
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step preparation of a hydrogel evaporator by in-liquid pulsed discharge for efficient solar desalination†
Tianyu
Sun
,
Yanbin
Xin
 *,
Jiabao
Sun
,
Bing
Sun
and
Xinfei
Fan
*
*,
Jiabao
Sun
,
Bing
Sun
and
Xinfei
Fan
*
College of Environmental Science and Engineering, Dalian Maritime University, Dalian 116026, China. E-mail: xinyb33@dlmu.edu.cn; fxf0909@dlmu.edu.cn
First published on 12th May 2025
Abstract
The usage of solar energy to achieve seawater desalination is emerging as an alternative to solve the problem of energy and fresh water shortage. In this work, a photothermal hydrogel is prepared by direct in-liquid pulsed discharge plasma, by which the cross-linking of the hydrogel was promoted rapidly by self-initiated active substances (such as ·OH, ·O, etc.). The prepared photothermal hydrogel has great evaporation performance and the evaporation rate in water can reach 1.316 kg m−2 h−1 and an energy utilization efficiency of up to 89.57%. Besides, the prepared photothermal hydrogel exhibits stable evaporation characteristics under extreme conditions, and has high interception of salts, heavy metals, dyes and oils. The results show that the photothermal hydrogel prepared by in-liquid pulsed discharge plasma has broad application prospects in seawater desalination.
Broader contextSolar-powered interfacial water evaporation offers a sustainable strategy for clean water production and energy conversion. However, challenges remain in achieving rapid fabrication, operational stability and simultaneous removal of contaminants. Recent advances in photothermal materials, particularly hydrogels, have attempted to address these limitations through innovative synthesis methods. While plasma technology holds great promise for tailoring material properties, its application to hydrogel design remains underexplored. This study bridges this gap by utilizing in-liquid pulsed discharge plasma for the rapid synthesis of high-performance hydrogels that provide scalable solutions for desalination and wastewater remediation. The development of a photothermal hydrogel via pulsed discharge plasma is a breakthrough in solar-powered evaporation technology. This study addresses these limitations by employing in-liquid pulsed discharge plasma to rapidly synthesize photothermal hydrogels. The optimized parameters achieved a record evaporation rate of 1.316 kg m−2 h−1 under solar irradiation with an energy utilization efficiency of 89.57%. The hydrogel demonstrated exceptional contaminant retention (>99%) for salts, dyes, heavy metals, and oils, enabling efficient seawater purification. This work emphasizes the critical role of plasma technology in advancing hydrogel synthesis, providing a sustainable platform for clean water production and energy conversion, as well as contributing to non-thermal plasma research. |
1. Introduction
Adequate fresh water resources are an important condition for human survival and development.1 The traditional desalination methods are mainly divided into two categories: the thermal method and membrane method.2 However, the thermal method has the disadvantages of low efficiency, high energy consumption, large equipment footprint, etc. Most of the energy consumed for desalination belongs to non-renewable fossil energy sources such as petroleum and coal, and their combustion also produces greenhouse gases, resulting in secondary pollution. The membrane method also has disadvantages such as difficulty in dealing with fouling, easy contamination, and high preparation cost.3 In recent years, solar-powered desalination, particularly solar-driven interfacial evaporation (SIE), has attracted much attention due to its low cost, non-reliance on electricity, simple structure and environmental friendliness, and is considered to be one of the most promising technologies for solving the problem of water scarcity.4–7Typically, the evaporation performance of SIE is dominated by an evaporator. Among various evaporator materials, hydrogels are widely used in solar evaporation systems to improve the evaporation rate and conversion efficiency due to their large specific surface area, good mechanical properties and recyclability. In most of the existing work, hydrogel solar evaporators have been characterized by good mechanical properties,8 photothermal conversion efficiency,9 and high evaporation rates.8,10,11 For example, Zhao et al. proposed a double-face arch hydrogel evaporator with enhanced water transport.11 By tailoring the water transport path, the evaporator forms a unique interconnected semi-crescent water film on the evaporation surface, benefiting from the advantages of the double-face hydrogel. The inclusion of a concentrated brine drainage system in the DDFA-TPIH5 evaporator enhances its performance and stability, offering a novel solution to the issue of salt crystallization in arched evaporators. Additionally, Farahpour and Azizian explored more environmentally friendly hydrogel materials by combining a low-cost, bio-based basil seed hydrogel with activated carbon to synthesize activated carbon-basil seed hydrogel composites. They characterized these composites as cost-effective, environmentally friendly, and efficient solar evaporator systems.7
However, the existing hydrogel preparation methods mainly rely on the addition of a large number of chemical reagents, which subsequently harms the environment. Moreover, hydrogel preparation conditions are harsh, and the preparation process is complex, time-consuming and energy-consuming. This has prevented hydrogels from being widely used in desalination. Therefore, it is of great significance to explore a green, low-cost and high-efficiency hydrogel preparation method for its practical application.
