
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amorphous anion skeletons induce rapid and cation-selective ion flux towards stable aqueous zinc–iodine batteries†
Zhenjing
Jiang
ab,
Zijuan
Du
c,
Kailin
Luo
a,
Yanfei
Zhang
d,
Hang
Yang
 b,
Wei
Zhang
b,
Ruwei
Chen
b,
Jie
Chen
b,
Zhe
Cui
b,
Fuhan
Cui
a,
Rui
Pan
a,
Guoju
Zhang
a,
Shuangying
Lei
a,
Litao
Sun
a,
Kuibo
Yin
*a and
Guanjie
He
*b
b,
Wei
Zhang
b,
Ruwei
Chen
b,
Jie
Chen
b,
Zhe
Cui
b,
Fuhan
Cui
a,
Rui
Pan
a,
Guoju
Zhang
a,
Shuangying
Lei
a,
Litao
Sun
a,
Kuibo
Yin
*a and
Guanjie
He
*b
aSEU-FEI Nano-Pico Center, Key Laboratory of MEMS of Ministry of Education, Southeast University, Nanjing, 210096, P. R. China. E-mail: yinkuibo@seu.edu.cn
bChristopher Ingold Laboratory, Department of Chemistry, University College London, London, WC1H 0AJ, UK. E-mail: g.he@ucl.ac.uk
cState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan, 430070, P. R. China
dSchool of Materials Science and Engineering, Qilu University of Technology, Jinan, 250353, China
First published on 17th June 2025
Abstract
The aqueous zinc–iodine battery is considered a promising technology for large-scale energy storage due to its high safety, large energy density, and easy accessibility. However, its development suffers from two challenges: parasitic side reactions on Zn anodes and polyiodide shuttling effects. To overcome them, we designed an artificial protective layer on the Zn anode based on amorphous zeolite-like Na2Zn2(TeO3)3, whose crystalline counterpart possesses periodic ion channels and an anion skeleton. It not only preserves the original coordination environments and pore structures of the Na2Zn2(TeO3)3 crystal, but also exhibits broadened ion channels and shortened ion diffusion pathways. Combined with the superior structural stability of the amorphous Na2Zn2(TeO3)3, the Zn anode can cycle stably for 2790 h at 1 mA cm−2 with a low overpotential. Meanwhile, the Zn2(TeO3)32− anion skeleton can also repel I−-species and SO42− anions from the anode surface, thus enabling outstanding Zn plating/stripping reversibility and excellent cycling ability of the full cells coupled with different cathodes. Significantly, the capacity retention of the high mass loading zinc–iodine pouch cell was 92.7% after 600 cycles. This work provides a novel strategy to achieve high-performance zinc–iodine batteries, which has great promise for practical applications.
Broader contextAqueous zinc–iodine batteries are highly safe, have large volumetric energy densities, and are cost-effective, making them well-suited for grid-scale energy storage devices. However, their practical implementation is significantly hampered by rampant Zn dendrite growth, polyiodide shuttling, and interfacial side reactions. To solve this dilemma, we provide a facile strategy, constructing a cation-selective artificial layer on Zn anodes based on the amorphous Na2Zn2(TeO3)3. It possesses periodic ion channels and an anion skeleton, while exhibiting broadened ion channels and shortened ion diffusion pathways compared to the crystalline counterpart. Benefitting from the rapid and cation-selective ion flux enabled by the amorphous Na2Zn2(TeO3)3 layer, a high stability of the Zn platting/striping process is achieved, especially in terms of the superior cycle performance of the high loading zinc–iodine pouch battery. This work emphasizes the importance of structural innovation of the interface material in promoting aqueous zinc-ion batteries. |
Introduction
In the backdrop of the global energy crisis, the green transformation of energy through developing sustainable energy sources, e.g., wind and solar energy, has become urgent.1–3 The wide application of reliable grid-scale energy storage instruments is indispensable to enhance their utilization.4 Lithium-ion batteries are the mainstream electrochemical energy storage technology because of their substantial energy density and low self-discharge rate.5 However, the limited lithium resources and high flammability of electrolytes severely hinder their commercial application in stationary energy storage systems. Therefore, it is crucial to develop other energy storage technologies that feature abundant resources and high safety for substitution, e.g., aqueous aluminum-ion, aqueous sodium-ion, and aqueous magnesium-ion batteries.6–9 Aqueous zinc-ion batteries are considered a promising energy storage technology due to their high volumetric capacity of 5188 mA h cm−3, low redox potential (−0.72 V vs. standard hydrogen electrode), and abundance of the anode materials.10,11 Furthermore, the non-flammable and environmentally friendly aqueous electrolytes also enable easy fabrication, making them particularly suitable for grid-scale energy storage.12 For cathode materials, iodine (I2) has garnered widespread attention due to its high theoretical capacity and abundance of raw materials, particularly the high discharge potential plateau of 1.38 V vs. Zn2+/Zn, enabling batteries to achieve a high energy density of 220 W h kgiodine−1 (based on the reversible conversion between I− and I0).13,14 However, the practical application of zinc–iodine batteries is hindered by uncontrollable parasitic reactions on the Zn metal anode (e.g., hydrogen evolution reaction, corrosion, and dendrite growth) and the shuttle effect of the iodine cathode.15–17 Consequently, the battery would generally cycle with low coulombic efficiency (CE) and suffer from rapid short-circuiting.Numerous efforts have been made to enhance the practical feasibility of aqueous zinc–iodine batteries, including polyiodide shuttling elimination, iodine species confinement, and Zn metal anode protection.18–20 Among them, protecting the Zn metal anode is rather crucial as the I− species in the electrolyte also accelerate the performance decay of the anode. However, the simultaneous realization of fast Zn2+ ion transport and long-term cycling stability of the battery remains a significant challenge.21,22 The most common approach is using ion-selective membranes that allow positively charged Zn2+ ions to pass through while preventing the diffusion of high-concentration I3− from the I2 cathode to the Zn anode.16,23,24 For example, Nafion membranes are widely applied in zinc–iodine batteries because of their reliable cation-selective performance, attributed to the electrostatic repulsion effect generated by rich immobilized sulfonic acid groups.25 Nevertheless, Nafion membranes typically have narrow and torturous ion transport channels, thus generally delivering sluggish electrochemical reaction kinetics due to the anion-repelled ion migration behavior.26 On the other hand, the pore engineering of porous polyolefin/carbon and metal–organic frameworks (MOFs, e.g., UiO-66 and Zn-BTC with diameters of 0.6 and 0.5 nm, respectively) has also been reported to effectively block the large hydrated I3− ions.27,28 In addition, they can also exclude water molecules of hydrated Zn2+ ions and uniformize Zn2+ ion flux at the interface to stabilize the Zn anode. Regretfully, the structural stability of the ion diffusion channels of MOFs and other porous materials in aqueous electrolytes needs further improvement. In particular, the charge/discharge processes would generate an alkaline environment at the anode interface due to the violent HER, thus accelerating the failure and blockage of those functional interlayers.29 That is, searching for an interfacial material with wide ion transport channels and high structural stability, capable of long-term blockage of I3− and rapid Zn2+ ion flux, is essential for the application of zinc–iodine batteries in grid-scale energy storage applications.
