior adhesion between the synthesized binders and Pt surface immediately formed even after a short contact time (5 s). The lower cohesive force of DPTA631 is probably due to the stronger bidentate hydrogen bonding compared to the π–π stacking of At groups. Furthermore, the differences in the macroscopic lap shear strength of DPA64 (9.11 ± 2.04 kPa) and DPTA631 (7.36 ± 1.73 kPa) statistically were non-significant (P = 0.406, Fig. 6e). The adhesion and cohesion properties of the synthesized binder seem to be strong enough to immobilize Pt/C onto carbon current collectors for a significant operation time.35 The immobilization of Pt/C on the carbon current collector was analyzed using SEM (Fig. 5b and c). After 100 charge/discharge cycles in an aqueous environment, the polymer binder and Pt catalyst (Pt peak) remained intact. These results, together with the electrochemical performance (Fig. 2 and 4), indicate that the slightly lower adhesion performance of DPTA631 is negligible and completely overcompensated by its better electrical conductivity compared to that of DPA64.
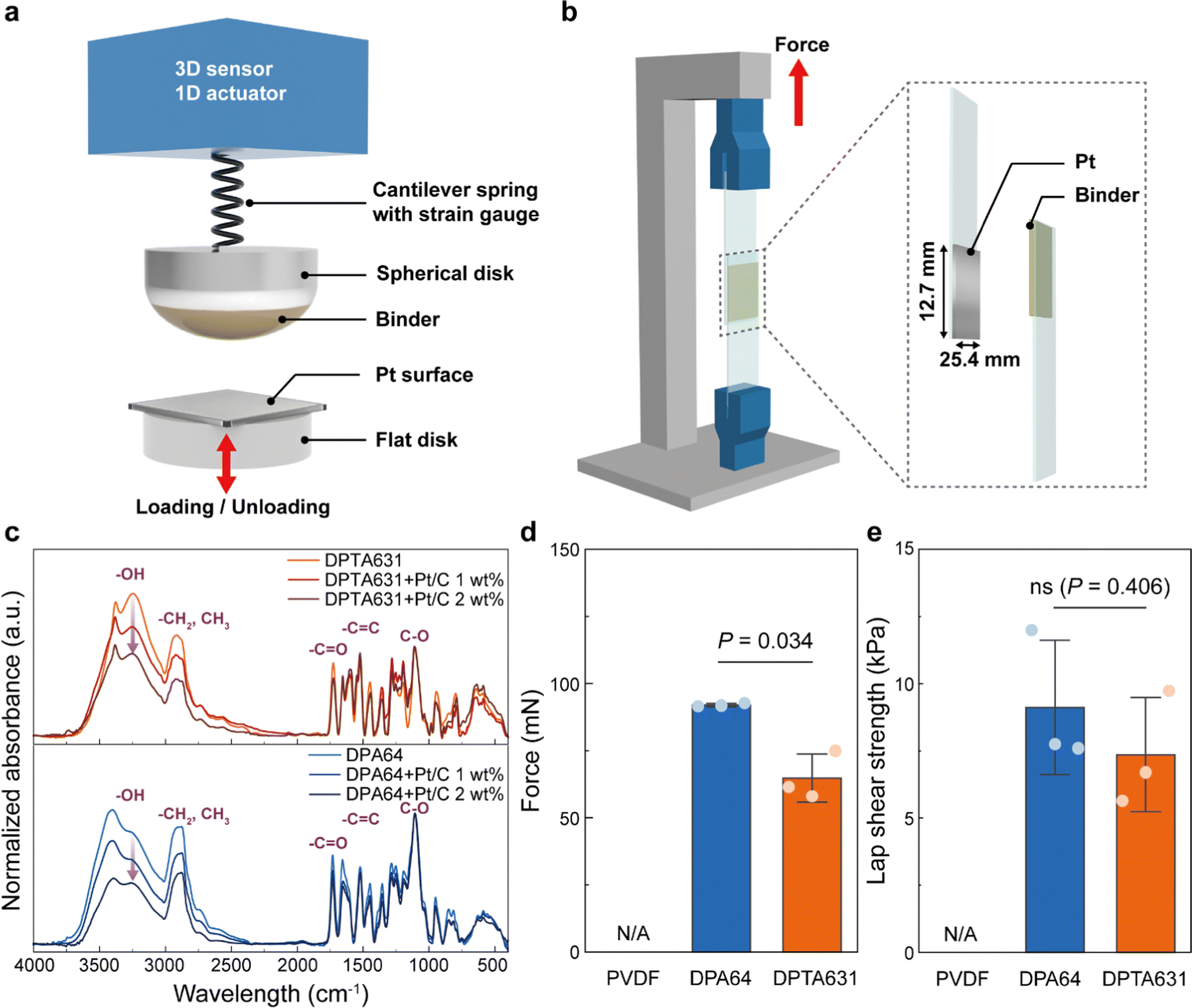 | ||
| Fig. 6 Adhesive properties of the synthesized binders. (a) microscale force measurement experimental set-up using an SFA. (b) Schematic illustration of the UTM experimental set-up for measuring the lap shear strength. (c) FT-IR spectra of the synthesized binder (DPTA631 or DPA64) and mixture (binder and Pt/C). (d) Adhesive performances at the microscale (surface forces apparatus, SFA) and (e) macroscale (lap shear strength). Values represent mean ± s.d. (n = 3 independent samples). Statistical significance and P values were determined by a two-sided t-test; ns means non-significant. | ||
Conclusions
The performance of commercialized polymer binders (e.g., PVDF, Nafion, and PAA) for aqueous metal–air batteries is limited owing to their poor underwater adhesion, low electrolyte wettability, and electrical conductivity. In this study, a novel aqueous polymer binder was developed by copolymerizing monomers with catechol (for underwater adhesion), PEG (for electrolyte wettability), and At groups (for electrical conductivity). Compared with DPA64 (which was previously shown to significantly enhance the performance of SABs compared to PVDF), the newly developed DPTA631 polymer binder significantly improved the electrochemical performance of SABs (increasing the energy