Low-temperature plasma has low gas temperature (100–1000 K) and high electron temperature (1000–100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 K), and simultaneously produces a large amount of highly active substances, such as free radicals, photons, and high-energy electrons.12 Also, it is easy to operate and control, so it is widely used in material synthesis, material modification, and nanotechnology.13,14 Among numerous forms of discharge plasma, in-liquid discharge plasma can directly produce a large number of reactive substances (·OH, ·O, ·H, etc.) in liquid, which can be used to initiate radical reactions in chemical synthesis. Few studies exist on hydrogel preparation by in-liquid discharge plasma, and there are only some reports on the preparation of hydrogels by glow discharge in liquid.15,16 Lu et al. prepared a novel acrylic acid/vermiculite hydrogel by using thiosalicylic acid (MBA) as a cross-linking agent and in-liquid glow discharge plasma as the initiator. It has a three-dimensional structure with a large number of pores, which can be used as an adsorbent for the efficient removal of cationic dyes from aqueous solutions, but its preparation time is over 4 h.15 Yu et al. used in-liquid glow discharge plasma as the initiator and MBA as the cross-linking agent to obtain a poly(2-acrylamide-2-methyl-1-propanesulfonic acid-coacrylamide) highly absorbent hydrogel, whose optimal equilibrium swelling can be up to 652.6 g g−1 with high pH and salt sensitivity, but it also requires long stirring time over 3 h after plasma treatment.16 In-liquid pulsed discharge plasma combines the advantages of pulsed discharge and in-liquid discharge, which is characterized by high plasma density, large spatial distribution, and high efficiency of electron and free radical generation.17,18 These characteristics are conducive to the rapid preparation of hydrogels. However, research on in-liquid pulsed discharge plasma for hydrogel preparation is limited. Therefore, it is of great significance to explore in-liquid pulsed discharge to realize the efficient and environmentally friendly preparation of hydrogels.
000 K), and simultaneously produces a large amount of highly active substances, such as free radicals, photons, and high-energy electrons.12 Also, it is easy to operate and control, so it is widely used in material synthesis, material modification, and nanotechnology.13,14 Among numerous forms of discharge plasma, in-liquid discharge plasma can directly produce a large number of reactive substances (·OH, ·O, ·H, etc.) in liquid, which can be used to initiate radical reactions in chemical synthesis. Few studies exist on hydrogel preparation by in-liquid discharge plasma, and there are only some reports on the preparation of hydrogels by glow discharge in liquid.15,16 Lu et al. prepared a novel acrylic acid/vermiculite hydrogel by using thiosalicylic acid (MBA) as a cross-linking agent and in-liquid glow discharge plasma as the initiator. It has a three-dimensional structure with a large number of pores, which can be used as an adsorbent for the efficient removal of cationic dyes from aqueous solutions, but its preparation time is over 4 h.15 Yu et al. used in-liquid glow discharge plasma as the initiator and MBA as the cross-linking agent to obtain a poly(2-acrylamide-2-methyl-1-propanesulfonic acid-coacrylamide) highly absorbent hydrogel, whose optimal equilibrium swelling can be up to 652.6 g g−1 with high pH and salt sensitivity, but it also requires long stirring time over 3 h after plasma treatment.16 In-liquid pulsed discharge plasma combines the advantages of pulsed discharge and in-liquid discharge, which is characterized by high plasma density, large spatial distribution, and high efficiency of electron and free radical generation.17,18 These characteristics are conducive to the rapid preparation of hydrogels. However, research on in-liquid pulsed discharge plasma for hydrogel preparation is limited. Therefore, it is of great significance to explore in-liquid pulsed discharge to realize the efficient and environmentally friendly preparation of hydrogels.
2. Experimental section
2.1 Materials
The materials and reagents used in the experiment are polyvinyl alcohol (PVA, Shanghai Macklin Biochemical Co., Ltd), glutaric dialdehyde (GA, 50% in H2O, Tianjin Zhiyuan Chemical Reagent Co., Ltd), KPS (Shanghai Macklin Biochemical Co., Ltd), L-ascorbic acid (Shanghai Macklin Biochemical Co., Ltd), graphite (Shanghai Macklin Biochemical Co., Ltd), sulfuric acid (H2SO4, 98%, Tianjin Kemiou Chemical Reagent Co., Ltd), potassium permanganate (Nanjing Chemical Reagent Co., Ltd), hydrogen peroxide (Tianjin Damao Chemical Reagent Factory), hydrochloric acid (Tianjin Kemiou Chemical Reagent Co., Ltd), methylene blue (Tianjin Beichen Founder Reagent Factory), methyl orange (Shanghai Macklin Biochemical Co., Ltd), dimethyl silicone oil (Tianjin Kemiou Chemical Reagent Co., Ltd), span 80 (Tianjin Fuchen Chemical Reagents Factory), and sodium hydroxide (NaOH, Tianjin Kemiou Chemical Reagent Co., Ltd). Nickel(II) sulfate (NiSO4·6H2O), cadmium(II) chloride (CdCl2), chromium(III) nitrate nonahydrate (Cr(NO3)3·9H2O), cupric(II) chloride (CuCl2·2H2O), and lead(II) acetate trihydrate (Pb(CH3COO)2·3H2O) were provided by Sinopharm Chemical Reagent Co., Ltd. Seawater was collected from Qixianling (Dalian, China). All solutions were prepared using deionized water.2.2 Preparation of materials
 | ||
| Fig. 1 Schematic diagram of a hydrogel precursor prepared from a needle-plate electrode. | ||
The prepared hydrogel precursor was placed in 2 mM ascorbic acid solution and heated to 90 °C for 3 h. After reduction, it was rinsed with deionized water to remove the residual ascorbic acid. After the hydrogel was fully absorbed, it was refrigerated for freezing treatment, and then thawed in 30 °C deionized water, and the freeze–thaw process was repeated five times to obtain the rGO/PVA hydrogel.