Zemannite crystals are typically consistent with A2−x[M2(TeO3)3]·nH2O (A = Na, K, Rb, Cs; M = Mn, Co, Ni, Cu, Zn). They are made up of an anionic [M2(TeO3)3]2− framework and A cations and are permeated by large and hexagonal channels (about 0.82 nm) extending along the [001] direction.30,31 Because of the larger structural frameworks and channel size, we chose Na2Zn2(TeO3)3 crystals to construct a protective layer on the Zn metal anode, which has the potential to facilitate the Zn2+ ion transport process.31 After heat treatment, the crystalline Na2Zn2(TeO3)3 is confirmed to be transformed into an amorphous one with preserved coordination environments and pore structures. The amorphous Na2Zn2(TeO3)3 shows an enhanced structural stability and shortened ion diffusion pathway (along the lone pairs of electrons of the TeO3 units), thereby enabling the Zn‖Zn symmetric cell a lifespan of 2790 h with a low and stable overpotential. In addition, the presence of the Zn2(TeO3)32− anion skeleton can also repulse SO42− and I3− anions at the anode interface, thus effectively suppressing the corrosion and surface passivation of the Zn anode (Scheme 1). Consequently, the amorphous Na2Zn2(TeO3)3 interlayer enables a high CE of 99.6% of the Zn‖Cu half-cell, while the Zn‖MnO2 full-cells with limited Zn (N/P ratio of 2.6, close to the practical application level) could cycle stably for more than 620 cycles with little capacity decay. In particular, when assembled into a high loading Zn‖I2 pouch cell, they could still cycle stably 600 times with a capacity retention of 92.7%, which is among the most stable cycling performances to date. That is, the amorphous Na2Zn2(TeO3)3 interlayer can not only push the aqueous zinc-ion batteries towards practical application but also promote the development of other electrochemical devices based on the selective ion diffusion mechanism.
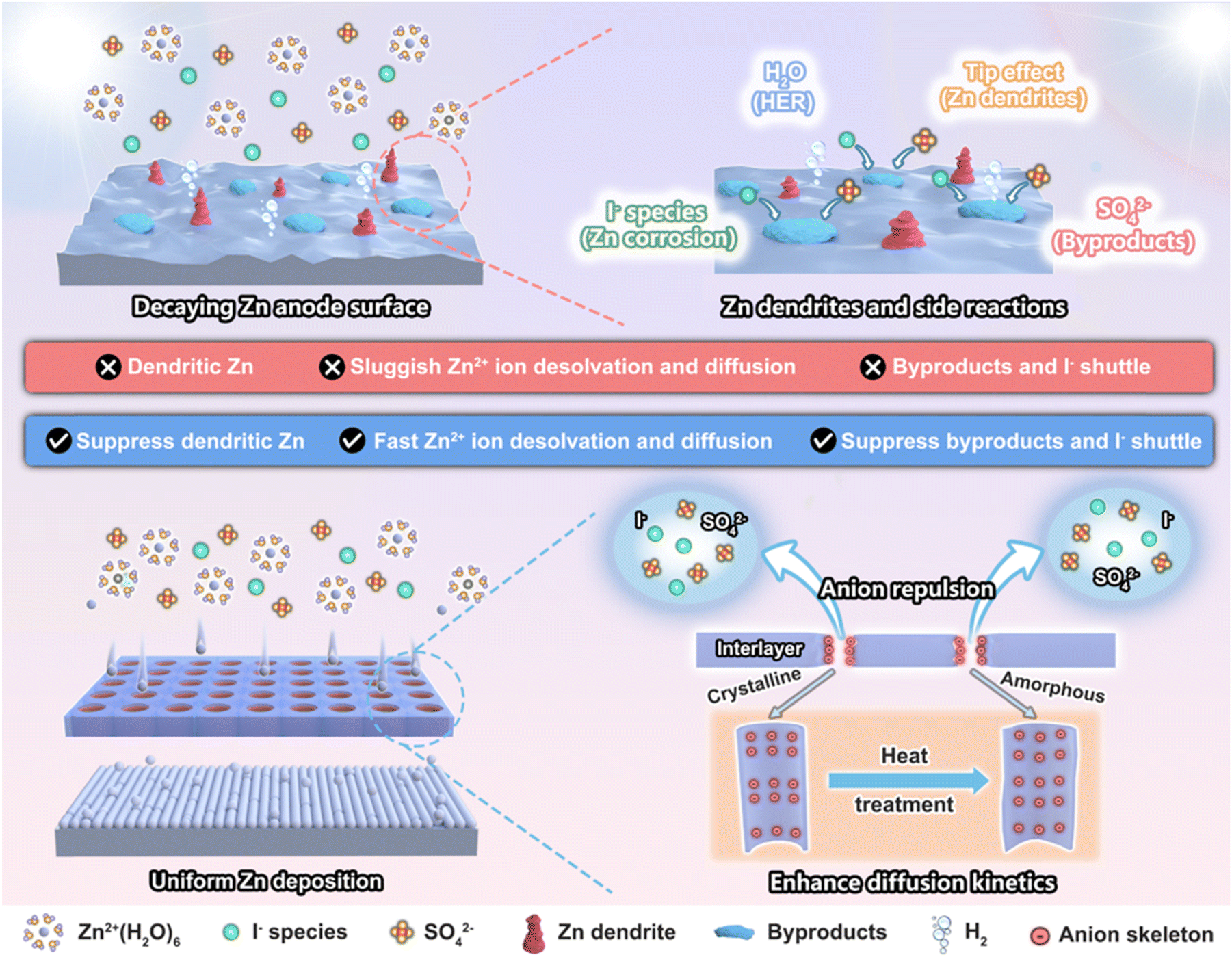 | ||
| Scheme 1 The role of the artificial interlayer based on the anion skeleton in inhibiting Zn dendrite and other side reactions on the Zn anode and its amorphous transformation in enhancing ion diffusion kinetics. | ||
Results and discussion
Characterization of crystalline and amorphous samples
The crystalline Na2Zn2(TeO3)3 (named CNZT) powder was prepared through a facile solvothermal method and then heated at 453 K under an Ar atmosphere to acquire its amorphous counterpart, which is named ANZT. As exhibited in Fig. 1a, the X-ray powder diffractometer (XRD) patterns show that all diffraction peaks of the as-prepared CNZT sample resemble the Na2Zn2(TeO3)3 (ICSD no. 97-003-3638). Those diffraction peaks disappeared after heat treatment, indicating the crystalline NZT powder was transformed into an amorphous one. Both the CNZT and ANZT samples are highly stable, as their crystalline and amorphous states were preserved after ball milling with PVDF followed with a drying process at 375 K for 24 h. To clarify the crystalline-amorphous transform progress of CNZT powder, the differential scanning calorimetry (DSC) measurements were carried out. Clearly, the weight loss of the sample begins at 383 K, which is accompanied by the endothermic peak in the DSC up-scan curve (Fig. 1b). That is, the amorphous transformation process is mainly induced by the loss of water molecules in the channels of CNZT samples.32 | ||
| Fig. 1 (a) XRD patterns of CNZT powder, ANZT powder, CNZT interlayer, and ANZT interlayer. (b) DSC up-scan and weight loss curves of the CNZT powder. (c) Raman spectra of CNZT and ANZT powders. (d) High resolution XPS spectra of Te 3d of ANZT and CNZT powders. SEM images of (e) CNZT and (f) ANZT powders and the corresponding HRTEM images with SAED results. SEM images of the prepared (g) CNZT and (h) ANZT interlayers. (i) The contact angles of different Zn anodes to the 2 M ZnSO4 electrolyte. | ||
To detect the structure evolution of the crystalline sample during the heat-treatment process, the Raman spectra of both the CNZT and ANZT powders were acquired. As shown in Fig. 1c, the bands at 441 cm−1, 665 cm−1, and 782 cm−1 belong to the bending motion of O in the Zn–O–Te/Te–O–Te asymmetric stretching, and the Te–O− stretching, respectively.33 In addition, their Fourier transform infrared spectroscopy (FTIR) spectra (Fig. S1a, ESI†) also show identical peak locations of about 590 cm−1 and 643 cm−1, corresponding to the Zn–O and Te–O bonds, respectively.34 Meanwhile, in the high-resolution XPS O 1s and Te 3d spectra, signal peaks of CNZT and ANZT exhibit similar locations and shapes, indicating the same coordination environment around their O and Te atoms (Fig. 1d and Fig. S1b, c, ESI†). It is worth mentioning that these XPS signal peaks are slightly blue-shifted in the ANZT compared to the CNZT, implying the longer Te–O and Zn–O bonds in the ANZT sample, which will be discussed later. The BET analysis also shows that the CNZT and ANZT powders possess similar BET surface area, pore volume, and pore distribution (Fig. S2, ESI†). These above results all confirm the chemical composition and coordination environment of CNZT is preserved during the heat treatment, which can be confirmed by the inductively coupled plasma (ICP) results in Table S1 (ESI†).