efficiency (∼7%) and maximum power (∼22%), and decreasing the overpotential (∼52%)) and further mitigated carbon corrosion in the air electrode. The superior properties of the DPTA631 binder are attributed to (i) the high crystallinity of the At group in water (confirmed by GI-WAXD and POM), (ii) the strong underwater adhesion of catechol groups (confirmed by SFA, lap shear test, and FE-SEM), and (iii) the excellent hydrophilic properties of PEG (confirmed by water contact angle). We believe that the synthesized binder can be applied to various energy storage systems (e.g., redox flow batteries, aqueous rechargeable batteries, LIB, and fuel cells) to improve their electrochemical performance.Experimental
Materials
Dopamine hydrochloride, 2,2′-azobis(2-methylpropionitrile) (AIBN), poly(ethylene glycol)monomethyletheracrylate (PEG-acrylate, Mw = 480 g mol−1), and poly(ethylene glycol)diacrylate (PEGDA, Mw = 550 g mol−1) were purchased from Sigma-Aldrich. Methacrylic anhydride and triethylamine were purchased from TCI Chemicals. HPLC-grade tetrahydrofuran (THF), ethanol (EtOH), and dimethylformamide (DMF) solvents were purchased from J. T. Baker Chemicals. All the reagents were used without further purification. Sodium trifluoromethanesulfonate (NaCF3SO3, Sigma-Aldrich), tetraethylene glycol dimethyl ether (TEGDME, Alfa Aesar), and 20 wt% platinum carbon (Pt/C, Alfa Aesar) were used for electrode preparation.Synthesis of a multi-functional binder (DPTA631)
Polymerization of dopamine-m-acrylamide (DMA), 9-(acryloyloxy)butyl anthracene-9-carboxylate (At-acrylate), PEG-acrylate, and PEGDA was carried out in DMF. DMA (110.50 mg, 0.6 mmol) and At-acrylate (34.84 mg, 0.1 mmol) were dissolved in 400 μL of DMF, and PEG-acrylate (192.00 mg, 0.3 mmol) and PEGDA (11.5 mg, 0.02 mmol) were added into a polymerization ampoule. 0.5 mmol% AIBN and RAFT agent (dibenzyl trithiocarbonate) were dissolved in DMF (0.1 mg μL−1) and then added into the polymerization ampoule. After removing the oxygen through three freeze–thaw cycles, polymerization was carried out at 70 °C for 24 h. After polymerization, DPTA631 was precipitated twice from diethyl ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (2:1 volume ratio), followed by centrifugation to purify the polymer, which was then dried in a vacuum oven at 40 °C for 5 days. Approximately 280 mg of yellow product was obtained (Table S1, ESI†).