2.3 Characterization
A digital oscilloscope (TDS2024B, Tektronix Inc.) was used to monitor the current–voltage characteristics during discharge. A high-speed camera (HiSpecl 2G Color, Fastec Imaging) was used to monitor the time-resolved properties of the in-liquid discharge plasma. A multichannel spectrum analyzer (C7473, Hamamatsu) was used to monitor the active substances generated during the discharge process. A scanning electron microscope (GeminiSEM 300, ZEISS) was used to characterize the surface of the hydrogel evaporator, and cross-section morphology pores. A Raman spectrometer (DXR 2xi, Thermo Fisher Scientific) was used to analyze the materials. A rheometer (ARES-G2, TA) was used to determine the rheological properties of the hydrogel samples and to obtain the correlation curves of the energy storage modulus and loss modulus. An infrared thermal imager (Uti220a, UNI-T) was used to record the temperature changes at the hydrogel evaporation interface during evaporation. Inductively coupled plasma chromatography (ICP-MS 7700ce, Agilent) was used to determine the changes in ion concentrations of seawater and simulated heavy metal polluted seawater before and after seawater desalination in order to assess the effectiveness of seawater desalination.2.4 Solar evaporation experiment
The solar vapor generation experiments were conducted with a xenon lamp light source (CEL-HXF300-T3, Beijing Zhongjiao Jinyuan Technology Co., Ltd) having an A.M 1.5 G filter as a simulated solar light source at a light intensity of one sun illumination (1 kW m−2). Solar flux was determined using a solar power meter (TES-1333, TES Electrical Electronic Corp.). The hydrogel samples can be self-floated by placing the hydrogels with a thickness of 5 mm in glass test tubes filled with deionized water (or seawater) at a room temperature of 25 °C and relative humidity of 45%. The glass test tubes were 1 cm × 1 cm × 15 cm square test tubes. The mass of water lost was measured by using an electronic balance (PTX-FA210, Huazhi (Fujian) Electronic Technology Co., Ltd) with an accuracy of 0.0001 g. The position was adjusted so that the simulated sunlight always shone vertically on the surface of the evaporated material. The electronic balance collects data every 0.5 s and transfers it to the computerized recording software. Unless otherwise stated, mass change represents the change in the mass of the evaporator for 1 h of evaporation. In the illustrations of this paper, the evaporation of pure water refers to the evaporation of water under solar illumination in the absence of hydrogels, which is used to compare the contribution of hydrogels to the evaporation process. The evaporation rate and energy utilization efficiency are calculated as follows. | (1) |
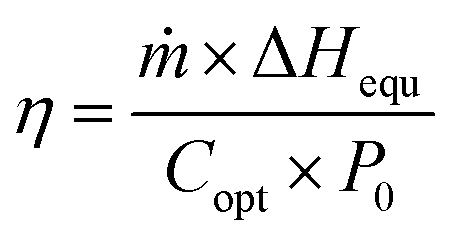 | (2) |
In order to determine the evaporated area, we performed an equivalent conversion of the evaporated area with reference to the simple method proposed by Tahzibi and Azizian for estimating the evaporated area.19 By considering bulk water and water covered with an interfacial support in the dark, both of them receive the same energy power from the environment (Uin). Water and hydrogel samples with equal evaporated areas were placed in a closed container protected from light at a humidity of 45% and a temperature of 25 °C, the weight loss of both was measured for a certain period of time, and the evaporated area of the hydrogel was calculated by using formula (3).
 | (3) |
Since the evaporation time (t) for both systems is the same, after simplification of eqn (4), the following equation is derived:
 | (4) |
3. Results and discussion
3.1 Characteristics of in-liquid pulsed discharge plasma
By comparing the evaporation mass changes in four common carbon-based materials (graphite, carbon nanotubes, GO and rGO) for doping, rGO is selected as the photothermal conversion material in this work (Fig. S1†). The process of plasma generation is essential to explore the preparation of hydrogels by the plasma method. Since the color of the solution changes to dark brown after hydrogel doping with GO, which masks the bubbles generated by the discharge, we chose to film the discharge process without the addition of GO. The initiation process of in-liquid pulsed discharge plasma can be considered as the process of bubble breakdown. Fig. 2 illustrates the temporal and spatial variation characteristics of the plasma discharge. Under stronger electric field conditions, the liquid is locally heated to produce small bubbles, the gas inside the bubbles is ionized, and the plasma is triggered inside the bubbles. At the same time, as the bubble region continues to grow, the plasma volume increases until the bubble bursts and the plasma disappears. | ||
| Fig. 2 The time–space characteristics of plasma discharges. | ||
In addition to probing the plasma generation process, the current–voltage characteristics of the discharge process were analyzed. As in-liquid pulsed discharge has short rise time and pulse width, the instantaneous strong electric field does not accelerate the ions but only accelerates the electrons, thus forming high-energy electrons that collide with the surrounding particles to form a plasma channel.20Fig. 3a shows the current–voltage waveforms for discharge in a premixed solution. At the instant of breakdown (at 0 ms), a plasma region was formed between the needle and the plate electrodes at a peak voltage of 24 kV. At the same time, the current reaches a peak, with amplitudes of up to 75 A. After about 10 min of discharge, the premixed solution gels, and the form of discharge changes from corona discharge to spark discharge. Fig. 3b shows the current–voltage waveforms after the formation of the hydrogel precursor. The discharge occurs in a localized region. Both current and voltage show violent waveform oscillations, with the maximum peak occurring in the first peak and the peak voltage dropping from 24 kV to 20 kV.