In addition, the scanning electron microscope (SEM) images and energy dispersive spectrometer (EDS) results (Fig. S3, ESI†) show both the CNZT and ANZT samples consist of small particles with the uniformly distributed Na, O, Te, and Zn elements. Furthermore, those small particles in ANZT are slightly agglomerated during the amorphization process. The selected-area electron diffraction (SAED) results confirm the highly ordered single-crystalline hexagonal arrangement as characterized by the Na2Zn2(TeO3)3 (210) plane with a crystalline interplanar spacing of 0.308 nm in the high-resolution transmission electron microscopy (HRTEM) image, as shown in Fig. 1e. However, for the ANZT sample, no periodic lattice fringes are observed (Fig. 1f), which is in good agreement with the XRD results.
Both the prepared CNZT and ANZT interlayers on the Zn anode deliver smooth and compact morphology, thereby ensuring the good protection effect for the Zn metal anode (Fig. 1g and h). Meanwhile, their side-view SEM images were acquired to determine the thickness of the prepared CNZT and ANZT interlayers (Fig. S4, ESI†), which further prove that both the prepared interlayers are compact. The electrolyte wettability of those interlayers between the Zn metal anode and the electrolyte was also essential to the interfacial ion transport process. As displayed in Fig. 1i, both the CNZT (67.3°) and ANZT (65.8°) interlayers exhibit relatively low contact angles to 2 M ZnSO4 electrolyte, which of the bare Zn anode is 99.8°, thus facilitating Zn2+ ion transport kinetics at the anode interface.35 The hardness of the CNZT and ANZT interlayers are also compared by nanoindentation measurement. As shown in Fig. S5 (ESI†), the interlayer with CNZT shows low hardnesses of 148.1, 168.3, and 99.3 GPa at the three selected positions. In contrast, the amorphous one delivers high hardnesses of 185.2, 197.8, and 200.3 GPa, thereby enabling the stability of the prepared interface during cycling.36 In this work, the thickness of the used Zn anode is 50 μm unless otherwise noted.
The enhanced ion transport kinetics in the ANZT interlayer
The detailed local structures of CNZT and ANZT samples were studied through classical molecular dynamic (MD) simulations. Based on the corresponding structural models, the radial distribution function (RDF) curves were acquired. Clearly, the Te–O coordination distance increased from 1.85 Å (CNZT) to 1.95 Å (ANZT), as displayed in Fig. 2a. According to the structural model of Na2Zn2(TeO3)3 crystal, the ion transport channels are formed by the alternating connection of TeO3 and ZnO4 units. Therefore, the increased Te–O distance in ANZT indicates the enlargement of the radius of those ion transport channels. In addition, the contained Na+ ions in the hexagonal channels extending along the [001] direction that consist of the anionic [Zn2(TeO3)3]2− framework can be exchanged with the external metal ions.30 Therefore, we propose that the Zn2+ ions in the electrolyte will also replace the Na+ ions in those channels during cycling (Fig. S6c, ESI†). To confirm this, the high-resolution Na 1s XPS spectra of CNZT and ANZT interlayers before and after cycles were acquired and compared. As shown in Fig. S6d (ESI†), the peaks of the Na 1s XPS spectra of the ANZT and CNZT interlayers located at about 1071 eV (belong to the Na+ ions), both disappear after cycling, implying the loss of Na+ ions in the channels. | ||
| Fig. 2 (a) The radial distribution function (RDF) curves of the Te–O distance of the CNZT and ANZT samples that are based on classical molecular dynamics simulation, insert: corresponding structural model of the Na2Zn2(TeO3)3 crystal. (b) Calculated binding energies between the O, Te, and Zn sites of CNZT toward the adsorbed Zn atom with the corresponding charge difference pictures. (c) The RDF and coordination number curves of the O–O distance of CNZT and ANZT samples (based on classical molecular dynamics simulation). (d) The mean square displacement (MSD) results of Zn2+ ions in the channels of the CNZT and ANZT samples. (e) A schematic illustration of the reason for the faster Zn2+ ion transport kinetics in the ANZT compared to the CNZT samples. (f) The calculated activation energy for hydrated Zn2+ ions in the electrolyte to deposit on bare Zn, CNZT@Zn, and ANZT@Zn anodes. (g) Temperature-varied 23Na NMR spectra of the ANZT and CNZT samples. (h) The full width at half maximum of the peaks in the 23Na NMR spectra of different samples. | ||
On the other hand, the Zn2+ ion diffusion path in the channels is determined by the DFT calculation (Fig. 2b). Specifically, the O site (−1.35 eV) in the channel shows the strongest interaction with the adsorbed Zn atom, which for the Te site and Zn site to Zn atoms are −0.59 eV and −0.16 eV, respectively. Consequently, the Zn2+ ion is energetically preferred to transport along the O site to the O site in the channel.37 However, according to the structural model of CNZT, the arrangement of O atoms along the channels’ wall are not even. Specifically, the Zn2+ ion diffuses to an adjacent O atom at a distance of 2.78 Å, and then needs to diffuse an additional 4.85 Å to reach the third O atom, thus hindering the Zn2+ ion's transport process. As shown in the RDF curves in Fig. 2c, the O–O coordination distance between the first and second coordination layers of ANZT slightly decreased compared to that of the CNZT sample. Meanwhile, the O–O coordination number increases from 6.6 (CNZT) to 8.1 (ANZT). As a result, based on the mean square displacements (MSD, Fig. 2d) curves, the calculated Zn2+ ion diffusion coefficient in the channel of CNZT (3.2 × 10−5 A2 ps−1) is obviously lower than the Zn2+ ions in ANZT (5.3 × 10−5 A2 ps−1). Based on their structure models, Fig. 2e schematically describes the randomly arrangement of O atoms on the wall of channel in ANZT sample, thus reducing the diffusion distance of Zn2+ ion.
In this way, the calculated activation energy for Zn2+ ions in the electrolyte to deposit on the ANZT@Zn anode is the smallest (25.58 kJ mol−1, Fig. 2f and Fig. S6e–g, ESI†). The activation energies on the CNZT@Zn anode and the bare Zn anode are 28.36 and 33.73 kJ mol−1, respectively. Furthermore, the temperature-varied nuclear magnetic resonance (NMR) measurement was also conducted to study the ion transport kinetics in the channels of the CNZT and ANZT samples. The full width at half maximum (FWHM) of the signal of NMR spectra is closely associated with the ion diffusion coefficient.38 Here, the 23Na NMR spectrum is used to substitute the 67Zn NMR spectrum to evaluate the ion diffusion kinetics because of its higher sensitivity to the NMR measurement. As shown in Fig. 2g and h, the FWHM values of ANZT are less influenced by the test temperatures (increasing from 0 to 80 °C) compared to the CNZT, indicating a larger ion diffusion coefficient.38 These above results confirm that the amorphous Na2Zn2(TeO3)3 could effectively enhance the Zn2+ ion transport kinetics by enlarging the diameter of channels and reducing the distance for ion diffusion. Note that the signal peak of the 23Na NMR spectrum of the CNZT layer almost disappeared after one Zn plating/stripping cycle (Fig. S7a, ESI†), further confirming the Na+ ion in the channel is replaced by the transported Zn2+ ion in electrolyte. Consequently, the Zn metal anode with the ANZT interlayer shows the highest Zn2+ ion transference number of 0.87 (which of bare Zn and CNZT@Zn anodes are 0.58 and 0.75, respectively, Fig. S7b–f, ESI†). Therefore, the ANZT interlayer enables cells with the fastest Zn plating/stripping kinetics (the CV curves of half-cells) and the smallest Zn plating/stripping overpotential (the rate performance of symmetry cells, Fig. S8, ESI†) compared to others electrodes.