:hexane (2:1 volume ratio), followed by centrifugation to purify the polymer, which was then dried in a vacuum oven at 40 °C for 5 days. Approximately 280 mg of yellow product was obtained (Table S1, ESI†).
Preparation of electrodes
The SAB consisted of an air electrode as the cathode, a Na/C composite as the anode, catholyte seawater (Ilsan Beach in Ulsan, South Korea), and an anolyte. The overall assembly process was performed in an argon atmosphere glove box. The fabrication of an air electrode using a binder solution containing Pt/C catalyst can be divided into three steps. First, 10 mg of polymer binder and 40 mg of Pt/C catalyst were dissolved in 3 mL of EtOH by sonication at room temperature for 15 min. The carbon current collector was then immersed in the polymer binder solution and sonicated for 5 min on each side. Finally, the air electrode was dried in a convection oven at 90 °C for 1 d. Coin cells containing the Na/C composite were fabricated for the anode compartment. The coin cell compartments and flow zig for the air battery were purchased from Four To One; 1 M NaCF3SO3 in TEGDME was used as the anolyte. The Na/C composite was prepared, followed by steeping carbon cloth in melted sodium metal and punching. A 0.8 mm-thick NASICON-type solid electrolyte (Na3Zr2Si2PO12) with a diameter of 16 mm was mounted on an anode top holder. A stainless-steel spacer and spring were used for the contact.Characterization
Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded using a 400 MHz NMR spectrometer (AVANCE III HD, Bruker, USA) with DMF-d7 as a solvent. Fourier-transform infrared (FT-IR) spectra were recorded using an FT-IR spectrometer (FT/IR-4600, JASCO, Japan) in the transmittance mode. Glass transition temperature was measured using differential scanning calorimetry (DSC, Q200, TA Instrument, USA) at a scanning rate of 10 °C min−1 (from −80 °C to 150 °C) under a nitrogen atmosphere. The absorbance of the synthesized binders was recorded using a double-beam UV-vis spectrophotometer (UV-2600, Shimadzu). The scanned wavelength range was 200–450 nm. Morphological observation and energy dispersive spectroscopy (EDS) were performed using a scanning electron microscope (SEM, SU8220, Hitachi High-Technologies, Japan). Two-dimensional grazing incidence wide-angle X-ray diffraction (GI-WAXD) patterns were performed using a PLS-II 6D UNIST-PAL beamline with an X-ray energy of 11.564 keV at the Pohang Accelerator Laboratory (PAL), Korea. The angle of incidence was fixed at 0.12° to enhance the diffraction intensity and reduce substrate scattering.For morphological analysis of carbon felt samples coated with a polymer layer, transmission electron microscopy (TEM) characterization was performed using high-resolution TEM (HR-TEM) and high-angle annular dark-field (HAADF) imaging in the scanning TEM mode with a JEM-2100F (JEOL). Sample preparation for TEM analysis was carried out using a dual-beam focused ion beam (FIB) system (Helios 450HP, FEI) to investigate the Pt/polymer layer deposited on the carbon felt samples. To mitigate beam-induced damage during the FIB process, carbon and Pt protective layers were previously applied to the carbon felt composite. Crystallographic analysis of DPTA631 samples in TEM was also carried out using HR-TEM (JEM-2100F, JEOL). To prepare the samples, a solution of DPTA631 dissolved in a mixture of EtOH and tetrahydrofuran was carefully mounted onto a copper (Cu) grid in the form of a small droplet. The grid was then left to dry under ambient conditions for a day, allowing DPTA631 to recrystallize. Once the recrystallization process was complete, the Cu grid containing the recrystallized DPTA631 samples was inserted into the HR-TEM for detailed structural and crystallographic observation.
Water contact angles were measured using a KRÜSS DSA100 instrument. All polymer solutions (10 mg mL−1, THF) were spin-coated (1000 rpm for 1 min) onto glass and dried in a vacuum dryer at room temperature for 1 h. DI water (6 μL) droplets were deposited onto polymer-coated substrates. The contact angles were obtained as the average values of five measurements at different positions. X-ray diffraction (XRD) was performed using a Bruker D8-Advance film diffractometer equipped with a Vantec-1 detector (Cu Kα radiation). The XRD samples were prepared by dissolving polymers in EtOH (2 wt%), followed by drop casting them onto glass and dried in a 40 °C vacuum oven overnight. Note that the polymers are fully soluble in EtOH at 2 wt% (Fig. S20, ESI†) due to their hydrophilicity and low crosslinking density.