 | ||
| Fig. 3 In-liquid pulsed discharge current–voltage waveforms: (a) corona discharge before glue formation and (b) spark discharge after glue formation. | ||
The in-liquid corona discharge process produces both physical and chemical effects. Physical effects include ultrasonic cavitation, shock waves and ultraviolet light. Chemical effects refer to the production of a large number of reactive free radicals and active substances in solution. Physical effects such as ultraviolet light, heat, and shock waves generated during the discharge process can be utilized to break the peroxide bonds of KPS, generating strongly oxidizing sulfate radicals (SO4−·).21 The chemical effect generates ·H, ·O, ·OH and other free radicals at the same time, and a variety of active particles trigger the reaction of free radicals in the solution to promote the cross-linking of the hydrogel. Fig. 4 shows the atomic emission spectra of the hydrogel preparation process, from which it can be observed that Hα (n = 3 → n = 2), O ((3p5P0) → (3s5S0)), and O ((3p3P) → (3s3S0)) are produced during the discharge process in this system. The SO4−· produced by KPS in the presence of plasma is a polyatomic radical, which is not detectable in the emission spectrum. In the control experiment without the addition of KPS, the hydrogel still did not crosslink successfully with a discharge time up to 30 min, proving that SO4−· plays a crucial role in the crosslinking of the hydrogel. Also, as the hydrogel matrix PVA is an alcohol, the hydroxyl groups burst and were not monitored during the discharge.
 | ||
| Fig. 4 Atomic emission spectra of (a) the PVA hydrogel and the (b) rGO/PVA hydrogel preparation process. | ||
In order to examine the performance of plasma-prepared hydrogels, two preparation methods, the plasma method and the widely used heating synthesis method, were compared. The hydrogels prepared by the heated synthesis method were also subjected to freeze–thaw treatment, and the two methods differed only in the preparation of the hydrogel precursors. The heated synthesis method was prepared by heating at 80 °C for 2 h, and the plasma method was obtained by in-liquid pulsed discharge plasma. The hydrogel properties obtained by the two preparation methods with the same volume were evaluated by hydrogel water loss weight. During an evaporation time of 1 h, the water loss of the hydrogel prepared by the conventional heating method was 0.1286 g, while the water loss of the hydrogel prepared by the plasma method was 0.2323 g (Fig. 5). This reflects the advantages of hydrogels prepared by the plasma method. It is proved that plasma as an initiator in the process of hydrogel preparation can increase the binding sites and play an important role in the construction of hydrogel crosslinked polymer networks.22 Compared with the chemical cross-linking method, the water loss of hydrogels prepared by the plasma method is faster, which may be due to the active particles generated by the plasma treatment, which can promote hydrogel cross-linking more effectively.
 | ||
| Fig. 5 Mass change of hydrogels prepared by the heated synthesis method and plasma method by evaporation for 1 h. | ||
3.2 Optimization of hydrogel preparation parameters
In order to optimize the hydrogel properties, the experimental parameters during hydrogel preparation were optimized. First, the effect of discharge frequency on the evaporation rate was explored. As Fig. 6a shows, the highest evaporation rate appears at a discharge frequency of 30 Hz. The higher the pulse frequency under the same voltage condition, the more energy injected into the reactor per unit time, which can produce more active species and promote the polymerization of the crosslinked network, and the cross-linking rate of the hydrogel can also be further accelerated. However, with higher discharge frequency, the chance of collision between the active species and PVA per unit time is too high, and the hydrogel is hit by active ions again when it is not fully crosslinked, the degree of crosslinking decreases, and the surface area of the hydrogel decreases (Fig. 6b). The hydrogel reaches the fastest evaporation rate and the highest evaporation surface area at 30 Hz, and the energy utilization efficiency can reach 85.5% (Fig. 6c). | ||
| Fig. 6 Effect of discharge frequency on the hydrogel: (a) evaporated mass change, (b) discharge gelation time and evaporated surface area, and (c) evaporation rate, energy efficiency and dark evaporation rate. | ||
Then, the effect of peak voltage on the evaporation rate of hydrogels was explored, and hydrogels with different peak voltages were prepared for water evaporation tests. As Fig. 7a shows, the evaporation rate shows an initial increase and then a subsequent decrease with increasing peak voltage. When the peak voltage is low, the number of active species is limited, so its polymerization speed is slow, crosslinking requires longer discharge gelation time, and it is difficult for the hydrogel network to form. With increasing peak voltage, more active species are generated during the discharge process, which increases the polymerization rate and forms a stable polymer structure. At peak voltages greater than 24 kV, a further increase in active species causes self-crosslinking among the polymer end groups and over-polymerization of the hydrogel network, resulting in an increasingly compact polymer network structure, reduced hydrogel porosity, and a significant reduction in the evaporated surface area (Fig. 7b and c).