The electrochemical performance of cells with different anodes
To further determine the superiority of the ANZT interlayer, the ANZT@Zn, ANZT@Cu, CNZT@Zn, CNZT@Cu, bare Zn, and bare Cu electrodes were assembled into Zn‖Cu half-cells and Zn‖Zn symmetric cells to carry out the electrochemical measurement. For the Zn‖Cu half-cells, the bare Cu electrode delivers a low average CE (ACE) of 94.8% at the current density of 1 mA cm−2 (Fig. 3a). Surprisingly, the CNZT and ANZT interlayers significantly promote the reversibility of the half-cell, as the ACE reached 98.6% and 99.2%, respectively. The corresponding charge/discharge curves of the half-cell with bare Cu electrode shows the gradually increasing voltage polarizations during cycling, and then failed after 100 cycles, as displayed in Fig. 3b. In striking contrast, the voltage polarization of the half-cell with the ANZT interlayer is almost unchanged even after 500 cycles, emphasizing its outstanding cyclic stability (Fig. 3c). For the cell with the CNZT@Cu electrode, its polarization significantly increases after 200 cycles due to the fast failure of the interlayer (Fig. S9a, ESI†). The smallest corrosion current and the lowest onset potential for the HER explain the superior cyclic stability of the Zn‖ANZT@Cu half-cell, including the high CE at 5 mA cm−2 (Fig. S9b–d, ESI†). In addition, the long-term cycling stability of the Zn‖Zn symmetric cells with or without the CNZT and ANZT interlayers was evaluated. As displayed in Fig. 3d, under a current density of 1 mA cm−2 with a capacity of 1 mA h cm−2, the ANZT@Zn anode realizes the longest lifespan of 2790 h, as well as the lowest overpotential among the three Zn anodes. For the CNZT@Zn and bare Zn anodes, they exhibit shorter cyclic lifespans of 962 and 142 h, respectively. In addition, these cells were also tested under high current densities of 9 mA cm−2 (with a capacity of 4.5 mA cm−2 and 10 μm Zn foil, Fig. S9e, ESI†) and 20 mA cm−2 with a capacity of 5 mA h cm−2. As displayed in Fig. 3e, the bare Zn anode merely sustains for 33 h, while the CNZT@Zn and ANZT@Zn anodes can cycle for 450 and 800 h. In short, as shown in Fig. 3f and Table S2 (ESI†), when compared to the recently reported Zn anode modification strategies, the ANZT interlayer shows the most superior performance.39 | ||
| Fig. 3 (a) CE test of the Zn‖Cu half-cells with bare Cu, CNZT@Cu, and ANZT@Cu electrodes (1 mA cm−2/1 mA h cm−2). Charge–discharge curves of the Zn‖Cu half-cells with (b) bare Cu and (c) ANZT@Cu electrodes at the 5th, 20th, 50th, 100th, 200th, and 500th cycles, respectively. Cycle performance of the Zn‖Zn symmetric cell cycled at a current density of (d) 1 mA cm−2 with a capacity of 1 mA h cm−2 and (e) 20 mA cm−2 with a capacity of 5 mA h cm−2. (f) Comparison of the cyclic lifespan of the ANZT@Zn anode with recently reported anode-modified strategies. Cycling performance of the Zn‖MnO2 cells at the current densities of (g) 1 A g−1 and (h) 0.5 A g−1 (10 μm Zn foil, with a low N/P ratio of 2.6). (i) Cycling performance of the Zn‖NH4V4O10 cells with bare Zn and ANZT@Zn anodes at a current density of 1 A g−1. | ||
To determine the potential of the designed ANZT artificial interlayer in practical applications, the Zn‖MnO2 and Zn‖NH4V4O10 cells with the ANZT@Zn anode were tested. The CV curves of the ANZT@Zn‖MnO2 cell exhibit a smaller polarization than the bare Zn‖MnO2 cell, indicating its faster electrochemical reaction kinetics (Fig. S10a, ESI†). Similarly, the charge/discharge curves of the full-cell with the ANZT@Zn anode show a much lower voltage polarization of 305 mV compared to the cell with bare Zn anode (373 mV) and a better rate performance (Fig. S10b and c, ESI†). On the other hand, the long-term cycling stability of those full cells was also tested. The ANZT@Zn‖MnO2 cell prevents a discharge capacity of 108.9 mA h g−1 after cycling for 1500 cycles under a current density of 1 A g−1 with a capacity retention of 79.2% (Fig. 3g). In contrast, the bare Zn‖MnO2 cell undergoes fast decay of its discharge capacity after 482 cycles and then fails after 610 cycles due to the formation of Zn dendrites (Fig. S10d, ESI†). Furthermore, the Zn‖MnO2 cells were also tested under the conditions closed to practical application (the N/P ratio reduces to 2.6, Fig. 3h). The ANZT interlayer still endows an excellent cycling performance of the Zn‖MnO2 cell. In detail, the cell with the ANZT@Zn anode maintains stable cycling for 600 times, which of the bare Zn anode is 142 times. Meanwhile, the Zn‖NH4V4O10 cells with different Zn anodes were tested at the current density of 1 A g−1 to confirm the universality of the designed ANZT interlayer. As shown in Fig. 3i, the ANZT@Zn‖NH4V4O10 cell maintains stable cycling for about 1000 cycles and delivers a discharge capacity retention of 80.1% (336.3 mA h g−1). For the bare Zn‖NH4V4O10 cell, although it delivers a similar initial discharge capacity as the ANZT@Zn anode, its discharge capacity suddenly reduces after 56 cycles and fails after 513 cycles.