The mesomorphic properties were analyzed using polarized optical microscopy (POM, BX53M, Olympus) on a LINKAM stage (LTS420). The electrical conductivity was measured using four-point probe measurement equipment (CMT2000N, Advanced Instrument Technology Corp.). The 14 φ carbon current collector was immersed in 10 mg mL−1 (polymers/EtOH) in solution, sonicated at room temperature for 5 min, and dried at 40 °C overnight in a vacuum oven.
DEMS analysis for real-time observation of gas production was utilized during the galvanostatic charging of a specially designed, airtight Swagelok-type cell containing 2465 coin-type cells.67 The cells for DEMS were directly assembled in the ambient atmosphere. DEMS measurements reflect the average CO2 evolution rate (y-axis unit, nmol min−1), determined by collecting the CO2 gas produced over a 5-minute interval before analysis. CO2 (m/z = 44) gas emissions were observed consistently during the initial galvanostatic charging cycle conducted at 0.25 mA cm−2 and a temperature of 25 °C, monitoring every five minutes.
Electrochemical analysis
All coin-type cells underwent galvanostatic charge/discharge tests and power density evaluations using a battery testing system (WBCS3000L32, WonAtech). The charge and discharge characteristics of the cells were tested at a current density of 0.25 mA cm−2. Cyclic voltammetry (CV) was performed using a three-electrode system to evaluate the oxidative characteristics of PVDF, DPA64 and DPTA631 and using a potentiostat or an electrochemical analyzer (CHI660, CH Instruments Electrochemical Workstation). CV was performed in a 0.1 M solution of tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) in acetonitrile at a scan rate of 5 mV s−1 at room temperature. The redox potential of the Fc/Fc+ internal reference was 0.09 V vs. SCE. EIS was performed using a potentiostat. Seawater from Ilsan Beach, Ulsan, Republic of Korea, was used after vacuum filtration.Adhesion evaluation
Statistical analysis
MATLAB (v.R2024a) was used to assess the statistical significance of all comparisons. Data distribution was assumed to be normal for all parametric tests but was not formally tested. The homogeneity of variance was confirmed through the F-test between two sample groups. Two-sided Student's t-test and Welch's t-test were conducted to compare two sample groups with homoscedasticity and heteroscedasticity, respectively.Author contributions
Conceptualization: J. H., M.-J. B., and D. W. L. Data curation: J. H., M. H. M., J. H. H., S. C., S.-J. J., S. L., J. P., Y.-R. K., H. J., N. J. K., and M.-J. B. Formal analysis: J. H., Y. K., H.-W. L., H. K., T. J. S., S. J. K., M.-J. B., and D. W. L. Investigation: J. H., J. H. H., T. J. S., M.-J. B., and D. W. L. Methodology: J. H., M.-J. B., and D. W. L. Resources: Y. K., H.-W. L., H. K., T. J. S., S. J. K., and D. W. L. Funding acquisition: M.-J. B. and D. W. L. Supervision: Y. K., H.-W. L., H. K., T. J. S., S. J. K., and D. W. L. Validation: J. H. and D. W. L. Visualization: J. H. Writing – original draft: J. H., M.-J. B., and D. W. L. Writing – review and editing: J. H., M. H. M., J. H. H., S. C. S.-J. J., S. L., J. P., Y.-R. K., H. J., N. J. K., Y. K., H.-W. L., H. K., T. J. S., S. J. K., M.-J. B., and D. W. L.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was funded by the Nano & Material Technology Development Program (RS-2024-00408845) and the Basic Science Research Program (RS-2023-NR077057 and RS-2023-00242584) through the National Research Foundation (NRF) of Korea and Ministry of Science and ICT. This study contains the results obtained by using the equipment of UNIST Central Research Facilities (UCRF). Experiments at the Pohang Accelerator Laboratory (PAL) 6D beamline were supported in part by MSIT, POSTECH, and UNIST central research facilities (UCRF).Notes and references
- S. K. Das, S. Lau and L. A. Archer, J. Mater. Chem. A, 2014, 2, 12623–12629 RSC.
- S. Ha, J.-K. Kim, A. Choi, Y. Kim and K. T. Lee, ChemPhysChem, 2014, 15, 1971–1982 CrossRef CAS PubMed.