 | ||
| Fig. 7 Effect of peak voltage on the hydrogel: (a) evaporated mass change, (b) discharge gelation time and evaporated surface area, and (c) evaporation rate, energy efficiency and dark evaporation rate. | ||
3.3 Characterization of photothermal hydrogels
The present photo-hydrothermal gel forms a dual network structure through plasma-triggered chemical cross-linking and physical cross-linking via freeze–thaw cycles. In the chemical cross-linking network, the plasma-generated reactive radicals activate KPS to generate SO4−·, which promotes the cross-linking of PVA and GA. In the physical cross-linking network, five freeze–thaw cycles induce PVA molecular chain crystallization, forming micron-sized pores (Fig. 8a–c) that facilitate rapid water transport. The morphology of the prepared hydrogel is observed by SEM that displays a micrometer-scale pore structure and rough surface, which is conducive to the realization of continuous water supply and surface absorption of light. During the evaporation, the hydrophilicity (Fig. S2†) and the water transport channels of the hydrogel help to supplement water to the evaporation interface maintaining continuous evaporation operation. Fig. 8d shows the degree of graphitization of graphene in the hydrogel before and after reduction using Raman spectroscopy. The ID:IG ratio of the GO/PVA hydrogel is 0.851, and the ID:IG ratio of the reduced rGO/PVA hydrogel is 0.972, indicating that new defects were formed by the reduction of GO.23 The increase in the ID:IG ratio indicates that the reduction has restored the crystalline structure of graphene to a certain extent of order, proving that GO was partially reduced to rGO.
 | ||
| Fig. 8 Morphological and structural characterization of hydrogels. rGO/PVA hydrogels: (a) surface and (b and c) cross-section SEM images. (d) Raman spectra of GO/PVA and rGO/PVA hydrogels in the range of 400–2000 cm−1. | ||
Since hydrogels are viscoelastic materials, rGO penetrating the skeleton alters energy storage and energy dissipation. As can be observed in Fig. 9, the G′ values of both hydrogels are higher than the G′′ values, confirming that the hydrogels present a solid state and form a cross-linked polymer backbone. The G′ values of the rGO/PVA hydrogels were lower than those of the PVA gels, indicating that the incorporation of graphene into the crosslinked polymer backbone reduces the number of crosslinking sites. In contrast, the lower G′′ values of rGO/PVA hydrogels indicate that there is an effective limitation of slip between polymer chains by rGO, which reduces the friction of molecules within the hydrogel material.24 These results indicate that rGO was inserted into the molecular web of the PVA network.
 | ||
| Fig. 9 Dynamic mechanical analysis of the energy storage modulus (G′) and loss modulus (G′′) of the hydrogel. | ||
Photo-thermal hydrogels act as solar absorbers, and the strength of their photoconversion ability is important for the evaporation rate of the hydrogels. The hydrogel has a high light absorption rate and absorbs sunlight and converts it into heat, which is used to achieve surface evaporation. The higher the surface temperature of the hydrogel, the greater the driving force for evaporation behavior. Fig. 10 shows that the surface temperature of the hydrogel increases the fastest in the first 5 min of the evaporation process, from a room temperature of 25 °C to 38.4 °C, and the surface temperature of the hydrogel is stabilized at about 48 °C in 50 min, which proves the excellent photo-thermal conversion ability of the hydrogel. Meanwhile, the hydrogel has a low thermal conductivity, which effectively prevents the transfer of heat from the evaporating surface to the bottom water and helps to concentrate the heat on the evaporating surface during the evaporation process (Table S1†). The results show that hydrogels are capable of achieving good photothermal conversion for the evaporation process.