Study of Zn deposition morphology and interface stability
The analysis of Zn deposition morphology was carried out to clarify the origin of the excellent stability of the Zn metal anode that enabled by the ANZT interlayer. The focused ion beam scanning electron microscopy (FIB-SEM) technology is powerful in studying the micromorphology of deposited Zn metal on Cu electrodes. For the cross-section SEM image of FIB-milled bare Cu electrode (after 2.5 mA h cm−2 Zn metal deposited, Fig. 4a), the nucleation and growth of Zn are uneven, thus accelerating the growth of Zn dendrite. In contrast, as shown in Fig. 4b, the Zn deposition morphology on the ANZT@Cu electrode is uniform and tightly attached to the substrate. Furthermore, the Zn deposition morphologies on different Zn anodes during the Zn plating/stripping cycling were investigated. After 50 cycles at the current density of 5 mA h cm−2 with a capacity of 2.5 mA h cm−2, the surface of cycled bare Zn anode is uneven and accompanied by the formation of Zn dendrites (Fig. 4c). Meanwhile, the corresponding enlarged SEM image shows that the orientation of deposited Zn plates is random, causing the incoherent distribution of electric field and ion flux at the anode interface. In contrast, as displayed in Fig. 4d, the surface of the cycled ANZT@Zn anode is still smooth, and the enlarged SEM images show that most of the deposited Zn metal is oriented with the (101) plane, thus delivering a dense Zn deposition morphology. The corresponding side-view SEM images also confirm that the Zn deposition on the bare Zn anode is loose and accompanied by many byproducts, whereas the Zn deposition on the ANZT@Zn anode is remarkably even and compact (Fig. S11a and b, ESI†). In addition, the in situ optical microscopy measurement was used to further detect the Zn deposition morphology evolution in a large view. Obviously, as displayed in Fig. 4e, some Zn dendrites form and grow on the bare Zn anode with the continuing Zn deposition process. For the ANZT@Zn anode, the uniform Zn deposition morphology can be maintained for over 15 minutes. This morphology is highly dependent on the stable Zn2+ ion channels and their suppression of the two-dimensional diffusion of Zn2+ ions on the anode surface (Fig. S11c, ESI†).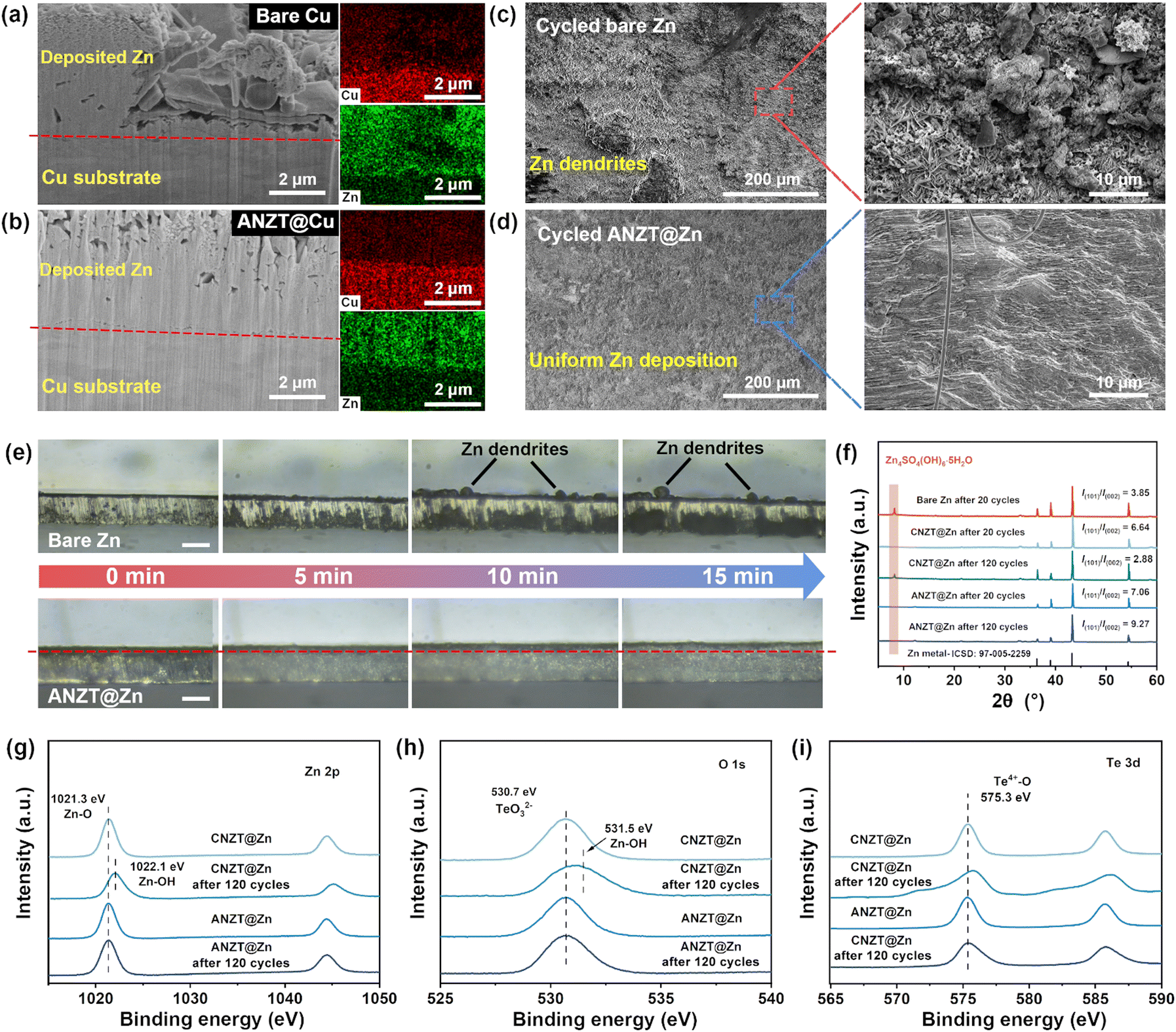 | ||
| Fig. 4 Side-view SEM images of FIB-milled (a) bare Cu and (b) ANZT@Cu electrodes with 2.5 mA h cm−2 Zn metal deposition (at 5 mA cm−2) and the corresponding EDS mapping results. SEM images of (c) bare Zn and (d) ANZT@Zn electrodes after 50 cycles at 5 mA h cm−2/2.5 mA h cm−2, and the enlarged SEM images are inserted on the right. (e) In situ optical microscopy measurement records the Zn deposition morphology on bare Zn and ANZT@Zn anodes (100 μm Zn foil) at a current density of 30 mA cm−2, scale bar: 100 μm. (f) XRD patterns of bare Zn, CNZT@Zn, and ANZT@Zn anodes after different cycle numbers (in symmetric cells at 1 mA cm−2/1 mA h cm−2), with calculated I(002)/I(101) values. High resolution XPS spectra of (g) Zn 2p, (h) O 1s, and (i) Te 3d of CNZT and ANZT interlayers after 120 cycles (in symmetric cells at 1 mA cm−2/1 mA h cm−2). | ||
The XRD patterns of all the bare Zn, CNZT@Zn, and ANZT@Zn anodes after 20 cycles were acquired and displayed in Fig. 4f. Specifically, a diffraction peak at around 8.7° is observed from the bare Zn anode after 20 cycles, indicating the formation of Zn4(SO4)4(OH)6·5H2O.20 For comparison, this diffraction peak cannot be found on the cycled CNZT@Zn and ANZT@Zn anodes, implying that the side reactions on them are effectively suppressed. Besides, the values of I(101)/I(002) of bare Zn, CNZT@Zn, and ANZT@Zn are 3.85, 6.64, and 7.08, respectively, indicating that the NZT interlayers can realize the Zn deposition along the Zn (101) plane. Therefore, a compact Zn deposition morphology is achieved on the ANZT@Zn surface (as confirmed by the SEM image), which also effectively reduces the contact area between the Zn anode and electrolyte to inhibit the side reactions.40 As determined by the DFT calculation (Fig. S11d, ESI†), the Zn (101) plane shows the highest adsorption energy of 6.54 eV to the TeO32− group, which of the Zn (002) and Zn (100) planes are 1.63 and 2.77 eV. Therefore, the TeO32− group that fixed on the ion channels of the ANZT interlayer can guide the Zn metal deposition along the Zn (101) plane.41
When cycled for 120 times, the diffraction peaks of Zn4(SO4)4(OH)6·5H2O are still not observed from the XRD pattern of the ANZT@Zn anode, and the I(101)/I(002) value increases to 9.27. However, for the CNZT@Zn anode, a weak peak of the byproduct is observed in the XRD pattern, and the I(101)/I(002) value reduces to 2.88. That is, the CNZT interlayer gradually lost its ability to protect the Zn anode during cycling. To clarify this, XPS measurement of the CNZT and ANZT interlayers was conducted to discuss their structure evolution during cycling. As shown in Fig. 4g, the peak located at 1021.3 eV in the Zn 2p XPS spectra belongs to the Zn–O bond, which in the ANZT interlayer is almost unchanged before and after 120 cycles. For the CNZT interlayer, this peak shifts to a higher binding energy of 1022.2 eV after 120 cycles, which belongs to the Zn–OH bond.42 As a supplement, the high-resolution O 1s XPS spectra are also acquired, and the peak at 530.5 eV belongs to the anionic [Zn2(TeO3)3]2− skeleton (Fig. 4h). The location of this peak of the ANZT interlayer is almost unchanged before and after 120 cycles, verifying the local structure of the anionic [Zn2(TeO3)3]2− framework is preserved. This peak slightly broadens due to the residue of electrolyte. However, a significantly broad and blue shift happens to the O 1s spectrum of the CNZT interlayer after 120 cycles, indicating severe structural destruction and side reactions during cycling. In addition, the Te 3d XPS spectrum of the ANZT interlayer exhibits the same peak location (575.3 eV, belonging to the Te4+–O bond, Fig. 4i) before and after 120 cycles, confirming the Te–O bond of the anionic [Zn2(TeO3)3]2− framework is preserved during cycling. In contrast, the Te 3d XPS spectrum of CNZT after 120 cycles shows a broad peak which shifts towards a higher binding energy, indicating the coordination environment of the Te atom is significantly changed. These characterizations all determine the superior structural stability of the ANZT interlayer, thereby exhibiting the stable charge transfer resistance of the symmetric cell during cycling (Fig. S12, ESI†).