- S. H. Sahgong, S. T. Senthilkumar, K. Kim, S. M. Hwang and Y. Kim, Electrochem. Commun., 2015, 61, 53–56 CrossRef CAS.
- S. M. Hwang, J.-S. Park, Y. Kim, W. Go, J. Han, Y. Kim and Y. Kim, Adv. Mater., 2019, 31, 1804936 CrossRef PubMed.
- Y. Chen, D. Ma, K. Ouyang, M. Yang, S. Shen, Y. Wang, H. Mi, L. Sun, C. He and P. Zhang, Nano-Micro Lett., 2022, 14, 154 CrossRef CAS PubMed.
- H. Kim, J.-S. Park, S. H. Sahgong, S. Park, J.-K. Kim and Y. Kim, J. Mater. Chem. A, 2014, 2, 19584–19588 RSC.
- J.-K. Kim, E. Lee, H. Kim, C. Johnson, J. Cho and Y. Kim, ChemElectroChem, 2015, 2, 328–332 CrossRef CAS.
- Y. Zhang, S. T. Senthilkumar, J. Park, J. Park and Y. Kim, Batteries Supercaps, 2018, 1, 6–10 CrossRef.
- J. K. Kim, F. Mueller, H. Kim, D. Bresser, J.-S. Park, D.-H. Lim, K.-T. Kim, S. Passerini and Y. Kim, NPG Asia Mater., 2014, 6, e144 CrossRef CAS.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- S. Yang, P. He and H. Zhou, Energy Storage Mater., 2018, 13, 29–48 CrossRef.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- J. G. Vos, Z. Liu, F. D. Speck, N. Perini, W. Fu, S. Cherevko and M. T. M. Koper, ACS Catal., 2019, 9, 8561–8574 CrossRef CAS.
- W. Lee, J. Park, J. Park, S. J. Kang, Y. Choi and Y. Kim, J. Mater. Chem. A, 2020, 8, 9185–9193 RSC.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320 RSC.
- R. Cao, J.-S. Lee, M. Liu and J. Cho, Adv. Energy Mater., 2012, 2, 816 CrossRef CAS.
- R. Wu, Y. Xue, B. Liu, K. Zhou, J. Wei and S. H. Chan, J. Power Sources, 2016, 330, 132–139 CrossRef CAS.
- C. Lee, T.-U. Wi, W. Go, M. F. Ragman, M. T. McDowell, Y. Kim and H.-W. Lee, J. Mater. Chem. A, 2020, 8, 21804–21811 RSC.
- J. Kim, J. Choi, K. Park, S. Kim, K. W. Nam, K. Char and J. W. Choi, Adv. Energy Mater., 2022, 12, 2103718 CrossRef CAS.
- L. Hencz, H. Chen, H. Y. Ling, Y. Wang, C. Lai, H. Zhao and S. Zhang, Nano-Micro Lett., 2019, 11, 17 CrossRef CAS PubMed.
- G. Zhou, K. Liu, Y. Fan, M. Yuan, B. Liu, W. Liu, F. Shi, Y. Liu, W. Chen, J. Lopez, D. Zhuo, Z. Zhao, Y. Tsao, X. Huang, Q. Zhang and Y. Cui, ACS Cent. Sci., 2018, 4, 260–267 CrossRef CAS PubMed.
- W. We, W. Li, L. Tang, T. Yang, W. Zhang, Y. Zhang and Z. Chen, Adv. Energy Mater., 2024, 14, 2400101 CrossRef.
- N. K. Wagh, S. S. Shinde, C. H. Lee, S.-H. Kim, D.-H. Kim, H.-D. Um, S. U. Lee and J.-H. Lee, Nano-Micro Lett., 2022, 14, 190 CrossRef CAS PubMed.
- Z. Chen, M. Lu, Y. Qian, Y. Yang, J. Liu, Z. Lin, D. Yang, J. Lu and X. Qiu, Adv. Energy Mater., 2023, 13, 2300092 CrossRef CAS.
- K. K. Halder, M. Tomar, V. K. Sachdev and V. Gupta, Mater. Res. Express, 2019, 6, 075324 CrossRef CAS.
- European Chemicals Agency (ECHA). Report on the restriction of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in the European Union. 2023 Search PubMed.
- X. Lim, Nature, 2023, 620, 24–27 Search PubMed.
- A. H. Hofman, I. A. Hees and J. Yang, Adv. Mater., 2018, 30, 1704640 CrossRef PubMed.