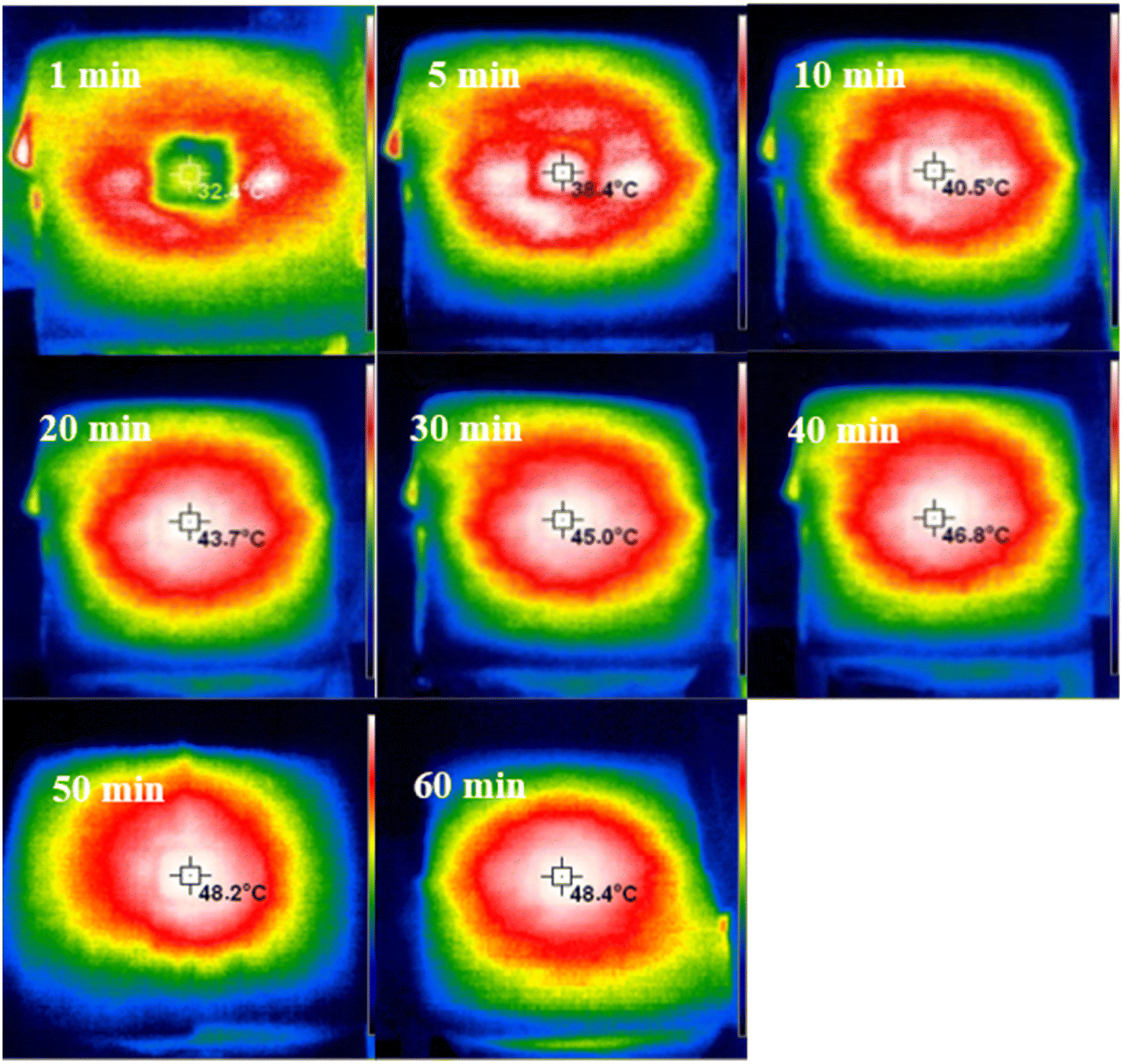 | ||
| Fig. 10 Surface temperature during desalination of the hydrogel in 3.5% NaCl solution. | ||
3.4 Evaluation of desalination performance and environmental tolerance
To investigate the universality of hydrogels to seawater worldwide, artificial seawater with different salinities was used for evaporation tests (Fig. 11). The 0% NaCl solution represents deionized water, the 0.8% NaCl solution represents the world's lowest salinity seawater, the 3.5% NaCl solution represents the world's average salinity seawater, and the 21% NaCl solution represents the highest salinity seawater in the world. The overall hydrogel water loss mass change tends to decrease with increasing salt concentration. This may be due to the fact that the interaction between the polymer network and the salt ions is enhanced with increasing salt concentration, and the high enthalpy of evaporation of the salt solution requires more heat to be gathered for evaporation. | ||
| Fig. 11 Effects of NaCl concentration on the mass change of artificial seawater over 60 min of evaporation of the hydrogel. | ||
Meanwhile, the evaporation in 3.5% NaCl solution was continuous for 8 h, and the evaporation was basically stable without a salt crystallization phenomenon (Fig. 12). Even if salt crystallization occurs, salt dissolution can be achieved very quickly by means of its own water transport channels (Fig. S3†). The above demonstrates that the hydrogel can operate stably for a long period of time and that it has the potential for long-term application in desalination.
 | ||
| Fig. 12 Changes in water loss mass of the hydrogel by continuous evaporation in 3.5% NaCl solution for 8 h. | ||
The primary goal of desalination is to separate the various salt ions in seawater, thereby acquiring fresh water. Since it was difficult for the artificial seawater configured in the laboratory to simulate the complexities in real seawater, real seawater was used for solar desalination and the desalination effect was quantitatively analyzed in this section. The salinity of real seawater was 3.19%, which was slightly lower than the world average seawater salinity. The water loss mass change of real seawater under one sun illumination was 0.2273 g, which was similar to that in 3.5% NaCl solution, proving that 3.5% NaCl solution was representative of simulated seawater. After desalination, the concentration of the four main ions decreased by 3–4 orders of magnitude, proving that the hydrogel can effectively realize solar desalination of seawater, and the interception rates were all above 99.99% (Fig. 13).
 | ||
| Fig. 13 Changes in concentration of the four major ions in real seawater before and after desalination. | ||
Considering the environmental tolerance of hydrogels in the actual desalination process, a variety of contaminated (e.g., dyes, heavy metals, acids, alkalis, oils, etc.) artificial seawater was simulated for desalination treatment. Cationic dye methylene blue and anionic dye methyl orange at 10 mg L−1 were used as simulating dyes. Five heavy metal salts were used to simulate seawater containing Cu2+, Cr3+, Ni2+, Pb2+, and Cd2+. Sulfuric acid and sodium hydroxide were used to adjust the acidity and alkalinity of seawater. The simulated oil contaminated seawater was obtained by adding silicone oil (1 g) and Span 80 (0.1 g) to 100 mL of 3.5% NaCl solution and sonication for 12 h.