Anion rejection behaviour of the designed interlayer
The negatively charged [Zn2(TeO3)3]2− skeleton of the ANZT interlayer can also reject the SO42− and I3− anions from the Zn anode surface to avoid the byproducts formation and shuttle effect. In this way, the interlayer can prevent the fast capacity decay when coupled with the iodine cathode. To determine its ability in rejecting SO42− ions, the in situ Raman measurement was carried out on the bare Cu and ANZT@Cu mesh electrodes in a custom mold. As shown in Fig. 5a, during the Zn plating process, a strong peak occurs at about 980 cm−1 in the Raman spectra of the bare Cu mesh electrode, which belongs to the vibration of SO42−.16 However, for the ANZT@Cu mesh electrode, only a weak peak can be observed from the Raman spectra during the Zn deposition process, implying the side reactions are effectively suppressed (Fig. 5b). As a supplementary investigation, Ar+-etching XPS measurements were also adapted to study the depth distribution of S element on different Zn anodes after 50 cycles in the symmetry cells. As displayed in Fig. 5c, the signal peak of the S element could be detected even after 150 nm of Ar+-etching from the bare Zn anode surface. In contrast, on the surface of the ANZT@Zn anode, this peak cannot be observed in the S 2p XPS spectrum after Ar+-etching for merely 50 nm. In addition, the corresponding SEM images and EDS results of the two Zn anodes after cycling were acquired. The content of the S element on the bare Zn anode is 5.6%, which on the ANZT@Zn anode reduced to 0.9% (Fig. S13, ESI†). All the above results verify the excellent ability of the ANZT interlayer in expelling the SO42− anions from the Zn anode surface to avoid the formation of byproducts. Consequently, their Ar+-etching high-resolution XPS spectra (O 1s and Zn 2p) show that the considerable amounts of oxygen-containing byproducts were detected on bare Zn after 150 nm of etching, while cannot be detected on the ANZT@Zn anode after 50 nm of etching (Fig. S14, ESI†). | ||
| Fig. 5 In situ Raman spectra of (a) bare Cu mesh and (b) ANZT@Cu mesh during the Zn plating/stripping process. (c) The Ar+-etching high-resolution XPS spectra of S 2p of bare Zn and ANZT@Zn anodes after 50 cycles in the symmetric cells (at 5 mA cm−2/2.5 mA cm−2). (d) Optical images of H-type glass mold with anolyte (left, 0.5 M KIO3 and 0.1 M I2), catholyte (0.5 M KIO3), and the GF separator with/without the ANZT coating layer. (e) The Ar+-etching high-resolution XPS spectra of I 3d of bare Zn and ANZT@Zn anodes after 100 cycles in the Zn‖I2 cells (at 1 A g−1). (f) Electrochemical aging test (from 0.5 to 5 h standing after full charge state) for Zn‖I2 batteries with bare Zn (upper) and ANZT@Zn (lower) anodes. (g) The cycling performance of Zn‖I2 cells with bare Zn and ANZT@Zn anodes at a current density of 2 A g−1. (h) The cycling performance of Zn‖I2 pouch cells with different Zn anodes at a current density of 1 A g−1 with the high cathode mass loading of 9.31 (bare Zn) and 9.75 (ANZT@Zn) mg cm−2. | ||
The iodine-diffusion, Raman, and UV/vis measurements were carried out to confirm the ability of the ANZT interlayer to repel the iodine compound in the electrolyte during cycling. First, we prepared an ANZT layer on a glass matrix, then transferred and attached it to the surface of the glass fiber (GF) separator, named ANZT@GF separator. Those separators were fixed at the middle joint of the H-type glass mold (left: 0.5 M KIO3 + 0.1 M I2; right: 0.5 M KIO3), and left for 6 min, 6 h, 1 day, and 2 days for photographing (Fig. 5d). Clearly, for the H-type glass mold with the GF separator, the iodine-containing yellow solution on the left permeates into the bottom of the right transparent solution after standing for just 6 min. And the solution on the right gradually turns yellow from the bottom up after standing for 2 days. Whereas, for the mold with the ANZT@GF separator, the solution on the right remains transparent even after standing for 2 days because only small amounts of I-contained species permeate through. This result can also be confirmed by the Ar+-etching XPS of the bare Zn and ANZT@Zn anode after 100 cycles in the Zn‖I2 cell. The high-resolution XPS spectra of I 3d of the cycled bare Zn anode delivers an obvious signal peak even after etching for 200 nm (Fig. 5e). In contrast, benefitting from the protection effect of the ANZT layer, the signal peak of I 3d can hardly be detected on the ANZT@Zn anode surface. Furthermore, the ex situ Raman and UV/vis were also used to determine the accumulation of polyiodide compounds on these Zn anode surfaces during the continuous charge/discharge process. As displayed in Fig. S15 (ESI†), the concentration of I3− (the peak located at 103 cm−1 in the Raman spectra) on the bare Zn anode is significantly increased as the cycle number is increased.41 In contrast, the concentration of I3− on the Zn anode under the ANZT interlayer remains at a low concentration. In addition, the concentration of I− (the peak located at 220 nm in the UV/vis spectra) on the bare Zn anode also significantly increases during cycling, which on the ANZT anode is almost unchanged.43 In other words, the ANZT interlayer can effectively eliminate the I3− accumulation on Zn anode (its accumulation would cause the incomplete conversion of I2/I− and the corrosion of Zn anode), thereby stabilizing the Zn‖I2 battery.