- J. H. Ryu, S. Hong and H. Lee, Acta Biomater., 2015, 27, 101–115 Search PubMed.
- S. Yu and C. Cha, Macromol. Res., 2023, 31, 427–441 CrossRef CAS.
- Q. Zhao, D. W. Lee, B. K. Ahn, S. Seo, Y. Kaufman, J. N. Isarelachvili and J. H. Waite, Nat. Mater., 2016, 15, 407–412 CrossRef CAS PubMed.
- S. M. Hodgson, S. A. McNelles, L. Abdullahu, I. A. Marozas, K. S. Anseth and A. Adronov, Biomacromolecules, 2017, 18, 4054–4059 CrossRef CAS PubMed.
- S. Ko, M.-J. Baek, T.-U. Wi, J. Kim, C. Park, D. Lim, S. J. Yeom, K. Bayramova, H. Y. Lim, S. K. Kwak, S. W. Lee, S. Jin, D. W. Lee and H.-W. Lee, ACS Mater. Lett., 2022, 4, 831–839 CrossRef CAS.
- T. Zheng, T. Zhang, M. S. de la Fuente and G. Liu, Eur. Polym. J., 2019, 114, 265–270 CrossRef CAS.
- Y. Chen, C. Li, X. Xu, M. Liu, Y. He, I. Murtaza, D. Zhang, C. Yao, Y. Wang and H. Meng, ACS Appl. Mater. Interfaces, 2017, 9, 7305–7314 CrossRef CAS PubMed.
- X. Zhu, Y. Li, Z. Wu, C. Lin, D. Ma, Z. Zhao and B. Z. Tang, J. Mater. Chem. C, 2021, 9, 5198–5205 RSC.
- Y.-H. Chen, V. S. Nguyen, H.-H. Chou, Y. S. Tingare, T.-C. Wei and C.-Y. Yeh, ACS Appl. Energy Mater., 2020, 3, 5479–5486 CrossRef CAS.
- H. Cha, J. Li, Y. Li, S.-O. Kim, Y.-H. Kim and S.-K. Kwon, Macromol. Res., 2020, 28, 820–825 CrossRef CAS.
- E. Briqueleur, M. Dolle and W. G. Skene, J. Mater. Chem. C, 2024, 12, 6805–6815 RSC.
- P. Ranque, C. George, R. K. Dubey, R. van der Jagt, D. Flahaut, R. Dedryvère, M. Fehse, P. Kassanos, W. F. Jager, E. J. R. Sudhölter and E. M. Kelder, ACS Appl. Energy Mater., 2020, 3, 2271–2277 CrossRef CAS PubMed.
- M.-J. Baek, J. Choi, T.-U. Wi, H. Y. Lim, M. H. Myung, C. Lim, J. Sung, J.-S. Park, J. H. Park, Y. H. Shim, J. Park, S. J. Kang, Y. Kim, S. Y. Kim, S. K. Kwak, H.-W. Lee and D. W. Lee, J. Mater. Chem. A, 2022, 10, 4601–4610 RSC.
- T. Wright, Y. Petel, C. O. Zellman, E. R. Sauve, Z. M. Hudson, C. A. Michal and M. O. Wolf, Chem. Sci., 2020, 11, 3081–3088 RSC.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C.-H. Tan, S. D. Dimitrov, Z. Shang, N. Gaspaini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS PubMed.
- Z. Li, X. Xu, W. Zhang, X. Meng, W. Ma, A. Yartsev, O. Inganas, M. R. Andersson, R. A. J. Hanssen and E. Wang, J. Am. Chem. Soc., 2016, 138, 110935–10944 Search PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- S. T. Senthilkumar, W. Go, J. Han, L. P. Thuy, K. Kishor, Y. Kim and Y. Kim, J. Mater. Chem. A, 2019, 7, 22803–22825 RSC.
- S. Kim, D. Kim, Y. Kim and J. Park, J. Energy Storage, 2025, 106, 114805 CrossRef.
- X. Bi, R. Wang, Y. Yuan, D. Zhang, T. Zhang, L. Ma, T. Wu, R. Shahbazian-Yassar, K. Amine and J. Lu, Nano Lett., 2020, 20, 4681–4686 CrossRef CAS PubMed.
- Y. Kim, J. Jung, H. Yu, G.-T. Kim, D. Jeong, D. Bresser, S. J. Kang, Y. Kim and S. Passerini, Adv. Func. Mater., 2020, 30, 2001249 CrossRef CAS.