The characteristic peaks of methylene blue and methyl orange dyes appeared at 664 nm and 464 nm, respectively, before desalination. No obvious peaks were observed in the desalinated water after desalination, indicating that the concentration of the dyes in the desalinated water was extremely low (Fig. 14a and b). Desalinated water collected from heavy metal contaminated seawater was tested by ICP, and its heavy metal ion concentration decreased by about 6–7 orders of magnitude compared to that before desalination (Fig. 15a). More than 99% of the heavy metal ions are trapped in the bottom water by the hydrogel. The interception of heavy metal ions in the hydrogel is mainly attributed to the hydroxyl group in PVA. The oxygen in the pendant hydroxyl group contains a lone pair of electrons, which can form a stable chelating bond with the empty orbitals of the metal ions, preventing heavy metal ions from escaping from the water. The hydroxyl group can also form complexes with heavy metal ions, thus allowing heavy metals to be effectively retained.25
 | ||
| Fig. 14 UV absorption wavelength scans of (a) methylene blue and (b) methyl orange in simulated contaminated seawater before and after desalination (inset shows a digital photograph before and after desalination). | ||
 | ||
| Fig. 15 Evaluation of ion interception capacity of the hydrogel. (a) Concentration of heavy metal ions before and after desalination of heavy metal polluted seawater; (b) mass changes of hydrogels evaporated in seawater contaminated with different heavy metals for 1 h; (c) mass change of hydrogels evaporated in seawater polluted with different pH values for 1 h; comparison of pH before and after desalination of seawater at (d) pH = 1 and (e) pH = 13. | ||
As shown in Fig. 15b, the change in the evaporated mass of seawater containing CuCl2, Cr(NO3)3·4H2O, NiSO4·6H2O, (CH3COO)2Pb·3H2O and CdCl2 is almost identical to that in a 3.5% NaCl solution under one sun illumination. However, the evaporation of seawater containing (CH3COO)2Pb·3H2O was inhibited, and the mass of evaporated water was slightly reduced. This was primarily due to lead acetate dissolving in water, where the acetate bound to hydrogen ions from water ionization, and the lead ions bound to the hydroxide ions generated by water ionization, leading to the formation of a Pb(OH)2 precipitate. This caused the solution to become turbid, blocking the water transport channels of the hydrogel during evaporation and significantly reducing the evaporation rate.
The evaporation of the hydrogel is almost unchanged over a wide range of acid–base intervals (Fig. 15c). The desalinated water in strongly acidic and alkaline seawater was neutral, indicating that the hydrogel effectively retained the acidic and alkaline ions in solution (Fig. 15d and e).
In the actual desalination operation, oil leakage from oil rigs in the sea may be encountered, and the leaked oil mixed with seawater may affect the evaporation rate of the hydrogel and the desalination effect. Therefore, simulated oil-containing seawater was prepared for evaporation tests and the desalinated water state was observed under a microscope. A simulated oil-containing seawater mixture was obtained by adding silicone oil (1 g) and Span 80 (0.1 g) to 100 mL of 3.5% NaCl solution and mixing with ultrasound for 12 h. After desalination, the seawater became clear from turbidity, and the oil-containing seawater showed a uniform oil-in-water distribution under the microscope. No oil droplets were found in the desalinated water, which proved that the hydrogel could also realize effective interception of oil (Fig. 16). In summary, the hydrogel prepared in this paper has wide application potential in the practical application of seawater desalination.
 | ||
| Fig. 16 Microscope images of oily seawater before and after desalination (insets are digital photographs). | ||
To further investigate the feasibility of the hydrogel in practical desalination applications, a homemade, simple outdoor collection device was used for desalination experiments. The outdoor seawater desalination experiment was conducted at Dalian Maritime University on April 8, 2024, with evaporation lasting for 9 h (8:00–17:00). Solar light intensity, ambient temperature, and evaporation rate were recorded every hour. As shown in Fig. 17, the average light intensity was 519 W m−2, with the highest intensity occurring at 12:00, reaching 804 W m−2, and the hourly mass change reaching 3.415 g. Compared to the indoor experiments, the mass change was lower, primarily due to reduced light intensity and increased humidity in the evaporation unit, which hindered water evaporation. The results indicate that the average daily water production of the rGO/PVA hydrogel can reach 10.25 kg m−2, which can meet the daily drinking water needs of 3–4 people, demonstrating the hydrogel's potential for large-scale desalination water production.
 | ||
| Fig. 17 Mass change, outdoor solar irradiation and environmental temperature of hydrogels in different time periods in an outdoor evaporation experiment. | ||
4. Conclusion
In this work, photothermal gel was innovatively prepared by in-liquid pulsed discharge plasma. The hydrogel properties were characterized and its ability to capture fresh water in seawater and contaminated wastewater was focused on. The following conclusions were specifically obtained:(1) The discharge showed typical corona discharge characteristics, and many free radicals such as ·H and ·O were generated during the discharge process, which triggered the free radical reaction in the solution and promoted the cross-linking of the hydrogel. The reaction parameters were optimized and the water evaporation rate was up to 1.316 kg m−2 h−1.
(2) The GO in the hydrogel was partially reduced to rGO and successfully inserted into the hydrogel polymer network structure. The hydrophilic properties and longitudinal channels of the hydrogel ensure its continuous operation. Meanwhile, the hydrogel has good photothermal conversion, thermal stability and low thermal conductivity, which are conducive to the realization of a high evaporation rate and high energy utilization efficiency of the hydrogel.