The Zn‖I2 full-cell with bare Zn and ANZT@Zn anodes were assembled and tested to evaluate the superiority of the ANZT interlayer. As shown in the CV curve (Fig. S16, ESI†), both the cells with bare Zn and ANZT@Zn anodes exhibit one paired reduction and oxidation peaks. The smaller polarization between redox peaks and the higher current of the cell with the ANZT@Zn anode compared to bare Zn imply faster electrochemical reaction kinetics and higher iodine utilization.44,45 Consequently, it delivers an excellent rate performance under the step-varied current densities from 0.2 to 2 A g−1. Meanwhile, the aging test (Fig. 5f) also confirms the superior performance of the ANZT interlayer in stabilizing the Zn‖I2 cell, as the cell only exhibits a slight decay of the discharge capacity after aging for 5 h. In addition, as presented in Fig. 5g, under the current density of 2 A g−1, the discharge capacity of the Zn‖I2 cell with the bare Zn anode decays at 580 cycles, and the cell failed after 4060 cycles. In contrast, the cell with the ANZT@Zn anode maintains stable cycling for more than 5500 cycles with a capacity retention of 99.3%. Furthermore, these Zn anodes and I2 cathodes were assembled into a pouch cell and tested under conditions close to those of practical applications, as shown in Fig. 5h. When the cathode mass loading arrived at 9.75 mg cm−2, after activation for 10 cycles, the ANZT@Zn‖I2 pouch cell still maintained stable cycling for 600 times with a capacity retention of 92.7% at a current density of 1 A g−1. However, the bare Zn‖I2 pouch cell shows a fast capacity decay and then failed after 98 cycles, which is ascribed to the magnification of both the polyiodide shuttling and side reactions in the pouch cell compared to the coin cell.46,47 It is worth mentioning that the cyclic ability of the assembled pouch cell is superior to recently reported high mass loading pouch cells with different types of cathode materials, including I2, VO2, LiMnO2O4, and NaV3O8 (Table S3, ESI†).
Conclusions
In this work, the construction of an amorphous zeolite-like Na2Zn2(TeO3)3 interlayer enhances the electrochemical kinetics of the Zn metal anode. Meanwhile, it realizes the excellent cycling lifespan and reversibility of the Zn anode and the excellent cycling ability of the zinc–iodine/MnO2/NH4V4O10 batteries. Through elaborate structural characterization, the coordination environment and pore structure of the crystalline Na2Zn2(TeO3)3 sample is confirmed to be preserved in its amorphous counterpart. Besides, based on the MD simulation and ssNMR measurement, both the increased distance of the Te–O bond and the reduced distance between the O atom of the first and second coordination layers help the fast Zn2+ ion transport process. Meanwhile, the constructed interlayer realizes the closely arranged Zn deposition morphology along the Zn (101) plane, thus effectively inhibiting the Zn dendrite and water-induced parasitic reactions. It is worth mentioning that the Zn2(TeO3)32− anion skeleton is verified to repel the SO42− and I− anions from the Zn anode surface, thus suppressing the shuttle effect of the I2 cathode and inhibiting the corrosion/byproducts on the Zn metal anode. Consequently, the ANZT interlayer achieves a long lifespan of Zn anodes and high Zn deposition/stripping reversibility, including superior electrochemical performances when coupling the modified Zn anode with MnO2, NH4V4O10, and I2 cathodes. In short, our work provides a deep insight into the ion transport mechanism of porous materials and its multiple protective effects for the Zn metal anode to promote the development of high-performance aqueous zinc-ion batteries.Author contributions
Conceptualization: Z. J., G. H. Experimental design and investigation: Z. J., Z. D., Y. Z., K. L. Data analyses: Z. J., R. C., H. Y., J. C., Z. C., F. C. Material characterization: Z. J., W. Z., R. P., G. Z. Theoretical simulation: Z. J., S. L. Writing – original draft: Z. J., L. S. Writing – review & editing: Z. J., K. Y., G. H. All authors contributed to discussions of the research.Conflicts of interest
The authors declare no competing interests.Data availability
All data supporting this article have been included as part of the ESI.†Acknowledgements
This work was supported by the National Key R&D Program of China (2024YFA1207900), the National Natural Science Foundation of China (12174050 and T2321002), the Natural Science Foundation of Jiangsu Province (BK20231411), the funding support from China Scholarship Council/University College London for the joint PhD scholarship, the Engineering and Physical Sciences Research Council (EPSRC, EP/027433/3), the Royal Society (IES\R2\212115; IEC\NSFC\211019), and UK Research and Innovation(UKRI) under the UK government's Horizon Europe funding (101077226; EP/Y008707/1).References
- K. C. Yazzie, K. Whyte, S. Begay, J. Glavin, T. Jones, K. Leni-Konig, C. Pratte, D. Madden, D. Reicher and C. B. Field, Science, 2024, 384, 6692 CrossRef PubMed.
- E. Aramendia, P. E. Brockway, P. G. Taylor, J. B. Norman, M. K. Heun and Z. Marshall, Nat. Energy, 2024, 9, 803–816 CrossRef.
- J. Zheng and L. A. Archer, Sci. Adv., 2021, 7, eabe0219 CrossRef CAS PubMed.
- Y. Liu, X. Lu, F. Lai, T. Liu, P. R. Shearing, I. P. Parkin, G. He and D. J. L. Brett, Joule, 2021, 5, 2845–2903 CrossRef CAS.
- Y. Jin, K. Liu, J. Lang, D. Zhuo, Z. Huang, C. Wang, H. Wu and Y. Cui, Nat. Energy, 2018, 3, 732–738 CrossRef CAS.
- S. W. D. Gourley, R. Brown, B. D. Adams and D. Higgins, Joule, 2023, 7, 1 CrossRef.
- Q. Zhao, M. J. Zachman, W. I. Al Sadat, J. Zheng, L. F. Kourkoutis and L. Archer, Sci. Adv., 2018, 4, eaau8131 CrossRef CAS.
- H. Wu, J. Hao, Y. Jiang, Y. Jiao, J. Liu, X. Xu, K. Davey, C. Wang and S. Qiao, Nat. Commun., 2024, 15, 575 CrossRef CAS PubMed.
- K. W. Leong, W. Pan, X. Yi, S. Luo, X. Zhao, Y. Zhang, Y. Wang, J. Mao, Y. Chen, J. Xuan, H. Wang and D. Y. C. Leung, Sci. Adv., 2023, 9, eadh1181 CrossRef CAS PubMed.
- Z. Cai, H. Wang, T. Wu, H. Ji, Y. Tang, Q. Zhang, Z. Peng and H. Wang, Mater. Today Energy, 2024, 43, 101592 CrossRef CAS.
- S. Chen, D. Ji, Q. Chen, J. Ma, S. Hou and J. Zhang, Nat. Commun., 2023, 14, 3526 CrossRef CAS PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- J. Yang, T. Xiao, T. Xiao, J. Li, Z. Yu, K. Liu, P. Yang and H. Fan, Adv. Mater., 2024, 36, 2313610 CrossRef CAS.
- J. Hao, S. Zhang, H. Wu, L. Yuan, K. Davey and S. Qiao, Chem. Soc. Rev., 2024, 53, 4312–4332 RSC.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1727–1742 CrossRef.
- W. Zong, J. Li, C. Zhang, Y. Dai, Y. Ouyang, L. Zhang, J. Li, W. Zhang, R. Chen, H. Dong, X. Gao, J. Zhu, I. P. Parkin, P. R. Shearing, F. Lai, K. Amine, T. Liu and G. He, J. Am. Chem. Soc., 2024, 146, 21377–21388 CrossRef.
- L. Zhang, H. Guo, W. Zong, Y. Huang, J. Huang, G. He, T. Liu, J. Hofkens and F. Lai, Energy Environ. Sci., 2023, 16, 4872–4925 RSC.
- T. Li, X. Li, H. Yang, Y. Zhou, X. Li, M. Su, A. Dou, P. Zhang, X. Wu, A. Naveed, J. Sumner and Y. Liu, Mater. Today Energy, 2024, 40, 101513 CrossRef CAS.
- J. Hu, Z. Zhang, T. Deng, F. Cui, X. Shi, Y. Tian and G. Zhu, Adv. Mater., 2024, 36, 2401091 CrossRef CAS PubMed.