- S. M. Hwang, W. Go, H. Yu and Y. Kim, J. Mater. Chem. A, 2017, 5, 11592–11600 RSC.
- P. He, Y. Wang and H. Zhou, J. Power Sources, 2011, 196, 5611 CrossRef CAS.
- P. Mandal, K. Stokes, G. Hernandez, D. Brandell and J. Mindemark, ACS Appl. Energy Mater., 2021, 4, 3008–3016 CrossRef CAS.
- S.-J. Park, H. Zhao, G. Ai, C. Wang, X. Song, N. Yuca, V. S. Battaglia, W. Yang and G. Liu, J. Am. Chem. Soc., 2015, 137, 2565–2571 CrossRef CAS PubMed.
- P. Ranque, C. Georage, R. K. Dubey, R. Jagt, D. Flahaut, R. Dedryvère, M. Fehse, P. Kassanos, W. F. Jager, E. J. R. Sudhölter and E. M. Kelder, ACS Appl. Energy Mater., 2020, 3, 2271–2277 CrossRef CAS PubMed.
- H. Zhao, Y. Wei, C. Wang, R. Qiao, W. Yang, P. B. Messersmith and G. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 5440–5446 CrossRef CAS PubMed.
- J. Kim, M.-S. Kim, Y. Lee, S.-Y. Kim, Y.-E. Sung and S. H. Ko, ACS Appl. Mater. Interfaces, 2022, 14, 17340–17347 CrossRef CAS PubMed.
- D. Zhang, S. Yokomori, R. Kameyama, C. Zhao, A. Ueda, L. Zhang, R. Kumai, Y. Murakami, H. Meng and H. Mori, ACS Appl. Mater. Interfaces, 2021, 13, 989–998 CrossRef CAS PubMed.
- V. A. Nguyen and C. Kuss, J. Electrochem. Soc., 2020, 167, 065501 CrossRef CAS.
- Z. Peng, L. Ye and H. Ade, Mater. Horiz., 2022, 9, 577–606 RSC.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- N. Sinha, G. Ray, S. Godara, H. Yadav and S. Bhandari, Physica B, 2015, 470, 15–20 CrossRef.
- J. J. B. Tol, G. Vantomme, A. R. A. Palmans and E. W. Meijer, Macromolecules, 2022, 55, 6820–6829 CrossRef PubMed.
- J. Ryu, H. Lee, H. Kim and D. Sohn, Macromolecules, 2018, 51, 7745–7755 CrossRef CAS.
- Y. Liu, X. An, K. Yang, J. Ma, J. Mi, D. Zhang, X. Cheng, Y. Li, Y. Ma, M. Liu, F. Kang and Y.-B. He, Energy Environ. Sci., 2024, 17, 344–353 RSC.
- A. M. Fraind, G. Sini, C. Risko, L. R. Ryzhkov, J.-L. Brédas and J. D. Tovar, J. Phys. Chem. B, 2013, 117, 6304–6317 CrossRef CAS PubMed.
- S. J. Kang, T. Mori, S. Narizuka, W. Wilcke and H.-C. Kim, Nat. Commun., 2014, 5, 3937 CrossRef CAS PubMed.
- P. Sharma, J. Han, J. Park, D. Y. Kim, J. Lee, D. Oh, N. Kim, D.-H. Seo, Y. Kim, S. J. Kang, S. M. Hwang and J.-W. Jang, JACS Au, 2021, 1, 2339–2348 CrossRef CAS PubMed.
- H. Park, M. Kang, D. Lee, J. Park, S. J. Kang and B. Kang, Nat. Commun., 2024, 15, 2952 CrossRef CAS PubMed.
- H. Lee, T. Han, K. Y. Cho, M.-H. Ryou and Y. M. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 21366–21372 CrossRef CAS PubMed.
- J. Saiz-Poseu, J. Mancebo-Aracil, F. Nador, F. Busqué and D. Ruiz-Molina, Angew. Chem., Int. Ed., 2019, 58, 696–714 CrossRef CAS PubMed.
- B. K. Ahn, D. W. Lee, J. N. Israelachvili and J. H. Waite, Nat. Mater., 2014, 13, 867–872 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary notes, Fig. S1–S20, Scheme S1, Tables S1–S3 and Supplementary references. See DOI: https://doi.org/10.1039/d5ee01350j |
| This journal is © The Royal Society of Chemistry 2025 |