(3) Operational stability was maintained in 3.5% NaCl solution with high interception of salts, dyes, heavy metals and oils (>99%).
The work first realized the rapid preparation of photo-thermal hydrogels by in-liquid pulsed discharge plasma, which can shorten the hydrogel synthesis time, reduce the input of the initiator and the pollution by chemical substances in the environment. The photothermal hydrogel prepared in this work achieved a high evaporation rate and provided a new idea for hydrogel preparation. The prospect of a wide range of applications for photo-hydrothermal gels was confirmed.
Data availability
All data are available within this manuscript and the ESI file.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [grant number 12475258], the Natural Science Foundation of Liaoning Province, China [grant number 2023-MS-127], the Young Elite Scientists Sponsorship Program by CIN [grant number YESSCIN2022008], and the Fundamental Research Funds for the Central Universities [grant number 3132023503].References
- M. Salehi, Environ. Int., 2022, 158, 106936 CrossRef PubMed.
- A. A. Salehi, M. Ghannadi-Maragheh, M. Torab-Mostaedi, R. Torkaman and M. Asadollahzadeh, Sep. Purif. Rev., 2020, 50, 380–399 CrossRef.
- L. Zang, C. Finnerty, S. Zheng, K. Conway, L. Sun, J. Ma and B. Mi, Water Res., 2021, 198, 117135 CrossRef CAS PubMed.
- W. Lei, S. Khan, L. Chen, N. Suzuki, C. Terashima, K. Liu, A. Fujishima and M. Liu, Nano Res., 2020, 14, 1135–1140 CrossRef.
- X. Zhou, F. Zhao, Y. Guo, B. Rosenberger and G. Yu, Sci. Adv., 2019, 5, eaaw5484 CrossRef CAS PubMed.
- J. Sun, Y. Xin, B. Sun, Q. Yang and X. Fan, Chem. Eng. J., 2024, 497, 154621 CrossRef CAS.
- M. Farahpour and S. Azizian, J. Ind. Eng. Chem., 2025, 141, 285–296 CrossRef CAS.
- H. M. Wilson, H. W. Lim and S. J. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 47800–47809 CrossRef CAS PubMed.
- Y. Guo, F. Zhao, X. Zhou, Z. Chen and G. Yu, Nano Lett., 2019, 19, 2530–2536 CrossRef CAS PubMed.
- Y. Guo, X. Zhou, F. Zhao, J. Bae, B. Rosenberger and G. Yu, ACS Nano, 2019, 13, 7913–7919 CrossRef CAS PubMed.
- Z. Zhao, J. Li, F. Wang, P. Tan, L. Wang, B. Wang, J. Jin, X. Wang, W. Zhang and C. Wang, J. Energy Chem., 2025, 104, 540–550 CrossRef CAS.
- B. Sun, Discharge Plasma in Liquid and its Applications, Press of Science, 2013 Search PubMed.
- J. Sun, Y. Xin, T. Sun, B. Sun and X. Fan, J. Environ. Chem. Eng., 2023, 11, 110411 CrossRef CAS.
- J. Sun, Y. Xin, Z. Li, B. Sun and X. Fan, Chem. Eng. J., 2024, 484, 149669 CrossRef CAS.
- Q. Lu, J. Zheng, J. Yu, S. Yang, D. Ma, W. Yang and J. Gao, Adv. Polym. Technol., 2018, 37, 996–1007 CrossRef CAS.
- J. Yu, H. Zhang, Y. Li, Q. Lu, Q. Wang and W. Yang, Colloid Polym. Sci., 2016, 294, 257–270 CrossRef CAS.
- Y. Xin, B. Sun, X. Zhu, Z. Yan, Y. Liu and H. Liu, Appl. Energy, 2016, 168, 122–129 CrossRef CAS.
- Y. Xin, B. Sun, X. Zhu, Z. Yan, H. Liu and Y. Liu, Appl. Energy, 2016, 181, 75–82 CrossRef CAS.
- H. Tahzibi and S. Azizian, Langmuir, 2025, 41, 999–1004 CrossRef CAS PubMed.
- F. Wang, Z. Wang, L. Dong, H. Liu, L. Yang and Y. Wang, Adv. Funct. Mater., 2024, 34, 2403606 CrossRef CAS.
- B.-T. Zhang, Y. Zhang, Y. Teng and M. Fan, Crit. Rev. Environ. Sci. Technol., 2015, 45, 1756–1800 CrossRef CAS.
- R. Liang, D. Zhang, J. Guo, S. Bian, C. Yang, A. Lusi, W. Zhang and F. Huang, Int. J. Mol. Sci., 2024, 25, 2418–2436 CrossRef CAS PubMed.
- M. Yi, Z. Shen, X. Zhang and S. Ma, J. Phys. D: Appl. Phys., 2013, 46, 025301 CrossRef.
- F. Zhao, X. Zhou, Y. Shi, X. Qian, M. Alexander, X. Zhao, S. Mendez, R. Yang, L. Qu and G. Yu, Nat. Nanotechnol., 2018, 13, 489–495 CrossRef CAS PubMed.
- C. Zhang, X. Chen, B. Cui, L. Chen, J. Zhu, N. Bai, W. Wang, D. Zhao, Z. Li and Z. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 26303–26313 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5el00014a |
| This journal is © The Royal Society of Chemistry 2025 |