- Y. Lyu, J. A. Yuwono, P. Wang, Y. Wang, F. Yang, S. Liu, S. Zhang, B. Wang, K. Davey, J. Mao and Z. Guo, Angew. Chem., 2023, 135, e202303011 CrossRef.
- X. Cai, X. Wang, Z. Bie, Z. Jiao, Y. Li, W. Yan, H. Fan and W. Song, Adv. Mater., 2024, 36, 2306734 CrossRef CAS PubMed.
- Z. Cao, H. Zhang, B. Song, D. Xiong, S. Tao, W. Deng, J. Hu, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2023, 33, 2300339 CrossRef CAS.
- F. Wang, J. Tseng, Z. Liu, P. Zhang, G. Wang, G. Chen, W. Wu, M. Yu, Y. Wu and X. Feng, Adv. Mater., 2020, 32, 200028 Search PubMed.
- Z. Li, X. Wu, X. Yu, S. Zhou, Y. Qiao, H. Zhou and S. Sun, Nano Lett., 2022, 22, 2538–2546 CrossRef CAS PubMed.
- C. Xie, Y. Liu, W. Lu, H. Zhang and X. Li, Energy Environ. Sci., 2019, 12, 1834–1839 RSC.
- K. Schmidt-Rohr and Q. Chen, Nature Mater., 2008, 7, 75–83 CrossRef CAS PubMed.
- W. Xin, J. Xiao, J. Li, L. Zhang, H. Peng, Z. Yan and Z. Zhu, Energy Storage Mater., 2023, 56, 76–86 CrossRef.
- Y. Wang, Y. Liu, H. Wang, S. Dou, W. Gan, L. Ci, Y. Huang and Q. Yuan, J. Mater. Chem. A, 2022, 10, 4366–4375 RSC.
- C. Zhou, L. Longley, A. Krajnc, G. J. Smales, A. Qiao, I. Erucar, C. M. Doherty, A. W. Thornton, A. J. Hill, C. W. Ashling, O. T. Qazvini, S. J. Lee, P. A. Chater, N. J. Terrill, A. J. Smith, Y. Yue, G. Mali, D. A. Keen, S. G. Telfer and T. D. Bennet, Nat. Commun., 2018, 9, 5042 CrossRef PubMed.
- R. Miletich, Monatsh. Chem., 1995, 126, 417–430 CrossRef CAS.
- F. Eder, A. Marsollier and M. Weil, Mineral. Petrol., 2023, 117, 145–163 CrossRef CAS.
- F. Eder, B. Stöger and M. Weil, Cryst. Mater., 2022, 237, 329–341 CAS.
- H. Ticha, J. Schwarz and L. Tichy, Mater. Chem. Phys., 2019, 237, 121834 CrossRef CAS.
- G. Lakshminarayana, K. M. Kaky, S. O. Baki, A. Lira, P. Nayar, I. V. Kityk and M. A. Mahdi, J. Alloys Compd., 2017, 690, 799e816 CrossRef.
- Z. Jiang, Z. Du, R. Pan, F. Cui, G. Zhang, S. Lei, G. He, K. Yin and L. Sun, Adv. Energy Mater., 2024, 14, 2402150 CrossRef CAS.
- R. Chen, W. Zhang, C. Guan, Y. Zhou, I. Gilmore, H. Tang, Z. Zhang, H. Dong, Y. Dai, Z. Du, X. Gao, W. Zong, Y. Xu, P. Jiang, J. Liu, F. Zhao, J. Li, X. Wang and G. He, Angew. Chem., 2024, 136, e202401987 CrossRef.
- R. Guo, X. Liu, F. Xia, Y. Jiang, H. Zhang, M. Huang, C. Niu, J. Wu, Y. Zhao, X. Wang, C. Han and L. Mai, Adv. Mater., 2022, 34, 2202188 CrossRef CAS PubMed.
- P. Zuo, C. Ye, Z. Jiao, J. Luo, J. Fang, U. S. Schubert, N. B. McKeown, T. Liu, Z. Yang and T. Xu, Nature, 2023, 617, 299–305 CrossRef CAS PubMed.
- (a) Q. H. Cao, Y. Gao, J. Pu, X. Zhao, Y. Wang, J. Chen and C. Guan, Nat. Commun., 2023, 14, 641 CrossRef CAS PubMed; (b) Z. Wei, S. Wang, D. Li, S. Yang, S. D. Guo, G. M. Qu, Y. H. Yang and H. Li, Energy Environ. Sci., 2024, 17, 5440 RSC; (c) T. Li, S. Hu, C. Wang, D. Wang, M. Xu, C. Chang, X. Xu and C. Han, Angew. Chem., Int. Ed., 2023, 62, e202314883 CrossRef CAS PubMed; (d) Y. Gao, Q. Cao, J. Pu, X. Zhao, G. Fu, J. Chen, Y. Wang and C. Guan, Adv. Mater., 2023, 35, 2207573 CrossRef CAS; (e) J. Zhang, W. Huang, L. Li, C. Chang, K. Yang, L. Gao and X. Pu, Adv. Mater., 2023, 35, 2300073 CrossRef CAS PubMed; (f) Z. Zheng, D. Ren, Y. Li, F. Kang, X. Li, X. Peng and L. Dong, Adv. Funct. Mater., 2024, 34, 2312855 CrossRef CAS; (g) H. Tian, J. Yang, Y. Deng, W. Tang, R. Liu, C. Xu, P. Han and H. Fan, Adv. Energy Mater., 2023, 13, 2202603 CrossRef CAS; (h) W. Guo, L. Xu, Y. Su, Z. Tian, C. Qiao, Y. Zou, Z. Chen, X. Yang, T. Cheng and J. Sun, ACS Nano, 2023, 18, 10642–10652 CrossRef PubMed.
- Z. Cheng, K. Wang, J. Fu, F. Mo, P. Lu, J. Gao, D. Ho, B. Li and H. Hu, Adv. Energy Mater., 2024, 14, 2304003 CrossRef CAS.
- R. Al-Gaashani, S. Radiman, A. R. Daud, N. Tabet and Y. Al-Douri, Ceram. Int., 2013, 39, 2283–2292 CrossRef CAS.
- S. Zhang, J. Hao, H. Wu, Q. Chen, C. Ye and S. Qiao, Adv. Mater., 2024, 36, 2404011 CrossRef CAS PubMed.
- M. Kim and J. Yeon, J. Radioanal. Nucl. Chem., 2021, 330, 469–473 CrossRef CAS.
- L. Zhang, J. Huang, H. Guo, L. Ge, Z. Tian, M. Zhang, J. Wang, G. He, T. Liu, J. Hofkens, D. J. L. Brett and F. Lai, Adv. Energy Mater., 2023, 13, 2203790 CrossRef CAS.
- Y. Ouyang, W. Zong, X. Gao, S. Leong, J. Chen, Y. Dai, H. Dong, I. Phang, P. Shearing, G. He, Y. Miao, T. Liu and X. Ling, Angew. Chem., Int. Ed., 2025, e202504965, DOI:10.1002/anie.202504965.
- L. Zhang, H. Ding, H. Gao, J. Gong, H. Guo, S. Zhang, Y. Yu, G. He, T. Deng, I. P. Parkin, J. Hofkens, X. Fan, F. Lai and T. Liu, Energy Environ. Sci., 2025, 18, 2462 RSC.
- L. Zhang, K. Luo, J. Gong, Y. Zhou, H. Guo, Y. Yu, G. He, J. Gohy, I. P. Parkin, J. Hofkens, Q. He, T. Liu, K. Müllen and F. Lai, Angew. Chem., Int. Ed., 2025, e202506822, DOI:10.1002/anie.202506822.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ee02454d |
| This journal is © The Royal Society of Chemistry 2025 |